

Abstract
HtaA is a heme-binding protein that is part of the heme uptake system in Corynebacterium diphtheriae. HtaA contains two conserved regions (CR1 and CR2). It has been previously reported that both domains can bind heme; the CR2 domain binds hemoglobin more strongly than the CR1 domain. In this study, we report the biophysical characteristics of HtaA-CR2. UV-visible spectroscopy and resonance Raman experiments are consistent with this domain containing a single heme that is bound to the protein through an axial tyrosine ligand. Mutants of conserved tyrosine and histidine residues (Y361, H412, and Y490) have been studied. These mutants are isolated with very little heme (≤ 5%) in comparison to the wild-type protein (~20%). Reconstitution after removal of the heme with butanone gave an alternative form of the protein. The HtaA-CR2 fold is very stable; it was necessary to perform thermal denaturation experiments in the presence of guanidinium hydrochloride. HtaA-CR2 unfolds extremely slowly; even in 6.8 M GdnHCl at 37 °C, the half-life was 5 h. In contrast, the apo forms of WT HtaA-CR2 and the aforementioned mutants unfolded at much lower concentrations of GdnHCl, indicating the role of heme in stabilizing the structure and implying that heme transfer is effected only to a partner protein in vivo.
Keywords: heme, hemin, hemoglobin, Raman, guanidinium, Corynebacterium
Graphical abstract
The C. diphtheriae heme uptake protein HtaA-CR2 has an axial tyrosine. Reconstitution with hemin after unfolding gave a different form of the protein, as revealed by UV-visible and Raman spectroscopy. Thermal and chemical unfolding experiments showed that hemin plays a significant role in the stability of the protein.
1. Introduction
Corynebacterium diphtheriae is an important Gram-positive human pathogen, causing upper respiratory tract infections in humans [1, 2]. Although vaccination is widely available in many countries, diphtheria infections still occur in parts of the world with low vaccine coverage [3]. Analysis of the full genome sequence [2] is consistent with a number of iron uptake systems in this organism. The iron and heme transporter systems are predicted to be coordinately expressed with the toxin [4–7]. This, along with the iron requirement for virulence, suggests that iron acquisition is a key component for pathogenicity. Approximately 80% of heme in vivo is found in hemoglobin (Hb) [8], leading to the role of this prosthetic group as a significant source of iron.
Gram-positive pathogens in general use heme as a key source of the iron required for survival and proliferation. Heme uptake pathways have been the subject of recent reviews [8–14]. The majority of Gram-positive bacteria studied to date employ a series of NEAr-iron Transporter (NEAT) domains in many of the proteins involved in heme transport [14–16]. These domains are generally located in cell membrane-anchored proteins and serve to extract heme from Hb and haptoglobin-hemoglobin (Hp-Hb) and transfer it to additional heme-binding proteins, with final import through an ABC transporter. Classical NEAT domains comprise about 125 amino acids with a β-strand secondary structure; the canonical examples have a conserved YXXXY heme-binding motif, in which the N-terminal tyrosine of the sequence serves as an axial ligand to the heme and the C-terminal tyrosine is a hydrogen-bond partner to the first. On the opposite side of the heme ring, a loop starts with a serine which hydrogen bonds to one of the heme propionates. The first system to be studied in detail was the iron-regulated surface-determinant (isd) pathway in Staphylococcus aureus [17]. Of the proteins in this pathway, IsdA, IsdB, IsdC and IsdH bind heme in NEAT domains; leading references can be found in recent work [18–22]. The heme uptake system in Bacillus anthracis also uses a series of NEAT domains for heme uptake [23, 24]. In terms of the reliance on the NEAT architecture, uptake pathways studied to date involving Isd proteins in Streptococcus lugdunenesis [25] and Listeria monocytogenes [26, 27], Hal [28] and BslK [29] in B. anthracis, and IlsA in Bacillus cereus [30, 31] all use a NEAT-domain strategy [14]. Heme uptake in Streptococcus pyogenes has also been studied in some detail. Here, the heme is abstracted from Hb via Shr (with two NEAT domains) [32–34] and then passed to Shp, a bis-methionine heme binding protein whose structure resembles the NEAT fold [35].
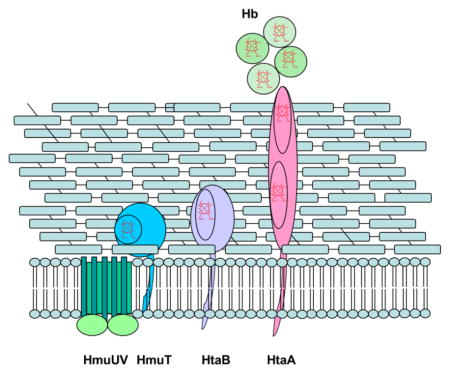
C. diphtheriae is distinct from the other Gram-positive bacteria cited above in that the major heme acquisition pathway studied to date does not employ NEAT domains. The hmu gene cluster comprises two heme-binding surface-anchored proteins, HtaA and HtaB, along with a heme binding lipoprotein (HmuT) and an ABC-type heme transporter (HmuUV) [7, 36–39] (Fig. 1). HtaA is a 61-kDa, membrane-anchored protein containing two conserved regions, CR1 and CR2, with 50% sequence similarity [37]; enzyme-linked immunosorbent assay (ELISA) experiments showed that HtaA binds Hb and is required for heme uptake from Hp-Hb [7]. HtaB, a 36 kDa protein, has one CR domain that binds heme, but not Hb [37]. HtaA can transfer hemin to HtaB in vitro [37, 38]. HmuT, a lipoprotein anchored to the cytoplasmic membrane, is thought to receive its hemin from either HtaB or HtaA and transfer it to the transmembrane permease HmuU, followed by translocation across the membrane in a process driven energetically by ATP hydrolysis catalyzed by the ATPase, HmuV [36]. Deletion of the htaA gene, the hmuTUV genes or the complete hmu gene cluster resulted in significantly decreased growth in comparison to the wild-type (WT) strain when the cells were grown in the presence of Hb as the sole iron source. Recent studies have shown that heme uptake from Hp-Hb necessitates the presence of one or two additional proteins, ChtA and ChtC, each of which has a single CR domain [7].
Fig. 1.

Schematic of heme uptake proteins in the hmu pathway of C. diphtheriae.
HtaA-CR1 and HtaA-CR2 are of interest because they take a position in the overall heme uptake strategy similar to that of the NEAT domains, yet have no significant sequence or structural homology to these more well-studied structures. Both HtaA-CR1 and HtaA-CR2 bind hemin; they bind Hb with less affinity than the complete HtaA, with the CR2 domain binding Hb more strongly than CR1 [37]. The domains bind hemin-albumin (HSA), Hp-Hb, and myoglobin, with the binding to HtaA-CR2 being stronger than that to HtaA-CR1 in each case [7].
Sequence alignment of selected Corynebacterium CR domains shows that one histidine and two tyrosine residues are fully conserved (Y361, H412 and Y490 in HtaA-CR2) (Fig. S1) [37]. Mutation of these conserved residues in HtaA-CR2 showed that all three are important for heme binding and uptake. For heme binding, the order was WT > Y490A > H412A Y361A. HtaA-CR2 Y361A was isolated with the lowest heme loading of the three mutants. Y361A also bound less Hb than either H412A or Y490A; the single amino acid substitutions Y361A and H412A in the full-length HtaA reduced Hb binding by almost 90%. In line with the hypothesis that Y361 is an axial ligand, mutation of this residue to alanine completely abolished the ability of the cells to use heme or Hb as iron sources. In addition, the GST-tagged CR2 Y361A mutant did not bind Hp-Hb, in contrast to the WT domain, which had an EC50 of 200 nM, nor did it bind heme-HSA, while the WT domain had an EC50 of 350 nM [7].
Given the crucial role of HtaA in heme acquisition by this important pathogen and, specifically, the second conserved domain with its presumably novel structural features, we have undertaken a study of the biophysical characteristics of HtaA-CR2. Resonance Raman (rR) spectroscopy was used to probe structural and electronic features of the bound heme along with binding aspects of the heme pocket. The stability of the protein fold was investigated via chemical and thermal denaturation techniques.
2. Experimental
2.1. Site-directed mutagenesis
The HtaA-CR2 was previously constructed in pET24a(+) [36]. Recombinant HtaA-CR2 mutants were made by site-directed mutagenesis by using a QIAprep Spin Miniprep Kit according to the manufacturer’s instructions. The primers were from Sigma Aldrich for Y361A, H412A and Y490A substitutions (Table S1). Primers (500 ng) were mixed with 100 ng of pET24a(+) template plasmid in the reaction mixture. DpnI restriction endonuclease was used to remove the methylated template. The mutated plasmids were isolated from the reaction mixture and transformed into the competent E. coli strain BL21(DE3). Sequence analysis was used to confirm the mutations.
2.2. Expression and purification
Wild type and the mutant proteins were expressed and purified using the same protocol. Terrific Broth (TB) with the inclusion of 50 μg/ml kanamycin was used as the media. A small-scale culture was incubated at 37 °C/220 rpm for 16 h. This culture (10 ml) was inoculated into a large-scale culture (500 ml) and incubated at 37 °C/220 rpm until the OD600 reached 0.8. The cultures were induced by adding 50 μM isopropyl-β-D-thiogalactopyranoside (IPTG) and allowed to grow for 16 h. The cells were harvested by centrifugation at 3100 rpm/4 °C for 30 min. The pellets were collected and stored in a -80 °C freezer overnight.
The cell pellet was re-suspended in buffer A (100 mM Tris-HCl, 150 mM NaCl, pH 8.0). The cells were lysed on ice for 30 min in a buffer A solution containing 10 mM MgCl2, 0.1 mM phenylmethanesulfonyl fluoride (PMSF), 0.2 mg/mL lysozyme, 5 μg/mL DNase I (from bovine pancreas, Roche) and 5 μg/mL RNase A (from bovine pancreas, Roche). The cells were treated with a cell disrupter, and cell debris was removed by centrifugation at 6500 rpm/4 °C for 30 min.
A GE Healthcare ÄTA fast protein liquid chromatography instrument (FPLC) was used for purification at 4 C. The HtaA-CR2 solution was loaded onto a Strep-Tactin Superflow column (5 mL, IBA BioTAGnology) which had been equilibrated with buffer A. Buffer A [5 column volumes (CV)] was used to wash the unbound material. The Strep-Tactin bound HtaA-CR2 protein was eluted with a linear gradient of 10 CV of buffer B (100 mM Tris-HCl, 150 mM NaCl, 2.5 mM d-desthiobiotin, pH 8.0). The purity of the fractions was tested by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Heme loading was approximately WT (20%), Y361A (1%), H412A (2%), and Y490A (5%).
2.3. Addition of hemin to apoHtaA-CR2
Four methods were used to add hemin to apoHtaA-CR2 in an effort to increase the heme loading. The first set of experiments involved preparation of fully apoHtaA-CR2 via the butanone extraction method [40]. An HtaA-CR2 solution at a pH value of 2.0 was prepared by adding 1 M HCl dropwise. Equal amounts of the acidic HtaA-CR2 solution and ice-cold 2-butanone were mixed. The mixture was vortexed for approximately 30 s and set on ice. After a 30 min incubation, layers of aqueous apoprotein at the bottom and organic heme-butanone at the top were formed. The bottom solution was removed by a pipette and dialyzed against 20 mM Tris-HCl pH 7.0 at 4 °C overnight. A stock hemin solution was prepared by dissolving hemin in DMSO using the literature extinction coefficient [41]. The concentration of apoprotein was determined by using an ExPASy extinction coefficient [42] of 24,075 M−1cm−1 at 280 nm. The apoHtaA-CR2 solution was titrated with the hemin solution by adding 1 μl aliquots; the solution was stirred for 30 min after each addition. At the end of the titration, the stoichiometric ratio of hemin and apoprotein was 1:1. Reconstituted HtaA-CR2 was dialyzed against 50 mM Tris-HCl pH 7.0 overnight. Reconstitution of the WT protein by this method gave a hemin-bound form that was spectrally distinct from the as-isolated protein.
The second set of experiments involved addition of hemin to the as-isolated protein (approximately 20% heme loaded). The protein was prepared in 50 mM Tris-HCl pH 7.0. A solution of hemin in DMSO was added in increments to a final ratio of hemin to apoHtaA-CR2 of ~ 1.5. The protein solution was equilibrated 30 min after each addition of hemin.
The third set of experiments involved hemin transfer from metHb on a column. Strep-tag purified HtaA-CR2 (a sample of 1 ml of about 70% apoprotein, 0.01 μmol apoHtaA-CR2, ASoret:A280 ~ 0.6) in buffer A was loaded onto a 5 mL Strep-Tactin column. A metHb solution (equine, Sigma-Aldrich, 200 μM hemin) was prepared in buffer A (2 ml, approximately 0.4 μmol metHb subunits, 40-fold excess hemin) and loaded onto the column. The column was allowed to stand for 19 h at 4 °C. Excess Hb was removed by washing the column with 40 CV of buffer A. Fractions were checked by UV-visible spectroscopy to make sure the eluent was free of hemin and protein. Bound HtaA-CR2 was eluted with 100% buffer B. Approximately 0.0082 μmol holoHtaA-CR2 (ASoret:A280 ~ 2.0) was obtained.
The fourth set of experiments involved attempted transfer from Hb in solution. Partially heme-loaded HtaA-CR2 (11.5 μM protein, 5.6 μM hemin) was titrated with human metHb. Additions of metHb to the initial solution (5.6 μM hemin, 0 μM from Hb), brought the total concentration of hemin to 9.6 μM (4 μM from Hb) and 12.1 μM (6.5 μM from Hb). Because the major bands of the optical absorbance spectra overlap, Soret-excited (406.7 nm excitation) rR spectra were also recorded in order to track whether rR features of metHb disappeared in favor of those for holoHtaA-CR2.
2.4. Purification of apo- and holoHtaA-CR2
In another approach to isolating the holoprotein, a solution of Strep-tag purified HtaA-CR2 (ASoret:A280 ~ 0.7) was initially passed through a 5-mL HiTrap Butyl FF hydrophobic interaction column (Fischer Scientific). The Butyl FF column was equilibrated with 5 CV of 20 mM NaH2PO4 at pH 7.0 followed by 5 CV of the binding buffer [20 mM NaH2PO4 and 1.5 M (NH4)2SO4 at pH 7.0]. The sample was loaded and the column was washed with 5 CV of binding buffer. The elution was carried out with a decreasing linear gradient of ammonium phosphate (1.5 - 0 M). A single chromatographic peak was observed. Fractions showing heme absorbance were pooled to give a sample with a ASoret:A280 ~ 0.9 (holoprotein recovery of 85%). After concentration and buffer exchange using an Amicon filter, the sample was passed through a Phenyl FF column using the same elution conditions. The chromatogram showed two overlapping peaks. The majority of the holoprotein was found at the start of the second peak (ASoret:A280 ~ 2.0). The latter fractions from this peak were mainly apoprotein. This elution order is consistent with the apoprotein being more hydrophobic than the holoprotein. The fractions that contained significant holoprotein were pooled and concentrated. After passing through both hydrophobic columns, the ASoret:A280 ratio increased from the as-isolated value of ~ 0.7 to ~ 2.0. Approximately 40% of the holoprotein was recovered from the initial sample after the two columns. Variations on this separation showed that although the Butyl FF column did not give a significant separation, protocols in which this step was omitted resulted in a lower final ASoret:A280 ratio.
2.5. UV-visible and circular dichroism absorbance spectroscopy
UV-visible absorbance spectra were recorded either with a single beam (Varian Cary 50 Bio) or a dual beam scanning spectrophotometer (OLIS-14). The measurements were made using a quartz cuvette having a 1.0 cm path length. Circular dichroism (CD) spectra were recorded using a Jasco J-810 spectropolarimeter using quartz Suprasil cuvettes with a 1 mm path length. Protein samples were recorded in 10 mM KH2PO4 buffer at pH 7.0; concentrations were adjusted to 10 μM. The final spectra represent an average of 5 scans.
2.6. Time scale of unfolding
The UV-visible spectrum of HtaA-CR2 was monitored over time at 37 °C as a function of the GdnHCl concentration from 6.6 to 7.4 M. The protein and the GdnHCl solutions were prepared in 50 mM Tris-HCl, pH 7.0. The GdnHCl concentration was determined by the refractive index method [43]. Absorbance measurements were recorded every 10 min for 24 h. The absorbance at 405 nm (A405) vs. time was fit to a single-term exponential function: equation (1)
| (1) |
where At is the absorbance at any time during the unfolding reaction, A0 is the initial absorbance, A∞ is the absorbance of the completely unfolded protein, (A0 A∞) is the total A for complete unfolding, and kU is the unfolding rate constant. These experiments were run on as-isolated holo/apo mixtures. A sample with 7.0 M GdnHCl (35% holo/65% apo) was dialyzed after unfolding to evaluate the reversibility of the process. Approximately 40% of the protein was recovered, perhaps because the apoprotein precipitated (apoprotein precipitation was also observed in other experiments). The recovered protein was about 25% heme loaded. The spectrum of this holoprotein was similar, but not identical, to that of the original as-isolated holoprotein.
2.7. Thermal unfolding of HtaA-CR2 in presence of GdnHCl
Thermal unfolding of HtaA-CR2 was carried out with the UV-visible spectrophotometer equipped with a TC125 Quantum Northwest temperature controller. A screw-top quartz cuvette with 1-cm path length was used. Holo HtaA-CR2 in 50 mM Tris-HCl pH 7.0 was prepared in GdnHCl concentrations of 1.5, 1.75, 2.0, 2.5 and 3.0 M. The thermal unfolding was carried out from approximately 45 to 90 °C. The spectrum was recorded every 1 °C after a 1 min incubation at each temperature.
Kaleidagraph (version 4.01, Synergy Software) was used to fit the data using a two-state protein unfolding model equation (2) [44]:
| (2) |
where A is the absorbance at any temperature along the unfolding curve, AF is the absorbance of the folded state, AU is the absorbance of the unfolded state, mF is the slope of A vs. T for the folded state, mU is the analogous slope for the unfolded state, Tm is the temperature at which the protein is half unfolded, ΔHm is the enthalpy of unfolding, R is the ideal gas constant, and T is the temperature (Kelvin).
2.8. Guanidinium-induced unfolding of HtaA-CR2 mutants
The guanidinium denaturation of HtaA-CR2 mutants (Y361A, H412A, and Y490A, all almost entirely in their apo forms) were investigated by following emission spectra on a Perkin Elmer LS fluorescence spectrophotometer. Apo WT protein was isolated by chromatographic separation on a hydrophobic column. Its chemically-induced unfolding was followed using the single-cuvette titration technique [45–48]. The protein in 50 mM Tris-HCl pH 7.0 was excited at 285 nm and the emission spectrum was monitored between 300 and 500 nm. The emission intensity decreased as a function of GdnHCl concentration.
The unfolding curves were analyzed using equation (3) [43]:
| (3) |
where A is the absorbance at any denaturant concentration along the fitted denaturation curve, AF is the absorbance of the folded state, AU is the absorbance of the unfolded state, m is the slope at the midpoint, mF is the slope of the folded state, mU is the slope of the unfolded state, [D] is the concentration of GdnHCl, [D]½ is the concentration of GdnHCl at the midpoint of the unfolding curve, R is the ideal gas constant, and T is the temperature (Kelvin).
2.9. Resonance Raman spectroscopy
Resonance Raman scattering was excited with either 406.7- or 413.1-nm emission from a Kr+ laser, or 441.6 nm from a HeCd laser. The laser beam was focused to a line parallel to the spectrograph entrance slit. Scattered light collected in the 135° backscattering geometry (f/1) was passed through a polarization filter, holographic notch filter, and polarization scrambler. The laser image was then focused on the entrance slit of a 0.67-m, f/4.7 Czerny-Turner spectrograph fitted with 1200 and 2400 g/mm gratings and a LN2-cooled CCD detector (1340×400 array of 20×20 μm pixels, 26.8×8.0 mm2 image area). The spectrometer was calibrated against the Raman shifts of toluene, dimethylformamide, acetone, and methylene bromide. Spectra were recorded at ambient temperature from samples contained in spinning 5-mm NMR tubes. UV-visible absorbance spectra were recorded from the rR samples before and after spectral acquisition to assess whether sample integrity had been compromised by exposure to the laser beam. Laser power at the samples ranged from 5 to 10 mW.
2.10 Electrospray ionization (ESI) mass spectrometry
ESI spectra were obtained using a Waters Micromass Q-TOF spectrometer in the positive mode. The fully holoprotein was prepared by adding of hemin to the as-isolated protein (approximately 20% heme loaded) as previously described. The protein solution (10 μM) was then dialyzed against 20 mM ammonium acetate, pH 6.8. Spectra were recorded (flow rate 10 μl/min) at the following collision energy voltages: 5, 10, 15, 20, 25, 30, and 40 V. All other parameters were held constant (capillary voltage 3000 V, cone voltage 18 V, extraction voltage 1.5 V, desolvation gas temperature 100 °C and source temperature 80 °C). Deconvolution of the charged state was performed using the MaxEnt program with the MassLynx software. Peaks were rounded to the nearest Dalton; peak heights were used to calculate holoprotein percentages.
3. Results
3.1. HtaA-CR2 spectroscopy
The UV-visible absorbance spectrum of as-isolated ferric HtaA-CR2 is shown in Fig. 2A. The Soret maximum is at 405 nm, and bands are seen in the visible region at 502, 537, and 624 nm. The charge-transfer (CT) band at 624 nm is characteristic of high-spin heme, common for axial tyrosine ligation [49, 50]. The ferric protein was reduced smoothly, albeit slowly, with dithionite ion (S2O42−) to give the ferrous species with a Soret at 424 nm and α/β bands at 544 and 569 nm. The reduction took ca. 30 min at pH 7.0 and ambient temperature without significant side products, as evinced by the isosbestic behavior of the visible absorbance spectra in Fig. S2. The addition of CO gave the ferrous carbonyl species with a Soret band at 415 nm (Fig. 2A).
Fig. 2.

A) UV-visible spectra of ferric (black —), ferrous (
 ), and ferrous carbonyl (
), and ferrous carbonyl (
 ) complexes of HtaA-CR2. The ferric and ferrous carbonyl samples were in 50 mM Tris-HCl pH 7.0; the ferrous HtaA-CR2 was in 50 mM Tris-HCl pH 7.0. B) High-frequency rR spectra of a) ferric HtaA-CR2, 406.7 nm excitation, b) ferrous HtaA-CR2, 413.1 nm excitation, and c) ferrous-CO HtaA-CR2 complex, 413.1 nm excitation, under the same buffer conditions listed in A. Laser powers incident on the sample for a and b were 5 - 8 mW while c was acquired with 2.5 mW.
) complexes of HtaA-CR2. The ferric and ferrous carbonyl samples were in 50 mM Tris-HCl pH 7.0; the ferrous HtaA-CR2 was in 50 mM Tris-HCl pH 7.0. B) High-frequency rR spectra of a) ferric HtaA-CR2, 406.7 nm excitation, b) ferrous HtaA-CR2, 413.1 nm excitation, and c) ferrous-CO HtaA-CR2 complex, 413.1 nm excitation, under the same buffer conditions listed in A. Laser powers incident on the sample for a and b were 5 - 8 mW while c was acquired with 2.5 mW.
Soret-excited rR spectra were recorded from the ferric, ferrous and ferrous carbonyl heme complexes of HtaA-CR2. Fig. 2B shows spectra in the frequency region of the porphine in-plane skeletal modes. The largest band in this region arises from the totally symmetric core breathing mode, ν4, which occurs near 1370 cm−1 for the ferric oxidation state and ferrous π-acid complexes, such as heme carbonyls [51–53]. The ν4 band in ferrous hemes falls near 1360 cm−1 [54]. The ν4 frequencies are consistent with ferric (a), ferrous (b) and ferrous carbonyl (c) hemes, respectively. The ν3 bands fall in Raman shift regions characteristic of both oxidation and spin states of the iron center, with those of ferric (1490 cm−1) and ferrous (1473 cm−1) HtaA-CR2 falling at frequencies consistent with pentacoordinate, high-spin (5cHS) iron centers [51–53]. The 1501-cm−1 ν3 band in the HtaA-CR2-CO spectrum is typical of the low-spin (LS) heme carbonyl complexes [54].
The spectral features of ferric HtaA-CR2 are essentially independent of pH between 5.8 and 9.5, as seen in the similarity among the rR spectra in Fig. S3. Furthermore, there were no 18O- or 2H-sensitive bands in the low-frequency spectrum at pH 9.8 (data not shown), as would be expected if a hemin hydroxide were forming in alkaline solution [53]. The νFe–CO and νC–O regions of the heme carbonyl spectrum are shown in Fig. 3. Isotope sensitivity of the 535-, 572- and 1949-cm−1 Raman shifts in the 13CO isotopolog allow their assignments to the νFe–CO, δFeCO and νC–O modes, respectively. The backbonding correlation plot in Fig. 3C shows that the as-isolated HtaA-CR2-CO is homogeneous and falls on the line correlating heme carbonyls having a charge neutral trans O-bound ligand [55].
Fig. 3.

Resonance Raman characterization of the ferrous HtaA-CR2 carbonyl complex. A) Low frequency region and B) C-O stretching region of the rR spectra of the isotopomers of HtaA-CR2-CO recorded using 413.1 nm: HtaA-CR2-12CO (top), HtaA-CR2-13CO (middle) and difference [HtaA-CR2-12CO - HtaA-CR2-13CO] (bottom) spectra. The ferrous carbonyl complex was prepared in 50 mM Tris-HCl pH 7.8. C) The νFe–CO/νC–O correlation plot of ferrous carbonyl proteins with the as-isolated HtaA-CR2-CO indicated by a solid red star. Reconstituted HtaA-CR2-CO is a mixture of two heme carbonyl species, indicated by open red stars. One is near the point for the as-isolated protein, and the other is near the black line for heme carbonyls having trans imidazole ligands. The green line is the least squares line for six-coordinate Fe–CO adducts in which the proximal ligand is thiolate or imidazolate and the blue line represents a compilation of model complexes and heme proteins, wherein the ligand trans to CO is likely to charge neutral and coordinated through an oxygen atom [55].
3.2. Approaches to holoprotein
As-isolated, HtaA-CR2 was only ~20% heme loaded. As more fully heme-loaded protein was desirable for the biophysical studies, various approaches were used to increase the amount of holoprotein in the sample. Passing the protein through two hydrophobic columns resulted in protein that was about 95% heme loaded. However, loss of protein in this lengthy procedure led to additional efforts to prepare samples with high heme loading. Full-length HtaA can take up hemin from Hb (metHbA) as previously demonstrated by UV-visible spectroscopy [37]. Those experiments involved binding of 100 μl of 30 μM as-isolated Strep-tag-labeled HtaA to a column, incubation with an equivalent volume of 30 μM Hb for 30 min, and isolation of the resulting HtaA. The heme loading had increased. An ELISA assay showed that both HtaA and the CR2 domain were able to bind Hb.
Transfer of hemin from metHb to HtaA-CR2 on a preparative scale was examined in this study as a means of generating more completely heme-loaded HtaA-CR2. As-isolated protein was loaded on a Strep-Tactin column. A metHb solution containing 40× excess of hemin was loaded into the column void volume and remained in contact with the HtaA-CR2-loaded resin for 19 h. After isolation, the amount of holoHtaACR-2 had increased approximately two-fold. It is clear from this significant increase in cofactor loading that hemin is passed from metHb to HtaA-CR2 when the HtaA-CR2 is bound to the affinity column.
The hemin transfer reaction was also examined in solution with 5.9 μM apoHtaA-CR2 (in a mixture with 5.6 μM holoprotein) and human metHbA (concentrations of 0, 4.0 and, 6.5 μM heme). Fig. S4 shows the UV-visible absorbance (A) and Soret-excited rR spectra (B) of those reaction mixtures. Examination of the starting HtaA-CR2 Raman spectrum reveals ν3 at 1490 cm−1 and a relatively large ν3:ν4 intensity ratio, consistent with the hemin being 5cHS with an O-bound axial ligand. If titration with metHbA were to transfer hemin efficiently to HtaA-CR2, a simultaneous and proportionate increase in all its band intensities would be expected. However, the ν3:ν4 intensity ratio decreased with each addition of metHbA while the porphine core marker bands reporting oxidation and spin states shifted monotonically toward their metHbA frequencies. Additionally, the UV-visible absorbance spectra contained bands attributable to a mixture of metHbA and hemin-loaded HtaA-CR2. Thus, significant transfer of hemin from metHbA to HtaA-CR2 was not observed in solution under dilute and near-stoichiometric conditions.
Finally, hemin loading could also be effected by titrating the as-isolated protein with hemin itself in DMSO to give a sample that was ~50% heme loaded. The spectra of holoHtaA-CR2 obtained via all of these procedures are shown in Fig. S5. The spectra are similar, but not identical. This observation may reflect mixtures of more than one conformation of the protein. Some of the HtaA-CR2 may also contain protoporphyrin rather than hemin. This has been observed in previous studies involving isolation of recombinant heme proteins [56, 57]. Consistent with this idea, an electrospray mass spectrum of HtaA-CR2 showed peaks for both protoporphyrin and hemin in the low molecular weight region and an HtaA-CR2 solution extracted with butanone showed a fluorescence peak appropriate for protoporphyrin [58] (data not shown).
3.3. Reconstitution after full unfolding: A second form of HtaA-CR2
The classic approach to reconstitution of a heme protein is to remove all tetrapyrroles using the butanone extraction method [40] and then reconstitute the apoprotein with hemin dissolved in DMSO. The UV-visible absorbance spectrum of HtaA-CR2 reconstituted in this manner was distinct from that of the as-isolated protein, with its Soret band being shifted to higher energy, the appearance of a shoulder at 370 nm, and broadening and shifting of the α/β bands (Fig. S5). Additionally, whereas the as-isolated protein could be reduced with dithionite at pH 7, the reconstituted protein could only be reduced under an atmosphere of CO. This method has been used to prepare ferrous heme carbonyls of proteins having highly negative reduction potentials; e.g., WT Serratia marcescens HasA-CO (heme reduction potential of − 550 mV) and HasA mutants with reduction potentials of approximately − 350 mV and lower [54]. The fact that this technique was necessary to reduce reconstituted HtaA-CR2 is consistent with it having a significantly negative reduction potential, which is in turn consistent with an axial tyrosinate ligand in the ferric state. CD spectra of the apoHtaA isolated via hydrophobic interaction chromatography and via butanone treatment were also different (Fig. S6), in line with the observations on the holoprotein.
Although the UV-visible spectra of the as-isolated and protein reconstituted after full unfolding were different, the Soret-excited rR spectrum of reconstituted ferric HtaA-CR2 (data not shown) was similar to that of the as-isolated protein, exhibiting features characteristic of a 5cHS form. Moreover, the large I(ν3):I(ν4) ratio of 0.70 [53] and its lack of spectral sensitivity to changes in pH argue for the hemin being bound to the protein through an O-bound axial ligand, such as the phenol side chain of Tyr [39]. By contrast, the rR spectrum of the ferrous carbonyl form of reconstituted HtaA-CR2 was distinct from that of its as-isolated counterpart. The correlation plot (Fig. 3C) reveals two forms of the reconstituted HtaA-CR2-CO complex; one very close to the as-isolated point with νFe–CO and νC–O frequencies of 536 and 1947 cm−1, respectively, and the second with frequencies of 497 and 1947 cm−1, putting it on the plot near the correlation line for a trans histidine ligand. This indicates that there are two forms of ferrous-CO species, one with trans histidine and the other with a trans O-bound ligand such as tyrosine or water. These spectral and chemical differences indicate that addition of hemin to apoHtaA-CR2 created via butanone extraction yields a different form of the holoprotein than that isolated after purification. For consistency, the biophysical studies reported herein were all performed on samples of the as-isolated HtaA-CR2, without unfolding of the protein or addition of hemin in the laboratory.
3.4. Time scale of protein unfolding in chemical denaturation
HtaA-CR2 is quite stable to treatment with denaturing agents and heat. GdnHCl-induced unfolding of HtaA-CR2 was not observed at room temperature over many hours for denaturant concentrations up to 4 M. Even at 7.0 M GdnHCl, the protein was not completely unfolded after 24 h at ambient temperature. The unfolding was then evaluated as a function of GdnHCl at 37 °C. At each concentration, the unfolding curve was well modeled by a single-term exponential decay function, suggesting a single first-order unfolding process (Fig. 4). At the indicated GdnHCl concentrations, the unfolding half-lives were: 6.6 M (330 min), 6.8 M (300 min), 7.0 M (260 min), 7.2 M (250 min), and 7.4 M (215 min) GdnHCl. Fig. S7 shows the dependence of the unfolding rate constant, ku, on the concentration of GdnHCl. A rate constant at 37 °C in the absence of denaturant of 1.3 ± 0.1 × 10−6 s−1 (t1/2 = 144 h) was calculated by extrapolation to the y-intercept.
Fig. 4.

Time scale of WT HtaA-CR2 unfolding in the presence of 6.8 M GdnHCl. The unfolding reaction was carried out in 50 mM Tris-HCl pH 7.0. The data were fit using the single-term exponential decay function shown in Eq. 1.
3.5. Thermal unfolding of HtaA-CR2 in the presence of GdnHCl
Thermal denaturation was monitored in the presence of increasing amounts of GdnHCl. As the temperature was increased, the Soret absorbance gradually decreased and shifted about 12 nm to higher energy (data not shown). The protein solutions were cooled to room temperature at the end of each experiment. Around 30% of the original Soret absorbance was recovered in solutions of 1.5 and 1.75 M GdnHCl. No recovery was observed in solutions in which the concentration of GdnHCl was greater than 1.75 M. Because the protein was so stable (i.e., not fully reaching equilibrium at each temperature), only apparent Tm values could be calculated. For example, at 1.5 M denaturant, the apparent Tm was 77 °C (Fig. 5). Extrapolation of a plot of Tm vs. the concentration of GdnHCl to zero denaturant concentration suggests an apparent intrinsic Tm of 84 °C for HtaA-CR2 (Fig. S8).
Fig. 5.

Soret absorbance of HtaA-CR2 at 405 nm in 1.5 M GdnHCl as a function of temperature. Points are experimental values of A405; the solid line shows the nonlinear least squares fit of the Soret absorbance to the function in Eq. 2. The fit yielded a value of 77 °C for Tm.
3.6. Guanidinium-induced unfolding of apoHtaA-CR2 and its mutants
Fluorescence spectroscopy was used to gain insight into the stability of the apoprotein forms of WT HtaA-CR2 and its mutants. Tryptophan emission was monitored as a function of the concentration of GdnHCl. Fig. 6 shows a representative example of the unfolding curve of apo Y490A. The midpoint denaturation concentrations were 1.34 ± 0.02 M for apo WT HtaA-CR2 and 1.25 ± 0.02, 1.28 ± 0.01, 1.41 ± 0.01 M for Y361A, H412A and Y490A, mutants, respectively.
Fig. 6.

Tryptophan emission of Y490A apoHtaA-CR2 as a function of GdnHCl concentration. The reaction was in 50 mM Tris-HCl pH 7.0. Data points were recorded after chemical equilibration (5 - 20 min). Solid points are photon counts per second at 340 nm, and the solid line is the fit using the function in Eq. 3.
3.7 ESI Mass spectrometry
The stability of the holoprotein was also evaluated using mass spectrometry. The mass spectrum of a fully holo WT sample was recorded as a function of collision voltage from 5 - 40 V. The observed percentage of holoprotein decreased from approximately 70% to approximately 20% (Fig. 7). The as-isolated mutants had less than 5% heme, and so were not studied with this method.
Fig. 7.

Electrospray ionization mass spectrometry detection of heme-bound holoHtaA-CR2 as a function of collision energy voltage. The protein solution was prepared in 20 mM ammonium acetate, pH 6.8.
4. Discussion
4.1. HtaA-CR2 axial ligands
Sequence alignment indicates three probable axial ligands that are completely conserved in HtaA Corynebacterium sequences: Tyr361, His412, and Tyr490. Previous experimental data indicate that Y361 is more likely than Y490 to be the axial ligand [37]. Sequence alignment of a variety of homologous proteins is in line with this expectation, as the region around Y361 shows greater conservation than the region around Y490. Optical spectroscopy of WT HtaA-CR2 shows three bands in the α/β region. This three-banded pattern, with a band near 630 nm, is characteristic of a tyrosine axial ligand as found in various heme uptake proteins in a variety of species (Table S2).
Consistent with the UV-visible spectral pattern, several aspects of the rR spectrum support a 5cHS hemin with an axial Tyr ligand. First, the ν3 and ν4 frequencies of 1490 and 1372 cm−1 are indicative of ferric 5cHS hemin [51–53]. Second, the high I(ν3):I(ν4) ratio is characteristic of an O-bound axial ligand. Although this spectral fingerprint is well established in a number of 5cHS Tyr-bound hemins [39, 59, 60], the possibility of a 5cHS aqua or hydroxo complex could not be excluded based solely on the spectrum of the WT protein. Therefore, rR spectra of HtaA-CR2 were recorded at high pH and pD in 18OH2 and OD2 to determine whether there were any isotope-sensitive bands in the νFe OH region of the spectrum. The lack of 18O- or 2H-sensitive bands further supports the conclusion that the O-bound axial ligand is endogenous. Third, the insensitivity of the HtaA-CR2 spectrum to pH is consistent with an axial Tyr ligand [53]. Finally, ferric hemin protein complexes having axial Tyr ligands are well known for their negative reduction potentials. Those complexes are generally either slow to reduce in the presence of S2O42− or simply do not reduce unless a strongly-coordinating ligand that favors Fe(II) over Fe(III), such as CO, is present to drive the reduction [54]. HtaA-CR2 exhibits this behavior, being only slowly reduced in the presence of excess S2O42− at pH 7 (Fig. S2).
The observation of Allen and Schmitt that mutation of H412 makes as much of a difference in heme binding as mutation of Y361 [37] is consistent with H412 playing a role in the heme pocket. In the current study, run on a larger scale with FPLC purification of the proteins, the Y361A and H412A mutants bound only 1–2% heme; Y490A was somewhat more heme loaded (consistent with this residue not being involved directly with heme binding). Many proteins in heme-uptake pathways with tyrosine as an axial ligand have a hydrogen-bonding partner in the binding pocket [39]. In some instances mutation of the hydrogen-bonding partner has been shown to have a very large effect on heme binding, e.g., removal of the H-bonding partner H83 in the S. marcescens hemophore HasA resulted in a loss in heme affinity of more than 2500-fold [61]. In other instances, the effect is small, e.g., mutation of the H81 hydrogen-bonding partner in Pseudomonas aeruginosa HasA did not change the heme association constant [62]. These data are consistent with many factors being important in controlling heme binding, including van der Waals, salt bridging, π-stacking and other hydrogen-bonding interactions. Even though a given interaction may not play a decisive role in controlling the heme affinity, it may be mechanistically important in the transfer of the heme to the cognate acceptor.
4.2. Reconstituted HtaA-CR2
The significant 370-nm absorbance in the UV-visible spectrum of HtaA-CR2 reconstituted with hemin itself is consistent with binding of a π-stacked heme dimer. Although we have not pursued structural characterization of this complex, the suggestion of complex with dimeric heme has considerable precedence. Hemin readily forms π-stacked as well as μ-oxo dimers in solution [63, 64]. X-ray data have shown that Yersinia pestis HmuT can bind heme as a π-stacked dimer; this form of the holoprotein had an absorbance band at 373 nm in solution [65]. X-ray data has also shown that Mycobacterium tuberculosis MhuD has the ability to bind π-stacked heme, although this complex is enzymatically inactive [66]. Finally, π-stacked dimers have been found at the interface between two heme trafficking proteins in the crystal structures of ChaN from Campylobacter [67], IsdH from S. aureus [68], and Shp from S. pyogenes [35].
4.3 Heme transfer from Hb to HtaA-CR2
Hemoglobin serves as a source of heme for C. diphtheriae; mutations in the htaA gene reduce the ability of the organism to employ Hb as an iron source [36]. Hemin can be transferred from Hb to HtaA as shown previously on a small scale [37] and in this work as a possible preparative procedure for holoHtaA-CR2.
In contrast, significant hemin transfer from Hb to HtaA-CR2 was not observed at lower concentrations of the proteins in solution. These experiments were run over ~ 2 h with an initial ratio [metHb]/[apoHtaA-CR2] of 0.7 and a final ratio of 1.1 (6.5 μM hemin in Hb). In comparison, the affinity column experiments were run for 19 h with a [metHb]/[apoHtaA-CR2] ratio of approximately 40. As the value of Kd for tetrameric metHbA is 1.5×10−6 M [69, 70] both experimental conditions (6.5 μM and 200 μM hemin in metHb) give rise to mixtures of dimer of αβ (1.2 and 8.3 μM αβ dimer, respectively) and (αβ)2 tetramer [1.0 and 46 μM (αβ)2 tetramer, respectively]. The role of the dimer is significant because the hemin dissociation rate constants for the α chain are 0.6 and 0.3 h−1 in the dimer and tetramer, respectively; the corresponding rate constants for the β chain are 15 and 1.5 h−1 [70]. The tetramer to dimer conversion is rapid (1–10 sec−1) and autoxidation of Hb is facile outside erythrocytes [70]. Although hemin transfer may occur within the complex (the Kd value of HbA:HtaA-CR2 is approximately 300 nM [38]), these rate constants for loss of hemin from Hb are fast enough to be consistent with passive loss of the prosthetic group from either the tetramer or the dimer followed by HtaA-CR2 uptake from solution on the time scales of these experiments. It is possible that the heme transfer observed in the affinity column experiments can be largely explained by longer contact times with the Hb and differences in the [αβ]/[apoHtaA] ratios. The microenvironment on the column may also be conducive to heme transfer. By contrast, the spectroscopically (UV-vis and rR) tracked reactions carried out at lower metHb concentration in solution were too slow to affect a similar passive transfer during the window of observation.
4.4. Comparison of the holo and apo forms of HtaA-CR2
As noted above, holoHtaA-CR2 is exceptionally stable, with unfolding incomplete even after 24 h in 7 M GdnHCl at 37 °C. In stark contrast, apoHtaA-CR2 unfolded readily, with a midpoint for unfolding of 1.34 ± 0.02 M at room temperature. The mutants behaved similarly, with D1/2 values ranging only from 1.25 to 1.41 M for the Y361A, H412A and Y490A mutants. Thus, heme plays a deterministic role in the stability of the protein fold.
The importance of the full HtaA-CR2 structure for binding the heme tightly is also seen in the fact that all three mutants were isolated with at most 5% bound heme, in comparison with the WT protein, which had about 20% heme loading. Mutation of axial ligands need not lead to such a dramatic loss in heme binding for b-type heme uptake proteins. For example, as-isolated holoprotein with mutated axial ligands can be purified for C. diphtheriae HmuT [39], P. aeruginosa HasA [60], S. pyogenes SiaA/HtsA [71] and S. pyogenes Shp [72].
4.5. Thermal and chemical denaturation
HtaA-CR2 exhibits very high stability. Even in 6.8 M GdnHCl at 37 °C, the half-life for protein denaturation was 5 h. The only other non-hyperthermophilic heme proteins of which we are aware that are approximately as stable as HtaA-CR2 are the Class III plant peroxidases [73, 74]. For example, soybean peroxidase has a half-life for denaturation of 3.3 h at 25 °C in 6.8 M GdnHCl [73]. Horseradish peroxidase unfolds approximately 300 times more quickly under these conditions.
The origin of hyperstability is multifaceted and includes hydrogen bonding, internal packing of hydrophobic residues, optimization of salt bridges, and factors that restrict flexibility (including dimerization) [75–77]. Significant stability in heme transfer proteins presumably protects the organism from the deleterious effects of reactive oxygen species produced in the presence of free hemin and also allows the hemin to be transferred to a specific partner protein. Proteins in heme uptake pathways may have evolved very stable heme-bound folds so that heme cannot be released spontaneously or during non-specific encounters with other bio-macromolecules. This stability may reflect a mechanistic requirement for heme transfer to be “triggered” by the formation of a highly specific protein-protein complex with the appropriate heme-accepting partner wherein the heme would be available for transfer. A number of studies have led to the conclusion that transient protein-protein complexes are involved in heme transfer including investigations of the pathways from Shp to HtsA in S. pyogenes [78, 79]; in the Isd proteins of S. aureus [80–82]; from IsdX1 to IsdC and IsdX2 [83] and BslKN to IsdC [29] in B. anthracis; and from Hbp1 to Hbp2 in L. monocytogenes [27].
4.6 Mass spectrometry
When the collision voltage was increased from 5 - 40 V, holo HtaA-CR2 lost about 70% of its heme. By comparison, previous studies on HmuT, another protein in the pathway, showed only 10% heme loss over this collision voltage range [39]. HmuT had a Tm of 67 °C in buffer without denaturant while HtaA had essentially the same Tm (specifically, 69 °C) only in buffer with 3.0 M GdnHCl. The gas phase and solution phase data do not show a direct correlation, but give complementary information on protein stability, as also observed in previous studies on myoglobin and cytochrome b5 [84].
5. Conclusions
The UV-visible absorbance spectra of as-isolated ferric HtaA-CR2 as well as the Raman spectra, including placement on the ferrous-CO back bonding plots, are consistent with a tyrosine axial ligand. In that the H412A mutant also has very low heme loading, and shows significant reduction of Hb binding compared to WT, this residue may play a role in the heme pocket, perhaps as the hydrogen-bonding partner to Y361 or elsewhere near the heme. Reconstitution of WT HtaA-CR2 with hemin gave a different form of the protein. Transfer of hemin from Hb depended on the conditions of the experiment, being favored in on-column protocols at hundreds of micromolar metHb subunit concentrations. HtaA-CR2 is highly stable. In chemical denaturation experiments, no unfolding was observed even up to 4 M GdnHCl over 24 h at room temperature. At higher concentrations of GdnHCl (6.6–7.4 M), a single unfolding process was observed at 37 °C. The protein was also stable to heat; it was necessary to perform thermal denaturation experiments in the presence of GdnHCl (1.5–3.0 M). In contrast, the apo forms of the WT protein and the mutants unfolded readily, suggesting that the bound heme is playing a significant role in the stability of the protein. The very high stability of the holoprotein may protect the bacteria against adventitious loss of heme during the uptake process. HtaA-CR2 joins other heme uptake proteins, such as many of those with NEAT domains, in using a tyrosine as the axial ligand. Changes in the hydrogen bonding of an axial tyrosine may be employed to promote heme transfer to the next protein in the pathway.
Supplementary Material
Highlights.
The heme in HtaA-CR2 is coordinated by an axial tyrosine
Mutation of the axial ligand and a potential hydrogen-bonding partner His reduced heme binding
Reconstitution of the protein after full unfolding significantly altered the heme binding pocket
HtaA-CR2 is very stable towards heat and treatment with guanidinium hydrochloride
Bound heme plays an essential role in the stability of HtaA-CR2
Acknowledgments
Funding Sources
This work was supported by the National Institutes of Health Grants AI072719 (to K.R.R.), GM094039 (to G.L.R.) the Research Corporation (to D.W.D.) and the Molecular Basis of Disease Program at Georgia State University (R.C.U.)
We thank John H. Dawson and Daniel P. Collins for contributions to the preliminary data. We also thank Siming Wang for technical assistance with the mass spectrometry experiments and insightful discussions.
Abbreviations
- ABC
transporter
- ATP-binding
cassette transporter
- CD
circular dichroism
- CO
carbon monoxide
- CR
conserved region
- CT
charge-transfer
- CV
column volume
- DMSO
dimethyl sulfoxide
- ELISA
enzyme-linked immunosorbent assay
- ESI
electrospray ionization
- FPLC
fast protein liquid chromatography
- GdnHCl
guanidinium hydrochloride
- Hb
hemoglobin
- Hp
haptoglobin
- HS
high spin
- HSA
human serum albumin
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- LS
low-spin
- PMSF
phenylmethanesulfonyl fluoride
- rR
resonance Raman spectroscopy
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TB
Terrific Broth
- WT
wild-type
- 5cHS
pentacoordinate, high-spin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Collier RJ. Toxicon. 2001;39:1793–1803. doi: 10.1016/s0041-0101(01)00165-9. [DOI] [PubMed] [Google Scholar]
- 2.Trost E, Blom J, Soares SD, Huang IH, Al-Dilaimi A, Schroder J, Jaenicke S, Dorella FA, Rocha FS, Miyoshi A, Azevedo V, Schneider MP, Silva A, Camello TC, Sabbadini PS, Santos CS, Santos LS, Hirata R, Mattos-Guaraldi AL, Efstratiou A, Schmitt MP, Hung TT, Tauch A. J Bacteriol. 2012;194:3199–3215. doi: 10.1128/JB.00183-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagner KS, White JM, Lucenko I, Mercer D, Crowcroft NS, Neal S, Efstratiou A. Emerg Infect Dis. 2012;18:217–225. doi: 10.3201/eid1802.110987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmitt MP. Infect Immun. 1997;65:4634–4641. doi: 10.1128/iai.65.11.4634-4641.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drazek ES, Hammack CA, Schmitt MP. Mol Microbiol. 2000;36:68–84. doi: 10.1046/j.1365-2958.2000.01818.x. [DOI] [PubMed] [Google Scholar]
- 6.Schmitt MP, Drazek ES. J Bacteriol. 2001;183:1476–1481. doi: 10.1128/JB.183.4.1476-1481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allen CE, Schmitt MP. J Bacteriol. 2015;197:553–562. doi: 10.1128/JB.02413-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farrand AJ, Skaar EP. In: Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine. Ferreira GC, Kadish KM, Smith KM, Guilard R, editors. Vol. 26. Heme Biochemistry, World Scientific; Hackensack, NJ: 2014. pp. 317–377. [Google Scholar]
- 9.Benson DR, Rivera M. Met Ions Life Sci. 2013;12:279–332. doi: 10.1007/978-94-007-5561-1_9. [DOI] [PubMed] [Google Scholar]
- 10.Contreras H, Chim N, Credali A, Goulding CW. Curr Opin Chem Biol. 2014;19:34–41. doi: 10.1016/j.cbpa.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodgers KR, Lukat-Rodgers GS. In: Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine, Vol. 30: Heme Proteins, Part II. Ferreira GC, Kadish KM, Smith KM, Guilard R, editors. World Scientific; Hackensack, N.J: 2014. pp. 251–309. [Google Scholar]
- 12.Wandersman C, Delepelaire P. Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine, Vol 26: Heme Biochemistry. In: Ferreira GC, Kadish KM, Smith KM, Guilard R, editors. Handbook of Porphyrin Science. 2014. pp. 191–222. [Google Scholar]
- 13.Wilks A, O'Neill MJ. In: Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine, Vol 26: Heme Biochemistry. Ferreira GC, Kadish KM, Smith KM, Guilard R, editors. World Scientific; Hackensack, NJ: 2014. pp. 267–315. [Google Scholar]
- 14.Sheldon JR, Heinrichs DE. FEMS Microbiol Rev. 2015;39:592–630. doi: 10.1093/femsre/fuv009. [DOI] [PubMed] [Google Scholar]
- 15.Andrade MA, Ciccarelli FD, Perez-Iratxeta C, Bork P. Genome Biol. 2002;3:RESEARCH0047. doi: 10.1186/gb-2002-3-9-research0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Honsa ES, Maresso AW, Highlander SK. PLoS One. 2014;9 doi: 10.1371/journal.pone.0104794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazmanian SK, Skaar EP, Gaspar AH, Humayun M, Gornicki P, Jelenska J, Joachmiak A, Missiakas DM, Schneewind O. Science. 2003;299:906–909. doi: 10.1126/science.1081147. [DOI] [PubMed] [Google Scholar]
- 18.Zhu H, Li DF, Liu MY, Copie V, Lei BF. PLoS One. 2014;9 doi: 10.1371/journal.pone.0100744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pishchany G, Sheldon JR, Dickson CF, Alam MT, Read TD, Gell DA, Heinrichs DE, Skaar EP. J Infect Dis. 2014;209:1764–1772. doi: 10.1093/infdis/jit817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moriwaki Y, Terada T, Tsumoto K, Shimizu K. PLoS One. 2015;10 doi: 10.1371/journal.pone.0145125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sjodt M, Macdonald R, Spirig T, Chan AH, Dickson CF, Fabian M, Olson JS, Gell DA, Clubb RT. J Mol Biol. 2016;428:1107–1129. doi: 10.1016/j.jmb.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tiedemann MT, Heinrichs DE, Stillman MJ. J Am Chem Soc. 2012;134:16578–16585. doi: 10.1021/ja305115y. [DOI] [PubMed] [Google Scholar]
- 23.Honsa ES, Maresso AW. Biometals. 2011;24:533–545. doi: 10.1007/s10534-011-9413-x. [DOI] [PubMed] [Google Scholar]
- 24.Honsa ES, Fabian M, Cardenas AM, Olson JS, Maresso AW. J Biol Chem. 2011;286:33652–33660. doi: 10.1074/jbc.M111.241687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zapotoczna M, Heilbronner S, Speziale P, Foster TJ. J Bacteriol. 2012;194:6453–6467. doi: 10.1128/JB.01195-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao Q, Jiang X, Moore KJ, Shao Y, Pi H, Dubail I, Charbit A, Newton SM, Klebba PE. Mol Microbiol. 2011;80:1581–1597. doi: 10.1111/j.1365-2958.2011.07667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malmirchegini GR, Sjodt M, Shnitkind S, Sawaya MR, Rosinski J, Newton SM, Klebba PE, Clubb RT. J Biol Chem. 2014;289:34886–34899. doi: 10.1074/jbc.M114.583013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balderas MA, Nobles CL, Honsa ES, Alicki ER, Maresso AW. J Bacteriol. 2012;194:5513–5521. doi: 10.1128/JB.00685-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarlovsky Y, Fabian M, Solomaha E, Honsa E, Olson JS, Maresso AW. J Bacteriol. 2010;192:3503–3511. doi: 10.1128/JB.00054-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daou N, Buisson C, Gohar M, Vidic J, Bierne H, Kallassy M, Lereclus D, Nielsen-LeRoux C. PLoS Pathog. 2009;5 doi: 10.1371/journal.ppat.1000675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Segond D, Khalil EA, Buisson C, Daou N, Kallassy M, Lereclus D, Arosio P, Bou-Abdallah F, Le Roux CN. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1003935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lei BF, Smoot LM, Menning HM, Voyich JM, Kala SV, Deleo FR, Reid SD, Musser JM. Infect Immun. 2002;70:4494–4500. doi: 10.1128/IAI.70.8.4494-4500.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ouattara M, Cunha EB, Li X, Huang YS, Dixon DW, Eichenbaum Z. Mol Microbiol. 2010;78:739–756. doi: 10.1111/j.1365-2958.2010.07367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ouattara M, Pennati A, Devlin DJ, Huang YS, Gadda G, Eichenbaum Z. Arch Biochem Biophys. 2013;538:71–79. doi: 10.1016/j.abb.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 35.Aranda R, Worley CE, Liu M, Bitto E, Cates MS, Olson JS, Lei BF, Phillips GN. J Mol Biol. 2007;374:374–383. doi: 10.1016/j.jmb.2007.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Allen CE, Schmitt MP. J Bacteriol. 2009;191:2638–2648. doi: 10.1128/JB.01784-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen CE, Schmitt MP. J Bacteriol. 2011;193:5374–5385. doi: 10.1128/JB.05508-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen CE, Burgos JM, Schmitt MP. J Bacteriol. 2013;195:2852–2863. doi: 10.1128/JB.00244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Draganova EB, Akbas N, Adrian SA, Lukat-Rodgers GS, Collins DP, Dawson JH, Schmitt MP, Rodgers KR, Dixon DW. Biochemistry. 2015;54:6598–6609. doi: 10.1021/acs.biochem.5b00666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teale FW. Biochim Biophys Acta. 1959;35:543. doi: 10.1016/0006-3002(59)90407-x. [DOI] [PubMed] [Google Scholar]
- 41.Collier GS, Pratt JM, De Wet CR, Tshabalala CF. Biochem J. 1979;179:281–289. doi: 10.1042/bj1790281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. In: The Proteomics Protocols Handbook. Walker JM, editor. Humana Press; Totowa, N.J: 2005. pp. 571–607. [Google Scholar]
- 43.Pace CN, Scholtz JM. In: Protein Structure: A Practical Approach, The Practical Approach Series. Creighton T, editor. Oxford University Press; Oxford: 1997. pp. 299–321. [Google Scholar]
- 44.Swint L, Robertson AD. Protein Sci. 1993;2:2037–2049. doi: 10.1002/pro.5560021205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arnesano F, Banci L, Bertini I, Koulougliotis D. Biochemistry. 1998;37:17082–17092. doi: 10.1021/bi981546p. [DOI] [PubMed] [Google Scholar]
- 46.Wittung-Stafshede P. Biochim Biophys Acta. 1999;1432:401–405. doi: 10.1016/s0167-4838(99)00116-8. [DOI] [PubMed] [Google Scholar]
- 47.Andersen NH, Nørgaard A, Jensen TJ, Ulstrup J. J Inorg Biochem. 2002;88:316–327. doi: 10.1016/s0162-0134(01)00358-0. [DOI] [PubMed] [Google Scholar]
- 48.Roncone R, Monzani E, Labo S, Sanangelantoni AM, Casella L. J Biol Inorg Chem. 2005;10:11–24. doi: 10.1007/s00775-004-0606-4. [DOI] [PubMed] [Google Scholar]
- 49.Du J, Sono M, Dawson JH. Coord Chem Rev. 2011;255:700–716. doi: 10.1016/j.ccr.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tiedemann MT, Stillman MJ. J Porph Phthalo. 2011;15:1134–1149. [Google Scholar]
- 51.Sharma KD, Andersson LA, Loehr TM, Terner J, Goff HM. J Biol Chem. 1989;264:12772–12779. [PubMed] [Google Scholar]
- 52.Jin YY, Nagai M, Nagai Y, Nagatomo S, Kitagawa T. Biochemistry. 2004;43:8517–8527. doi: 10.1021/bi036170g. [DOI] [PubMed] [Google Scholar]
- 53.Eakanunkul S, Lukat-Rodgers GS, Sumithran S, Ghosh A, Rodgers KR, Dawson JH, Wilks A. Biochemistry. 2005;44:13179–13191. doi: 10.1021/bi050422r. [DOI] [PubMed] [Google Scholar]
- 54.Lukat-Rodgers GS, Rodgers KR, Caillet-Saguy C, Izadi-Pruneyre N, Lecroisey A. Biochemistry. 2008;47:2087–2098. doi: 10.1021/bi7019518. [DOI] [PubMed] [Google Scholar]
- 55.Linder DP, Silvernail NJ, Barabanschikov A, Zhao JY, Alp EE, Sturhahn W, Sage JT, Scheidt WR, Rodgers KR. J Am Chem Soc. 2014;136:9818–9821. doi: 10.1021/ja503191z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pluym M, Vermeiren CL, Mack J, Heinrichs DE, Stillman MJ. J Porph Phthalo. 2007;11:165–171. [Google Scholar]
- 57.Sudhamsu J, Kabir M, Airola MV, Patel BA, Yeh SR, Rousseau DL, Crane BR. Protein Expr Purif. 2010;73:78–82. doi: 10.1016/j.pep.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ricchelli F, Jori G, Gobbo S, Tronchin M. Biochim Biophys Acta. 1991;1065:42–48. doi: 10.1016/0005-2736(91)90008-v. [DOI] [PubMed] [Google Scholar]
- 59.Ho WW, Li HY, Eakanunkul S, Tong Y, Wilks A, Guo ML, Poulos TL. J Biol Chem. 2007;282:35796–35802. doi: 10.1074/jbc.M706761200. [DOI] [PubMed] [Google Scholar]
- 60.Jepkorir G, Rodriguez JC, Rui H, Im W, Lovell S, Battaile KP, Alontaga AY, Yukl ET, Moenne-Loccoz P, Rivera M. J Am Chem Soc. 2010;132:9857–9872. doi: 10.1021/ja103498z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deniau C, Gilli R, Izadi-Pruneyre N, Létoffé S, Delepierre M, Wandersman C, Briand C, Lecroisey A. Biochemistry. 2003;42:10627–10633. doi: 10.1021/bi030015k. [DOI] [PubMed] [Google Scholar]
- 62.Kumar R, Matsumura H, Lovell S, Yao HL, Rodriguez JC, Battaile KP, Moenne-Loccoz P, Rivera M. Biochemistry. 2014;53:2112–2125. doi: 10.1021/bi500030p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Asher C, de Villiers KA, Egan TJ. Inorg Chem. 2009;48:7994–8003. doi: 10.1021/ic900647y. [DOI] [PubMed] [Google Scholar]
- 64.Kuter D, Venter GA, Naidoo KJ, Egan TJ. Inorg Chem. 2012;51:10233–10250. doi: 10.1021/ic301154e. [DOI] [PubMed] [Google Scholar]
- 65.Mattle D, Zeltina A, Woo JS, Goetz BA, Locher KP. J Mol Biol. 2010;404:220–231. doi: 10.1016/j.jmb.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 66.Graves AB, Morse RP, Chao A, Iniguez A, Goulding CW, Liptak MD. Inorg Chem. 2014;53:5931–5940. doi: 10.1021/ic500033b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chan ACK, Lelj-Garolla B, Rosell FI, Pedersen KA, Mauk AG, Murphy MEP. J Mol Biol. 2006;362:1108–1119. doi: 10.1016/j.jmb.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 68.Watanabe M, Tanaka Y, Suenaga A, Kuroda M, Yao M, Watanabe N, Arisaka F, Ohta T, Tanaka I, Tsumoto K. J Biol Chem. 2008;283:28649–28659. doi: 10.1074/jbc.M803383200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Edelstein SJ, Rehmar MJ, Olson JS, Gibson QH. J Biol Chem. 1970;245:4372–4381. [PubMed] [Google Scholar]
- 70.Hargrove MS, Whitaker T, Olson JS, Vali RJ, Mathews AJ. J Biol Chem. 1997;272:17385–17389. doi: 10.1074/jbc.272.28.17385. [DOI] [PubMed] [Google Scholar]
- 71.Sook BR, Block DR, Sumithran S, Montañez GE, Rodgers KR, Dawson JH, Eichenbaum Z, Dixon DW. Biochemistry. 2008;47:2678–2688. doi: 10.1021/bi701604y. [DOI] [PubMed] [Google Scholar]
- 72.Ran YC, Malmirchegini GR, Clubb RT, Lei BF. Biochemistry. 2013;52:6537–6547. doi: 10.1021/bi400965u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amisha Kamal JKA, Behere DV. Biochem Eng J. 2008;38:110–114. [Google Scholar]
- 74.Bernardes A, Textor LC, Santos JC, Cuadrado NH, Kostetsky EY, Roig MG, Bavro VN, Muniz JRC, Shnyrov VL, Polikarpov I. Biochimie. 2015;111:58–69. doi: 10.1016/j.biochi.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 75.Vieille C, Zeikus GJ. Microbiol Mol Biol Rev. 2001;65:1–43. doi: 10.1128/MMBR.65.1.1-43.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sanchez-Ruiz JM. Biophys Chem. 2010;148:1–15. doi: 10.1016/j.bpc.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 77.Wijma HJ, Floor RJ, Janssen DB. Curr Opin Struct Biol. 2013;23:588–594. doi: 10.1016/j.sbi.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 78.Liu MY, Lei BF. Infect Immun. 2005;73:5086–5092. doi: 10.1128/IAI.73.8.5086-5092.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nygaard TK, Blouin GC, Liu MY, Fukumura M, Olson JS, Fabian M, Dooley DM, Lei BF. J Biol Chem. 2006;281:20761–20771. doi: 10.1074/jbc.M601832200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Muryoi N, Tiedemann MT, Pluym M, Cheung J, Heinrichs DE, Stillman MJ. J Biol Chem. 2008;283:28125–28136. doi: 10.1074/jbc.M802171200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Villareal VA, Spirig T, Robson SA, Liu M, Lei B, Clubb RT. J Am Chem Soc. 2011;133:14176–14179. doi: 10.1021/ja203805b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abe R, Caaveiro JMM, Kozuka-Hata H, Oyama M, Tsumoto K. J Biol Chem. 2012;287:16477–16487. doi: 10.1074/jbc.M112.346700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fabian M, Solomaha E, Olson JS, Maresso AW. J Biol Chem. 2009;284:32138–32146. doi: 10.1074/jbc.M109.040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hunter CL, Mauk AG, Douglas DJ. Biochemistry. 1997;36:1018–1025. doi: 10.1021/bi961993+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.