

Abstract
Pathogens deliver complex arsenals of translocated effector proteins to host cells during infection, but the extent to which these proteins are regulated once inside the eukaryotic cell remains poorly defined. Among all bacterial pathogens, Legionella pneumophila maintains the largest known set of translocated substrates, delivering over 300 proteins to the host cell via its Type IVB, Icm/Dot translocation system. Backed by a few notable examples of effector–effector regulation in L. pneumophila, we sought to define the extent of this phenomenon through a systematic analysis of effector–effector functional interaction. We used Saccharomyces cerevisiae, an established proxy for the eukaryotic host, to query > 108,000 pairwise genetic interactions between two compatible expression libraries of ~330 L. pneumophila‐translocated substrates. While capturing all known examples of effector–effector suppression, we identify fourteen novel translocated substrates that suppress the activity of other bacterial effectors and one pair with synergistic activities. In at least nine instances, this regulation is direct—a hallmark of an emerging class of proteins called metaeffectors, or “effectors of effectors”. Through detailed structural and functional analysis, we show that metaeffector activity derives from a diverse range of mechanisms, shapes evolution, and can be used to reveal important aspects of each cognate effector's function. Metaeffectors, along with other, indirect, forms of effector–effector modulation, may be a common feature of many intracellular pathogens—with unrealized potential to inform our understanding of how pathogens regulate their interactions with the host cell.
Keywords: effector, genetic interaction, Legionella, metaeffector, structure‐function
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Genetics, Gene Therapy & Genetic Disease; Microbiology, Virology & Host Pathogen Interaction
Introduction
The concept of effector‐based modulation of host pathways is central to the current molecular understanding of microbial pathogenesis. In this view, effector activity is directed against host factors and is regulated by changes to effector expression or through modulation of translocation efficiency. There are, however, several ways that translocated proteins might functionally interact once inside the host (Shames & Finlay, 2012). The regulatory complexity provided by effector–effector interactions may add another layer to the program of pathogenesis, helping balance host perturbation with the maintenance of a replicative niche. Pathogens that strongly rely on their hosts for replication and translocate large numbers of proteins are candidates for possessing such effector complexity (Ensminger, 2016). The intracellular bacterial pathogen L. pneumophila uses over 300 Icm/Dot‐translocated substrate [IDTS; Table EV1, for review, see Ensminger (2016); Franco et al (2009); Isaac and Isberg (2014)] to grow within protozoan and mammalian hosts (Rowbotham, 1980; Fields, 1996; Molmeret et al, 2005). A few notable examples of effector–effector functional interaction have been documented in L. pneumophila, in which effectors directly or indirectly regulate the activity of each other inside the eukaryotic cell (Kubori et al, 2010; Neunuebel et al, 2011; Tan & Luo, 2011; Tan et al, 2011; Havey & Roy, 2015; Jeong et al, 2015).
Effector–effector interactions have largely been identified through individual studies of effector function; nevertheless, several early examples provide useful insight into what we might expect to find within a more complete set of such interactions. These interactions could take the form of direct effector–effector suppression, such as the case with the L. pneumophila IDTS LubX. LubX is an E3 ubiquitin ligase that targets another translocated substrate (SidH) for proteasomal degradation during the late stages of intracellular replication and is the founding member of a group of proteins known as “metaeffectors”—so named because it is an “effector of effectors” (Kubori et al, 2010). In addition to this rare example of direct effector–effector interaction, other translocated substrates are known to indirectly antagonize one another by targeting the same host proteins or pathways with counteracting activities. For instance, the L. pneumophila IDTS AnkX adds a phosphocholine group to a host protein, Rab1 (Mukherjee et al, 2011), while another IDTS, Lem3, removes it as part of a regulatory cascade (Tan et al, 2011). Similarly, SidM adds an AMP moiety to Rab1 (Muller et al, 2010) that is removed by the antagonizing IDTS SidD (Neunuebel et al, 2011; Tan & Luo, 2011). SidJ was also recently shown to be a functional antagonist for the SidE family of effectors (Havey & Roy, 2015; Jeong et al, 2015), which have a unique non‐canonical ubiquitination activity through their mono‐ADP‐ribosyltransferase motif that does not require components of the E1, E2, and E3 ubiquitin enzyme cascade (Qiu et al, 2016). SidJ releases SidE and its paralogs from the Legionella‐containing vacuole membrane, though the exact mechanism of the SidJ‐dependent release remains unknown (Jeong et al, 2015). As this phenomenon remained to be explored systematically, we screened over 108,000 pairwise effector–effector genetic interactions between two libraries of ~330 effectors co‐expressed in Saccharomyces cerevisiae—an established high‐throughput proxy for the eukaryotic cell.
Results
Widespread functional antagonism between IDTS
To systematically map IDTS functional interactions, we sought a high‐throughput, genetically tractable proxy for the diverse eukaryotic hosts within which the bacteria normally replicate. Because IDTS largely targets highly conserved eukaryotic pathways (a consequence of their dependence on diverse protozoa in the environment), the expression of several individual IDTS within the yeast S. cerevisiae is known to cause cell growth defects (Campodonico et al, 2005; Shohdy et al, 2005; de Felipe et al, 2008; Heidtman et al, 2009; Xu et al, 2010; Belyi et al, 2012; Guo et al, 2014; Nevo et al, 2014). Over 200 IDTS remained uncharacterized in yeast, so we generated a library of 330 yeast strains in which each strain expresses one IDTS (or putative IDTS) on a high‐copy HIS3 plasmid under control of a galactose‐inducible promoter. We measured their growth by high‐density spot size and liquid growth curves (Appendix Fig S1A, Table EV1). In these assays, 227 proteins conferred a moderate to severe growth defect in yeast when expressed, presenting a clear opportunity to identify effector–effector interaction by screening for suppression of these growth defects.
In order to obtain an IDTS‐wide genetic interaction map, we modified the automated, high‐density replica plating approaches previously developed for analyzing S. cerevisiae double mutants through synthetic genetic array (SGA) analysis (Tong et al, 2001). Instead of looking at double mutants, however, we used yeast genetics to systematically assess the effects of effector co‐expression on yeast growth (Fig 1A, see also Materials and Methods). Critically, a previously characterized yeast library (Heidtman et al, 2009 and Appendix Fig S1B) had properties (galactose‐inducible, URA+, MATα) that made it inherently compatible with the HIS+, MATa library described above (Appendix Fig S1A, Table EV1). After extending the Heidtman collection by over 200 clones (Table EV7), we used a pinning robot to mate each strain from this collection to an ordered array of our HIS3 plasmid effector library (laid out in quadruplicate in a 1,536‐spot pinning density). We selected for diploids on media lacking uracil and histidine and then replica plated them onto selective medium containing galactose to induce expression from both plasmids. After 2 days of incubation, we used automated image analysis to quantify the spot size of strains expressing each of the > 108,000 possible pairwise combinations between these libraries (Fig 1A).
Figure 1. Effector–effector functional interactions.
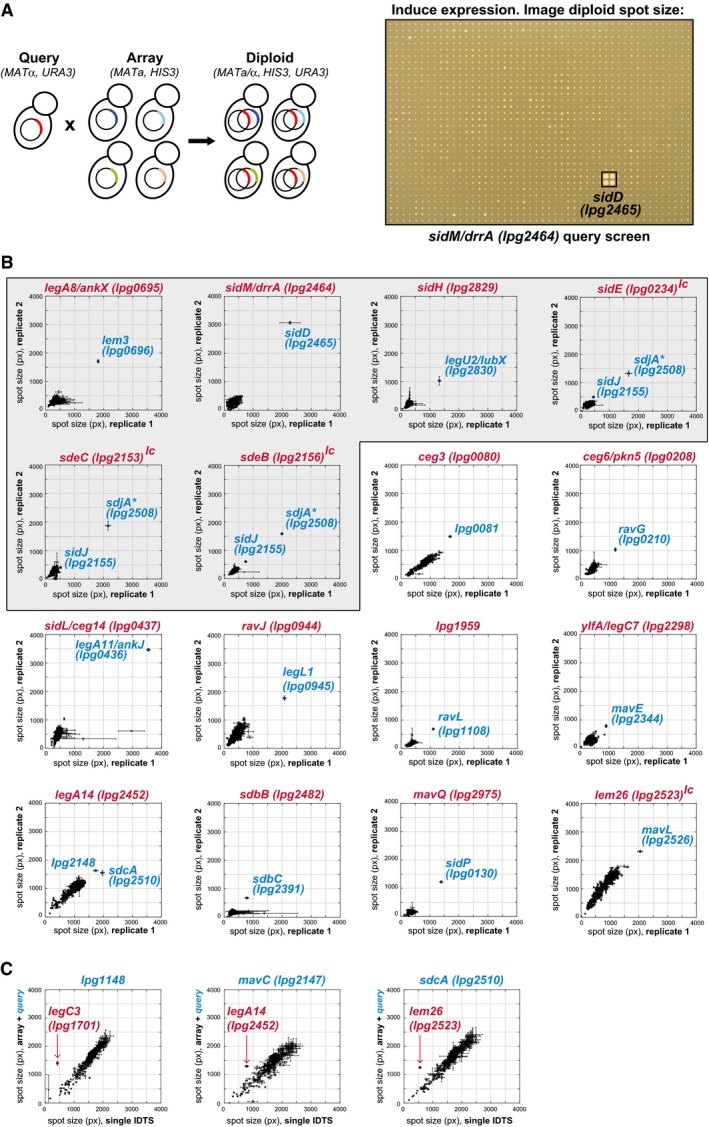
- High‐throughput effector–effector suppression profiling in yeast. Strains carrying an individual IDTS expressed from an inducible plasmid are mated into a 384‐array of strains carrying a compatible library of 330 IDTSs. The diploid array is pinned onto galactose medium to induce expression and spot size measured through image analysis. A 1,536‐spot array (SidM query) after 2 days of growth is shown.
- Suppression of growth defects through inter‐effector antagonism. Screening the > 108,000 pairwise IDTS interactions possible between the libraries reveals sixteen suppression profiles. Spot sizes of strains co‐expressing the IDTS query clone (in red) and each of the 330 effectors within the compatible arrayed IDTS library were quantified for two independent biological replicate screens and plotted on separate axes. Error bars represent standard deviation (SD) of spot size between quadruplicate spots measured within each biological replicate (see Materials and Methods). Bona fide suppressors show consistent divergence from the population following a diagonal line, while spurious yeast suppressor mutations that sometimes appear for toxic queries (e.g., SidL/Ceg14 and SdbB) occur in one replicate with a large SD. The shaded box contains all previously known suppression pairs, plus the novel antagonist, SdjA (*) of SidE, SdeB, and SdeC. “lc” = screened using a low‐copy query due to an inability to clone the IDTS in high‐copy vectors.
- Suppression of array strain growth defects by specific queries reveals additional instances of effector inactivation. Array strains were mated to create either query‐IDTS‐containing diploids (y‐axis) or empty vector‐IDTS diploids (x‐axis). In this representation, suppression of a growth inhibitory array strain is revealed as a deviation above the diagonal. Error bars represent the SD of spot size between quadruplicate spots within each dataset. (Note that the larger spread of diploid spot sizes in panel C reflects that, unlike in panel B, these queries are not toxic on their own.)
Using this approach, we identified all previously described instances of effector–effector antagonism (Fig 1B, shaded), as well as several IDTS whose growth inhibition in yeast was suppressed by one or more previously unknown antagonists. Due to technical reasons, growth suppression was not always observed in both directions (when the identity of an array and query strain were reversed). While infrequent, such instances are likely due to the potential of epitope tags and spontaneous mutations within the yeast genome to mask some interactions. As such, we also looked for additional suppressors in which expression of a query gene was able to suppress growth inhibition caused by one of the IDTS on the array (Fig 1C, Appendix Fig S2). In total, eighteen IDTS suppress the deleterious effects of one or more of the other effectors within the eukaryotic cell. Notably, all of the effector–effector interactions that we observe occur between experimentally validated translocated substrates (Table EV1). While each of the previously characterized effector–effector functional interactions involved effectors chromosomally adjacent to one another (or the broad‐acting paralogs of SidJ and SidE), several of our newly identified pairs are genomically unlinked.
Identification of inter‐substrate physical interaction
Effector–effector suppression could reflect counteracting activities on a shared host target (or pathway) or it could reflect the direct modulation of one IDTS by the activity of another. To identify instances of direct suppression, we examined all 23 effector–effector suppression pairs for physical interactions using the yeast two‐hybrid (Y2H) assay (Fields & Song, 1989; Fig 2A) and used an established high‐throughput mammalian system, LUMIER (Barrios‐Rodiles et al, 2005; Taipale et al, 2012) to measure all possible physical interactions between the cohort of functional antagonists and their cognate IDTS (Fig 2B). A previous study comparing Y2H, LUMIER, and other assays showed that each technology captures only a subset of true complexes (Braun et al, 2009), justifying the application of multiple approaches. In total, we identified nine novel effector–effector complexes (Table EV2), not including the interaction between LubX and SidH that was previously identified using purified proteins (Kubori et al, 2010) and by affinity purification (Quaile et al, 2015). This is not surprising, as LubX targets SidH for degradation—and raises the likely possibility that even more effector–effector complexes may be identified through additional methods.
Figure 2. Effector–effector physical interactions in yeast and mammalian cells.
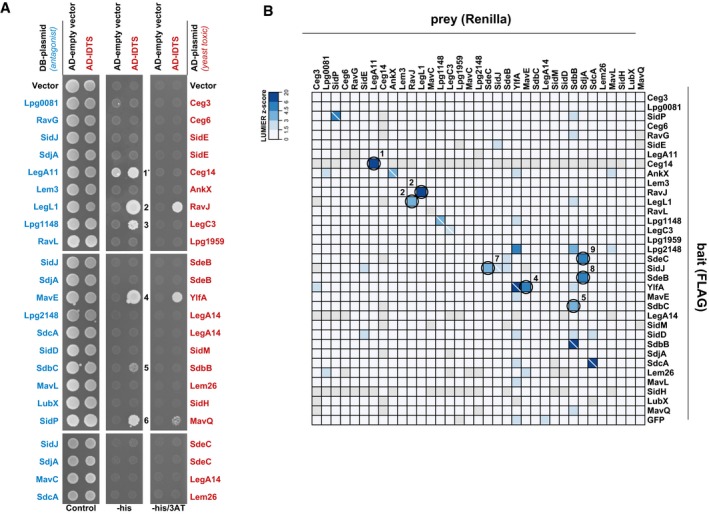
- Direct physical interactions between effectors as revealed by yeast two‐hybrid (Y2H) assay. For each of the 23 effector–effector suppression pairs, the rescuer was fused to the Gal4 DNA‐binding domain (DB) and its cognate toxic IDTS was fused to the Gal4‐activating domain (AD). DB‐IDTS with an AD‐empty vector control or AD‐IDTS were tested for growth on control medium or medium lacking histidine −/+ 3AT. Physical interactions drive HIS3 expression and confer growth under −his (low stringency) or −his +3AT (high stringency) conditions. Autoactivation of DB‐IDTS (the ability to grow on selective conditions without a DB‐AD complex) was observed for only one IDTS, LegA11.
- The LUMIER assay was used to detect physical interactions in mammalian cells. FLAG‐V5‐tagged bait proteins (y‐axis) and Renilla luciferase‐tagged prey proteins (x‐axis) were co‐transfected into HEK293T cells. Lysates were added to anti‐FLAG‐treated 384‐well plates. After washing, co‐precipitation is detected as a luminescent signal. Shown are luminescence Z‐scores calculated over all Renilla luciferase‐tagged preys. FLAG‐V5‐bait proteins that were not expressed were filtered from the dataset and are shown in gray. A white backslash indicates a homo‐dimer interaction. Circles indicate functionally antagonistic pairs with a Z‐score > 3. Numbers indicate the 9 novel effector–effector complexes that were identified using the Y2H and LUMIER methods (Table EV2).
Though SidJ was classified as a metaeffector of SidE and its paralogs due to its impact on their localization during infection (Jeong et al, 2015), previous studies were unable to show a direct interaction between SidJ and SdeA (Havey & Roy, 2015; Jeong et al, 2015). Notably, we detect a SdeC and SidJ complex in mammalian cells, suggesting that it is possible that SidJ regulates at least one SidE family representative through a direct physical interaction. Interestingly, while SidJ suppresses the activity of SidE, SdeB, and SdeC in our assays, its paralog SdjA (Liu & Luo, 2007) is a much more potent regulator of these SidE‐family members. We detect complexes of SdjA with SdeB and SdeC by the LUMIER assay, suggesting further attention should be placed upon the functional overlap and potential diversification of each of the SidJ and SidE paralogs.
A multifunctional metaeffector SidP links its cognate effector, MavQ, to PIP modulation
We were surprised that one of the putative metaeffectors identified in our screen, SidP, has an established role as a phosphatidylinositol 3‐phosphate (PI3P) phosphatase (Toulabi et al, 2013). In addition to this canonical role against the host, SidP also inactivates an effector of unknown function, MavQ (Fig 1B) and does so via a physical interaction—as shown by both Y2H (Fig 2A) and AP‐MS (Table EV3).
SidP has three distinct domains: an N‐terminal phosphatase domain along with insertion (I) and C‐terminal domains (Toulabi et al, 2013) each of thus far unknown functions (Fig 3A). Catalytically inactive SidP mutants retained the ability to suppress MavQ, indicating that the mechanism of SidP metaeffector activity is independent of its role as a PI3P phosphatase against the host. In contrast, the previously uncharacterized C‐terminal domain of SidP was both necessary and sufficient for MavQ inactivation and binding (Fig 3B and Appendix Fig S3B).
Figure 3. A link between the uncharacterized IDTS MavQ, SidP, and PIP modulation.
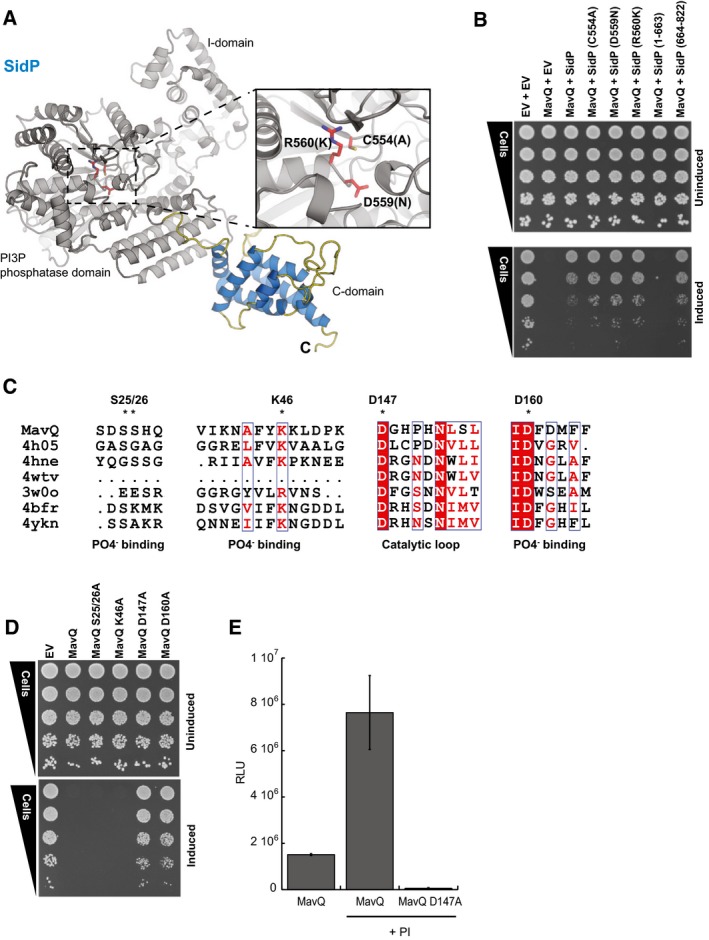
- The C‐terminal domain of SidP is required and sufficient for MavQ inactivation. SidP phosphatase mutants (C554A, D559N, and R560K, see inset A) and SidP fragments (1–663 and 664–822) were tested for the ability to inactivate MavQ in a yeast spot dilution assay on glucose (uninduced, upper panel) or galactose (induced, lower panel).
- HHpred (Soding, 2005) suggests that the N‐terminal part of MavQ may share homology with several PI3 and PI4 kinases and aminoglycoside phosphotransferases (see Materials and Methods for more information). The phosphate binding and catalytic loop regions are shown with identical residues (red) and similar residues (white box) highlighted. The putative ATP binding site and catalytic aspartate residues are indicated (*).
- MavQ residues S25, S26, K46, D147, and D160, which correspond to PI4P kinase residues involved in ATP binding and catalysis, were mutated and tested in a yeast spot dilution. The D147A and D160A mutants abrogate yeast growth inhibition by MavQ. Each mutant was tested for expression and stability (Appendix Fig S3C).
- In vitro ATP‐to‐ADP conversion by MavQ or MavQ D147A was measured in the presence of PI micelles using the ADP‐Glo kinase assay (Zhou et al, 2014). Basal ATP‐to‐ADP conversion increases several fold in the presence of PI micelles. Mutation of D147 in MavQ ablates this activity as the mutant activity is significantly different from wild type as assessed by an unpaired, two‐tailed Student's t‐test (P‐value = 0.02, n = 2). Error bars indicate the SD.
Using a guilt‐by‐association approach, we asked whether MavQ also plays a role in the regulation of PI metabolism during infection. If so, SidP's metaeffector activity against MavQ could serve to coordinate the modulation of lipid metabolism during infection. HHpred analysis (Soding, 2005) of MavQ reveals a weak similarity to PIP kinases, suggesting several of its N‐terminal residues might be involved in ATP binding and catalysis (Fig 3C). To test this, we individually mutated MavQ residues corresponding to the active site residues in PIP kinases. D147A and D160A substitutions completely abrogate yeast growth inhibition (Fig 3D), consistent with their predicted role in the active site. Using an in vitro activity assay, we observe that wild‐type MavQ, but not the D147A mutant, possesses robust ATP hydrolysis activity (Fig 3E). Importantly, the addition of phosphoinositide to this reaction increased the activity of the wild‐type MavQ fivefold as would be expected of a PIP kinase. Furthermore, like SidP, we also observe robust MavQ binding to a broad range of PIP molecules using the protein–lipid overlay assay (Appendix Fig S3D). Taken together, these data support a model in which MavQ is a kinase with a role in PIP modulation. In this context, the inactivation of MavQ by the C‐terminal domain of SidP likely provides a molecular mechanism by which to directly coordinate the activities of these two proteins against host PIPs.
The metaeffector LegL1 functions by blocking the active site of its cognate effector, RavJ
Our discovery of several novel direct IDTS antagonists in the L. pneumophila genome also provided an opportunity to start to define the full scope of mechanisms that a bacterial pathogen can use to regulate its own effectors after release into the host cell. To that end, we performed detailed structure‐function analysis of a completely uncharacterized set of proteins: the putative metaeffector LegL1, a leucine‐rich repeat (LRR) protein (de Felipe et al, 2005), and its cognate effector, RavJ.
To define the mechanism of LegL1 inhibition of RavJ, we first solved the crystal structure of the N‐ and C‐terminal portions of RavJ spanning amino acid residues 1–228 and 251–371, respectively (Fig 4A, Table EV4). Despite no identifiable sequence similarity, the overall fold of the N‐terminal domain of RavJ resembled that of papain‐like cysteine protease family members (Fig 4A). Comparison of RavJ with its top structural homolog, human tissue transglutaminase (TGM2), identified three conserved residues (C101, H138, D170) along with another adjacent residue (W172) as a putative catalytic site (Appendix Fig S4A and B). The C‐terminal domain of RavJ did not reveal any structural similarity to known proteins or functional domains, but we identified a short conserved motif (H316‐WNRHH‐V322; Fig 4A) that is present in another IDTS, WipB, as well as several non‐Legionella proteins. To determine which of these structural elements are essential for effector function, we tested the effect of their ablation on yeast growth. Strikingly, single substitutions in each of the four N‐terminal catalytic site residues, as well as the C‐terminal loop mutant, abrogated the yeast growth inhibition of RavJ (Fig 4B).
Figure 4. RavJ modularity and inhibition by LegL1.
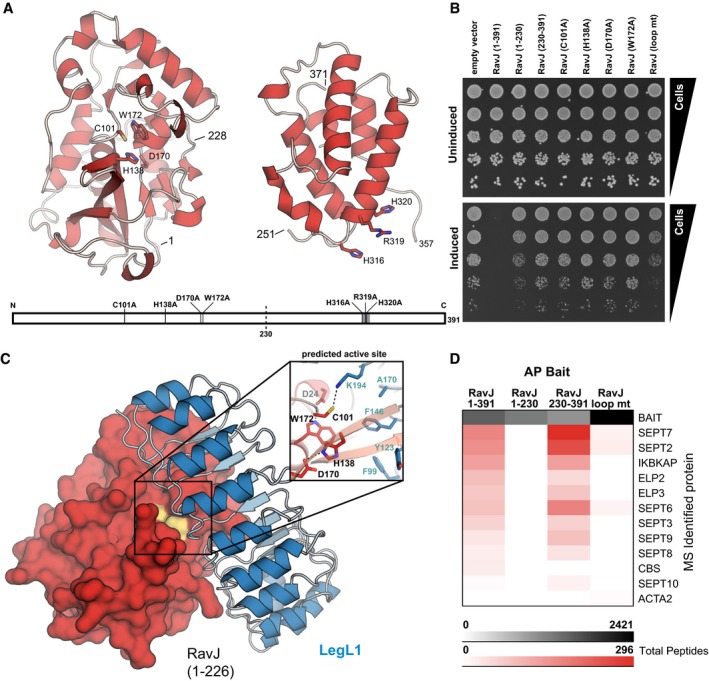
- Structures for amino acid residues 1–228 and 251–371 of RavJ were solved separately to a resolution of 1.3 and 1.9 Å, respectively. The N‐terminal structure contains a structurally conserved putative catalytic triad (C101/H138/D170). A conserved motif (highlighted in gray) is located in the C‐terminal domain with surface exposed residues H316, R319, and H320.
- While expression of full‐length RavJ showed a severe growth defect in a spot dilution assay, mutation of each of the putative catalytic residues or the adjacent W172 to alanine fully relieved toxicity as did substitutions in the conserved C‐terminal motif. Each mutant was tested for expression and stability (Appendix Fig S4C).
- LegL1 acts as a direct antagonist by blocking the active site of RavJ. The co‐crystal complex of LegL1 (blue) bound to RavJ (1–226, red) was solved to a resolution of 2.0 Å. LegL1 forms a canonical leucine‐rich repeat structure arching over the predicted active site of RavJ. The interface spans 1,240 Å2 and is reinforced by LegL1 K194 projecting into the RavJ active site where it forms a hydrogen bond with the predicted catalytic residue C101 (inset).
- Immobilized RavJ was incubated with U937 cell lysate and interacting proteins were identified by nLC‐MS/MS. Each column in the table represents the sum of the average total peptide counts for two replicates of affinity purification‐mass spectrometry. Septins and elongator complex proteins were identified only with full‐length RavJ and the (230–391) C‐terminal domain, suggesting that the C‐terminal domain is a substrate‐binding domain. Use of the otherwise full‐length loop mutant (H316A/R319A/H320A) as bait abrogated these interactions.
After using Y2H and gel filtration analysis to demonstrate a physical interaction between LegL1 and the N‐terminal domain of RavJ (Appendix Fig S5A–C), we cocrystallized and determined the crystal structure of this novel effector–effector complex (Fig 4C, Table EV4). The heterocomplex has a 1:1 stoichiometry, with LegL1 forming a canonical LRR horseshoe‐shaped structure arching over and interacting with the RavJ active site identified above. Such intimate contact suggests that LegL1 is able to block the activity of RavJ by sterically hindering its catalytic site. This structure, to our knowledge, represents the first metaeffector–effector complex structure, revealing a new archetype of metaeffector activity: non‐proteolytic inter‐substrate inhibition.
To confirm the modularity of RavJ suggested above, we used AP‐MS analysis to identify its host targets. The C‐terminal domain of RavJ was both necessary and sufficient to interact with several components of the eukaryotic septin and elongator complexes (Fig 4D). Mutation of the C‐terminal conserved motif (H316A|R319A|H320A, “loop mt”) was sufficient to ablate the interaction with both complexes—in agreement with our observation that this mutant and deletion of the entire C‐terminal domain cause a similar alleviation of the yeast growth defect. The RavJ‐septin AP‐MS data were confirmed by co‐IP (Appendix Fig S5D). Taken together, our data reveal RavJ as a two‐domain effector, in which the C‐terminal domain is responsible for host protein recognition, while the N‐terminal domain carries catalytic activity specifically inhibited by direct binding of the metaeffector LegL1.
The deubiquitinase LupA inactivates its cognate effector LegC3 through removal of ubiquitin
We next solved the crystal structure of another novel metaeffector, the previously uncharacterized Lpg1148, to reveal a domain (residues 123–304, Fig 5A, Table EV4) typical of eukaryotic ubiquitin proteases (UBP) involved in the deconjugation of ubiquitin or ubiquitin‐like proteins from their targets (Appendix Figs S6A and B). As Lpg1148 rescues the yeast growth defect caused by LegC3 (Fig 1C), we hypothesized that this inhibition may derive from the catalytic functionality revealed by our structure.
Figure 5. Metaeffector activity through deubiquitination.
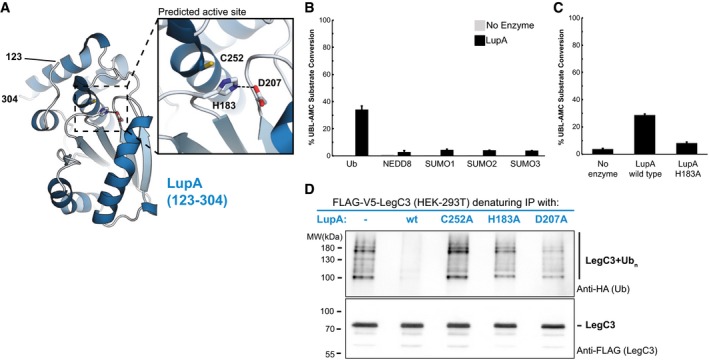
- The de novo crystal structure of the metaeffector LupA (123–304) was determined to 1.9 Å resolution and reveals that LupA belongs to the ubiquitin or ubiquitin‐like protease (UBP) family of proteins with a canonical cysteine protease triad (inset).
- LupA displays deubiquitinase activity in vitro. A fluorescence‐based assay was used to monitor the catalytic hydrolysis of ubiquitin and ubiquitin‐like proteins from a covalently linked fluorophore (AMC) after incubation with purified LupA or a no‐enzyme control. Substrates tested are displayed on the x‐axis, and % substrate conversion on the y‐axis. LupA activity is specific toward ubiquitin. The activity of LupA against ubiquitin‐AMC is significantly different than its activity against each of the other ubiquitin‐like substrates as assessed by unpaired, two‐tailed Student's t‐tests (Ub versus Nedd8: P‐value = 0.005; SUMO‐1: P‐value = 0.005; SUMO‐2: P‐value = 0.005; SUMO‐3: P‐value = 0.005; n = 2). The error bars indicate the SD.
- Mutation of a predicted catalytic residue (H183) almost completely abolishes the in vitro hydrolase activity. In the fluorescence‐based assay described above, the mutant activity is significantly reduced from the wild‐type enzyme activity as assessed by an unpaired, two‐tailed Student's t‐test (P‐value = 0.000009, n = 3). The error bars indicate the SD.
- LupA activity removes polyubiquitin linkages from its cognate IDTS, LegC3. Denaturing IPs of FLAG‐V5‐LegC3 expressed in HEK293T cells were analyzed by western blot and probed for ubiquitination during co‐expression of V5‐LupA or one of three catalytically impaired variants (see Appendix Fig S6C for input). While polyubiquitination of LegC3 was present when co‐expressed with a catalytically inactive LupA (or in the absence of LupA), no ubiquitination could be detected in the presence of wild‐type LupA, confirming its deubiquitination of LegC3 in vivo.
In vitro activity assays confirmed that Lpg1148 is a ubiquitin‐specific protease (Fig 5B) whose activity is abrogated by a mutation within the predicted catalytic triad (Fig 5C). We therefore hypothesized that Lpg1148 (hereafter referred to as Legionella ubiquitin‐specific protease A—LupA) removes a ubiquitin modification from LegC3 that otherwise supports its activity in a proteasomal‐independent manner. As a test of this hypothesis, we co‐transfected human HEK293T cells with LegC3 and either wild‐type or catalytically impaired variants of LupA. In accordance with our model for LupA metaeffector activity, we detected ubiquitinated species of LegC3 in the presence of catalytically inactive LupA variants but not in the presence of the wild‐type deubiquitinase (Fig 5D). These data reveal another novel mechanism of direct effector inactivation—the specific deubiquitination of a cognate effector—and suggest that LegC3 activity may depend upon modification by an endogenous (human) E3 ligase.
Evolutionary implications of direct effector–effector suppression
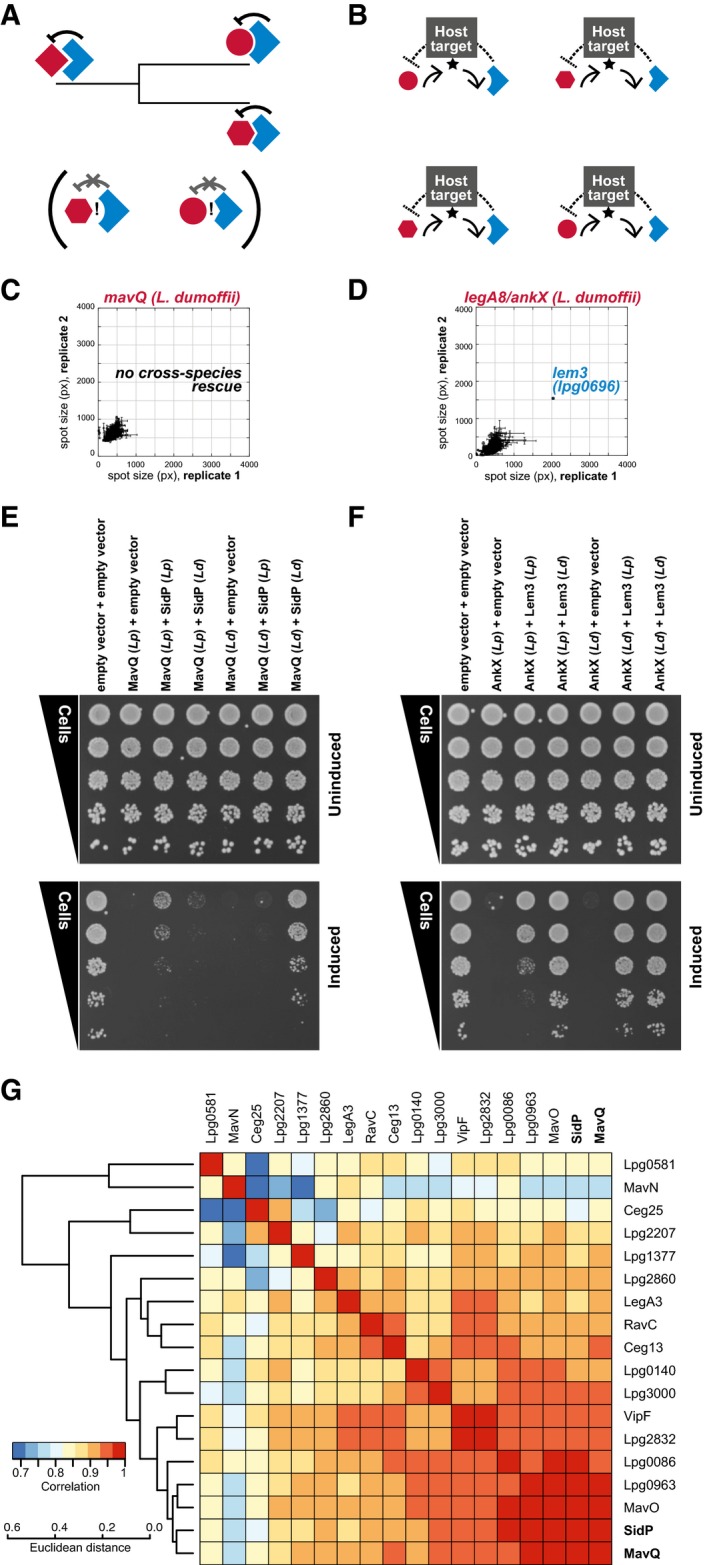
We next tested whether direct effector antagonists might be distinguished within pathogen arsenals based on the capacity for co‐evolution of the effector–effector interface. We reasoned that while direct antagonists and their cognate effectors are likely to co‐evolve during Legionella speciation (Fig 6A), effectors that antagonize one another indirectly through a common host target are under evolutionary pressure to maintain their individual activities against the conserved protozoan orthologs of that target (Fig 6B). One prediction of this model is that indirect functional antagonists from distinct Legionella species will maintain their ability to counteract one another's activity, whereas a direct antagonist from one species is likely to only suppress the activity of its cognate effector within that species.
Figure 6. The evolutionary constraints of inter‐substrate antagonism.
-
AFunctional antagonism by direct physical interactions between an antagonist (blue) and its bacterial target (red) provides an opportunity for co‐evolution of each direct antagonist and its cognate translocated substrate during bacterial speciation. A prediction of this model is that (i) direct antagonist activity is likely to be maintained within species, but (ii) co‐evolution is likely to modify the protein–protein interface such that cross‐species rescue between a direct antagonist and the ortholog of its cognate substrate is unlikely.
-
BIn contrast, indirect functional antagonism that occurs through counteracting activities on a shared host target is likely to be maintained cross‐species.
-
CConsistent with this model, direct antagonist activity is not maintained between orthologs derived from distinct Legionella species as revealed by screening the L. pneumophila library using an ortholog of SidP from L. dumoffii. Error bars represent the SD of spot size between quadruplicate spots measured within each biological replicate.
-
DIndirect antagonism is maintained, as revealed by the rescue of an L. dumoffii AnkX query with L. pneumophila Lem3 on the array. Error bars represent the SD of spot size between quadruplicate spots measured within each biological replicate.
-
E, FIntra‐species rescue is maintained in L. dumoffii for both types of antagonists. The antagonist pairs MavQ‐SidP (E) and AnkX‐Lem3 (F) from L. pneumophila (Lp) and L. dumoffii (Ld) were tested for intra‐ and inter‐species rescue by co‐expressing orthologs from the same species (Ld+Ld or Lp+Lp) and orthologs from different species (Lp+Ld or Ld+Lp) using a spot dilution assay on glucose (uninduced, upper panel) and galactose (induced, lower panel). Inter‐species rescue was only observed for AnkX and Lem3, consistent with the results in (A). Both pairs showed intra‐species rescue.
-
GGenomic analysis uncovers signatures of MavQ‐SidP co‐evolution. We examined the phylogenies of MavQ and SidP across 35 species, along with all the other proteins in our library with one and only one ortholog in the same set. (For technical issues shaping species choice, please see Materials and Methods). MirrorTree analysis was performed to generate inter‐protein correlation scores based on the phylogenetic trees of each of these IDTSs. Hierarchical clustering analysis of these scores places SidP and MavQ in a linked group of highly correlated phylogenies.
To test this hypothesis, we cloned orthologs of: (i) indirect antagonists AnkX and Lem3 and (ii) direct antagonists MavQ and SidP from Legionella dumoffii, a related species of Legionella that is also able to cause human disease (Qin et al, 2012). We observe no cross‐species rescue on the array between MavQ (L. dumoffii) and our L. pneumophila library (Fig 6C), consistent with a divergence of the MavQ‐SidP interface since these two Legionella species’ last common ancestor. In contrast, AnkX cloned from L. dumoffii was efficiently rescued by the L. pneumophila ortholog of Lem3 on the array (Fig 6D). As predicted, intra‐species rescue was maintained for both gene pairs (Fig 6E and F). Notably, Y2H assays confirmed a direct physical interaction between the L. dumoffii orthologs of MavQ and SidP (Appendix Fig S7), consistent with a conserved role of SidP as a direct antagonist of MavQ. (A technical aside: the standard Y2H assay relies on expression of a growth‐based reporter (HIS3) and resultant growth on histidine‐deficient media; thus, any examination of cross‐species physical interaction using this approach would be obscured by MavQ's unchecked growth inhibition.)
To look for further evidence that MavQ and SidP may have co‐evolved during Legionella speciation, we performed MirrorTree analysis (Pazos & Valencia, 2001) to compare the phylogenetic trees of MavQ, SidP, and the other effectors in our library for which pairwise orthologs could be identified across a shared set of 35 publically available species of Legionella (Burstein et al, 2016). Hierarchical clustering was next used to group each protein based on correlations of inter‐protein distance (Fig 6G). We observe clear linkage between the phylogenies of MavQ and SidP, which is consistent with our model that the physical constraints imposed by direct effector–effector interplay can serve as potent modulators of pathogen evolution.
Lem14 synergizes with SidP to inhibit yeast growth
In addition to capturing effector–effector suppression, our assay was also designed to detect synergy between effectors—in which two effectors inhibit yeast growth in concert. Two surprises from the screen were that only one set of effectors specifically synergized in a one‐to‐one fashion and that this synergy involved SidP, the bifunctional effector/metaeffector described above. In addition to its inhibitory effects on MavQ, SidP synergizes with another effector, Lem14 (Lpg1851), to cause a severe growth defect when co‐expressed in our yeast screen (Fig 7A and B). Strikingly, neither protein confers a measurable growth defect on its own (Fig 7B).
Figure 7. SidP and Lem14 display an aggravating interaction.
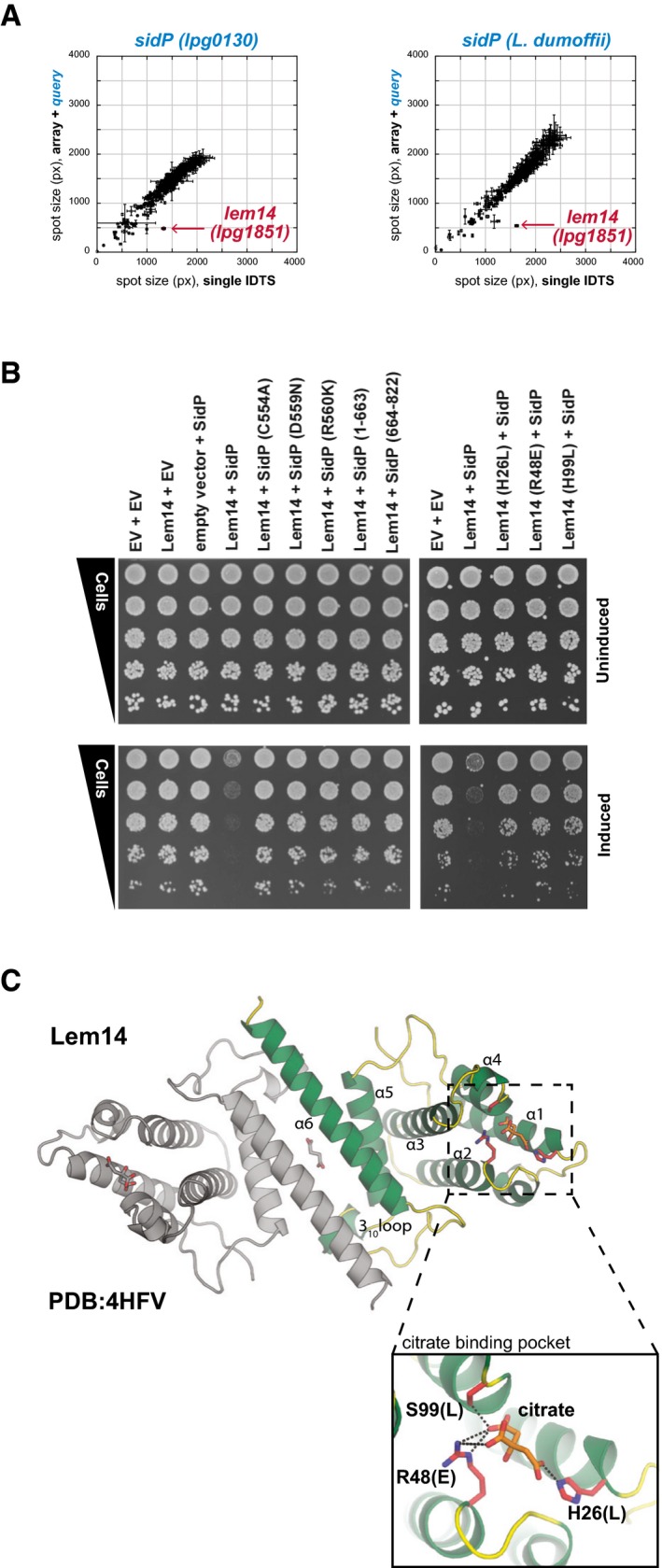
- Effector activation between Lem14 and SidP revealed by the effector–effector interaction screen. Each of the IDTS in the array was mated to create either query‐IDTS‐containing diploids (y‐axis) or vector‐IDTS controls (x‐axis). IDTS pairs that combine to cause a growth defect in yeast fall below the diagonal, as is the Lem14 array strain indicated in red. SidP and Lem14 are the only IDTS that display such a specific one‐to‐one phenotype in the screen. Querying the same array with the L. dumoffii ortholog of SidP also results in synergy with L. pneumophila Lem14 on the array; this cross‐species behavior may reflect an indirect functional interaction between the two proteins (see Fig 6). Error bars represent the SD of spot size between quadruplicate spots within each dataset.
- The synergistic effect of SidP and Lem14 is dependent on the SidP phosphatase activity and Lem14's charged pocket. Yeast strains co‐expressing (i) SidP phosphatase mutants and fragments with wild‐type Lem14 and (ii) co‐expressing wild‐type SidP with Lem14 mutants were tested in a yeast spot dilution assay as described previously. Co‐overexpression of wild‐type SidP and Lem14 caused a sicker than expected growth phenotype in yeast, but any SidP or Lem14 mutant alleviated this effect. Expression and stability of all mutants were tested (Appendix Figs S3A and S8B).
- The 1.9 Å crystal structure of Lem14 (4HFV) shows it to be a small, predominantly alpha‐helical protein forming an anti‐parallel dimer, interfacing via head‐to‐tail α6‐α6 hydrophobic interactions and interactions between the N‐terminal portion of α5 and the 310‐containing loop inserted between α5 and α6 of the neighboring chain. A citric acid molecule was found inside a positively charged pocket at the ends of α1–4.
While revealing the full mechanistic basis of SidP‐Lem14 synergy remained outside of the scope of this current study, the uniqueness of this pair and its linkage to our earlier work warranted at least some investigation. In contrast to the SidP‐MavQ interaction described above, the synergistic interaction between SidP and Lem14 does not appear to involve a physical interaction as tested by the Y2H assay (Appendix Fig S8) and LUMIER (Table EV5). Providing further support to an indirect synergistic interaction, we observe clear cross‐species synergy between L. pneumophila Lem14 and SidP from L. dumoffii in our screen (Fig 7A).
In notable contrast with SidP‐MavQ suppression, the synergistic interaction between SidP and Lem14 requires both the active and full‐length SidP protein (Fig 7B). To explore this further, we solved the crystal structure of Lem14, revealing a small, alpha‐helical protein that forms an anti‐parallel dimer (Fig 7C) and is structurally similar to another effector, LpiR1, whose molecular function remains elusive (Beyrakhova et al, 2016). Lem14 has a positively charged pocket that is required for SidP‐Lem14 synergy in yeast, as mutants that reverse the charge (Fig 7C, inset) alleviate the SidP‐Lem14 yeast growth inhibition (Fig 7B). Indeed, together with our suppression data, the observation of SidP‐Lem14 synergy makes SidP a remarkable hub of effector activity within the eukaryotic cell, likely linking SidP/MavQ host lipid modulation with the as‐of‐yet undiscovered function of Lem14.
Discussion
Our systematic pairwise effector–effector interaction screen has highlighted the complex hierarchy present in the effector arsenal of Legionella. We captured all previously known examples of effector–effector suppression and identified an additional seventeen effector–effector suppression pairs, including nine novel putative metaeffectors (Fig 8A). While the concept of “metaeffector” remains fluid, there is a clear need to distinguish this special class of proteins from equally informative effector–effector relationships like that of SidM and SidD, where counteracting activities on a shared target reverse the effects of each effector on the host cell. We previously proposed that a defining characteristic of a metaeffector should be a direct physical interaction between it and its cognate IDTS, following the established relationship between canonical effectors and their host targets (Ensminger, 2016).
Figure 8. A summary of effector–effector interactions revealed by our screen.
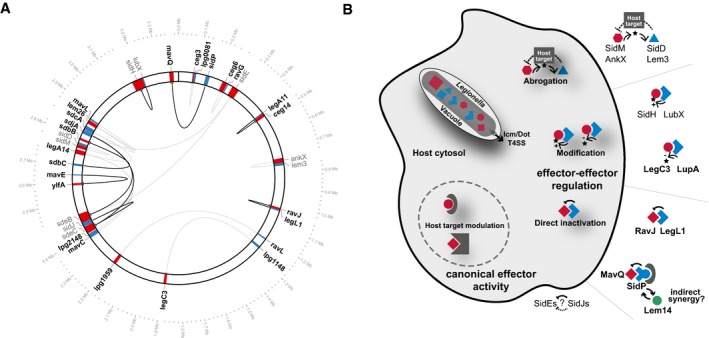
- Summary of all effector–effector suppression pairs and physical interaction data. Suppressing effectors (blue) and their cognate effectors (red) are shown at their genomic location. Previously identified effector–effector suppression pairs are shown in gray, and effector–effector suppression pairs identified in this study are labeled in black. Lines connect each pair (black: evidence for a physical interaction; dashed, gray: no evidence for a physical interaction). Excluding the multiple SidE/SidJ paralogs in the dataset, of the remaining effector–effector pairs six are immediately adjacent to one another on the chromosome, two are nearby (within 2–3 loci), and six are unlinked. The plot was generated using Circos v.0.69 (Krzywinski et al, 2009) with a 10× zoom at the effector genes shown.
- Several mechanisms of effector–effector suppression. After release into the host cell, translocated bacterial substrates (effectors) regulate one another through several different functional interactions: indirectly, through counteracting modification of a shared host target, or directly through either steric complex formation or direct modification of one effector by another.
Our functional characterization of several metaeffectors identified in this study provides a number of important insights: we demonstrate that a single protein can function as both a metaeffector (targeting other IDTS) and a canonical effector (targeting the host) [SidP], these interactions may place evolutionary constraints on the plasticity of effector arsenals, and mechanisms of effector–effector suppression are diverse (Fig 8B). In light of these findings, we expect that as additional mechanisms of metaeffector function are discovered, they are likely to derive from as broad a set of activities as classical effectors that target the host.
It was recently shown that the cognate effector of LupA, LegC3, interferes with vesicle trafficking (de Felipe et al, 2008) by inhibiting the formation of endogenous trans‐SNARE complexes during vacuolar fusion (Bennett et al, 2013) and forms a stable and functional SNARE acceptor complexes with two other IDTS YlfA/LegC7 and YlfB/LegC2 and the mammalian R‐SNARE VAMP4 (Shi et al, 2016). In light of our data showing that YlfA is also directly inactivated by another novel metaeffector, MavE (Figs 1B and 2A), one attractive model is that LupA and MavE act to prevent detrimental off‐target effects of their individual cognate effectors prior to complete SNARE complex formation. Indeed, such a model would explain why we observe yeast growth inhibition when we express either YlfA or LegC3 individually—and would reveal another important role for metaeffectors during infection.
By examining previously generated RNA‐seq data for Legionella pneumophila str. Philadelphia‐1 during intracellular replication in Acanthamoeba castellanii (Weissenmayer et al, 2011), we can begin to place several effector–effector interactions within the context of infection. Like the previously described SidH‐LubX interaction, four additional pairs exhibit differential expression patterns consistent with the cognate effector being held in check during the replicative (mid) stages of infection: LegA8(AnkX)‐Lem3, LegC3‐LupA, SdbB‐SdbC, and SidM(DrrA)‐SidD (Appendix Fig S9A). Four other effector–effector pairs exhibit differential expression patterns consistent with the cognate effector being held in check during the transmissive (early/late) stages of infection: Ceg3‐Lpg0081, SidE‐SdjA|SidJ, Lpg1959‐RavL, and LegA14‐MavC|Lpg2148|SdcA (Appendix Fig S9B). Notably, the rest of our pairs do not display differential transcriptional profiles across the infection stage (Appendix Fig S9C). For these, we propose other mechanisms of regulation, including post‐transcriptional regulation of protein levels, conditional‐specific regulation, chaperone‐based differential regulation of translocation to the host, or spatial regulation within the host cell in which effector activity is inhibited in only specific subcellular compartments. Indeed, a simple diffusion model could set up a spatial gradient of effector activity: close to the site of translocation, where effector and metaeffector concentration are highest, the two proteins would be more likely to physically interact. In this model, inactivation would predominate near the Legionella‐containing vacuole; as effector and metaeffector diffuse further into the host cell, the frequency of effector–metaeffector physical interaction would reduce, providing an opportunity for unchecked effector activity to dominate. To fully explore the regulatory network of effectors and metaeffectors during infection, detailed proteomic analysis of effector protein levels and localization within the host cell are obvious next steps for the field. Notably, the mechanisms of inhibition we describe are likely to be critically important for interpreting these results, as metaeffectors such as LegL1 that rely upon steric hindrance of their cognate effectors will require absolute protein levels greater than catalytic antagonists such as LupA.
Current models of Legionella pathogenesis tend to view the pathogen's remarkable number of IDTS as a redundant cohort in support of its broad natural host range (Ensminger, 2016). Direct and indirect effector–effector suppression may provide a further explanation for IDTS expansion—as a mechanism to provide regulatory complexity to the progression of intracellular replication (Jeong et al, 2015; Ensminger, 2016). Indeed, some effector–effector regulation is likely a common feature of intracellular pathogens, reflecting the delicate balance that must be maintained between host perturbation and homeostasis for the duration of each replicative cycle. Effector–effector interaction may also provide a mechanism to regulate the assembly or disassembly of multi‐effector complexes, or to compensate for a potentially leaky translocation system (Ensminger, 2016). To that end, the systematic identification of effector–effector interaction described herein should be extended to other pathogens of plants, animals, and humans—with the level of observed regulatory complexity providing key insights into how each pathogen balances the establishment and maintenance of its replicative niche.
Materials and Methods
Saccharomyces cerevisiae Icm/Dot‐translocated substrate (IDTS) overexpression strains
330 pDONR221‐IDTS constructs from a pDONR‐IDTS library encompassing the Icm/Dot‐translocated substrates first described by Losick et al (2010) and additional unpublished constructs (a kind gift from Ralph Isberg; see Table EV1) were used to clone IDTS into various yeast Gateway expression vectors (Alberti et al, 2007). To allow for expression of IDTS with an alternate start codon and to allow for assessment of expression of each IDTS by western blot, an N‐terminal 1×HA‐tag was introduced into pAG423GAL‐ccdB and pAG416GAL‐ccdB (Alberti et al, 2007). To make pAG423GAL‐HA‐ccdB and pAG416GAL‐HA‐ccdB, pre‐annealed oligos HA_pAGXXXgal_F (/5Phos/ctagtcttaccatgggttcttacccatacgatgttccagattacgcta) and HA_pAGXXXgal_R (/5Phos/ctagtagcgtaatctggaacatcgtatgggtaagaacccatggtaaga) were ligated into the SpeI site of pAG423GAL‐ccdB and pAG416GAL‐ccdB and the resulting clones were sequence‐verified.
To create the BY4741+pAG423GAL‐HA array strains, the pDONR‐IDTS collection was cloned into the high‐copy yeast expression vector pAG423GAL‐HA‐ccdB (GAL1 promoter, N‐terminal HA‐tag, and HIS3 selectable marker) using LR clonase II (Life Technologies) according to the manufacturer's instructions. pAG423GAL‐HA‐IDTS clones were verified by PCR and transformed to the S. cerevisiae strain BY4741 (MATa his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0; Brachmann et al, 1998) using the high‐efficiency 96‐well PEG/LiAc method (Gietz & Schiestl, 2007b).
The previously published L. pneumophila yeast expression library (Heidtman et al, 2009) in the high‐copy vector pYES2 NT/A (Life Technologies, GAL1 promoter, N‐terminal 6xHIS/Xpress tag, and URA3 selectable marker) in the S. cerevisiae strain BY4742 (MATα his3Δ1 leu2Δ0 met15Δ0 ura3Δ0; Brachmann et al, 1998; Table EV6, Appendix Fig S1B) was extended by yeast homologous recombination cloning or ligation cloning to a total of 333 IDTS or putative IDTS (see Table EV7 for primers and cloning methods). Yeast homologous recombination cloning into pYES2 NT/A was performed as described previously (Heidtman et al, 2009) with the following modification: primers were designed with a 40‐nt overlap with the vector sequence flanking the EcoRI‐XhoI sites in the pYES2 NT/A multiple cloning site. Briefly, the IDTS ORFs were PCR‐amplified from L. pneumophila str. Philadelphia‐1 genomic DNA [GenBank Accession AE017354 (Chien et al, 2004; Rao et al, 2013)]. The resulting PCR products were transformed to BY4742 with EcoRI‐XhoI‐digested pYES2 NT/A using the high‐efficiency PEG/LiAC method (Gietz & Schiestl, 2007a) and transformants were screened by PCR and sequence‐verified. Previously, six Icm/Dot‐translocated substrates (lpg1368, lpg1488, lpg1489, lpg2157, lpg2504, and lpg2862) could not be cloned by yeast homologous recombination and were designated non‐recombinant (NR; Heidtman et al, 2009). In our extension of the pYES2 NT/A collection, we identified an additional eight NR IDTS (lpg0090, lpg0693, lpg0234, lpg2153, lpg2461, lpg2519, lpg2523, and lpg2828). Since the inability to clone these NR IDTS ORFs into a high‐copy yeast expression vector is likely due to a combination of low‐level leaky expression and their high toxicity to yeast (Heidtman et al, 2009), they and five other IDTS that failed to PCR‐amplify or could not be screened in a high‐copy plasmid due to phenotype (see Table EV7) were cloned into the low‐copy pAG416GAL‐HA‐ccdB vector (GAL1 promoter, N‐terminal HA‐tag, URA3 selectable marker) from the pDONR‐IDTS collection using LR clonase II (Life Technologies) according to the manufacturer's instructions. These galactose‐inducible low‐copy plasmids were transformed to BY4742 using the high‐efficiency PEG/LiAC method (Gietz & Schiestl, 2007a).
Analysis of yeast growth defects
Liquid growth assays were performed in flat‐bottom, clear 96‐well plates (Greiner) sealed with adhesive plate seals (AB‐0580, AB‐gene) using a custom platform incorporating Tecan GENios plate readers (Tecan; Proctor et al, 2011). Freshly transformed BY4741 strains with pAG423GAL‐HA‐IDTS were transferred to a 96‐well plate with 100 μl SD‐his + 2% glucose using a 96‐floating pin tool (V&P Scientific) and grown overnight at 30°C. A fresh 96‐well plate with 100 μl SD‐his + 2% glucose or with 2% galactose was inoculated from the preculture plate using a 2‐μl pin tool (V&P Scientific) and grown at 30°C with continuous shaking. Yeast growth was monitored up to 20 h by measuring the OD595 every 15 min. The growth fitness of each strain was calculated as the ratio of the area under the curve (AUC) of a IDTS‐expressing strain over a pAG423GAL‐HA‐ccdB empty vector control after 20 h. The average AUC ratio and standard deviation of three independent replicates were calculated.
The BY4741 with pAG423GAL‐HA‐IDTS collection and the 126 strains of the original BY4742 with pYES2 NT/A‐IDTS collection (Heidtman et al, 2009) were arrayed in 384‐well format with the border and empty spots filled with empty vector control. The 384‐well format arrays were pinned in quadruplicate (1,536‐density) onto SD‐his + 2% galactose or SD‐ura + 2% galactose, respectively, using the ROTOR HDA pinning robot (Singer Instruments) and grown for 2 days at 30°C. The arrays were then imaged using a high‐resolution camera, and the spot sizes were quantified using SGAtools (http://sgatools.ccbr.utoronto.ca/; Wagih et al, 2013). Outlier spot sizes flagged by the Jackknife filter (JK) in SGAtools were removed, and the average and standard deviation of the remaining values were calculated and normalized to the average empty vector control (Tables EV1 and EV6, Appendix Fig S1). Five of the 330 array strains were filtered out after analysis because their identity could not be established, likely due to a mixture of clones within the spot on the array (lpg1154, lpg0403, lpg0195, lpg2147, and lpg2806). To estimate the total number of strains that have a growth defect in the BY4741 + pAG423GAL‐HA‐IDTS array, the empty vector spots of the inner rim of the border were averaged and compared to the average spot size of individual strains. In total, 227 strains have an (average strain spot size + stdDev) < (average empty vector spot size − StdDev).
Suppression profiling screen
Each haploid query strain (BY4742 + pYES2 NT/A‐IDTS) was individually mated to the BY4741 + pAG423GAL‐HA strain array, similar to procedures in the SGA protocol (Tong & Boone, 2006), to test for possible rescue of yeast toxicity by each of the array overexpression strains. A 3‐ml overnight culture of the IDTS query strains and an empty vector control query was spread onto a SD‐ura + 2% glucose agar PlusPlates (Singer Instruments) and grown overnight at 30°C. The arrayed library (330 strains overexpressing individual IDTS and empty vector controls in 384‐well density) was pinned in quadruplicate (1,536 density) onto SD‐his + 2% glucose plate using the ROTOR HDA (Singer instruments) and 384 short pads (Singer Instruments) and grown overnight at 30°C. To mate the query with the array strains, the freshly grown query lawn was transferred to an YPD plate using the 1,536 short pads (Singer Instruments) and the 1,536‐format array was pinned on top of the query spots. The strains were allowed to mate overnight at room temperature and subsequently pinned to media selecting for the presence of both plasmids (SD‐ura/his + 2% glucose) to recover diploid strains. The diploid plate was grown for 1 day at 30°C and transferred to SD‐ura/his + 2% galactose to induce expression of the query and array IDTS plasmids. These galactose plates were incubated at 30°C and imaged after 1, 2, and 3 days using a high‐resolution camera. Spot sizes were quantified using SGAtools (http://sgatools.ccbr.utoronto.ca/; Wagih et al, 2013), with the advanced option “keep large replicates”. The SGA output files used in our analysis—all query strains (at 2 days)—will be deposited into the Dryad Digital Repository.
In 16 suppression profiling screens, the growth inhibition caused by the query IDTS was rescued by the overexpression of IDTS on the array (Fig 1B). These screens were subsequently repeated to confirm rescue. The two replicate screens were then analyzed as follows: outlier spot sizes flagged by the Jackknife filter (JK) in SGAtools were removed and the average and standard deviation of the remaining values were calculated. (Five array strains were filtered out after analysis because their identity could not be established, as described above.) For the strains that suppressed toxicity of the query IDTS in both replicates, the plasmid was recovered and sequenced to confirm identity. Converse pairwise suppression where the yeast growth inhibition was suppressed by a query IDTS was confirmed by yeast spot dilution assay (see below). Three colonies from the array IDTS were sequenced to confirm identity, mated with the suppressing query IDTS or empty vector control, and the resulting diploid strains were used in the spot dilution assay (Appendix Fig S2).
Yeast two‐hybrid assays
To investigate a possible physical interaction between the two partners of the effector–effector suppression pairs, we used the Y2H assay (Dreze et al, 2010). The strain Y8800 (MATa leu2‐3,112 trp1‐901 his3‐200 ura3‐52 gal4∆ gal80∆ GAL2‐ADE2 LYS2::GAL1‐HIS3 MET2::GAL7‐lacZ cyh2R; Yu et al, 2008) and vectors with the DNA‐binding (DB) and transcription‐activating (AD) domain of Gal4 (pDEST‐DB, pDEST‐AD; Dreze et al, 2010) were a kind gift from N. Yachie and F. Roth (University of Toronto, Canada).
The suppressing IDTSs were cloned into Gateway destination plasmid pDEST‐DB (ADH1 promoter, CEN, LEU2 selectable marker, N‐terminal Gal4 DNA‐binding domain) by Gateway LR reaction using LR clonase II, transformed to Y8800 using the LiAc/PEG method (Gietz & Schiestl, 2007a), and plated onto SD‐leu + ade/2% glucose. The IDTSs with a yeast growth defect were cloned into pDEST‐AD (ADH1 promoter, CEN, TRP1 selectable marker, N‐terminal Gal4 transcription activation domain) by Gateway LR reaction using LR clonase II. Because the ADH1 promoter is a constitutive promoter, the AD‐IDTS fusions or empty vector control was transformed to Y8800 + pDEST‐DB‐antagonist IDTS, to ameliorate the toxicity of the pDEST‐AD‐IDTS, and plated onto SD‐leu/trp + ade/2% glucose. A single colony of Y8800 + pDEST‐DB‐IDTS/pDEST‐AD‐IDTS or pDEST‐AD‐empty vector strains was inoculated in a 96‐well plate with 100 μl SD −leu/trp + ade/2% glucose medium, grown overnight and back‐diluted 1:25 or 1:50 in fresh medium before spotting onto SD −leu/trp + ade/2% glucose and SD‐leu/trp/his, +ade/2% glucose with or without 1 mM 3‐amino‐1,2,4‐triazole (3‐AT; Bioshop) using the VP 407AH pin tool. The plates were grown for 2 or 3 days before imaging. Positive Y2H growth (growth on −his and –his + 1 mM 3AT conditions) was screened for cryptic autoactivator mutations in the pDEST‐DB plasmids by counterselecting for the pDEST‐AD on SD‐leu/his + ade/2% glucose and 1 mg/l cycloheximide as described (Dreze et al, 2010). All experiments were performed in triplicate.
LUMIER with bait concentration assay
The functional antagonist IDTSs were cloned into the Gateway destination vectors pcDNA3.1‐3XFLAG‐V5‐ccdB and pcDNA3.1‐Renilla‐ccdB (Taipale et al, 2012) from pDONR221 clones using Gateway technology. The LUMIER with BACON assay was performed as described previously (Taipale et al, 2012) with minor modifications. HEK293T cells were seeded into a 96‐well plate at 30,000 cells/well density in 100 μl DMEM supplemented with 10% fetal bovine serum, penicillin/streptomycin, and l‐glutamine and allowed to grow for 24 h before adding 50 μl OptiMEM (Life Technologies) with 100 μg/ml Lipofectamine 2000 (Life Technologies), 75 ng pcDNA3.1‐FLAG‐V5 bait, and 75 ng pcDNA3.1‐Renilla‐prey DNA. After 48 h, the cells were washed with 1× PBS and lysed in cold HENG buffer [20 mM Hepes‐KOH pH 7.9, 150 mM NaCl, 2 mM EDTA pH 8.0, 0.5% Triton X‐100, 5% glycerol, 1× EDTA‐free complete mini protease inhibitor cocktail (Roche)]. The lysate was transferred to white 384‐well LUMITRAC 600 high‐binding plates (Greiner) coated with anti‐FLAG‐M2 (Cat# F1804, Sigma‐Aldrich) and incubated for 3 h at 4°C. The 384‐well plates were washed with cold HENG buffer and with cold 0.5 M NaCl/1% Triton X‐100 HENG buffer before adding 2.5 μM coelenterazine h (Nanolight Technology) in Nanoluc assay buffer (20 mM Tris–HCl pH 7.5, 1 mM EDTA, 150 mM KCl, 0.5% Tergitol NP9). The luminescence signal was measured in a Synergy Neo plate reader (Biotec). The individual 3XFLAG‐V5‐bait concentration was determined by adding anti‐FLAG‐M2‐HRP (Cat# A8592, Sigma‐Aldrich). After 1 h incubation at room temperature, the plates were washed with PBS 0.05% Tween and the bait ELISA signal was measured using fivefold diluted Pico substrate (Thermo Scientific) in the above‐mentioned plate reader.
The LUMIER interaction scores were calculated as Z‐scores and filtered for detectable bait concentrations (Taipale et al, 2014). For each Renilla luciferase prey, luminescence signals were mean‐normalized and log2‐transformed and Z‐scores were calculated from the estimated mean and standard deviation for all datapoints (Table EV5). Bait ELISA signals below the mean plus 2× standard deviation of ELISA signal from no‐bait controls were removed from the final data set and are indicated as NA in Table EV5. The heatmap was generated using heatmap.2 without clustering in gplots v2.15.0 in R v3.1.1.
Yeast spot dilution assay
The growth defect of MavQ (Lpg2975), RavJ (Lpg0944) mutants, the rescue of MavQ by SidP (Lpg0130) mutants, and the synergy between SidP and Lem14 mutants were assayed by a yeast spot dilution assay. Overnight cultures were grown in appropriate selective medium with 2% glucose, normalized to OD600 of 1, and used to make dilution series with fivefold dilution steps. The dilutions were spotted onto selective media plates with 2% glucose or 2% galactose to induce expression, using the VP 407AH pin tool (V&P Scientific), and imaged after incubation for 2 days at 30°C.
To verify expression and stability of the MavQ, SidP, RavJ, and Lem14 mutants, strains were grown overnight in the SD‐his + 2% glucose, back‐diluted to OD600 = 1.5 in SD‐his + 2% galactose, and incubated at 30°C for 5 h. Six OD600 units of cells were harvested and lysed as described previously (von der Haar, 2007). Briefly, per three OD600 units 200 μl lysis buffer (0.1 M NaOH, 2% SDS, 2% β‐mercaptoethanol, 50 mM EDTA) is added and incubated at 90°C for 10 min followed by the addition of 5 μl 4 M acetic acid and a second incubation at 90°C for 10 min. 50 μl of loading buffer (0.05% bromophenol blue, 250 mM Tris–HCl pH 6.8, 50% glycerol) was added. The equivalent of 0.4 OD600 units of sample (RavJ and Lem14 mutants) or 0.8 OD600 units (SidP and MavQ mutants) was analyzed by SDS–PAGE and western blot using anti‐HA.11 (Cat# 901501, Clone 16B12, BioLegend Inc) or anti‐Xpress (R910‐25, ThermoFisher Scientific) antibodies.
Protein purification
Gene fragments corresponding to proteins RavJ (Lpg0944) residues 1–230, 230–391, LegL1 (Lpg0945) 1–296, LupA (Legionella ubiquitin‐specific protease A, Lpg1148) 1–373, SidP (Lpg0130), MavQ (Lpg2975) 1–871, and Lem14 (Lpg1851) 1–220 were PCR‐amplified from Legionella pneumophila str. Philadelphia‐1 genomic DNA and inserted into either plasmids p15TV‐LIC (Eschenfeldt et al, 2009) or pET28‐SBP‐TEV (Addgene plasmid #36943), providing N‐terminal 6xHIS‐TEV or 6xHIS‐SBP‐TEV epitope tags, respectively. Additional point mutants were prepared by QuikChange mutagenesis (Stratagene). Plasmids were sequenced and subsequently transformed to either BL21 Gold or BL21(RIL)DE3 E. coli for purification.
For selenomethionine enriched proteins, cultures for each of the proteins were grown in M9 SeMET High‐Yield growth medium (Shanghai Medicilon Inc.) at 37°C with shaking to an OD595 of 1.2 followed by a reduction in the temperature to 16°C and overnight induction of protein expression using 0.4 mM IPTG. For native proteins, cultures were grown in LB and expression was induced at an OD595 of 0.8 with 0.4 mM IPTG, with the exception of MavQ and MavQ D147A, which were grown in Studier auto‐induction medium ZYM‐5052 (Studier, 2005) at 20°C overnight. Cells were harvested by centrifugation at 9,300× g, resuspended in 50 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 5 mM imidazole, and lysed by sonication. Lysates were clarified by centrifugation at 21,000× g at 4°C and the supernatants incubated with Ni‐NTA agarose (Qiagen) at 4°C with gentle mixing for 4 h, followed by washing with 50 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 30 mM imidazole. Protein was eluted using 50 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 250 mM imidazole. Cleavage of the His‐tag prior to crystallization was performed by incubation of the purified protein with TEV protease followed by dialysis into 10 mM HEPES pH 7.5, 500 mM NaCl and finally removal of both TEV protease and the 6xHis‐tag by further incubation with Ni‐NTA agarose. Where required, proteins were further purified by size‐exclusion chromatography on Superdex 200 HiLoad 16/60 column (GE Healthcare Life Sciences) in 0.5 M NaCl, 10 mM HEPES pH 7.5, after which the proteins were concentrated to approximately 10 mg/ml using a centrifugal concentrator (Corning) with an appropriate molecular weight cutoff and either used immediately or flash‐frozen in liquid nitrogen for storage at −80°C.
Crystallization and structure determination
For Lem14 structure determination, selenomethionine‐substituted protein was crystalized at room temperature by hanging‐drop vapor diffusion, the final crystallization solution contained 0.2 M diammonium citrate, 20% PEG3350, 1/70 thermolysin, pH 5.0. For the LupA (1–373) structure determination, selenomethionine‐substituted protein was crystallized at room temperature by hanging‐drop vapor‐diffusion method and the successful condition contained 10 mM MgCl2, 4% sucrose and 1.6 M NH4 sulfate. For the RavJ (1–230) structure determination, native protein was crystalized at room temperature by sitting‐drop vapor diffusion and the final crystallization solution contained 1.6 M ammonium sulfate, 0.1 M NaCl, 0.1 M HEPES pH 7.5. For the RavJ (230–391) structure determination, selenomethionine‐substituted protein was crystalized at room temperature by hanging‐drop vapor diffusion and the final crystallization solution contained 1.6 M ammonium sulfate, 0.1 M NaCl, 0.1 M HEPES pH 7.5. Finally, for the RavJ (1–230 fragment)‐LegL1 complex structure, the selenomethionine‐containing protein sample of the RavJ (1‐230 fragment) was pre‐incubated with LegL1 native protein sample in 1:1 ratio for 1 h at 4°C in 0.1 M NaCl and 10 mM HEPES pH 7.5 buffer. This protein mixture was run through a Superdex 200 HiLoad 16/60 column (GE Healthcare Life Sciences) calibrated in the same buffer, and the fraction corresponding to the RavJ (1–230)‐LegL1 complex was collected and concentrated to 57 mg/ml. Diffraction quality crystals were obtained by mixing 0.5 μl of this protein complex sample in 1:1 ratio with crystallization solution containing 0.1 M NaCl, 100 mM Tris pH 8.5 and 26% (w/v) PEG3350 by hanging‐drop vapor‐diffusion method at room temperature. All crystals were cryo‐protected by immersion in paratone‐N oil before being flash‐frozen in liquid nitrogen.
The X‐ray diffraction data were collected at the 19‐BM and 19‐ID beamlines at the Structural Biology Center, Advanced Photon Source, Argonne National Laboratory, at the selenium absorption peak wavelength at 100 K (Rosenbaum et al, 2006). All datasets were processed with HKL‐3000 (Minor et al, 2006). The structures of RavJ (1–230), RavJ (230–391), the RavJ (1–230)—LegL1 complex and of LupA were determined using the SAD phasing method, and the structure of Lem14 was determined using the MAD phasing method using the program Shelx (Sheldrick, 2010). Model building was initially performed by ShelxE, and the initial models were further extended using the ARP/wARP web service (Langer et al, 2008), followed by manual adjustments. All structures were refined using the program Phenix.refine (Adams et al, 2010) and manually inspected and rebuilt using the program Coot (Emsley & Cowtan, 2004). Translation–libration–screw rotation (TLS) parameterization as defined by the TLSMD server (Painter & Merritt, 2006) was used for refinement of the complex. Geometric suitability for all structures was verified using the Phenix validation tools and the wwPDB validation server. The data collection and refinement statistics are summarized in Table EV4. Structural comparisons were performed using the program COFACTOR (Roy et al, 2012).
Homology modeling of SidP (Lpg0130)
Homology modeling of Lpg0130 was performed using Phyre2 (Kelley & Sternberg, 2009) in intensive mode. The resulting model was based on a single template (LLO_3270, PDB: 4JZA; Toulabi et al, 2013, 53% identical) with a 100% confidence score. 99% of residues were modeled at > 90% confidence. Only 10 residues were modeled using ab initio methods.
MavQ (Lpg2975) homology detection and HMM‐HMM alignment
The MavQ amino acid sequence was submitted to HHPred (Soding, 2005) with suggested default parameters (HMMdatabase: pdb70_05Jun16, MSAgeneration HHblits, Max 3 iterations, secondary structure scoring, local alignment). The N‐terminal part of MavQ may share some homology with several PI3 and PI4 kinases and aminoglycoside phosphotransferases [4h05, 4hne (Zhou et al, 2014), 4wtv (Klima et al, 2015), 3w0o (Iino et al, 2013), 4bfr (Certal et al, 2014), and 4ykn (Yang et al, 2015)]. The resulting HMM‐HMM alignments with MavQ were used to assist in determining potentially important residues. Residues S25/26, K46, D147A, and D160A were mutated to alanine using site‐directed mutagenesis.
In vitro determination of MavQ ATP hydrolysis activity
ATP‐to‐ADP conversion by MavQ was performed according to Zhou et al (2014). Briefly, PI diC8 dissolved in water was dried in a vacuum centrifuge, resuspended to a concentration of 7 mM in kinase buffer (20 mM Tris pH 7.5, 150 mM NaCl, 0.2% Triton X‐100, 1 mM EDTA, 20 mM MgCl2), and sonicated until translucent. 0.5 μg of MavQ or MavQ D147A (1 mg/ml) in kinase buffer was added to 5 μl of resuspended lipid micelles with a final volume of 9 μl. A third reaction without PI with wild‐type MavQ was also prepared. Reactions were started by the addition of 1 μl of 10 mM ultrapure ATP, incubated for 30 min at room temperature, and were performed in duplicate. ATP‐to‐ADP conversion was measured using ADP‐Glo kinase assay kit (Promega) according to the manufacturer's instructions. Luminescence was recorded using a Tecan plate reader with an integration time set to 750 ms.
AP‐MS analysis of MavQ‐SidP, LegL1‐RavJ, and RavJ‐host interactions
Legionella Lp03 lysate from exponential and post‐exponential cells for AP‐MS analysis of Mav‐SidP and LegL1‐RavJ interactions was prepared as described (Quaile et al, 2015). U937 lysate for RavJ‐ host interactions was prepared from U937 cells grown in RPMI 1640 supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were harvested by centrifugation (150× g, 5 min), washed once with PBS, and stored at −80°C. Per technical replicate, 1 × 108 frozen U937 cells were thawed in 1 ml of lysis buffer (50 mM HEPES pH 8.0, 150 mM NaCl, 5 mM EDTA, 5 mM DTT, 0.1% Nonidet P‐40, 1× EDTA‐free complete mini protease inhibitor cocktail (Roche)), lysed by five rounds of freeze/thaw in liquid nitrogen, and cleared by centrifugation (10 min, 16,000× g, 4°C). Lysate was depleted of endogenous biotin using 50 μl/ml washed streptavidin magnetic sepharose beads (New England Biolabs) for 45 min.
AP‐MS analysis was performed as described (Quaile et al, 2015). Briefly, 50 μl of streptavidin mag sepharose beads with 6XHis‐SBP tagged RavJ, LegL1, or MavQ were added to a 1‐ml aliquot of clarified, biotin‐depleted lysate (Lp03 or U937) and incubated with gentle mixing at 4°C for 3 h. Beads were washed twice with 1 ml lysis buffer, transferred to a fresh tube followed by a final wash with 500 μl lysis buffer. Bait and bound proteins were eluted with 100 μl of elution buffer (50 mM ammonium bicarbonate, 2.5 mM biotin) on ice for 10 min. One microgram of sequencing grade modified trypsin (Promega) was added to the eluate, and proteins were digested overnight at 37°C. Digestion was terminated by adding trifluoroacetic acid (TFA) to a final concentration of 0.2% (v/v), and peptides were desalted and concentrated using Agilent C18 OMIX tips according to the manufacturer's instructions and dried to completion in a vacuum centrifuge. Samples were resuspended in 0.1% formic acid and analyzed by mass spectrometry on an LTQ XL mass spectrometer (Thermo Scientific). Raw instrument data were converted to mzXML format using msconvert (Holman et al, 2014), and peptide spectra were searched using X! Tandem (Craig & Beavis, 2004). Protein/peptide identifications were further compared and analyzed using ProHits VM (Liu et al, 2012). A number of no‐bait controls, as well as several other IDTSs, were used to identify non‐specific binding proteins. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Vizcaino et al, 2013) and are summarized in Table EV3 (MavQ or LegL1 with Lp03 lysate) and Table EV8 (RavJ with U937 lysate).
Lipid overlay blots with SidP, MavQ, and Lem14
6xHisSBP‐tagged SidP, MavQ, and Lem14 were purified using the 6xHis‐tag, as described above, followed by an additional SBP‐tag purification step with streptavidin mag sepharose (GE Healthcare Life Sciences) and eluted in 50 mM HEPES pH 7.5, 300 mM NaCl, 2.5% glycerol, and 4 mM biotin. The lipid overlay blot was performed as described (Weber et al, 2013), with minor modifications. Briefly, the PIP strips (Echelon, P‐6001) were blocked in 3% fatty acid‐free BSA (Roche) in Tris‐buffered saline with 0.1% Tween 20 (blocking buffer) and incubated overnight in the dark at 4°C with 5 pmol/ml purified protein in a total volume of 10 ml blocking buffer. The PIP strips were washed with blocking buffer, incubated with mouse anti‐polyhis antibody (1:2,000, clone HIS1, Sigma‐Aldrich) in blocking buffer, washed, and incubated with anti‐mouse HRP antibody (7076, Cell Signaling Technologies) in blocking buffer.
Gel filtration chromatography of the RavJ‐LegL1 complex
One milligram of each protein was analyzed by gel filtration, in the combinations specified or alone using an AKTA Explorer 100 system (GE Healthcare Life Sciences) equipped with a Superdex 200 10/300 column (GE Healthcare Life Sciences). The column was equilibrated and run in 10 mM HEPES pH 7.5, 100 mM NaCl at a flow rate of 0.5 ml/min. Elution profiles were measured by absorbance at 280 nm. A280 data from multiple runs were normalized to 100% and aligned according to elution time.
In vitro determination of LupA (Lpg1148) hydrolase substrate specificity
To determine the hydrolase specificity of LupA, ubiquitin‐like substrates (ubiquitin, SUMO, NEDD8) derivatized with 7‐amido‐4‐methylcoumarin (AMC) on the carboxy‐terminus were employed. All substrates were purchased from Boston Biochem (USA) and used as per manufacturer's recommendations. Briefly, the assay was set up in a 96‐well plate format (Microfluor1 plates, ThermoScientific) in a 200 μl reaction volume. For initial substrate specificity tests, the purified Lpg1148 1–373 fragment was diluted into the reaction buffer (50 mM HEPES pH 7.8, 50 mM NaCl, 0.5 mM EDTA, 1 mM DTT) to a final concentration of 50 nM, and the reaction was initiated with 5 μl of substrate resulting in the following final concentrations: 300 nM Ubiquitin‐AMC, 190 nM SUMO‐3‐AMC, 180 nM SUMO‐2‐AMC, 120 nM SUMO‐1‐AMC, and 180 nM NEDD8‐AMC. Substrate hydrolysis was measured as fluorescence of released AMC (Ex380 nm, Em460 nm). The reaction was monitored in continuous mode using the SpectraMax Plate Reader (Molecular Devices) for 1 h at 23°C. Reaction conditions used to assess the effects of LupA active site mutant H183A on the ubiquitin‐AMC substrate were the same as described above, but was monitored for 1.5 h at 23°C. A reaction mixture containing no enzyme was used to monitor spontaneous degradation of the substrates. Potential non‐specific proteolysis carried over from enzyme purification was monitored by using identical experimental conditions with an unrelated protein (Lpg0439) purified under the same conditions as LupA (1–373).
In vivo deubiquitination of LegC3 by LupA (Lpg1148)
HEK293T cells were grown using standard techniques; incubated at 37°C, 5% CO2 in DMEM (Gibco) supplemented with 10% FBS (Gibco), and 1% penicillin/streptomycin (Sigma‐Aldrich). Cells were transfected with pRK5‐HA‐Ubiquitin (Addgene plasmid # 17608; Lim et al, 2005), pcDNA3.1‐3XFLAG‐V5‐LegC3 (see LUMIER assay), and either pcDNA3.1‐nV5‐DEST (Life Technologies) empty vector, LupA, LupA C252A, H183A, or D207A mutants as indicated, at approximately 60–70% cell confluence using Lipofectamine 3000 (Life Technologies) according to the manufacturer's instructions. After 24 h, cells were washed once with PBS, harvested by centrifugation, and frozen at −80°C.
Denaturing IPs were performed as described (Tansey, 2007) with minor modifications. Briefly, cell pellets from a 10‐cm plate were thawed into 200 μl TSD lysis buffer (50 mM Tris pH 7.5, 1% SDS, 5 mM DTT, 1× EDTA‐free complete mini protease inhibitor cocktail (Roche)) and heated at 98°C for 10 min. Lysates were clarified by centrifugation (18,000× g, 5 min), and 100 μl of supernatant was diluted with 1.2 ml of TNN immunoprecipitation (IP) buffer (50 mM Tris pH 7.5, 250 mM NaCl, 5 mM EDTA, 0.5% Nonidet P‐40). 130 μl of clarified lysate was retained for analysis of inputs. All subsequent incubation steps were performed at 4°C with gentle rotation. Lysates were precleared with 50 μl of washed protein G magnetic beads (NEB) to minimize non‐specific binding. The precleared lysates (without beads) were transferred to a fresh tube and incubated with 5 μg of mouse anti‐FLAG M2 antibody 1 h, after which 25 μl of protein G magnetic beads were added and incubated for 1 h. Beads were washed three times with IP buffer, resuspended in 30 μl 3× SDS–PAGE loading buffer, and heated at 98°C for 7 min.
26 μl of each IP was analyzed by SDS–PAGE and western blot and probed for HA‐Ub using mouse anti‐HA‐7 (1:20,000, Cat# H3663, Sigma‐Aldrich) in 1% BSA in PBS‐T and TrueBlot ULTRA anti‐mouse Ig HRP (1:2,000, Cat# 18‐8817‐31, Rockland Inc) in 5% milk, TBS‐T. The western blot was stripped by three 10‐min incubations in low pH glycine stripping buffer (0.1 M glycine, 20 mM magnesium acetate, 50 mM KCl, pH 2.2) and reprobed for FLAG‐V5‐LegC3 using mouse anti‐FLAG M2 (1:2,000, Cat# F1804, Sigma‐Aldrich) in TBS‐T and TrueBlot ULTRA anti‐mouse Ig HRP as above. 10 μl of clarified input lysates with 5 μl of 3× SDS–PAGE loading dye was analyzed by SDS–PAGE and western blot using mouse anti‐V5 antibody (1:2,000, Cat# R960, ThermoFisher Scientific) in 5% milk, TBS‐T, and anti‐mouse IgG HRP antibodies (1:2,000, Cat# 7076, Cell Signaling Technologies) in 5% milk, TBS‐T.
Legionella dumoffii ortholog identification and cross‐species rescue
An Ontario Legionella dumoffii strain (L. dumoffii str Hamilton, a kind gift from C. Guyard and Public Health Ontario) was subjected to Illumina MiSeq 250x250x8 (paired‐end) sequencing at the Donnelly Sequencing Centre, University of Toronto, as described previously (Rao et al, 2013). The resulting raw paired‐end sequence reads were deposited as Illumina FASTQ files to the NCBI sequence read archive (study accession: SRP051121). Contigs were assembled from these reads using the Velvet de novo assembler (Zerbino & Birney, 2008) and subjected to automatic annotation using Prokka (Seemann, 2014) with default settings. Orthologs for the L. pneumophila genes lpg2975, lpg0130, lpg0695, and lpg0696 were identified with PGAP (Zhao et al, 2012) using the MultiParanoid method with default settings. The L. dumoffii orthologs (Table EV9) were amplified by PCR from L. dumoffii str Hamilton genomic DNA and cloned by Gateway BP reactions with BP clonase (Life Technologies) to pDONR221‐ccdB (Life Technologies). The resulting pDONR‐IDTS plasmids were sequence‐verified and the yeast toxic IDTS orthologs, LdumoAE_01458 (mavQ) and LdumoAE_01458 (ankX), were cloned into pAG426GAL‐ccdB and pDEST‐AD, while the cognate IDTS orthologs, LdumoAE_01218 (sidP) and LdumoAE_01010 (lem3), were cloned into pAG423GAL‐HA‐ccdB and pDEST‐DB. Spot dilution assays and Y2H assays were performed as described above.
MirrorTree analysis
To examine evolutionary relationships between conserved IDTSs, genome sequences of 41 Legionella species described by Burstein et al (2016) were downloaded from NCBI. Based on the assigned Legionella ortholog groups (LOGs; Burstein et al, 2016), 36 of the 41 species, excluding L. adelaidensis, L. londiniensis, L. oakridgensis, L. maceachernii, and L. drancourtii, show one single ortholog (no paralogs predicted) each of SidP (Lpg0130) and MavQ (Lpg2975). Legionella cherrii was also excluded from the analysis due to an assembly gap in the SidP ortholog gene. Based on the similar criterion of single existence in all the remaining 35 Legionella species, 16 other conserved IDTSs were selected. Protein sequences of each IDTS in the 35 Legionella species were aligned using the ClustalW option in MEGA v6.0. These alignments were then input to MirrorTree software (Pazos & Valencia, 2001; software kindly provided by Florencio Pazos). Results were represented as co‐evolution correlation values between each two conserved IDTSs. Hierarchical clustering was then performed based on euclidean distance of correlation values. The heatmap was generated using heatmap.2 in gplots v2.15.0 in R v3.1.1.
The 35 species used in MirrorTree analysis are as follows: L. pneumophila, L. longbeachae, L. anisa, L. birminghamensis, L. bozemanii, L. brunensis, L. cincinnatiensis, L. drozanskii, L. dumoffii, L. erythra, L. feeleii, L. geestiana, L. gormanii, L. gratiana, L. hackeliae, L. israelensis, L. jamestowniensis, L. jordanis, L. lansingensis, L. micdadei, L. moravica, L. nautarum, L. parisiensis, L. quateirensis, L. quinlivanii, L. rubrilucens, L. sainthelensi, L. santicrucis, L. shakespearei, L. spiritensis, L. steelei, L. steigerwaltii, L. tucsonensis, L. waltersii, and L. worsleiensis.
Data accessibility
Coordinates of Lem14 (4HFV), RavJ 1–225 (4RXV), RavJ 251–371 (4RXI), the complex structure of RavJ (1–230) and full‐length LegL1 (4XA9), and LupA 123–403 (5DGG) have been deposited in the Protein Data Bank. SGA output files from the genetic interaction screen, including raw spot size information, have been deposited into the Dryad Digital Repository (doi: 10.5061/dryad.kj666).
Author contributions
MLU, AWE, and AS conceived and designed the screen for effector–effector interplay. AWE, AS, and MLU with input from other authors prepared the manuscript. AWE, AS, and MLU supervised the project. MLU performed the screen and targeted assays for effector–effector interactions with library construction assistance from RDL and AWE. CO assisted MLU with cross‐species analysis of effector–effector interactions. MLU performed the LUMIER assay with assistance from MT and ML. MM performed the in vitro assays of Lpg1148 deubiquitinase activity. ATQ performed the in vivo assays of this activity on LegC3, the assays on MavQ activity, and RavJ‐host interaction identification. ATQ and EE performed analytical gel filtration analysis. CR performed MirrorTree analysis. PJS, MEC, JO, BPN, KM assisted in the determination of RavJ, RavJ/LegL1 complex, LupA, and Lem14 crystal structures.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Table EV9
Review Process File
Acknowledgements
We thank members of the Ensminger and Savchenko laboratories, including V. Cartier‐Archambault for assistance with mining the published RNA‐seq data. We thank J. Claycomb, L. Cowen, J. Brumell, C. Gradinaru, and M. Machner for critical comments; C. Nislow for scientific advice; M. Meneghini and C. Boone for equipment use; and F. Roth and N. Yachie for Y2H strains and plasmids. We thank all members of the Structural Biology Center at Argonne National Laboratory, especially A. Joachimiak, for help with protein crystallography experiments. This work was supported by an operating grant from the Canadian Institutes of Health Research (MOP‐133406) to AWE, an infrastructure grant from the Canada Foundation for Innovation and the Ontario Research Fund (30364) to AWE; in part by a National Institutes of Health grant (GM094585) to AS through the Midwest Center for Structural Genomics and by the U. S. Department of Energy, Office of Biological and Environmental Research (contract DE‐AC02‐06CH11357); and by an operating grant from the Canadian Institutes of Health Research (MOP‐142384) to MT.
Mol Syst Biol. (2016) 12: 893
Contributor Information
Alexei Savchenko, Email: alexei.savchenko@utoronto.ca.
Alexander W Ensminger, Email: alex.ensminger@utoronto.ca.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Gitler AD, Lindquist S (2007) A suite of gateway cloning vectors for high‐throughput genetic analysis in Saccharomyces cerevisiae . Yeast 24: 913–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrios‐Rodiles M, Brown KR, Ozdamar B, Bose R, Liu Z, Donovan RS, Shinjo F, Liu Y, Dembowy J, Taylor IW, Luga V, Przulj N, Robinson M, Suzuki H, Hayashizaki Y, Jurisica I, Wrana JL (2005) High‐throughput mapping of a dynamic signaling network in mammalian cells. Science 307: 1621–1625 [DOI] [PubMed] [Google Scholar]
- Belyi Y, Tartakovskaya D, Tais A, Fitzke E, Tzivelekidis T, Jank T, Rospert S, Aktories K (2012) Elongation factor 1A is the target of growth inhibition in yeast caused by Legionella pneumophila glucosyltransferase Lgt1. J Biol Chem 287: 26029–26037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett TL, Kraft SM, Reaves BJ, Mima J, O'Brien KM, Starai VJ (2013) LegC3, an effector protein from Legionella pneumophila, inhibits homotypic yeast vacuole fusion in vivo and in vitro . PLoS ONE 8: e56798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyrakhova KA, van Straaten K, Li L, Boniecki MT, Anderson DH, Cygler M (2016) Structural and functional investigations of the effector protein LpiR1 from Legionella pneumophila . J Biol Chem 291: 15767–15777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR‐mediated gene disruption and other applications. Yeast 14: 115–132 [DOI] [PubMed] [Google Scholar]
- Braun P, Tasan M, Dreze M, Barrios‐Rodiles M, Lemmens I, Yu H, Sahalie JM, Murray RR, Roncari L, de Smet AS, Venkatesan K, Rual JF, Vandenhaute J, Cusick ME, Pawson T, Hill DE, Tavernier J, Wrana JL, Roth FP, Vidal M (2009) An experimentally derived confidence score for binary protein‐protein interactions. Nat Methods 6: 91–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein D, Amaro F, Zusman T, Lifshitz Z, Cohen O, Gilbert JA, Pupko T, Shuman HA, Segal G (2016) Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nat Genet 48: 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campodonico EM, Chesnel L, Roy CR (2005) A yeast genetic system for the identification and characterization of substrate proteins transferred into host cells by the Legionella pneumophila Dot/Icm system. Mol Microbiol 56: 918–933 [DOI] [PubMed] [Google Scholar]
- Certal V, Carry JC, Halley F, Virone‐Oddos A, Thompson F, Filoche‐Romme B, El‐Ahmad Y, Karlsson A, Charrier V, Delorme C, Rak A, Abecassis PY, Amara C, Vincent L, Bonnevaux H, Nicolas JP, Mathieu M, Bertrand T, Marquette JP, Michot N et al (2014) Discovery and optimization of pyrimidone indoline amide PI3Kbeta inhibitors for the treatment of phosphatase and tensin homologue (PTEN)‐deficient cancers. J Med Chem 57: 903–920 [DOI] [PubMed] [Google Scholar]
- Chien M, Morozova I, Shi S, Sheng H, Chen J, Gomez SM, Asamani G, Hill K, Nuara J, Feder M, Rineer J, Greenberg JJ, Steshenko V, Park SH, Zhao B, Teplitskaya E, Edwards JR, Pampou S, Georghiou A, Chou IC et al (2004) The genomic sequence of the accidental pathogen Legionella pneumophila . Science 305: 1966–1968 [DOI] [PubMed] [Google Scholar]
- Craig R, Beavis RC (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20: 1466–1467 [DOI] [PubMed] [Google Scholar]
- Dreze M, Monachello D, Lurin C, Cusick ME, Hill DE, Vidal M, Braun P (2010) High‐quality binary interactome mapping. Methods Enzymol 470: 281–315 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Ensminger AW (2016) Legionella pneumophila, armed to the hilt: justifying the largest arsenal of effectors in the bacterial world. Curr Opin Microbiol 29: 74–80 [DOI] [PubMed] [Google Scholar]
- Eschenfeldt WH, Lucy S, Millard CS, Joachimiak A, Mark ID (2009) A family of LIC vectors for high‐throughput cloning and purification of proteins. Methods Mol Biol 498: 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Felipe KS, Glover RT, Charpentier X, Anderson OR, Reyes M, Pericone CD, Shuman HA (2008) Legionella eukaryotic‐like type IV substrates interfere with organelle trafficking. PLoS Pathog 4: e1000117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Felipe KS, Pampou S, Jovanovic OS, Pericone CD, Ye SF, Kalachikov S, Shuman HA (2005) Evidence for acquisition of Legionella type IV secretion substrates via interdomain horizontal gene transfer. J Bacteriol 187: 7716–7726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S, Song O (1989) A novel genetic system to detect protein‐protein interactions. Nature 340: 245–246 [DOI] [PubMed] [Google Scholar]
- Fields BS (1996) The molecular ecology of Legionella . Trends Microbiol 4: 286–290 [DOI] [PubMed] [Google Scholar]
- Franco IS, Shuman HA, Charpentier X (2009) The perplexing functions and surprising origins of Legionella pneumophila type IV secretion effectors. Cell Microbiol 11: 1435–1443 [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH (2007a) High‐efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2: 31–34 [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH (2007b) Microtiter plate transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2: 5–8 [DOI] [PubMed] [Google Scholar]
- Guo Z, Stephenson R, Qiu J, Zheng S, Luo ZQ (2014) A Legionella effector modulates host cytoskeletal structure by inhibiting actin polymerization. Microbes Infect 16: 225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Haar T (2007) Optimized protein extraction for quantitative proteomics of yeasts. PLoS ONE 2: e1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havey JC, Roy CR (2015) Toxicity and SidJ‐mediated suppression of toxicity require distinct regions in the SidE family of Legionella pneumophila effectors. Infect Immun 83: 3506–3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidtman M, Chen EJ, Moy MY, Isberg RR (2009) Large‐scale identification of Legionella pneumophila Dot/Icm substrates that modulate host cell vesicle trafficking pathways. Cell Microbiol 11: 230–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman JD, Tabb DL, Mallick P (2014) Employing proteowizard to convert raw mass spectrometry data. Curr Protoc Bioinformatics 46: 13.24.1–13.24.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino D, Takakura Y, Fukano K, Sasaki Y, Hoshino T, Ohsawa K, Nakamura A, Yajima S (2013) Crystal structures of the ternary complex of APH(4)‐Ia/Hph with hygromycin B and an ATP analog using a thermostable mutant. J Struct Biol 183: 76–85 [DOI] [PubMed] [Google Scholar]
- Isaac DT, Isberg R (2014) Master manipulators: an update on Legionella pneumophila Icm/Dot translocated substrates and their host targets. Future Microbiol 9: 343–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong KC, Sexton JA, Vogel JP (2015) Spatiotemporal regulation of a Legionella pneumophila T4SS substrate by the metaeffector SidJ. PLoS Pathog 11: e1004695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4: 363–371 [DOI] [PubMed] [Google Scholar]
- Klima M, Baumlova A, Chalupska D, Hrebabecky H, Dejmek M, Nencka R, Boura E (2015) The high‐resolution crystal structure of phosphatidylinositol 4‐kinase IIbeta and the crystal structure of phosphatidylinositol 4‐kinase IIalpha containing a nucleoside analogue provide a structural basis for isoform‐specific inhibitor design. Acta Crystallogr D Biol Crystallogr 71: 1555–1563 [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubori T, Shinzawa N, Kanuka H, Nagai H (2010) Legionella metaeffector exploits host proteasome to temporally regulate cognate effector. PLoS Pathog 6: e1001216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G, Cohen SX, Lamzin VS, Perrakis A (2008) Automated macromolecular model building for X‐ray crystallography using ARP/wARP version 7. Nat Protoc 3: 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM (2005) Parkin mediates nonclassical, proteasomal‐independent ubiquitination of synphilin‐1: implications for Lewy body formation. J Neurosci 25: 2002–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Luo ZQ (2007) The Legionella pneumophila effector SidJ is required for efficient recruitment of endoplasmic reticulum proteins to the bacterial phagosome. Infect Immun 75: 592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zhang J, Choi H, Lambert JP, Srikumar T, Larsen B, Nesvizhskii AI, Raught B, Tyers M, Gingras AC (2012) Using ProHits to store, annotate, and analyze affinity purification‐mass spectrometry (AP‐MS) data. Curr Protoc Bioinformatics Chapter 8: Unit8 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losick VP, Haenssler E, Moy MY, Isberg RR (2010) LnaB: a Legionella pneumophila activator of NF‐kappaB. Cell Microbiol 12: 1083–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M (2006) HKL‐3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62: 859–866 [DOI] [PubMed] [Google Scholar]
- Molmeret M, Horn M, Wagner M, Santic M, Abu Kwaik Y (2005) Amoebae as training grounds for intracellular bacterial pathogens. Appl Environ Microbiol 71: 20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Liu X, Arasaki K, McDonough J, Galan JE, Roy CR (2011) Modulation of Rab GTPase function by a protein phosphocholine transferase. Nature 477: 103–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller MP, Peters H, Blumer J, Blankenfeldt W, Goody RS, Itzen A (2010) The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science 329: 946–949 [DOI] [PubMed] [Google Scholar]
- Neunuebel MR, Chen Y, Gaspar AH, Backlund PS Jr, Yergey A, Machner MP (2011) De‐AMPylation of the small GTPase Rab1 by the pathogen Legionella pneumophila . Science 333: 453–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevo O, Zusman T, Rasis M, Lifshitz Z, Segal G (2014) Identification of Legionella pneumophila effectors regulated by the LetAS‐RsmYZ‐CsrA regulatory cascade, many of which modulate vesicular trafficking. J Bacteriol 196: 681–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter J, Merritt EA (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr 62: 439–450 [DOI] [PubMed] [Google Scholar]
- Pazos F, Valencia A (2001) Similarity of phylogenetic trees as indicator of protein‐protein interaction. Protein Eng 14: 609–614 [DOI] [PubMed] [Google Scholar]
- Proctor M, Urbanus ML, Fung EL, Jaramillo DF, Davis RW, Nislow C, Giaever G (2011) The automated cell: compound and environment screening system (ACCESS) for chemogenomic screening. Methods Mol Biol 759: 239–269 [DOI] [PubMed] [Google Scholar]
- Qin T, Cui Y, Cen Z, Liang T, Ren H, Yang X, Zhao X, Liu Z, Xu L, Li D, Song Y, Yang R, Shao Z, Song Y (2012) Draft genome sequences of two Legionella dumoffii strains, TEX‐KL and NY‐23. J Bacteriol 194: 1251–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Sheedlo MJ, Yu K, Tan Y, Nakayasu ES, Das C, Liu X, Luo ZQ (2016) Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 533: 120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaile AT, Urbanus ML, Stogios PJ, Nocek B, Skarina T, Ensminger AW, Savchenko A (2015) Molecular characterization of LubX: functional divergence of the U‐box fold by Legionella pneumophila . Structure 23: 1459–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao C, Benhabib H, Ensminger AW (2013) Phylogenetic reconstruction of the Legionella pneumophila Philadelphia‐1 laboratory strains through comparative genomics. PLoS ONE 8: e64129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum G, Alkire RW, Evans G, Rotella FJ, Lazarski K, Zhang RG, Ginell SL, Duke N, Naday I, Lazarz J, Molitsky MJ, Keefe L, Gonczy J, Rock L, Sanishvili R, Walsh MA, Westbrook E, Joachimiak A (2006) The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J Synchrotron Radiat 13: 30–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowbotham TJ (1980) Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J Clin Pathol 33: 1179–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Yang J, Zhang Y (2012) COFACTOR: an accurate comparative algorithm for structure‐based protein function annotation. Nucleic Acids Res 40: W471–W477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069 [DOI] [PubMed] [Google Scholar]
- Shames SR, Finlay BB (2012) Bacterial effector interplay: a new way to view effector function. Trends Microbiol 20: 214–219 [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr D Biol Crystallogr 66: 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Halder P, Yavuz H, Jahn R, Shuman HA (2016) Direct targeting of membrane fusion by SNARE mimicry: convergent evolution of Legionella effectors. Proc Natl Acad Sci USA 113: 8807–8812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohdy N, Efe JA, Emr SD, Shuman HA (2005) Pathogen effector protein screening in yeast identifies Legionella factors that interfere with membrane trafficking. Proc Natl Acad Sci USA 102: 4866–4871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J (2005) Protein homology detection by HMM‐HMM comparison. Bioinformatics 21: 951–960 [DOI] [PubMed] [Google Scholar]
- Studier FW (2005) Protein production by auto‐induction in high density shaking cultures. Protein Expr Purif 41: 207–234 [DOI] [PubMed] [Google Scholar]
- Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S (2012) Quantitative analysis of HSP90‐client interactions reveals principles of substrate recognition. Cell 150: 987–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Tucker G, Peng J, Krykbaeva I, Lin ZY, Larsen B, Choi H, Berger B, Gingras AC, Lindquist S (2014) A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 158: 434–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Luo ZQ (2011) Legionella pneumophila SidD is a deAMPylase that modifies Rab1. Nature 475: 506–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Arnold RJ, Luo ZQ (2011) Legionella pneumophila regulates the small GTPase Rab1 activity by reversible phosphorylcholination. Proc Natl Acad Sci USA 108: 21212–21217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey WP (2007) Denaturing protein immunoprecipitation from Mammalian cells. CSH Protoc 2007: pdb prot4619 [DOI] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, Andrews B, Tyers M, Boone C (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368 [DOI] [PubMed] [Google Scholar]
- Tong AH, Boone C (2006) Synthetic genetic array analysis in Saccharomyces cerevisiae . Methods Mol Biol 313: 171–192 [DOI] [PubMed] [Google Scholar]
- Toulabi L, Wu X, Cheng Y, Mao Y (2013) Identification and structural characterization of a Legionella phosphoinositide phosphatase. J Biol Chem 288: 24518–24527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, O'Kelly G, Schoenegger A, Ovelleiro D, Perez‐Riverol Y, Reisinger F, Rios D, Wang R, Hermjakob H (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res 41: D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagih O, Usaj M, Baryshnikova A, VanderSluis B, Kuzmin E, Costanzo M, Myers CL, Andrews BJ, Boone CM, Parts L (2013) SGAtools: one‐stop analysis and visualization of array‐based genetic interaction screens. Nucleic Acids Res 41: W591–W596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber S, Dolinsky S, Hilbi H (2013) Interactions of Legionella effector proteins with host phosphoinositide lipids. Methods Mol Biol 954: 367–380 [DOI] [PubMed] [Google Scholar]
- Weissenmayer BA, Prendergast JG, Lohan AJ, Loftus BJ (2011) Sequencing illustrates the transcriptional response of Legionella pneumophila during infection and identifies seventy novel small non‐coding RNAs. PLoS ONE 6: e17570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ (2010) Inhibition of host vacuolar H+‐ATPase activity by a Legionella pneumophila effector. PLoS Pathog 6: e1000822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Medeiros PF, Raha K, Elkins P, Lind KE, Lehr R, Adams ND, Burgess JL, Schmidt SJ, Knight SD, Auger KR, Schaber MD, Franklin GJ, Ding Y, DeLorey JL, Centrella PA, Mataruse S, Skinner SR, Clark MA, Cuozzo JW et al (2015) Discovery of a potent class of PI3Kalpha inhibitors with unique binding mode via Encoded Library Technology (ELT). ACS Med Chem Lett 6: 531–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane‐Kishikawa T, Gebreab F, Li N, Simonis N, Hao T, Rual JF, Dricot A, Vazquez A, Murray RR, Simon C, Tardivo L, Tam S, Svrzikapa N, Fan C et al (2008) High‐quality binary protein interaction map of the yeast interactome network. Science 322: 104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wu J, Yang J, Sun S, Xiao J, Yu J (2012) PGAP: pan‐genomes analysis pipeline. Bioinformatics 28: 416–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li J, Yu H, Zhai Y, Gao Z, Liu Y, Pang X, Zhang L, Schulten K, Sun F, Chen C (2014) Molecular insights into the membrane‐associated phosphatidylinositol 4‐kinase IIalpha. Nat Commun 5: 3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Table EV9
Review Process File
Data Availability Statement
Coordinates of Lem14 (4HFV), RavJ 1–225 (4RXV), RavJ 251–371 (4RXI), the complex structure of RavJ (1–230) and full‐length LegL1 (4XA9), and LupA 123–403 (5DGG) have been deposited in the Protein Data Bank. SGA output files from the genetic interaction screen, including raw spot size information, have been deposited into the Dryad Digital Repository (doi: 10.5061/dryad.kj666).