

Abstract
We propose a new approach for the synthesis of biologically important nucleotides which includes a multi-enzymatic cascade conversion of D-pentoses into purine nucleotides. The approach exploits nucleic acid exchange enzymes from thermophilic microorganisms: ribokinase, phosphoribosylpyrophosphate synthetase, and adenine phosphoribosyltransferase. We cloned the ribokinase gene from Thermus sp. 2.9, as well as two different genes of phosphoribosylpyrophosphate synthetase (PRPP-synthetase) and the adenine phosphoribosyltransferase (APR-transferase) gene from Thermus thermophilus HB27 into the expression vectors, generated high-yield E. coli producer strains, developed methods for the purification of the enzymes, and investigated enzyme substrate specificity. The enzymes were used for the conversion of D-pentoses into 5-phosphates that were further converted into 5-phospho-α-D-pentofuranose 1-pyrophosphates by means of ribokinase and PRPP-synthetases. Target nucleotides were obtained through the condensation of the pyrophosphates with adenine and its derivatives in a reaction catalyzed by APR-transferase. 2-Chloro- and 2-fluoroadenosine monophosphates were synthesized from D-ribose and appropriate heterobases in one pot using a system of thermophilic enzymes in the presence of ATP, ribokinase, PRPP-synthetase, and APR-transferase.
Keywords: enzymatic nucleotide synthesis, ribokinase, phosphoribosylpyrophosphate synthetase, adenine phosphoribosyltransferase, thermophilic microorganisms, substrate properties
INTRODUCTION
5’-phosphorylated nucleosides are important metabolites of DNA and RNA biosynthesis, as well as co-substrates and cofactors of numerous biochemical reactions [1-3]. The important role these compounds play in a living cell underlies the interest in the synthesis of not only natural members of this class, but also their various analogues to directly affect the metabolism in normal and pathological conditions [4-8]. A large number of heterobase- and sugar-modified nucleosides are used as important antiviral and anticancer agents [9-13]. The effect of modified nucleosides is mediated through their intracellular conversion primarily into 5’-monophosphates and then usually into 5’-di- and triphosphates that act as antimetabolites. The first step in the metabolic activation of modified nucleosides – conversion into 5’-monophosphates – is known to determine the nucleoside’s biological properties. It should also be noted that heterobase- and/or sugar-modified nucleoside-5’-monophosphates are of considerable interest as starting materials for the chemical synthesis of phosphate derivatives (prodrugs) and enzymatic conversion into 5’-triphosphates for subsequent inclusion in oligonucleotides [14-16]. The development of effective biosynthetic approaches to the production of 5’-monophosphates of modified nucleosides draws the attention of researchers engaged in the development of highly efficient chemotherapeutic agents.
Mono- and multi-enzymatic synthesis of nucleoside 5’-mono- and 5’-triphosphates has been the subject of numerous publications [17-21]. Of these, we were particularly interested in the phosphoribosyltransferases that were recently successfully used in a cascade five-component synthesis of purine riboside-5’-monophosphates [22-24]. Nucleoside phosphorylases of thermophilic microorganisms are known to be less sensitive to the substrate structure [25, 26], which enables them to act at 70–80°C and significantly increases the enzymatic reaction efficiency due to the increased solubility of heterocyclic substrates [27]. All these data aroused our interest in the purification of the recombinant enzymes ribokinase, phosphoribosylpyrophosphate synthetase, and adenine phosphoribosyltransferase from thermophilic microorganisms and in the investigation of their substrate properties to determine the applicability of these enzymes in the cascade synthesis of purine nucleoside-5-monophosphates according to the Diagram in Fig. 1.
Fig. 1.

A diagram of the multi-enzymatic cascade synthesis of substituted adenosine-5’-monophosphates.
MATERIALS AND METHODS
Cloning
Genes TT_RS05985, TT_RS06430, and TT_RS06315 encoding TthPRPPS1, TthPRPPS2, and TthAPRT, respectively, were amplified on the genomic DNA template of the Thermus thermophilus HB27 strain by a polymerase chain reaction (PCR) using synthetic primers. The QT17_05185 gene encoding RK from Thermus sp. 2.9 was codon-optimized for expression in Escherichia coli and synthesized by a chemical and enzymatic method from overlapping oligonucleotides. All genes were cloned into the expression vector pET-23d+ at the NcoI and XhoI restriction endonuclease recognition sites.
Cultivation of producer strains
E. coli BL21(DE3), Rosetta (DE3), and C3029/pGTf2 strains were transformed with the produced expression vectors pER-PRPPS1-Tth, pER-PRPPS2-Tth, pER-APRT-Tth, and pER-RK-Tsp. Producer strains derived from E. coli BL21(DE3) and Rosetta(DE3) were cultured at 37 °C in a LB medium containing ampicillin (100 μg/mL). Cultivation of the producer strains derived from E. coli C3029/pGTf2 was carried out in a LB medium containing 50 μg/mL ampicillin, 20 μg/mL chloramphenicol, and 1 ng/mL tetracycline. After reaching a absorbance of A595 = 0.8, the cultures were added with IPTG to a final concentration of 0.4 mM and cultivation was continued at 23 and 37 °C. The cultivation duration varied from 4 to 16 h, depending on the strain. After culturing, the cell biomass was separated by centrifugation, homogenized at a 1:10 (w/v) ratio in a buffer solution (50 mM Tris-HCl, pH 8.5, 150 mM NaCl, 2 mM PMSF), and disrupted using a Labsonic P ultrasonicator (Sartorius, Germany) at 4°C for 10 min (cycle, 0.4 sec; amplitude, 30%). The amount of target enzymes in soluble and insoluble cell fractions was determined by densitometric analysis of electrophoretic gels using the ImageLab 5.0 software (Bio-Rad, USA) [28]. The producer strains containing the maximum amount of a targeted protein in the supernatant were chosen: E. coli BL21(DE3)/pER-APRT-Tth (culturing at 37 °C for 4 h after adding IPTG), E. coli Rosetta(DE3)/pER-PRPPS1-Tth (37 °C for 4 h), E. coli S3029/pGTf2/pER-PRPPS2-Tth (37 °C for 5 h), and E. coli S3029/pGTf2/pER-RK-Tsp (23 °C for 16 h). The strains were grown in 5–6 L of the culture medium.
Isolation and purification of TthPRPPS1,
Cell biomass of the producer strains TthPRPPS1, TthPRPPS2, and TspRK was re-suspended in a buffer solution (50 mM Tris-HCl, pH 8.7, 1 mM PMSF) at a 1:10 (w/v) ratio and disrupted using the Labsonic P ultrasonicator at +4 °C for 20 min (cycle, 0.5 sec; amplitude, 50%). Cell debris was removed by centrifugation at 12,000 rpm at +4 °C for 30 min on a Hermle Z383K centrifuge (HERMLE Labortechnik GmbH, Germany). In purification of TspRK, a clarified cell lysate was heat-treated at 65 °C for 10 min to precipitate contaminating proteins and DNA. The precipitate was removed by centrifugation. Further purification of the enzymes was carried out according to the same scheme. A clarified cell lysate was loaded onto a XK 16/20 column (GE Healthcare, USA) with Ni2+-IDA resin (Qiagen, Germany) pre-equilibrated with 50 mM Tris-HCl buffer, pH 8.7. Ballast proteins were removed by washing with 50 mM Tris-HCl buffer, pH 8.7, containing 50 mM imidazole. A target protein was eluted with a buffer solution of 50 mM Tris-HCl and 200 mM imidazole, pH 8.7. After affinity chromatography, fractions containing a target protein were added with EDTA to a concentration of 5 mM and concentrated using an Amicon 8200 200 mL stirred ultrafiltration cell (Millipore, USA) on an YM 10 kDa membrane (Millipore) when purified TthPRPPS1 and TthPRPPS2 and on an YM 30 kDa membrane (Millipore) when purified TspRK. Further purification was performed on a HiLoad 16/60 column with Superdex 200 resin (GE Healthcare) equilibrated with buffer containing 20 mM Tris-HCl, 1 mM ATP, 1 mM MgCl2, 5% glycerol, 0.04% NaN3, pH 8.5. Fractions containing a target protein were pooled and concentrated by ultrafiltration to a final concentration of 12 ± 1 mg/mL as previously described. The protein concentration was determined by the Bradford method using BSA as a standard [29]. Protein purity was determined by polyacrylamide gel electrophoresis under denaturing conditions [28]. Purified enzymes were stored at –80 °C.
Isolation and purification of TthAPRT
Cell biomass of the TthAPRT producer strain was disrupted according to the procedure described for the other enzymes. A clarified cell lysate was added with NaCl to a concentration of 300 mM and heat-treated at 65 °C for 10 min. After ballast protein precipitation by centrifugation, the lysate was applied to a PD-10 column with Sephadex G-25 Medium resin (GE Healthcare, USA) equilibrated with a buffer solution containing 20 mM Tris-HCl and 1.0 mM EDTA, pH 9.0. After desalting, the protein solution was loaded onto a XK 16/20 column with Q Sepharose XL (GE Healthcare, USA) equilibrated with the same buffer solution. A target protein was eluted with a linear concentration gradient of NaCl (0 to 400 mM). Fractions containing the target protein were pooled and loaded onto an XK 16/20 column with Phenyl Sepharose HP (GE Healthcare, USA) equilibrated with buffer containing 20 mM Tris-HCl, 1 M (NH4)2SO4, 1.0 mM EDTA, pH 7.6. TthAPRT was eluted with a linear gradient of (NH4)2SO4 (from 1 to 0 M). Fractions containing the target protein were pooled and concentrated by ultrafiltration on a PBGC 10 kDa polysulfone membrane to a final concentration of 5.0 ± 0.5 mg/mL as previously described. The final purification was performed on a HiLoad 16/60 column with Superdex 200 resin equilibrated with 20 mM Tris-HCl buffer, pH 8.0, containing 50 mM NaCl, 5% glycerol, and 0.04% NaN3. Fractions containing the target protein were pooled and concentrated by ultrafiltration to a final concentration of 12 ± 1 mg/mL. The protein purity and concentration were determined as described previously [28, 29]. The purified enzyme was stored at –80 °C.
Enzymatic activity assay
The TspRK activity was determined radiochemically based on the formation of D-ribofuranosyl-5-[32P]phosphate in the presence of [γ-32P]ATP. A reaction mixture (0.05 mL, 20 mM Tris-HCl, pH 8.0) contained a 0.4 mM disodium ATP salt, 1 mM D-ribose, 5 mM MgCl2, 50 mM KCl, 1 mM KH2PO4, 6 μCi of [γ-32P]ATP, and 0.15 μg of TspRK. The mixture was incubated at 75 °C. Then, 0.8 μL aliquots were taken at 10, 20, and 40 min, applied to plates with PEI-cellulose, and eluted with 0.5 M aqueous potassium dihydrogen orthophosphate. The amount of D-ribofuranosyl-5-[32P]phosphate was determined on a TRI-CARB 2100TR liquid scintillation counter (Packard BioScience Co.).
The TthPRPPS1 and TthPRPPS2 activity was determined in a reaction mixture containing a 1 mM disodium ATP salt, a 1 mM disodium salt of D-ribose-5 phosphate, 5 mM MgCl2, 10 mM KH2PO4, 20 mM Tris- HCl, pH 8.0, at 75 °C. TthPRPPS (0.75 μg) was added to 0.5 mL of the mixture.
The TthAPRT activity was determined in a reaction mixture containing 1 mM adenine, a 1 mM pentasodium salt of 5-phosphoribosyl-α-1-pyrophosphate (PRPP), 5 mM MgCl2, and 20 mM Tris-HCl, pH 8.0, at 75 °C. TthAPRT (0.125 μg) was added to 0.5 mL of the reaction mixture. The amount of the product (μM) formed for 1 min was taken as the activity unit.
Determination of ribokinase kinetic parameters
A reaction mixture (0.5 mL, 20 mM Tris-HCl, pH 8.0) contained (a) a disodium salt of ATP (0.01 to 0.6 mM) and D-ribose (1 mM) or (b) D-ribose (0.01 to 8.0 mM) and ATP (1 mM) to determine the KM and Vmax values for ATP and D-ribose, respectively, 5 mM MgCl2, 50 mM KCl, 10 mM KH2PO4, and 0.0175 μg of TspRK. Each experiment used 16 reaction mixtures. The mixtures were incubated at 75 °C for 12 min; each experiment was performed in triplicate. Then, the mean rate was determined in three experiments at the same enzyme concentration. Substrate and product concentrations were determined by HPLC under isocratic elution with 0.1 M KH2PO4 (water, pH 6.0, flow rate of 0.5 mL/min) with detection at 254 nm (Waters 2489 UV detector; Supelcosil LC-18-T column, 5 μm, 150 × 4.6 mm).
Determination of the kinetic parameters of PRPP-synthetases
The kinetic parameters of the enzymes were determined at a ATP concentration that varied in the range of 0.005 to 0.2 mM. A similar interval was used for D-ribose-5 phosphate. The remaining conditions were the same as those in the enzymatic activity assay. The reaction was conducted for 2 min in triplicate. Then, the mean rate was determined in three experiments at the same concentration. Concentrations of ATP and AMP were determined by HPLC under isocratic elution with 0.1 M KH2PO4 (pH 6.0, flow rate of 0.5 mL/min) with detection at 254 nm (Waters 2489 detector; Supelcosil LC-18-T column, 5 μm, 150 × 4.6 mm).
Determination of the kinetic parameters of APR-transferase
The kinetic parameters of APR-transferase were determined at an adenine concentration that varied in the range of 0.005 to 0.2 mM and a PRPP concentration that varied from 0.05 to 1.2 mM. The remaining conditions were the same as in the enzymatic activity assay. The reaction was conducted for 1 min in triplicate. The mean reaction rate was determined based on the results of three experiments at the same concentration. Adenine and AMP concentrations were determined by HPLC under isocratic elution with 36% aqueous methanol (flow rate of 0.5 mL/min) with detection at 254 nm (Waters 2489 detector; MZ PerfectSil 100 ODS-3 column, 5 μm, 150 × 4.6 mm).
The kinetic parameters were determined by a nonlinear regression analysis using the SciDAVis v.0.2.4 software. The apparent catalytic constant (kcat) was calculated per one subunit, whose weight was determined based on the amino acid sequence (32.0 kDa for TspRK, 34.5 kDa for TthPRPPS1, 34.6 kDa for TthPRPPS2, and 19.0 kDa for TthAPRT).
RESULTS AND DISCUSSION
To study the possibility of three-step nucleotide biosynthesis, we carried out comprehensive work on the production of recombinant enzymes of the nucleic acid metabolism (ribokinase, two PRPP-synthetases, and APR-transferase from T. thermophilus) and an investigation of their substrate properties
The QT17_05185 gene encoding RK from Thermus sp. 2.9 was generated by a chemical-enzymatic method. Genes TT_RS05985, TT_RS06430, and TT_RS06315 encoding TthPRPPS1, TthPRPPS2, and TthAPRT from T. thermophilus HB27, respectively, were amplified from the genomic DNA by PCR. All genes were cloned into the plasmid vector pET23d+. The resulting expression vectors pER-PRPPS1-Tth, pER-PRPPS2- Tth, and pER-RK-Tsp contained hybrid genes with a reading frame including sequences encoding enzymes and an affinity tag of six histidine residues. The expression vector pER-APRT1-Tth contained an unmodified sequence encoding TthAPRT.
The plasmids were used to transform the E. coli strains BL21(DE3), Rosetta(DE3), and C3029/pGTf2. We determined the culture conditions under which producer strains synthesized target enzymes in soluble form. Producers of soluble recombinant TspRK and TthPRPPS2 were derived from E. coli S3029/pGTf2. This strain was generated by transforming E. coli S3029 cells with the plasmid vector pGTf2 (Takara Bio Inc) carrying sequences encoding GroES-GroEL-Tig chaperones under the control of the Pzt-1 promoter. The choice of the E. coli Rosetta(DE3) strain for TthPRPPS1 biosynthesis was based on the fact that the TT_RS05985 gene encoding TthPRPPS1 contains codons rarely used in E. coli (one AGA, nine AGGs, 10 CGGs, one AUA, one CUA, and 15 CCCs). The Rosetta(DE) strain synthesizes tRNAs for these rare codons. Soluble recombinant TthAPRT was produced in the E. coli BL21(DE3) strain.
Isolation and purification of ribokinase and two PRPP-synthetases included metal chelate and gel filtration chromatography stages (Table 1). Heat treatment of the cell lysate during TspRK purification significantly enriched the target protein fraction due to aggregation of cellular proteins and DNA. A similar treatment of both TthPRPP-synthetases did not provide positive results, but it led to a loss of the target protein. Due to the high probability of proteolysis, metal chelate chromatography was conducted under cooling conditions, with addition of EDTA to the pooled fractions. Following the final purification step using gel filtration chromatography, all protein samples were concentrated.
Table 1.
Steps of the isolation and purification of ribokinase, PRPP-synthetases, and APR-transferase
| Purification step | Volume, mL | Protein concentration, mg/mL | Total protein, mg |
|---|---|---|---|
| Ribokinase from Thermus sp. 2.9 | |||
| 1. Ultrasonic disintegration | 150* | 8.3 | 1245 |
| 2. Heat treatment | 136 | 1.7 | 231.2 |
| 3. Metal chelate chromatography | 30 | 1.9 | 57 |
| 4. Concentrating | 9 | 6 | 54 |
| 5. Gel filtration chromatography | 28.5 | 1.28 | 36.4 |
| 6. Concentrating | 2.6 | 11.7 | 30.4 |
| PRPP-synthetase 1 from T. thermophilus HB27 | |||
| 1. Ultrasonic disintegration | 150** | 10 | 1500 |
| 2. Affinity chromatography | 100 | 0.8 | 80 |
| 3. Concentrating | 13 | 5.7 | 74.1 |
| 4. Gel filtration | 50 | 1 | 50 |
| 5. Concentrating | 4 | 12.3 | 49.2 |
| PRPP-synthetase 2 from T. thermophilus HB27 | |||
| 1. Ultrasonic disintegration | 130** | 11.2 | 1456 |
| 2. Affinity chromatography | 125 | 0.6 | 75 |
| 3. Concentrating | 10.3 | 6.2 | 63.9 |
| 4. Gel filtration | 37.5 | 1.2 | 45 |
| 5. Concentrating | 3.4 | 12 | 40.8 |
| APR-transferase from T. thermophilus HB27 | |||
| 1. Ultrasonic disintegration | 157*** | 6 | 942 |
| 2. Heat treatment | 149 | 1.3 | 193.7 |
| 3. Gel filtration | 258 | 0.7 | 181 |
| 4. Anion exchange chromatography | 72 | 1.2 | 86.4 |
| 5. Hydrophobic interaction chromatography | 73 | 0.8 | 58.4 |
| 6. Concentrating | 11 | 5 | 55 |
| 7. Gel filtration chromatography | 52 | 0.87 | 45.2 |
| 8. Concentrating | 3.4 | 12.5 | 42.5 |
*From 5.8 L of culture.
**From 5 L of culture.
***From 6 L of culture.
Isolation and purification of TthAPRT included heat treatment, desalting, concentrating, and anion exchange, hydrophobic interaction, and gel filtration chromatography steps (Table 1). As in the case of TspRK, the first step involved heat treatment of a clarified cell lysate with addition of a salt to the solution (up to 300 mM sodium chloride), which greatly accelerated the aggregation of contaminating cell proteins and DNA.
The developed techniques provided a yield of all recombinant thermophilic enzymes not less than 8–10 mg per 1 L of culture medium, with an electrophoretic purity of at least 95%.
Next, we determined the kinetic parameters and optimal activity conditions for the purified recombinant enzymes.
The specific activity of TspRK was determined by a radiochemical method based on the formation of D-ribofuranosyl-5-[32P]phosphate in the presence of [γ-32P] ATP. The amount of the product (μM) formed for 1 min was taken as the activity unit. The TspRK activity was 5.5 U/mg.
The specific activity of TthPRPPS1 and TthPRPPS2 was determined indirectly via AMP formation in a reaction mixture (according to HPLC) in the presence of two substrates: ATP and D-ribose-5-phosphate. The activity was 0.85 U/mg for the former enzyme and 11 U/mg for the latter enzyme. Given the large differences in the enzyme activity, using the second synthetase for nucleotide synthesis seems to be more rational, with allowance for lower protein consumption. The TthAPRT properties were studied in a model reaction of adenine with 5-phosphoribosyl-α-1-pyrophosphate. The AMP formation was monitored by HPLC. The specific enzyme activity was 8.8 U/mg.
A high concentration of ribose as a substrate was found to inhibit the enzymatic activity of TspRK. This effect was described for human ribokinase [30]. Therefore, the results of ATP experiments were analyzed using the Michaelis-Menten equation V = Vmax × S / (KM + S). In experiments with D-ribose, we used an equation allowing for the substrate inhibition effect due to the binding of a second molecule V = Vmax × S / (KM + S + S2 / Ki).
The kinetic parameters of natural substrates of the enzymes are listed in Table 2.
Table 2.
Kinetic parameters of the natural substrates of the studied enzymes
| Substrate | KM, μM | Ki, μM | Vmax, μM/min·mg | kcat, 1/s | kcat/KM, 1/M·s |
|---|---|---|---|---|---|
| Ribokinase from Thermus sp. 2.9 | |||||
| ATP | 75 ± 11 | – | 13 ± 1 | 6.8 ± 0.7 | 9.1 × 104 |
| D-ribose | 20 ± 6 | 1,700 ± 400 | 13 ± 2 | 7.1 ± 1.2 | 3.5 × 105 |
| PRPP-synthetase 1 from T. thermophilus HB27 | |||||
| ATP | 10 ± 2 | – | 0.71 ± 0.05 | 0.41 ± 0.03 | 4.3 × 104 |
| D-ribose-5 phosphate | 32 ± 6 | – | 0.85 ± 0.11 | 0.49 ± 0.06 | 1.5 × 104 |
| PRPP-synthetase 2 from T. thermophilus HB27 | |||||
| ATP | 12 ± 2 | – | 20 ± 2 | 11 ± 1 | 9.9 × 105 |
| D-ribose-5 phosphate | 40 ± 4 | – | 24 ± 2 | 14 ± 1 | 3.4 × 105 |
| APR-transferase from T. thermophilus HB27 | |||||
| Adenine | 13 ± 2 | – | 6.0 ± 0.4 | 1.9 ± 0.1 | 1.4 × 105 |
| PRPP | 179 ± 35 | – | 9.2 ± 1.1 | 2.9 ± 0.4 | 1.6 × 104 |
Figure 2, Figure 3, Figure 4 present the results of experiments on exploring optimal conditions for the activity of the recombinant enzymes.
Fig. 2.
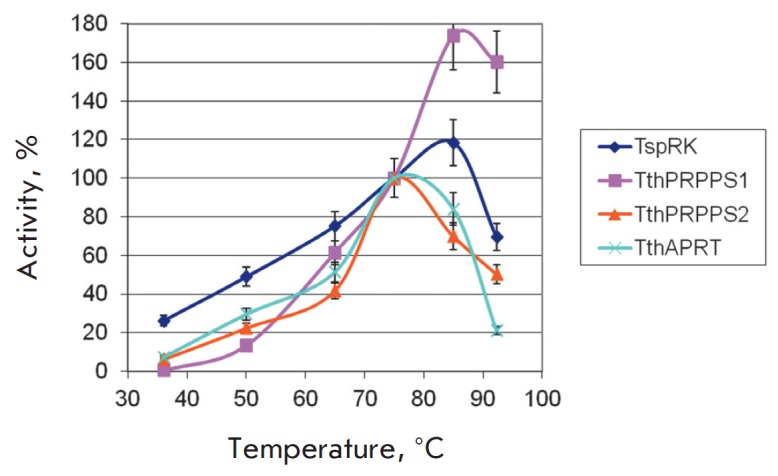
The temperature dependence of the enzyme activity. The activity at 75 °C was taken as 100%. Reactions were conducted in a temperature range from 36 to 92 °C in 0.05 mL of 20 mM Tris-HCl buffer, pH 8.0, containing: 1) 0.4 mM ATP, 1 mM ribose, 1 mM KH2PO4, 5 mM MgCl2, 50 mM KCl, 0.15 μg of TspRK, 2) 1 mM ATP, 1 mM D-ribose 5-phosphate, 5 mM MgCl2, 10 mM KH2PO4, 0.75 μg of TthPRPPS1 or TthPRPPS2, 3) 1 mM adenine, 1 mM PRPP, 5 mM MgCl2, 0.125 μg of TthAPRT.
Fig. 3.
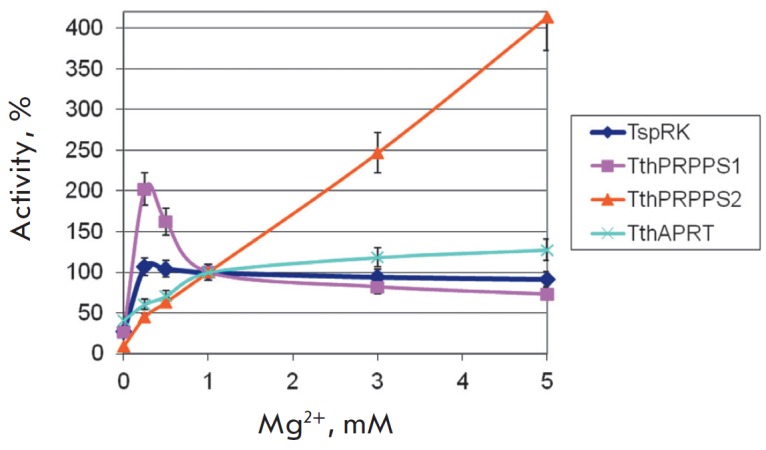
The dependence of enzyme activity on the magnesium ion concentration. The activity in the presence of 1 mM Mg2+ was taken as 100%. Reactions were conducted at a temperature of 75°C, at a MgCl2 concentration that varied from 0 to 5 mM, in 0.5 mL of 20 mM Tris-HCl buffer, pH 8.0, containing: 1) 0.4 mM ATP, 1 mM ribose, 1 mM KH2PO4, 50 mM KCl, 0.15 μg of TspRK, 2) 1 mM ATP, 1 mM D-ribose 5-phosphate, 10 mM KH2PO4, 0.75 μg of TthPRPPS1 or TthPRPPS2, 3) 1 mM adenine, 1 mM PRPP, 0.125 μg of TthAPRT.
Fig. 4.
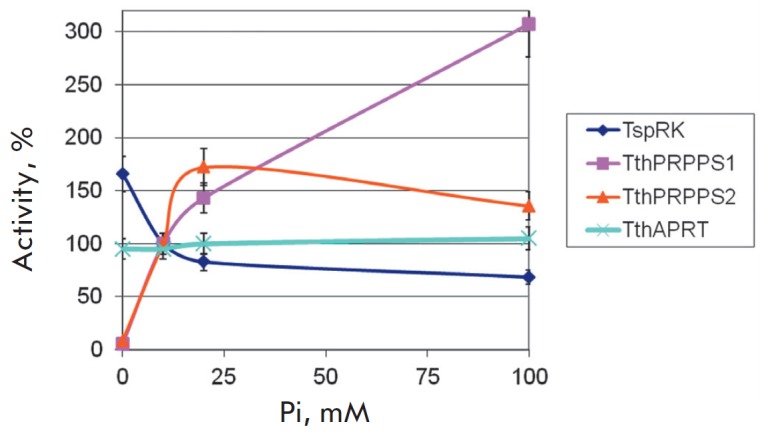
The dependence of enzyme activity on the inorganic phosphate concentration. The activity in the presence of 10 mM Pi was taken as 100%. Reactions were conducted at a temperature of 75°C, at a KH2PO4 concentration that varied from 0 to 100 mM, in 0.5 mL of 20 mM Tris-HCl buffer, pH 8.0, 5 mM MgCl2, containing: 1) 0.4 mM ATP, 1 mM ribose, 50 mM KCl, 0.15 μg of TspRK, 2) 1 mM ATP, 1 mM D-ribose 5-phosphate, 0.75 μg of TthPRPPS1 or TthPRPPS2, 3) 1 mM adenine, 1 mM PRPP, 0.125 μg of TthAPRT.
The studied enzymes were active in a broad temperature range. The maximum activity of TspRK and TthPRPPS1 was observed at 85°C, whereas the TthAPRT activity at this temperature was lower by 35%. Therefore, we decided to conduct further cascade syntheses at 75°C.
The activity of the enzymes in a buffer solution lacking magnesium ions was very low. Addition of magnesium chloride to a concentration of 0.25 mM led to a considerable increase in the activity.
At higher concentrations, the dependence of enzyme activity on the magnesium ion concentration was as follows: the activity decreased in the case of TspRK and TthPRPPS1, increased in TthAPRT, and drastically increased in TthPRPPS2. Thus, the cascade reactions involving TthPRPPS1 should be conducted at a magnesium chloride concentration of 0.25–0.5 mM; and those involving TthPRPPS2 – at 5 mM magnesium chloride.
The enzymes responded differently to the addition of potassium orthophosphate. The activity decreased in the case of TspRK, remained unchanged in TthAPRT, and significantly increased in both TthPRPPSs, with the TthPRPPS2 activity starting to decrease at a salt concentration of more than 25 mM. The optimum Pi concentration for conducting cascade reactions is 10– 25 mM.
The most crucial question remained the substrate specificity of each enzyme for different carbohydrates.
D-ribose and 2-deoxy-D-ribose are TspRK substrates (Fig. 5), with the activity toward deoxy-sugar being 57% of that for D-ribose. The activity toward D-arabinose and D-xylose was 2 times higher than the background activity, which may indicate that they can be used as substrates, but much less specific ones. Activity for D-glucose and D-lyxose was not detected.
Fig. 5.
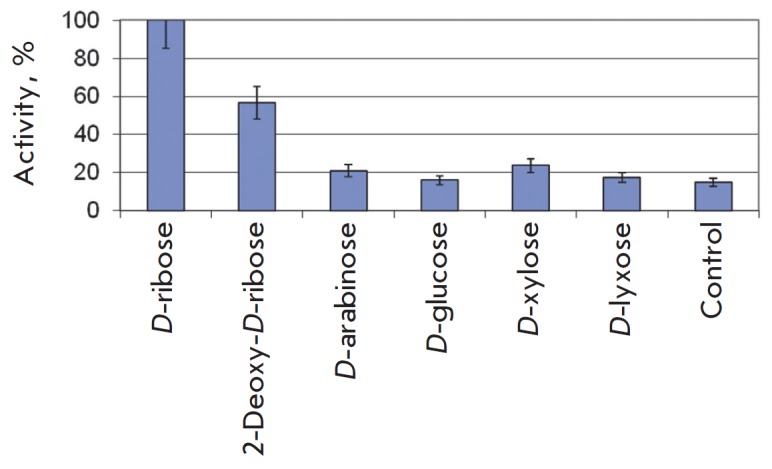
Enzymatic activity of ribokinase in the presence of different carbohydrates. Reactions were conducted at a temperature of 75°C in 0.05 mL of 20 mM Tris-HCl buffer, pH 8.0, containing 0.4 mM ATP, 1 mM carbohydrate (no carbohydrates in the control reaction), 1 mM KH2PO4, 5 mM MgCl2, 50 mM KCl, 0.15 μg of RK.
We tested the activity of both TthPRPPSs toward 2’-deoxy-ATP, GTP, and 2’-deoxy-GTP. The activity for 2’-deoxy-ATP was 80% of that for ATP. No activity toward GTP and 2’-deoxy-GTP was detected (data not shown). It may be that these TthPRPPSs are first-type enzymes [31]. All reactions were conducted under the same conditions as those for the activity assay: only the added substrate was varied. Both enzymes were able to catalyze the reaction with D-arabinose-5-phosphate (Fig. 6). However, activity for 2-deoxy-D-ribose-5 phosphate was not detected, indicating the importance of a hydroxyl group at the second sugar position to the enzymatic reaction.
Fig. 6.
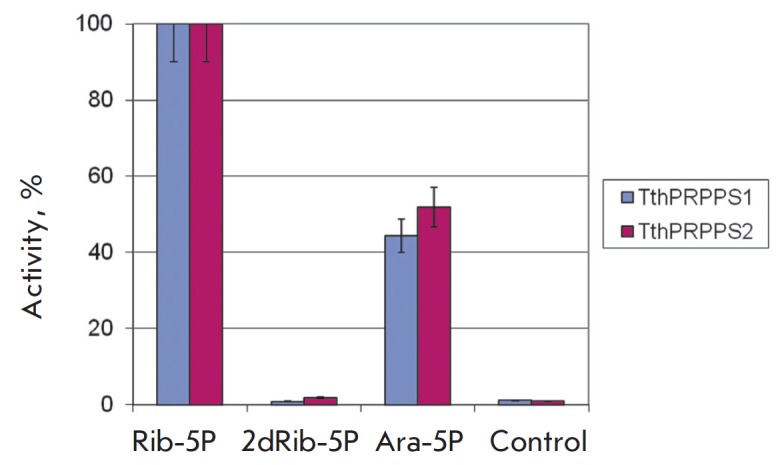
Substrate specificity of PRPP synthetases toward various carbohydrate-5-phosphates. Reactions were conducted at a temperature of 75 °C in 0.5 mL of 20 mM Tris-HCl buffer, pH 8.0, containing 1 mM ATP, 1 mM carbohydrate-5-phosphate (no carbohydrates in the control reaction), 5 mM MgCl2, 10 mM KH2PO4, 0.75 μg of PRPPS.
We conducted several reactions to test the applicability of different heterocyclic bases for nucleotide synthesis. The data on nucleotide synthesis using TthAPRT are shown in Table 3.
Table 3.
Substrates of APR-transferase
| Base | Conversion for 24 h, % | MS of product, [M+H]+ |
|---|---|---|
| 2,6-Diaminopurine | 16.8 | 363.0786 (calc. 363.0813) |
| 2-Chloroadenine | 97.6 | 382.0257 (calc. 382.0314) |
| 2-Fluoroadenine | 36.5 | 366.0564 (calc. 366.0611) |
| Adenine | 50.0 | 348.0677 (calc. 348.0704) |
| 2-Methoxyadenine | 60.9 | 378.0812 (calc. 378.0809) |
| N1-methyladenine | 78.2 | 362.0843 (calc. 362.0800) |
| N6-benzyladenine | 1.9 | 438.1117 (calc. 438.1173) |
| 2-Aminobenzimidazole | 0.1 | 346.0796 (calc. 346.0799) |
| 1,2,4-Triazole-3-carboxy-N-methylamide | 0 | – |
| Guanine | 0 | – |
| Hypoxanthine | 0 | – |
| 7-Deaza-2,6-diaminopurine | 0 | – |
Note. Reaction conditions: reaction mixtures (0.5 mL; 20 mM Tris-HCl, pH 8.0, 75 °C) contained 0.4 mM heterocyclic base, 0.4 mM PRPP, 0 to 5 mM MgCl2, 1.25 μg of TthAPRT.
The data in Table 3 demonstrate that TthAPRT is specific to 6-aminopurines. 2-Chloroadenine, N1-methyladenine, and 2-methoxyadenine proved to be good substrates. No enzymatic activity was detected in reactions with hypoxanthine, guanine, N6-benzyladenine, aminobenzimidazole, and 1,2,4-triazole-3-carboxy-N-methylamide. The equilibrium in reactions with 2-chloroadenine was strongly shifted towards nucleotide formation (98% after 1 day). The reaction was conducted at 75 °C, which was preferable in the case of 2-chloroadenine because of its poor solubility. 2,6-Diaminopurine also was a substrate, while no product was detected in the reaction with its 7-deaza analogue, suggesting the need for a nitrogen atom at the seventh position of the purine.
After studying the substrate specificity of TthAPRT, we conducted a cascade synthesis of 2Cl-AMP and 2F-AMP from appropriate heterocyclic bases and D-ribose. The results are shown in Fig. 7. The process of cascade nucleotide synthesis was monitored by a liquid chromatography-mass spectrometry analysis of the reaction mixture; samples were taken at 1, 2, 24 h from the start of the process. Figure 8 shows the mass spectra of the target products.
Fig. 7.
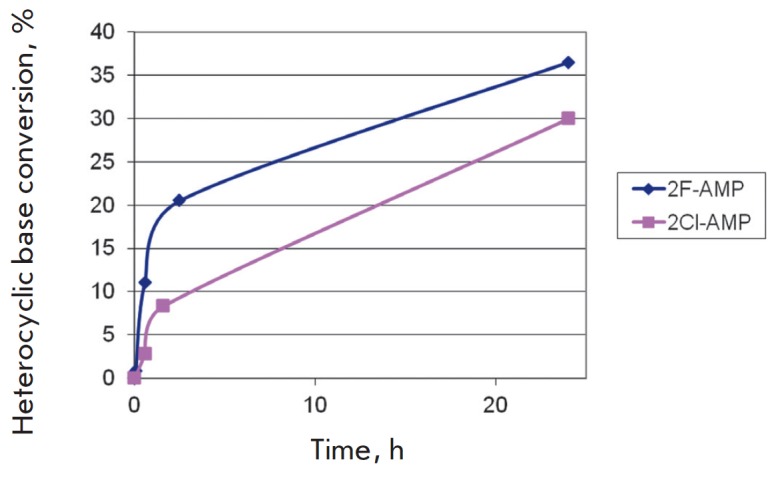
Cascade synthesis of 2Cl-AMP and 2F-AMP from appropriate heterocyclic bases and D-ribose. Reaction conditions: 0.5 mM D-ribose, 1 mM ATP, 0.4 mM heterocyclic base (2-chloro- or 2-fluoroadenine), 20 mM Tris-HCl (pH 8.0), 50 mM KCl, 1 mM MgCl2, 10 mM KH2PO4, 75°C, 0.3 μg of TspRK, 1.1 μg of TthPRPPS1, 0.25 μg of TthAPRT in 250 μL of the reaction mixture.
Fig. 8.
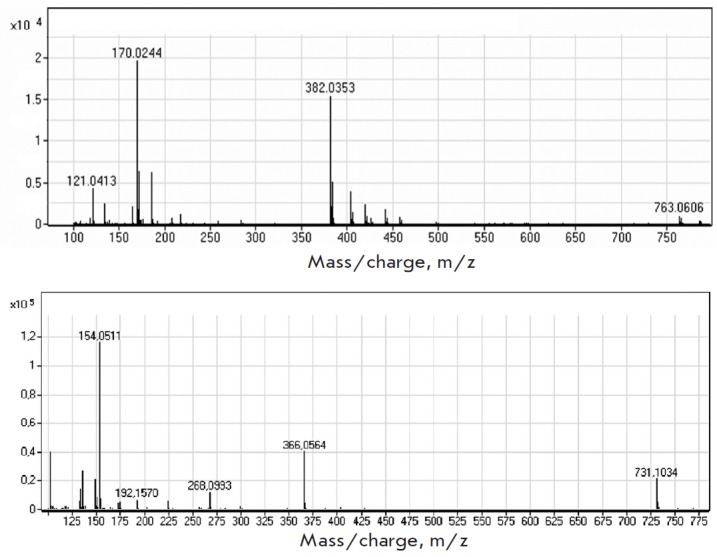
a) A mass spectrum of the 2Cl-AMP nucleotide produced by cascade synthesis:
Therefore, by the synthesis of 2-chloro (fluoro)-adenosine monophosphate (2Cl-AMP and 2F-AMP), we demonstrated the relevance of a thermophilic enzymatic system for the production of bioactive nucleotides.
CONCLUSION
Our findings indicate that a cascade of thermophilic enzymes of the nucleic acid metabolism (ribokinase; phosphoribosylpyrophosphate synthetase, and adenine phosphoribosyltransferase) can be used to produce modified nucleotides. This approach provides opportunities for the replacement of chemical methods of nucleotide synthesis with biocatalytic ones.
Acknowledgments
The authors are grateful to the Russian Science Foundation (project No. 14-50-00131) for financial support of this work.
Glossary
Abbreviations
- BSA
bovine serum albumin
- IPTG
isopropyl-β-D-1-thiogalactopyranoside
- PAAG
polyacrylamide gel
- MSF
phenylmethylsulfonyl fluoride
- PCR
polymerase chain reaction
- APR-transferase (TthAPRT)
adenine phosphoribosyltransferase from Thermus thermophilus
- LB
Luria-Bertani medium
- PRPP-synthetase (TthPRPPS)
phosphoribosylpyrophosphate synthetase from Thermus thermophilus
- RK (TspRK)
ribokinase from Thermus sp.
- 2Cl-AMP
2-chloroadenosine 5’-monophosphate
- 2F-AMP
2-fluoroadenosine 5’-monophosphate
- Pi
inorganic phosphate
- PRPP
5-phosphoribosyl-1-pyrophosphate
References
- 1.Nelson D.L., Cox M.M. Nucleotides and Nucleic Acids. Lehninger Principles of Biochemistry, 3rd ed. N.Y.: Worth Publ., 2000. 1255 p. 2000. Nucleotides and Nucleic Acids. [Google Scholar]
- 2.Bruce A. Molecular Biology of the Cell. N. Y.: Garland Publ., 2000. 974 p. 2000. Molecular Biology of the Cell. [Google Scholar]
- 3.Schärer O.D.. Angew Chem. Int. Ed. Engl. 2003;42(26):2946–2974. doi: 10.1002/anie.200200523. [DOI] [PubMed] [Google Scholar]
- 4.Jordheim L.P., Durantel D., Zoulim F., Dumontel C.. Nat. Rev. Drug Discov. 2013;12(6):447–464. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- 5.De Clercq E.. Rev. Med. Virol. 2009;19(5):287–299. doi: 10.1002/rmv.624. [DOI] [PubMed] [Google Scholar]
- 6.Diop-Frimpong B., Prakash T.P., Rajeev K.G., Manoharan M., Egli M.. Nucleic Acids Research. 2005;33(16):5297–5307. doi: 10.1093/nar/gki823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Famulok M., Hartig J.S., Mayer G.. J. Chem. Rev. 2007;107(9):3715–3743. doi: 10.1021/cr0306743. [DOI] [PubMed] [Google Scholar]
- 8.Peng C.G., Damha M.J.. Nucleic Acids Research. 2007;35(15):4977–4988. doi: 10.1093/nar/gkm520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Golden J., Motea E., Zhang X., Choi J.S., Feng Y., Xu Y., Lee I., Berdis A.J.. ACS Chem. Biol. 2013;8(11):2452–2465. doi: 10.1021/cb400350h. [DOI] [PubMed] [Google Scholar]
- 10.Lakshman M.K. Modified Nucleosides in Biochemistry, Biotechnology and Medicine. Weinheim: Wiley-VCH, 2008. 684 c. 2008. Modified Nucleosides in Biochemistry, Biotechnology and Medicine. [Google Scholar]
- 11.De Clercq E.. Annu. Rev. Pharmacol. Toxicol. 2011;51:1–24. doi: 10.1146/annurev-pharmtox-010510-100228. [DOI] [PubMed] [Google Scholar]
- 12.Cen Y., Sauve A.A.. Nucleos. Nucleotides. Nucleic Acids. 2010;29(2):113–122. doi: 10.1080/15257771003597758. [DOI] [PubMed] [Google Scholar]
- 13.Mikhailopulo I.A., Miroshnikov A.I., Mendeleev Commun. 2011;21:57–68. [Google Scholar]
- 14.Vineyard D., Zhang X., Donnelly A., Lee I., Berdis A.J.. Org. Biomol. Chem. 2007;5(22):3623–3630. doi: 10.1039/b712480e. [DOI] [PubMed] [Google Scholar]
- 15.Stein C.A., Cheng Y.C.. Science. 1993;261(5124):1004–1012. doi: 10.1126/science.8351515. [DOI] [PubMed] [Google Scholar]
- 16.Motea E.A., Lee I., Berdis A.J.. Nucleic Acids Research. 2012;40(5):2357–2367. doi: 10.1093/nar/gkr980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mikhailopulo I.A., Curr. Org. Chem. 2007;11(4):317–335. [Google Scholar]
- 18.Wintersberger E.. Biochem. Soc. Trans. 1997;25(1):303–308. doi: 10.1042/bst0250303. [DOI] [PubMed] [Google Scholar]
- 19.Tesmer J.J., Klem T.J., Deras M.L., Davisson V.J., Smith J.L.. Nat. Struct. Biol. 1996;3(1):74–86. doi: 10.1038/nsb0196-74. [DOI] [PubMed] [Google Scholar]
- 20.Kim M.J., Whitesides G.M., Appl. Biochem. Biotech. 1987;1(6):95–108. [Google Scholar]
- 21.Scism R.A., Stec D.F., Bachmann B.O.. Org. Lett. 2007;9(21):4179–4182. doi: 10.1021/ol7016802. [DOI] [PubMed] [Google Scholar]
- 22.Scism R.A., Bachmann B.O.. ChemBioChem. 2010;11(1):67–70. doi: 10.1002/cbic.200900620. [DOI] [PubMed] [Google Scholar]
- 23.Nagy M., Ribet A.M.. Eur. J. Biochem. 1977;77(1):77–85. doi: 10.1111/j.1432-1033.1977.tb11643.x. [DOI] [PubMed] [Google Scholar]
- 24.Lee D., Moffatt B.A., Physiologia Plantarum. 1993;87(4):483–492. [Google Scholar]
- 25.Zhou X., Mikhailopulo I.A., Cruz-Bournazou N., Neubauer P., J. Mol. Catalysis B: Enzymatic. 2015;115:119–127. [Google Scholar]
- 26.Zhou X., Szeker K., Jiao L.Y., Oestreich M., Mikhailopulo I.A., Neubauer P., Adv. Synthesis & Catalysis. 2015;357(6):1237–1244. [Google Scholar]
- 27.Taran S.A., Verevkina K.N., Feofanov S.A., Miroshnikov A.I.. Russ. J. Bioorg. Chem. 2009;35(6):739–745. doi: 10.1134/s1068162009060107. [DOI] [PubMed] [Google Scholar]
- 28.Laemmli U.K.. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 29.Bradford M.M.. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 30.Park J., van Koeverden P., Singh B., Gupta R.S.. FEBS Lett. 2007;581(17):3211–3216. doi: 10.1016/j.febslet.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 31.Kadziola A., Jepsen C.H., Johansson E., McGuire J., Larsen S., Hove-Jensen B.. J. Mol. Biol. 2005;354(4):815–828. doi: 10.1016/j.jmb.2005.10.001. [DOI] [PubMed] [Google Scholar]