

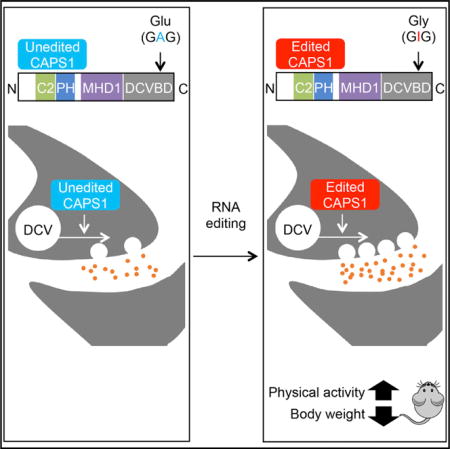
SUMMARY
Calcium-dependent activator protein for secretion 1 (CAPS1) plays a distinct role in the priming step of dense core vesicle (DCV) exocytosis. CAPS1 pre-mRNA is known to undergo adenosine-to-inosine RNA editing in its coding region, which results in a glutamate-to-glycine conversion at a site in its C-terminal region. However, the physiological significance of CAPS1 RNA editing remains elusive. Here, we created mutant mice in which edited CAPS1 was solely expressed. These mice were lean due to increased energy expenditure caused by physical hyperactivity. Electrophysiological and biochemical analyses demonstrated that the exocytosis of DCVs was upregulated in the chromaffin cells and neurons of these mice. Furthermore, we showed that edited CAPS1 bound preferentially to the activated form of syntaxin-1A, a component of the exocytotic fusion complex. These findings suggest that RNA editing regulates DCV exocytosis in vivo, affecting physical activity.
Graphical abstract
INTRODUCTION
Post-transcriptional modification is a mechanism by which vast repertoires of mature RNAs and their proteins are generated from a single gene. One type of post-transcriptional modification is adenosine-to-inosine RNA editing, which is catalyzed by ADAR1 (adenosine deaminase acting on RNA type1) and ADAR2 in mammals. These enzymes recognize double-stranded RNAs (dsRNAs) as targets (Behm and Öhman, 2016; Tomaselli et al., 2014). It is estimated that more than 85% of transcripts are edited (Athanasiadis et al., 2004). However, most of the editing sites are located in non-coding regions such as introns in pre-mRNAs or non-coding RNAs, including microRNAs, and their significance has been elucidated only in a limited number of cases (Kawahara et al., 2007a; Sakurai et al., 2014; Wulff et al., 2011). In contrast, although editing in coding regions is quite rare (55 sites [Li et al., 2009]), this modification can generate proteins that are not encoded directly by the genome, given that the translational machinery interprets inosine as if it were guanosine. It has been reported that the properties of neurotransmitter receptors and ion channels are modulated by amino acid conversion as a consequence of RNA editing (Bazzazi et al., 2013; Behm and Öhman, 2016; Bhalla et al., 2004; Brusa et al., 1995; Burns et al., 1997; Daniel et al., 2011; Morabito et al., 2010). However, only a few mouse models exist in which either edited or unedited proteins are expressed exclusively; these include the glutamate receptor GluA2 subunit and the serotonin 5-HT2C receptor (Brusa et al., 1995; Kawahara et al., 2008; Morabito et al., 2010). Therefore, little is known about the significance of the conversion of amino acid residues as a consequence of RNA editing in mammals.
Calcium-dependent activator protein for secretion 1 (CAPS1) and CAPS2 are members of the CAPS family, which is involved in vesicle exocytosis (Parsaud et al., 2013; Speidel et al., 2003). CAPS proteins play an indispensable role in the secretion of insulin and neurotransmitters, including catecholamines such as noradrenaline (NA) and dopamine, by regulating the exocytosis of dense-core vesicles (DCVs). Consequently, CAPS proteins are expressed in selective tissues, such as the pancreas, adrenal gland, and brain (Speidel et al., 2003, 2005, 2008). Knockout of CAPS1 in mice induced a reduction in DCV release, leading to neonatal death soon after birth (Farina et al., 2015; Speidel et al., 2005), which indicates that CAPS1 is required for efficient DCV exocytosis. Among the multiple steps required for exocytosis, CAPS1 promotes the priming reaction, which is a required step for the fusion of vesicles with the plasma membrane after docking has occurred (Farina et al., 2015). Membrane fusion is mediated by the trans-SNARE (Soluble NSF attachment protein receptor) complex, which is composed of the vesicle SNARE (v-SNARE) and target membrane SNARE (t-SNARE). CAPS1 promotes assembly of the trans-SNARE complex for vesicle exocytosis by binding directly through its C-terminal region to syntaxin-1, a t-SNARE protein (Hammarlund et al., 2008; Parsaud et al., 2013). Intriguingly, an RNA editing site was identified in the human Cadps gene, which encodes CAPS1, by using massively parallel target capture sequencing and then comparing the genomic DNA sequence with the sequence of RNAs from a single individual (Li et al., 2009). The identified RNA editing site corresponds to a glutamate (E) residue at position 1250 (1252 in mouse) in the C-terminal DCV-binding domain (DCVBD), which was identified originally as a domain essential for binding to DCVs and for NA secretion (Figure 1A; Figure S1A) (Grishanin et al., 2002). The E residue is converted to glycine (G) as a consequence of RNA editing, and this site is thereby termed the E/G site. However, the enzyme responsible for the editing, the secondary structure required for recognition by ADARs, and the physiological significance of the modification remain undetermined.
Figure 1. The Editing Frequency in CAPS1 mRNA and the Secondary Structure Required for Editing.
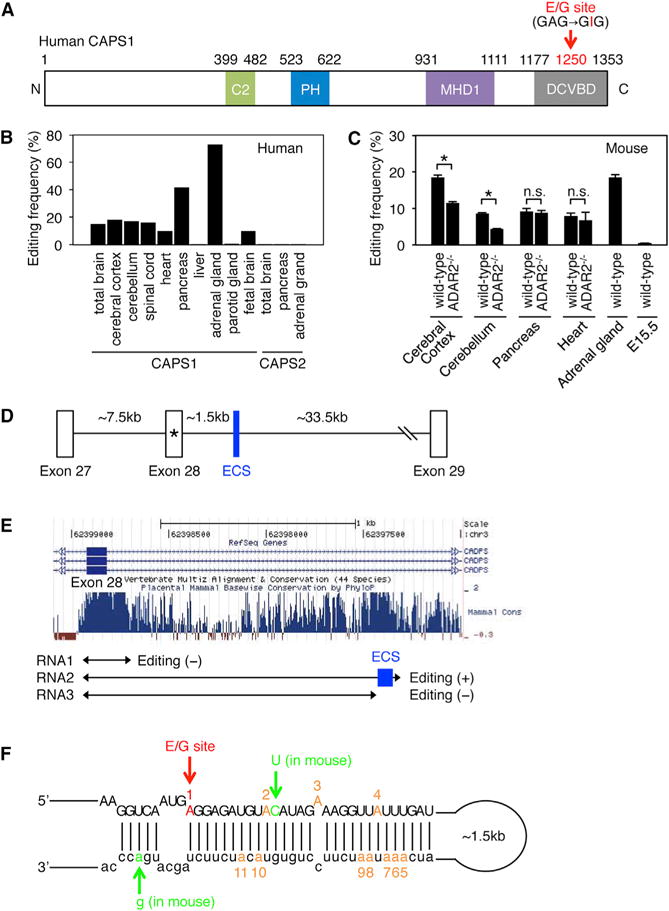
(A) Schematic diagram of human CAPS1. RNA editing in the dense core vesicle-binding domain (DCVBD) changes the codon for amino acid 1250 from GAG to GIG, which converts the glutamate at residue 1250 (E1250) to glycine (G). This E/G site is indicated with a red arrow (see Figure S1A). The position of each domain is assigned by referring to the longest isoform (NP_003707.2). C2, coiled-coil domain; PH, Pleckstrin homology domain; MHD1, Munc13-homology domain 1.
(B) The editing frequency of CAPS1 and CAPS2 mRNAs in various human tissues (see Figure S1B).
(C) The editing frequency of CAPS1 mRNA in various mouse tissues extracted from both wild-type and ADAR2−/− mice. Values are displayed as the mean ± SEM (n = 3, WT and 4, ADAR2−/−; unpaired t test, *p < 0.05). n.s., no significance.
(D) Schematic diagram of human chromosomal DNA encompassing exon 27 to exon 29 of the CADPS gene, which encodes CAPS1. The E/G site is designated with an asterisk. ECS, editing complementary sequence.
(E) The degree of conservation among mammals in the intronic region that is downstream of exon 28, which contains the E/G site, was analyzed using the UCSC genome browser. The regions transcribed to generate the three RNAs (RNA1, RNA2, and RNA3) that were used for the in vitro editing assays are shown below the graphs. The location of the identified ECS is highlighted in blue.
(F) The incomplete double-stranded RNA structure formed by the sequence around the E/G site (in red) and the ECS, which is required for CAPS1 RNA editing, is depicted. The nucleotides that differ between human and mouse are shown in green. The locations of an additional 10 editing sites observed in the in vitro editing assay are shown in orange with the appropriate number (see Figure S1C).
In this study, we examined the functional difference between edited and unedited CAPS1 using cultured cell lines and found that RNA editing accelerates DCV-mediated catecholamine secretion. Then, we created a mutant mouse model in which edited CAPS1 was expressed solely, to evaluate the physiological significance of CAPS1 RNA editing in vivo. Intriguingly, edited CAPS1 knockin (KI) mice were leaner and showed higher energy expenditure than wild-type mice due to increased physical activity, which was attenuated by the administration of dopamine D2 receptor antagonist. Electrophysiological and biochemical analyses of the mouse brains and adrenal glands demonstrated that DCV exocytosis was promoted by the RNA editing of CAPS1. This increased exocytosis was most likely due to an enhancement of the priming step through more efficient binding of edited CAPS1 to the activated form of syntaxin-1, at least in part.
RESULTS
ADAR1 and ADAR2 Coordinately Catalyze CAPS1 RNA Editing
To investigate the editing frequency of CAPS1 mRNA in vivo, we first obtained samples of total RNA derived from human tissues. The editing frequency at the E/G site was approximately 15%–20% in human spinal cord and brain tissues, regardless of the region examined (Figure 1B; Figure S1B). In contrast, the frequency was 42% in pancreas and the highest (70%) in the adrenal gland. The amino acid sequence around the editing site is conserved between CAPS1 and CAPS2 in mammals (Figure S1A). However, RNA editing was not detected in CAPS2 mRNA at the site corresponding to the E/G site of CAPS1 in brain, pancreas, or the adrenal gland (Figure 1B). Next, we examined the editing frequency of CAPS1 mRNA in mouse tissues. In contrast to the wide range of editing frequencies in human tissues, mouse CAPS1 mRNA was edited at the level of 10%–20%, regardless of the tissue examined (Figure 1C). No editing could be observed at embryonic stage E15.5 (Figure 1C), which indicated that the editing was developmentally regulated. To determine the enzymes responsible for CAPS1 RNA editing in vivo, we compared the editing frequency between wild-type (WT) and ADAR2−/− mice (Higuchi et al., 2000; Kawahara et al., 2007b). The editing level was reduced by 30% and 50% in cerebral cortex and cerebellum, respectively, in ADAR2−/− mice as compared with WT mice, whereas no significant alteration was observed in pancreas or heart (Figure 1C). Although it is difficult to examine the editing level of CAPS1 mRNA in ADAR1−/− mice due to embryonic lethality (Wang et al., 2000), this result suggests that ADAR2 is partially responsible for the editing in the brain, whereas ADAR1 probably plays the predominant role in editing the E/G site of CAPS1 mRNA.
Identification of the Conserved Secondary Structure Required for CAPS1 RNA Editing
Given that ADARs bind to dsRNAs, editing in certain exon regions requires the presence of an editing complementary sequence (ECS), which is usually located in either an upstream or downstream intron and forms a partial dsRNA structure (Gerber and Keller, 2001). To identify the ECS that is indispensable for human CAPS1 RNA editing, we examined the conserved sequences in the intron downstream of exon 28, which contains the E/G site, and found two candidate regions: one located just adjacent to exon 28, and the other located approximately 1.5 kb downstream of the exon (Figures 1D and 1E). To determine whether either region was the ECS, we prepared RNAs that contained the candidate regions (RNA1 and RNA2) by in vitro transcription (Figure 1E) and then performed in vitro editing assays with recombinant ADAR1 or ADAR2 protein as described previously (Kawahara et al., 2007b). We detected no editing with RNA1. However, when RNA2 was used as the substrate, ADAR1 edited the E/G site specifically within the exon, and also edited four sites in the intron, in a dose-dependent manner (Figure 1F; Figure S1C). In contrast, ADAR2 edited three adenosines in the exon and seven in the intron, in addition to editing the E/G site, in RNA2. We also performed in vitro editing assays with RNA3, which was slightly shorter than RNA2 and lacked a part of the candidate ECS region (Figure 1E). The results showed no editing, regardless of the ADAR used. We further confirmed that the corresponding sequence was absent in CAPS2 pre-mRNA, and that the predicted secondary structure was largely conserved between human and mouse (Figure 1F). Taken together, these results suggested that the ECS corresponds to the conserved region located approximately 1.5 kb downstream of exon 28 (Figures 1D and 1E).
CAPS1 RNA Editing Promotes Catecholamine Secretion from PC12 Cells
To evaluate the functional differences between the edited and unedited forms of CAPS1 in cultured PC-12 cells, which are derived from a pheochromocytoma of the rat adrenal medulla, we first established a stable cell line in which endogenous CAPS1 was reduced significantly by the stable expression of four small hairpin RNAs (shRNAs). We named this cell line shRNA_All-8 (Figure S2). Using shRNA_All-8 cells, we further established cell lines that stably expressed either the edited or unedited form of mCherry-mouse CAPS1 (Figure 2A). The expression construct for unedited CAPS1 lacked the ECS in the adjacent intronic region, and thus the resulting pre-mRNA was not subject to RNA editing in the established cell line. After confirming that the intracellular localization of edited and unedited CAPS1 did not differ, we monitored the amount of radiolabeled NA secreted from PC12 cells that solely expressed either edited or unedited CAPS1 during a period of 1 hr after depolarizing stimulation, as described previously (Parsaud et al., 2013). The data demonstrated that the cells that expressed edited CAPS1 secreted more NA than those that expressed unedited CAPS1 at 15 and 30 min post-stimulation, whereas no difference was observed at 60 min, when the secretion of radiolabeled NA probably reached the saturation (Figure 2B). These results suggest that edited CAPS1 can facilitate the release of NA more rapidly than unedited CAPS1.
Figure 2. CAPS1 RNA Editing Promotes Secretion of Noradrenaline from PC12 Cells.
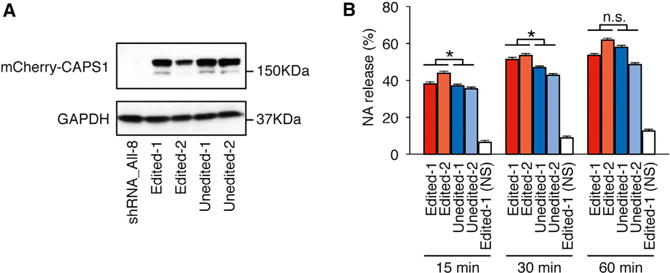
(A) Stable cell lines expressing either unedited or edited mCherry-mCAPS1 were established using shRNA_All-8 PC12 cells, in which endogenous CAPS1 was strongly knocked down (see Figure S2). The expression of exogenous mCherry-mCAPS1 was confirmed by western blot analysis using an anti-CAPS1 antibody in two lines (designated as 1 and 2) for the unedited and two for the edited forms. The expression of GAPDH is shown as a reference.
(B) The percentage efficiency of noradrenaline (NA) release from the cells expressing either unedited or edited mCherry-mCAPS1 was measured at 15, 30, and 60 min after stimulation with high K+ buffer. Values are displayed as the mean ± SEM (ten trials for each line; unpaired t test, *p < 0.05). n.s., no significance; NS, no stimulation.
Mutant Mice in which Edited CAPS1 Is Solely Expressed Exhibit Leanness
Given that the editing frequency at the E/G site tends to be higher in humans than in mice (Figures 1B and 1C), the role of edited CAPS1 might be more limited in mice but expanded during the course of evolution to become more significant in humans. Therefore, to investigate the role of CAPS1 RNA editing in vivo, we created mutant mice in which edited CAPS1 was solely expressed (edited CAPS1 KI mice) by inserting a point mutation at the E/G site (GAG → GGG) using the conventional homologous recombination technique (Figures S3A and S3B). We confirmed that this mutation did not affect the expression level of CAPS1 protein (Figure S3C). Homozygous edited CAPS1 KI (KI) mice were born with normal body weight but demonstrated a significant decrease in growth rate from 4 weeks after birth (Figure 3A). This reduction in body weight was sustained until adulthood, regardless of sex, with no difference in body length between KI and WT littermates. Male KI mice were approximately 20% leaner than their WT littermates at 12 weeks of age (Figures 3B and 3C; Figures S3D and S3E). Of note, heterozygous KI mice exhibited moderate leanness, which suggests that the level of CAPS1 RNA editing was correlated positively with the severity of the lean phenotype (Figure S3F). This leanness was attributed to a specific reduction in fat accumulation in white adipose tissue accompanied by a low concentration of plasma cholesterol (Figures 3D and 3E).
Figure 3. Edited CAPS1 KI Mice Exhibit Leanness.
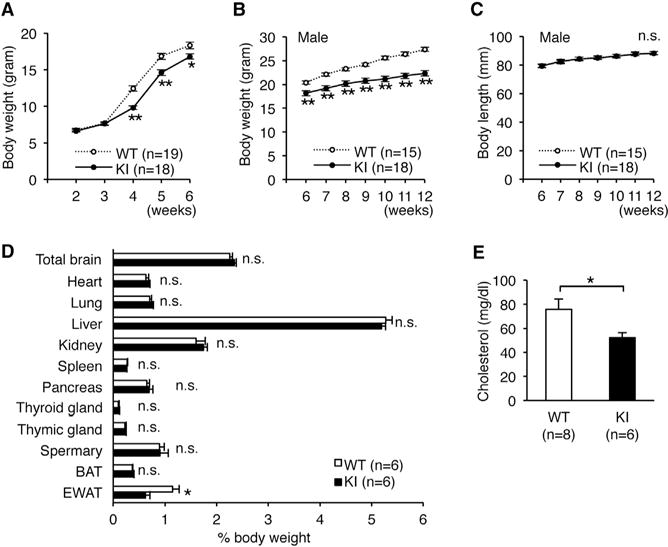
(A) The body weights of wild-type (WT; ten male and nine female) and homozygous KI (KI; eight male and ten female) mice were followed from 2 to 6 weeks after birth. The difference in sex distribution was not significant between the two groups (p = 0.43, chi-square test).
(B and C) Body weight (B) and body length (C) of male littermates of the WT and KI mice were followed from 5 to 12 weeks after birth (see Figure S3).
(D) The weights of various organs were compared between male WT and KI mice. BAT, brown adipose tissue; EWAT, epididymal white adipose tissue.
(E) Plasma cholesterol levels of male WT and KI mice are shown.
All data are presented as the mean ± SEM. Statistical differences were analyzed by the two-way factorial ANOVA with Tukey HSD post hoc test (A), multifactorial repeated-measures ANOVA with Tukey HSD post hoc test (B and C), Mann–Whitney test (D), and the unpaired t test (E) and are indicated by asterisks (*p < 0.05, **p < 0.01). n.s., no significance.
To investigate the cause of the lean phenotype of edited CAPS1 KI mice, we quantified metabolic rates by measuring oxygen consumption. Energy expenditure in KI mice was increased significantly as compared with WT mice during both light and dark periods (Figure 4A). In contrast, we could detect no significant alteration in food or water intake, or daily urine output (Figures 4B–4D). These results suggest that the leanness of edited CAPS1 KI mice was not due to hypophagia or dehydration, but rather due to increased energy expenditure. In addition, the expression levels of mitochondrial Uncoupled protein-1 (UCP-1) mRNA in brown adipose tissue and body temperature were not significantly different between edited CAPS1 KI and WT mice (Figures 4E and 4F). Therefore, the increased energy expenditure could not be attributed to excess wastage through nonshivering thermogenesis in brown adipose tissue.
Figure 4. Increased Energy Expenditure in Edited CAPS1 KI Mice.
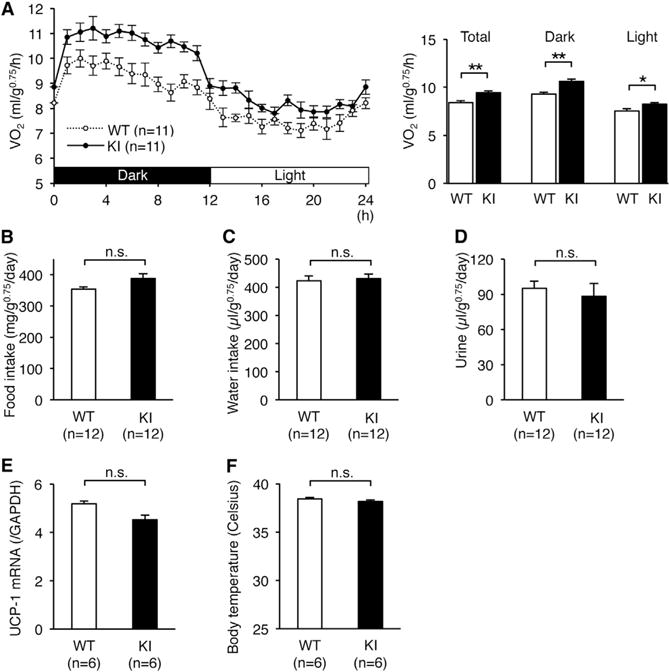
(A) The metabolic rates of male littermates of wild-type (WT) and homozygous KI (KI) mice were measured by oxygen uptake (VO2) during a period of 24 hr after habituation over 48 hr (left panel). Then, the metabolic rates during both the dark and light phases, as well as the total rates, were compared between the two groups (right panel). (B–F) Food intake (B), water intake (C), volume of urine (D), UCP-1 mRNA levels in brown adipose tissue (E), and body temperature (F) were compared between WT and KI mice.
All data are presented as the mean ± SEM. Statistical differences were analyzed by the unpaired t test (A–D) and Mann-Whitney test (E and F) and are indicated by asterisks (*p < 0.05, **p < 0.01). n.s., no significance.
Increased Physical Activity in Edited CAPS1 KI Mice
Next, we examined whether the increased energy expenditure was caused by an alteration in physical activity. As expected, the total locomotion counts of WT mice in the new environment decreased gradually during the 3 days tested, which suggests that the mice were habituated (Figure 5A). In contrast, the activity of KI mice did not decreased even after habituation. Next, we quantified daily activity after habituation. The total physical activity of KI mice was significantly higher than that of WT during both the dark and light cycles (Figure 5B). This hyperactive phenotype was attenuated by the administration of haloperidol, a dopamine D2 receptor antagonist (Figure 5C). In accordance, energy expenditure in KI mice was also reduced to the level of WT mice by the administration of haloperidol (Figure 5D). We could not detect any abnormalities in muscle strength, anxiety-related behavior, pain sensation, space perception, or short-or long-term memory as measured by the results of the wire hang test, elevated plus maze test, hot plate test, water maze learning test, Y maze test, and passive avoidance test (Figures S4A–S4F). Furthermore, we found no difference in the urinary concentration of catecholamine metabolites between KI and WT mice, which suggests that the peripheral catecholamine level was not highly elevated in KI mice (Figures S5A and S5B). Taken together, these findings suggested that the increased energy expenditure in edited CAPS1 KI mice could be attributed to increased physical activity, which was most likely mediated through the central dopamine pathway.
Figure 5. Edited CAPS1 KI Mice Exhibit Increased Physical Activity.
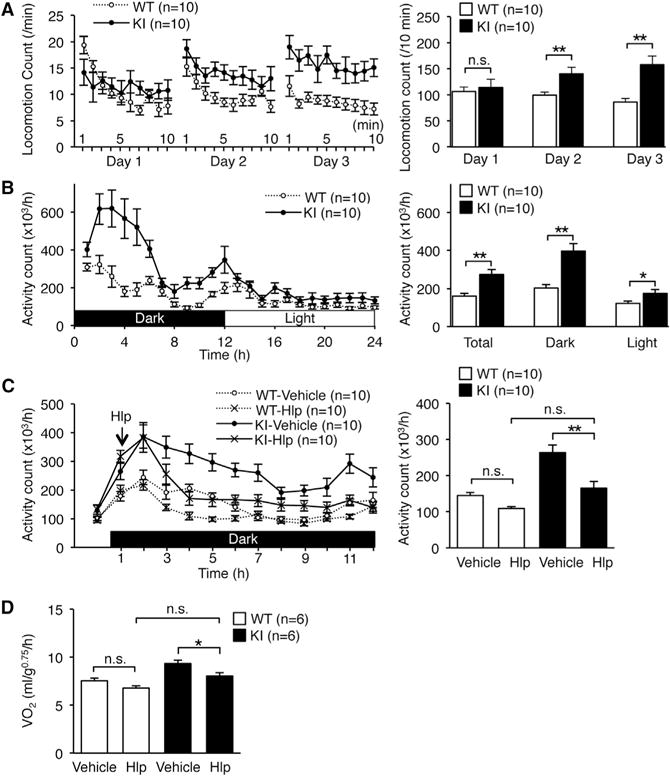
(A and B) Open-field tests (A) and circadian rhythm tests (B) were performed over the indicated time periods for male littermates of wild-type (WT) and homozygous KI (KI) mice (left panels). Then, the locomotion count (A) and the activity count (B) over the indicated time periods were compared between the two groups (right panels).
(C) Either haloperidol (Hlp; 0.5 mg/kg) or saline (vehicle) was injected intraperitoneally 1 hr after the dark cycle had started (indicated with an arrow on left panel). After waiting for an additional 1 hr, total activity during each hour was measured for the subsequent 10 hr and compared between WT and KI mice (right panel).
(D) Either haloperidol (Hlp; 0.5 mg/kg) or saline (vehicle) was injected intraperitoneally 1 hr after the dark phase had started. After waiting for an additional 1 hr, metabolic rates during each hour were measured for the subsequent 10 hr and compared between WT and KI mice. See also Figures S4 and S5.
All data were acquired from the indicated numbers of male mice and are presented as the mean ± SEM. Statistical differences were analyzed by multifactorial repeated-measures ANOVA with Tukey HSD post hoc tests (A), unpaired t tests (B), and one-way ANOVA with Tukey HSD post hoc tests (C and D) and are indicated by asterisks (*p < 0.05, **p < 0.01). n.s., no significance.
Enhanced DCV-Mediated Catecholamine Secretion in Edited CAPS1 KI Mice
Given that the secretion of catecholamines, including dopamine, is mediated by DCVs, and that edited CAPS1 can facilitate the release of NA more rapidly than unedited CAPS1 in PC12 cells (Figure 2B), we further examined whether CAPS1 RNA editing promoted DCV exocytosis in vivo. For this purpose, we first performed amperometric measurements using chromaffin cells, which demonstrated that the efficacy of vesicle exocytosis was enhanced significantly during depolarizing stimulation in edited CAPS1 KI mice (Figure 6A). Then, we measured intracellular Ca2+ concentrations during the stimulation and found no significant difference between KI and WT mice (Figure 6B). The results showed that increased Ca2+ influx did not cause the enhanced exocytosis. Furthermore, we could not detect any significant difference in adrenal catecholamine concentrations or the intracellular distribution of DCVs (Figures 6C and 6D; Figures S6A and S6B). Collectively, these findings suggest that the efficient DCV exocytosis in chromaffin cells of edited CAPS1 KI mice is achieved by most likely enhancing the priming step, but not the loading or docking step, by RNA editing. Finally, we investigated DCV-mediated secretion of catecholamines in neurons. Synaptosomes were prepared from the striatum of KI and WT mice, and the amount of radiolabeled dopamine that was secreted during a depolarizing stimulation was quantified. The results demonstrated that dopamine was secreted more efficiently from neurons in KI mice than in WT mice (Figure 6E), which further suggested that CAPS1 RNA editing promotes DCV-mediated secretion of catecholamines in vivo.
Figure 6. Increased DCV Exocytosis in Edited CAPS1 KI Mice.
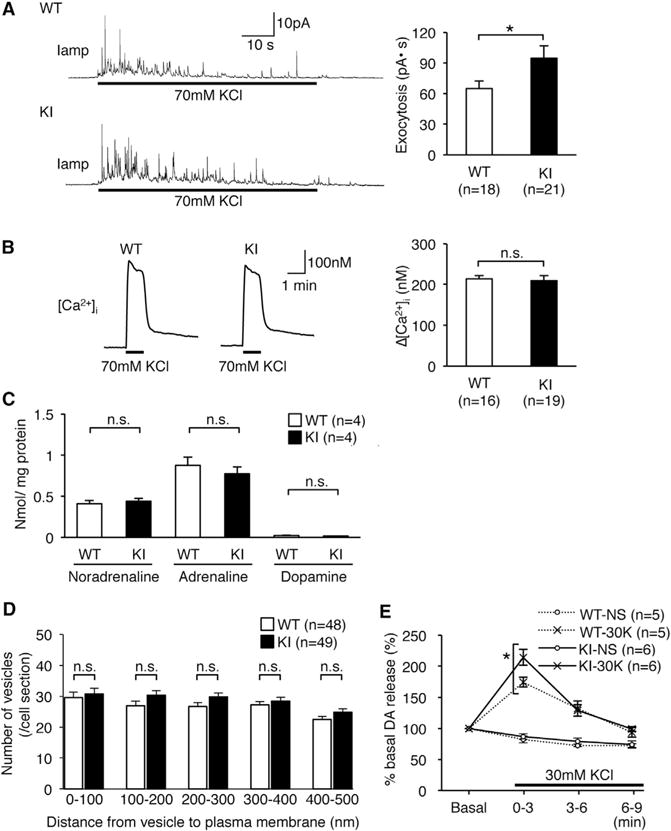
(A) Representative traces of amperometric measurements from individual chromaffin cells (left panel). Exocytosis was triggered during the period indicated by the black bars. Exocytotic responses were compared between wild-type (WT) and homozygous KI (KI) mice by measuring the total area under the curve during stimulation (right panel). All data were acquired from 18 and 21 cells isolated from three mice of the WT and KI, respectively.
(B) Representative traces of the measurements of the intracellular calcium concentration from individual chromaffin cells (left panel). KCl was applied during the period indicated by the black bars. The calcium influx was compared between WT and KI mice by subtracting the basal calcium concentration from the peak (right panel). All data were acquired from 16 and 19 cells isolated from three mice of the WT and KI, respectively.
(C) The concentration of three catecholamines in adrenal glands was compared between WT and KI mice (n = 4 for each group).
(D) The distribution of whole DCVs in individual chromaffin cells as determined from the electron microscopy images (see Figure S6). All data were acquired from a total of 48 cells from five WT mice and 49 cells from five KI mice.
(E) Dopamine secretion from synaptosomes derived from the striatum was measured every 3 min after stimulation with high K+ buffer (30K) and is shown as a percentage relative to the basal release, which is set as 100%. NS, no stimulation. All data are presented as the mean ± SEM. Statistical differences were analyzed by unpaired t tests (A–D) and Mann-Whitney tests (E) and are indicated by asterisks (*p < 0.05). n.s., no significance.
Edited CAPS1 Binds Preferentially to the Activated Form of Syntaxin-1
Finally, we sought to elucidate the mechanism by which the edited CAPS1 enhances DCV exocytosis. It has been reported that CAPS1 binds preferentially through its C-terminal region to the “open” conformation of syntaxin-1A, which is the activated form, and that deletion of the C-terminal region, including the E/G site, abolishes efficient NA secretion from PC12 cells (Grishanin et al., 2002; Hammarlund et al., 2008; Parsaud et al., 2013). Therefore, we examined whether RNA editing affected the ability of CAPS1 to bind syntaxin-1. First, we performed pull-down assays using human CAPS1 tagged with HaloTag, which was incubated with a mouse brain homogenate enriched with synaptosomal proteins. The data demonstrated that edited CAPS1 bound to syntaxin-1A more efficiently than unedited CAPS1 (Figures 7A and 7B). We also compared the binding efficiency of CAPS1 between constitutively “open” and “closed” conformations of syntaxin-1A by pull-down assays. This analysis showed that although both edited and unedited CAPS1 bound the “open” conformation of syntaxin-1A significantly more efficiently than the “closed” form, edited CAPS1 bound more strongly than the unedited protein to the former (Figures 7C and 7D). Taken together, these results suggested that edited CAPS1 can bind to the activated form of syntaxin-1 more efficiently than unedited CAPS1, leading to enhancement of the priming step, which could result in more efficient exocytosis.
Figure 7. RNA Editing Promotes the Binding of CAPS1 to the Activated Form of Syntaxin-1A.
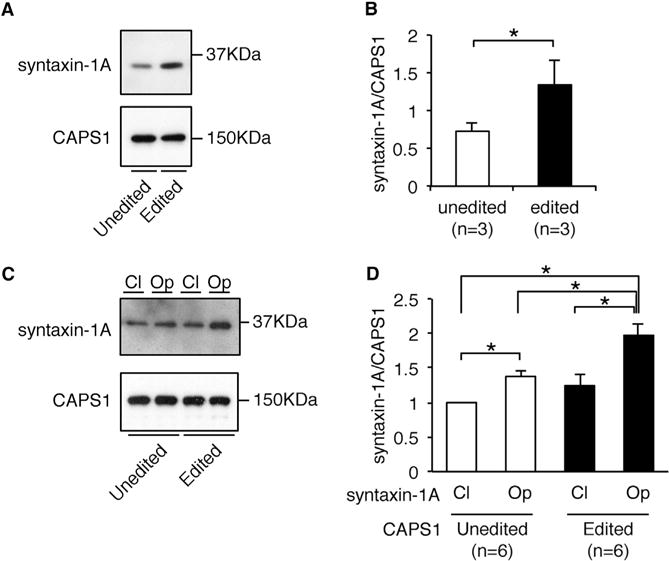
(A) The amount of endogenous syntaxin-1A in mouse brain homogenate that bound to either unedited or edited Halo-human CAPS1 in pull-down assays was analyzed by western blotting. The expression level of CAPS1 is shown as a reference.
(B) The relative amount of endogenous syntaxin-1A bound to CAPS1 was compared between the unedited and edited isoforms. Values are displayed as the mean ± SEM (n = 3; Mann-Whitney test, *p < 0.05).
(C) The amount of syntaxin-1A in either the closed (CI) or open (Op) conformation that bound to either unedited or edited Halo-human CAPS1 in pull-down (IP) assays was analyzed by western blotting. The expression of CAPS1 is shown as a reference.
(D) The relative amount of syntaxin-1A in either the closed (CI) or open (Op) conformation bound to either unedited or edited CAPS1 was compared. All values are displayed as the mean ± SEM (n = 6; one-way ANOVA with Wilcoxon test, *p < 0.05). The mean value of the relative amount of closed conformation syntaxin-1A bound to unedited CAPS1 was set as one.
DISCUSSION
The E/G site in CAPS1 mRNA was identified initially as one of 55 editing sites in coding regions, by massive parallel sequencing (Li et al., 2009). However, the effects of CAPS1 RNA editing have not been investigated previously, even in vitro. In the present study, we identified the secondary structure required for CAPS1 RNA editing and the enzymes responsible. We found that CAPS1 RNA editing promotes catecholamine secretion in cultured cells. Furthermore, we provide in vivo evidence that CAPS1 RNA editing regulates physical activity, by using mutant mice that express edited CAPS1 exclusively. These mice exhibited increased locomotion and energy expenditure, which could be attenuated by the suppression of the dopamine pathway. These phenotypic features were attributable to enhanced DCV exocytosis and contributed to the leanness of the mutant mice. We found that DCV exocytosis was also upregulated in the chromaffin cells of the adrenal gland in mutant mice, which might contribute to the lean phenotype, in part.
The editing frequency of CAPS1 mRNA in mice tended to be lower than that in humans, which is consistent with the much higher level of RNA editing in primates than in mice. This higher level is probably due to the presence of Alu elements in primates, which can be targeted preferentially by ADARs (Eisenberg et al., 2005). However, given that the editing levels of the conserved sites are largely preserved between human and mouse (Pinto et al., 2014), the physiological significance of CAPS1 RNA editing might have increased in some selected tissues, such as the adrenal gland, over the course of evolution. It is also a unique feature that the editing frequency of CAPS1 mRNA is higher in peripheral tissues than in neuronal tissues in human. Analysis of the editing frequency in ADAR2−/− mice suggested that both ADAR1 and ADAR2 participated in CAPS1 RNA editing in the brain. Indeed, coordinated regulation of RNA editing by ADAR1 and ADAR2 is sometimes observed for certain sites, such as the C and E sites in the neuron-specific 5-HT2C receptor (Hartner et al., 2004). However, if ADAR2 is the main enzyme responsible for CAPS1 RNA editing, we would expect that the editing frequency would be much higher in the brain than in other tissues, given that ADAR2 is highly expressed in the brain (Huntley et al., 2016). Considering that ADAR1 is ubiquitously expressed, and the editing frequency was not altered in the peripheral tissues of ADAR2−/− mice, we concluded that ADAR1 was probably the main enzyme responsible for CAPS1 RNA editing. Unfortunately, given that ADAR1−/− mice die at embryonic day (E) 11.5–12.5 (Wang et al., 2000) and that no editing of CAPS1 mRNA is detected at E15.5, it is impossible to investigate how RNA editing is altered in ADAR1−/− mice. It has recently been reported that mutant mice that express inactive ADAR1 die at E13.5, whereas the lethality can be rescued by concurrent deletion of a cytosolic sensor of dsRNA, MDA5, after which the mice can survive until adulthood (Liddicoat et al., 2015). Therefore, future analyses of these double mutant mice could determine whether ADAR1 is the major enzyme responsible.
The role of the C-terminal region of CAPS1 in exocytosis, especially in the priming step, remains controversial. The region that contains the E/G site was identified originally as a domain required for the association with DCVs (Grishanin et al., 2002). Subsequently, the C-terminal region that encompasses MDH1 (Munc13-homology domain) and DCVBD has been reported to bind to SNARE proteins and to promote SNARE-mediated vesicle fusion (Hammarlund et al., 2008; James et al., 2009; Parsaud et al., 2013). In contrast, a recent study demonstrated that the pleckstrin homology (PH) domain located in the middle of CAPS1, but not the C-terminal region, is essential for the priming of secretory vesicles by CAPS1 (Nguyen Truong et al., 2014). However, the present study demonstrated that a single point mutation in the DCVBD, leading to the exclusive expression of edited CAPS1, induced leanness and hyperactivity in mice. Furthermore, DCV exocytosis was enhanced in these mutant mice, which indicated that the E/G site-containing C-terminal region was physiologically significant and critical for exocytosis.
The edited CAPS1 KI mice exhibited increased physical activity, which was attenuated by the administration of the dopamine D2 receptor antagonist. We further demonstrated that synaptosomes derived from these mutant mice could secrete dopamine more efficiently than those from WT mice. Intriguingly, mutant mice that lack the dopamine transporter also exhibit a greater degree of hyperactivity and leanness (Giros et al., 1996), which suggests that the hyperactivity of the edited CAPS1 KI mice is mediated predominantly through the dopamine pathway. The extent to which CAPS1 RNA editing controls physical activity in humans and the WT mouse, in which 10%–20% of brain CAPS1 is edited, remains to be determined. It is possible that the editing frequency is higher in certain subtypes of neurons or the editing is dynamically regulated among individual neurons. Furthermore, it has been reported that the priming step of synaptic vesicle exocytosis is downregulated in the hippocampal glutamargic neurons of CAPS1 KO mice, although it remains unknown whether this affects physical activity (Jockusch et al., 2007). Consequently, further investigation is needed to elucidate the differences in editing frequency at the single cell level and to determine whether CAPS1 RNA editing modulates the exocytosis of synaptic vesicles in addition to DCVs.
Of note, the CAPS family is known to be involved in developmental disorders that affect physical activity. The locus of the CADPS2 gene, which encodes CAPS2, is located within the autism susceptibility locus 1 (AUTS1) (International Molecular Genetic Study of Autism Consortium, 2001), and it is reported to be mutated in patients with intellectual disability (Bailey et al., 1998). CAPS2−/− mice exhibit autistic-like phenotypes, including reduced activity in an open field in the presence of a novel object (Sadakata et al., 2007). Furthermore, a recent study reported a case of a proximal interstitial deletion of chromosome 3p that includes the CADPS gene locus (de la Hoz et al., 2015). Although it is unclear whether physical activity is affected, the deletion results in speech and social interaction difficulties and learning disability. These findings imply that dysregulation of CAPS1 RNA editing might be involved in some disorders that affect physical activity, and this requires further investigation.
EXPERIMENTAL PROCEDURES
Please see Supplemental Experimental Procedures for the full experimental procedures.
Mouse Administration
Mice were maintained on a 12-hr-light/dark cycle at a temperature of 23°C ± 1.5°C with a humidity of 45% ± 15%. All experimental procedures that involved mice were approved by the Institutional Animal Care and Use Committee of Osaka University and Institutional Animal Care and RIKEN Kobe Branch.
Generation of Edited CAPS1 KI Mice
The mutant mice carrying glycine at the E/G site in CAPS1 (accession number CDB1091K: http://www2.clst.riken.jp/arg/mutant%20mice%20list.html) were generated as described (Yagi et al., 1993) using TT2 embryonic stem (ES) cells that were derived from an F1 embryo between C57BL/6 and CBA (http://www.clst.riken.jp/arg/Methods.html). To generate a targeting vector, genomic fragments of the Cadps locus that contained the exon containing the E/G site were obtained from the RP23-466K8 BAC clone (BACPAC Resources) and inserted into the DT-A-pA/loxP/PGK-Neo-pA/loxP vector (http://www.clst.riken.jp/arg/cassette.html). An adenine-to-guanine substitution was introduced by site-directed mutagenesis so that the codon for glutamic acid (GAG) at the E/G site was replaced by one for glycine (GGG). Targeted ES clones were microinjected into ICR 8-cell stage embryos, and the injected embryos were transferred into pseudopregnant ICR females. The resulting chimeras were bred with C57BL/6 mice, and the germline transmission to heterozygous offspring was confirmed by Southern blotting using probes that targeted the positions indicated in Figure S3A and also by PCR. Then, the mutant mice were backcrossed with C57BL/6 mice more than six times for phenotypic analyses. Genotyping was performed by direct sequencing of PCR products that contained the E/G site using the sequencing primer mCAPS1-KI-Dw1 (5′-AGAGGGAGAGGAGAGATTC-3′). PCR products (306 bp) were amplified with the following primers: mCAPS1-KI-Fw1 (5′-ACTTTCGTCCGCCATTCTC-3′) and mCAPS1-KI-Dw2 (5′-CGTCCACATATCACACAGACT-3′).
In Vitro RNA Editing Assay for Human CAPS1 Pre-mRNA
The in vitro editing assay was performed as described previously (Kawahara et al., 2007b). In brief, the in vitro editing reaction mixture, which contained ~5 fmol of synthesized RNA and the indicated amount of recombinant ADAR1 or ADAR2 protein (Figure S1C), was incubated at 30°C for 1 hr. Then, the samples were treated with 2 × Proteinase K buffer (100 mM Tris-HCl [pH 7.5], 100 mM NaCl, and 20 mM MgCl2) that contained 2 mg/mL Proteinase K (Ambion) at 55°C for 30 min, and the RNA fragments were recovered by phenol extraction and 2-propanol precipitation with glycogen. After treatment with DNase I, the denatured RNAs were subjected to RT-PCR followed by direct sequencing. The editing frequency was determined as the % ratio of the “G” peak over the sum of the “G” and “A” peaks of the sequencing chromatogram.
[3H]NA Release Assays Using PC12 Cells
The release of [3H]NA was quantified as described previously with minor modifications (Parsaud et al., 2013). In brief, 2.0 × 105 shRNA_All-8 PC12-derived cells expressing either unedited or edited mCAPS1 stably were incubated in a 24-well plate with 1.0 μCi/mL [3H]NA (levo-[7-3H]-Norepinephrine; PerkinElmer) in the presence of 0.5 mM ascorbic acid for 16 hr. The radiolabeled cells were incubated with fresh DMEM for 5 hr to remove unincorporated [3H]NA. After the cells had been washed once with 200 μl of PSS buffer (145 mM NaCl, 5.6 mM KCl, 2.2 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose, and 15 mM HEPES [pH 7.4]), NA secretion was induced by incubation at 37°C in 200 μl of high K+ buffer 1 (HK1; 81 mM NaCl, 70 mM KCl, 2.2 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose, and 15 mM HEPES [pH 7.4]) for 15, 30, or 60 min. Then, the supernatant was removed immediately and collected into a 1.5-mL tube. The resultant pellets were lysed with 500 μl of 0.1% Triton X-100. Both the supernatant and the cell lysate (140 μl each) were mixed with 700 μl of MicroScint-20 cocktail (PerkinElmer), and the β activity derived from [3H] was quantified using a liquid scintillation counter (Micro Beta2 Plate Counter; PerkinElmer). The efficiency of [3H]NA release was calculated by dividing the amount of [3H]NA in the supernatant by the total amount of [3H] NA present in both the supernatant and the cells.
Metabolic Rate
The rate of oxygen consumption was measured by indirect calorimetry using a small animal metabolic measurement system (MK-5000RQ; Muromachi). An individual adult male mouse (9–14 weeks of age) was placed in the chamber under standard feeding conditions. After habituation over a period of 48 hr, the oxygen consumption rate was measured every 3 min for 24 hr. To evaluate the effect of haloperidol, a dopamine D2 receptor antagonist, on physical activity, an individual adult male mouse (9–20 weeks of age) was placed in the chamber as well. Then, either 0.5 mg/kg haloperidol (Sigma-Aldrich) dissolved in saline or an equal volume of saline (control) was injected intraperitoneally 1 hr after the dark cycle had started, while the rate of oxygen consumption was being measured. After waiting for an additional 1 hr, the oxygen consumption rate was measured for the subsequent 10 hr.
Behavioral Analyses
All the behavioral analyses were performed with the same groups of adult male littermates of WT and homozygous KI mice (11–19 weeks of age) by inserting an adequate time period between each test.
Open-Field Tests
The open-field test was conducted as described previously (Taniguchi et al., 2005). A cubic box made of transparent acrylic plates (30 × 30 × 30 cm) without a ceiling was housed in a ventilated soundproof chamber. An overhead incandescent light bulb provided room lighting that measured approximately 110 lux at the center of the test arena. In addition, a fan attached to the upper part of the wall at one end of the chamber provided a masking noise of 45 dB inside the chamber. On each X and Y lateral side of the open-field box, two infrared beams were attached 2 cm above the floor at a distance of 10 cm apart. A flip-flop circuit was set up between the two beams. The total number of circuit breaks was taken as a measure of locomotive behavior. Animals were allowed to explore freely in the open-field arena for 10 min.
Circadian Rhythm Tests
Circadian rhythm tests were conducted using a cubic box (45 × 45 × 45 cm) equipped with an infrared sensor on the ceiling. Each mouse was placed inside the box 5 hr after the start of the light cycle. After 9 hr of rest, the activity of the mouse was quantified by the infrared sensor every 1 s for 24 hr. To evaluate the effect of haloperidol, a dopamine D2 receptor antagonist, on physical activity, either 0.5 mg/kg haloperidol dissolved in saline or an equal volume of saline (control) was injected intraperitoneally 1 hr after the dark cycle had started. After waiting for an additional 1 hr, the total activity during each hour was determined for the subsequent 10 hr.
Amperometric Detection
Catecholamine release from each individual adrenal chromaffin cell was measured by amperometric recording as described previously (Ohta et al., 2010). The coverslip with an attached chromaffin cell was placed in the recording chamber mounted on the stage of an inverted microscope (Diaphot 300; Nikon). Cells were superfused at a flow rate of ~2 mL/min with standard external buffer (134 mM NaCl, 6 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 5 mM glucose, and 10 mM HEPES [pH 7.4]). The tip of the carbon fiber electrode (ProCFE; Dagan) was advanced gently to touch the surface of the chromaffin cell of interest using a micromanipulator (MHW-3; Narishige). The +600 mV was applied to the electrode under voltage-clamp conditions. The currents that resulted from the oxidation of catecholamines were recorded using an electrochemical detector (CHEM-CLAMP; Dagan). The data on the currents were digitized and analyzed using an A/D converter (PowerLab; AD Instruments) in conjunction with a personal computer. The recorded amperometric currents reflect vesicles that have been released close to the electrode and represent the fusion of exocytotic vesicles with the cell membrane. The responsiveness of the carbon fiber electrodes to catecholamine was examined before the experiment using 5 μM adrenaline (Daiichi Sankyo). High K+-induced changes in the amperometric current (Iamp) and the total charge were assessed by calculating the area under the curve (a time-integral; AUC) during the application of high K+ buffer 2 (HK2; 70 mM KCl, 70 mM NaCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 5 mM glucose, and 10 mM HEPES [pH 7.4], with NaOH). All experiments were carried out at room temperature.
Measurement of Intracellular Ca2+ Concentration
The intracellular Ca2+ concentration ([Ca2+]i) in each individual chromaffin cell was measured with the fluorescent Ca2+ indicator fura-2 by dual excitation, using a fluorescence-imaging system with controlled illumination and acquisition (Aqua Cosmos; Hamamatsu Photonics). After the cells had been incubated for 40 min at 37°C with 10 μM fura-2 AM (Molecular Probes) in standard external solution, a coverslip with fura-2-loaded cells was placed in an experimental chamber that was mounted on the stage of an inverted microscope (IX71; Olympus) equipped with an image acquisition and analysis system (Aqua Cosmos). Cells were illuminated every 5 s with light at 340 and 380 nm, and the respective fluorescence signals at 500 nm were detected. The fluorescence emitted was projected onto a charge-coupled device camera (ORCA-ER; Hamamatsu Photonics), and the ratios of the fluorescent signals (F340/F380) for [Ca2+]i were stored on the hard disk of a personal computer.
Ultrastructural Analyses
After overnight fasting, adult male mice (9 weeks of age) were anesthetized by intraperitoneal injection of a mixture of 0.3 mg/kg medetomidine, 4 mg/kg midazolam, and 5 mg/kg butorphanol. After perfusion of the fixing solution (2% glutaraldehyde and 2% paraformaldehyde), the adrenal gland was dissected and incubated in the fixing solution for 2 hr. Sections were prepared at the National Institute of Biomedical Innovation, Osaka, Japan, as follows. The sample was incubated in 2% osmium acid (Merck) for 1 hr at 4°C and then dehydrated with a graded series of ethanol (50%–100%) and QY-1 (Nisshin EM). The fixed adrenal gland was embedded in EPON812 (TAAB Laboratories Equipment) and incubated at 6°C for 1 hr and at 80°C for an additional 48 hr. Then, the embedded samples were sectioned on an ultramicrotome (EM UC6; Leica) at a thickness of 60 nm and mounted on grids. The sections were stained with uranyl acetate (Electron Microscopy Science) for 30 min and with lead stain solution (Lead citrate EM, Lead nitrate EM, and Lead acetate EM; TAAB Laboratories Equipment) for 10 min. Images were obtained using a transmission electron microscope (JEM-1400 plus; JEOL) at an acceleration voltage of 80 kV. The distribution of DCVs was measured as described previously with modifications (de Wit et al., 2009; Hammarlund et al., 2008; Liu et al., 2008). In brief, multiple digital images taken at 20,000 × magnification were merged using Microsoft ICE software (http://research.microsoft.com/en-us/um/redmond/projects/ice/) to cover the entire area of each cell. Then, the distance from the plasma membrane to the membrane of each granule that had a round, dense core, and a diameter of more than 40 nm was measured in each individual cell using ImageJ software (http://rsb.info.nih.gov/ij/).
Dopamine Release Tests
Dopamine release tests were conducted as described previously with modifications (Grady et al., 1992; Narboux-Nême et al., 2011). Synaptosomes prepared from mouse brain striatum were incubated in 200 μl of the perfusion buffer with 0.1 μM [3H]DA (3,4-[ring-2,5,6-3H]-dopamine; PerkinElmer) for 10 min at 37°C and centrifuged at 6,000 × g for 10 min at room temperature. The synaptosomal pellet was resuspended in 200 μl of the perfusion buffer and incubated for 10 min. Then, 100 μl of the synaptosomal suspension were loaded on a glass filter (grade GF/B; GE Healthcare) that had been fixed in a 3-piece filter funnel (GE Healthcare). The filter had been prewashed by loading 500 μl of the perfusion buffer onto the glass filter and vacuum filtering after incubation for 3 min; this was repeated six times. After loading of the synaptosomal suspension, 500 μl of the perfusion buffer were loaded onto the glass filter and vacuum filtered after incubation for 3 min. Then, an additional 1,500 μl of the perfusion buffer were loaded and vacuum filtered immediately. As a result, the [3H]DA released from the synaptosomes during the 3-min incubation was collected in a volume of 2,000 μl. Subsequently, 500 μl of either high K+ buffer 3 (HK3; 100.4 mM NaCl, 30.0 mM KCl, 3.2 mM CaCl2, 1.2 mM MgSO4, 25.0 mM HEPES [pH 7.4], 10.0 mM glucose, 1.0 mM ascorbate, and 0.01 mM pargyline) or the perfusion buffer was loaded and vacuum filtered after incubation for 3 min. Then, an additional 1,500 μl of HK3 or the perfusion buffer were loaded and vacuum filtered immediately. This procedure was repeated every 3 min three times. Finally, 200 μl of each filtrate was mixed with 800 μl of MicroScint-20 cocktail, and the β activity derived from [3H] was quantified by using liquid scintillation counting.
Statistical Analyses
The chi-square test, one-way ANOVA, two-way ANOVA, multifactorial repeated-measures ANOVA, Mann–Whitney test, or the unpaired t test was used as described in each figure legend. All p values were two sided. Statistical significance was set at *p < 0.05 and **p < 0.01.
Supplementary Material
Highlights.
CAPS1 RNA editing facilitates the rapid release of catecholamine
Edited CAPS1 promotes dense core vesicle exocytosis
CAPS1 RNA editing upregulates physical activity through the dopamine pathway
Sole expression of edited CAPS1 leads to leanness in mice
Acknowledgments
We thank Dr. K. Takahashi, A. Mieda-Sato, T. Sono, and H. Okada for technical assistance. We thank Drs. T. Sadakata (Gunma University) and T. Furuichi (Tokyo University of Science) for helpful discussion. This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan for Scientific Research on Innovative Areas (21112515, 15H01471) to Y.K. and for Scientific Research (B) (26292150) to T.O. and by grants from Takeda Medical Research Foundation and Suzuken Memorial Foundation to Y.K. as well as grants from the US National Institutes of Health (R01 GM040536, R01 CA17508), Ellison Medical Foundation (AG-SS-228-09), Macula Vision Research Foundation, and the Commonwealth Universal Research Enhancement Program of the Pennsylvania Department of Health to K.N.
Footnotes
ACCESSION NUMBERS
The accession number for the mutant mouse reported in this paper is RIKEN: CDB1091K (http://www2.clst.riken.jp/arg/mutant%20mice%20list.html).
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and six figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.10.073.
AUTHOR CONTRIBUTIONS
K.M. and Y.K. designed the study. T.O., H.N., and K.H. performed the electrophysiological experiments. N.D. performed behavioral analyses and interpreted the results. T.A. and Y.Y. generated the mutant mice. K.M. and Y.K. performed all other experiments with assistance from Y.T. and E.O. I.N., A.K., K.N., and Y.K. conceived and discussed the projects. K.N. contributed to essential reagents and materials. K.M. and Y.K. wrote the manuscript. All authors agree to the contents of the final manuscript.
References
- Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2:e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey A, Hervas A, Matthews N, Palferman S, Wallace S, Aubin A, Michelotti J, Wainhouse C, Papanikolaou K, Rutter M, International Molecular Genetic Study of Autism Consortium A full genome screen for autism with evidence for linkage to a region on chromosome 7q. Hum Mol Genet. 1998;7:571–578. doi: 10.1093/hmg/7.3.571. [DOI] [PubMed] [Google Scholar]
- Bazzazi H, Ben Johny M, Adams PJ, Soong TW, Yue DT. Continuously tunable Ca(2+) regulation of RNA-edited CaV1.3 channels. Cell Rep. 2013;5:367–377. doi: 10.1016/j.celrep.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behm M, Öhman M. RNA editing: A contributor to neuronal dynamics in the mammalian brain. Trends Genet. 2016;32:165–175. doi: 10.1016/j.tig.2015.12.005. [DOI] [PubMed] [Google Scholar]
- Bhalla T, Rosenthal JJC, Holmgren M, Reenan R. Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat Struct Mol Biol. 2004;11:950–956. doi: 10.1038/nsmb825. [DOI] [PubMed] [Google Scholar]
- Brusa R, Zimmermann F, Koh DS, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270:1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- Daniel C, Wahlstedt H, Ohlson J, Björk P, Öhman M. Adenosine-to-inosine RNA editing affects trafficking of the gamma-aminobutyric acid type A (GABA(A)) receptor. J Biol Chem. 2011;286:2031–2040. doi: 10.1074/jbc.M110.130096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Hoz AB, Maortua H, García-Rives A, Martínez-González MJ, Ezquerra M, Tejada MI. 3p14 de novo interstitial microdeletion in a patient with intellectual disability and autistic features with language impairment: A comparison with similar cases. Case Rep Genet. 2015;2015:876348. doi: 10.1155/2015/876348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Walter AM, Milosevic I, Gulyás-Kovács A, Riedel D, Sørensen JB, Verhage M. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell. 2009;138:935–946. doi: 10.1016/j.cell.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Eisenberg E, Nemzer S, Kinar Y, Sorek R, Rechavi G, Levanon EY. Is abundant A-to-I RNA editing primate-specific? Trends Genet. 2005;21:77–81. doi: 10.1016/j.tig.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Farina M, van de Bospoort R, He E, Persoon CM, van Weering JR, Broeke JH, Verhage M, Toonen RF. CAPS-1 promotes fusion competence of stationary dense-core vesicles in presynaptic terminals of mammalian neurons. eLife. 2015;4:e05438. doi: 10.7554/eLife.05438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AP, Keller W. RNA editing by base deamination: More enzymes, more targets, new mysteries. Trends Biochem Sci. 2001;26:376–384. doi: 10.1016/s0968-0004(01)01827-8. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Grady S, Marks MJ, Wonnacott S, Collins AC. Characterization of nicotinic receptor-mediated [3H]dopamine release from synaptosomes prepared from mouse striatum. J Neurochem. 1992;59:848–856. doi: 10.1111/j.1471-4159.1992.tb08322.x. [DOI] [PubMed] [Google Scholar]
- Grishanin RN, Klenchin VA, Loyet KM, Kowalchyk JA, Ann K, Martin TF. Membrane association domains in Ca2+-dependent activator protein for secretion mediate plasma membrane and dense-core vesicle binding required for Ca2+-dependent exocytosis. J Biol Chem. 2002;277:22025–22034. doi: 10.1074/jbc.M201614200. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Watanabe S, Schuske K, Jorgensen EM. CAPS and syntaxin dock dense core vesicles to the plasma membrane in neurons. J Cell Biol. 2008;180:483–491. doi: 10.1083/jcb.200708018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem. 2004;279:4894–4902. doi: 10.1074/jbc.M311347200. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- Huntley MA, Lou M, Goldstein LD, Lawrence M, Dijkgraaf GJ, Kaminker JS, Gentleman R. Complex regulation of ADAR-mediated RNA-editing across tissues. BMC Genomics. 2016;17:61. doi: 10.1186/s12864-015-2291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Molecular Genetic Study of Autism Consortium (IMGSAC) Further characterization of the autism susceptibility locus AUTS1 on chromosome 7q. Hum Mol Genet. 2001;10:973–982. doi: 10.1093/hmg/10.9.973. [DOI] [PubMed] [Google Scholar]
- James DJ, Kowalchyk J, Daily N, Petrie M, Martin TF. CAPS drives trans-SNARE complex formation and membrane fusion through syntaxin interactions. Proc Natl Acad Sci USA. 2009;106:17308–17313. doi: 10.1073/pnas.0900755106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jockusch WJ, Speidel D, Sigler A, Sørensen JB, Varoqueaux F, Rhee JS, Brose N. CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell. 2007;131:796–808. doi: 10.1016/j.cell.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007a;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007b;8:763–769. doi: 10.1038/sj.embor.7401011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Grimberg A, Teegarden S, Mombereau C, Liu S, Bale TL, Blendy JA, Nishikura K. Dysregulated editing of serotonin 2C receptor mRNAs results in energy dissipation and loss of fat mass. J Neurosci. 2008;28:12834–12844. doi: 10.1523/JNEUROSCI.3896-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, Zhang K, Gao Y, Church GM. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349:1115–1120. doi: 10.1126/science.aac7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Schirra C, Stevens DR, Matti U, Speidel D, Hof D, Bruns D, Brose N, Rettig J. CAPS facilitates filling of the rapidly releasable pool of large dense-core vesicles. J Neurosci. 2008;28:5594–5601. doi: 10.1523/JNEUROSCI.5672-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morabito MV, Abbas AI, Hood JL, Kesterson RA, Jacobs MM, Kump DS, Hachey DL, Roth BL, Emeson RB. Mice with altered serotonin 2C receptor RNA editing display characteristics of Prader-Willi syndrome. Neurobiol Dis. 2010;39:169–180. doi: 10.1016/j.nbd.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narboux-Nême N, Sagné C, Doly S, Diaz SL, Martin CB, Angenard G, Martres MP, Giros B, Hamon M, Lanfumey L, et al. Severe serotonin depletion after conditional deletion of the vesicular monoamine transporter 2 gene in serotonin neurons: Neural and behavioral consequences. Neuropsychopharmacology. 2011;36:2538–2550. doi: 10.1038/npp.2011.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Truong CQ, Nestvogel D, Ratai O, Schirra C, Stevens DR, Brose N, Rhee J, Rettig J. Secretory vesicle priming by CAPS is independent of its SNARE-binding MUN domain. Cell Rep. 2014;9:902–909. doi: 10.1016/j.celrep.2014.09.050. [DOI] [PubMed] [Google Scholar]
- Ohta T, Ohba T, Suzuki T, Watanabe H, Sasano H, Murakami M. Decreased calcium channel currents and facilitated epinephrine release in the Ca2+ channel beta3 subunit-null mice. Biochem Biophys Res Commun. 2010;394:464–469. doi: 10.1016/j.bbrc.2010.01.036. [DOI] [PubMed] [Google Scholar]
- Parsaud L, Li L, Jung CH, Park S, Saw NMN, Park S, Kim MY, Sugita S. Calcium-dependent activator protein for secretion 1 (CAPS1) binds to syntaxin-1 in a distinct mode from Munc13-1. J Biol Chem. 2013;288:23050–23063. doi: 10.1074/jbc.M113.494088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto Y, Cohen HY, Levanon EY. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol. 2014;15:R5. doi: 10.1186/gb-2014-15-1-r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadakata T, Washida M, Iwayama Y, Shoji S, Sato Y, Ohkura T, Katoh-Semba R, Nakajima M, Sekine Y, Tanaka M, et al. Autistic-like phenotypes in Cadps2-knockout mice and aberrant CADPS2 splicing in autistic patients. J Clin Invest. 2007;117:931–943. doi: 10.1172/JCI29031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai M, Ueda H, Yano T, Okada S, Terajima H, Mitsuyama T, Toyoda A, Fujiyama A, Kawabata H, Suzuki T. A biochemical landscape of A-to-I RNA editing in the human brain transcriptome. Genome Res. 2014;24:522–534. doi: 10.1101/gr.162537.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speidel D, Varoqueaux F, Enk C, Nojiri M, Grishanin RN, Martin TFJ, Hofmann K, Brose N, Reim K. A family of Ca2+-dependent activator proteins for secretion: Comparative analysis of structure, expression, localization, and function. J Biol Chem. 2003;278:52802–52809. doi: 10.1074/jbc.M304727200. [DOI] [PubMed] [Google Scholar]
- Speidel D, Bruederle CE, Enk C, Voets T, Varoqueaux F, Reim K, Becherer U, Fornai F, Ruggieri S, Holighaus Y, et al. CAPS1 regulates catecholamine loading of large dense-core vesicles. Neuron. 2005;46:75–88. doi: 10.1016/j.neuron.2005.02.019. [DOI] [PubMed] [Google Scholar]
- Speidel D, Salehi A, Obermueller S, Lundquist I, Brose N, Renström E, Rorsman P. CAPS1 and CAPS2 regulate stability and recruitment of insulin granules in mouse pancreatic β cells. Cell Metab. 2008;7:57–67. doi: 10.1016/j.cmet.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Doe N, Matsuyama S, Kitamura Y, Mori H, Saito N, Tanaka C. Transgenic mice expressing mutant (N279K) human tau show mutation dependent cognitive deficits without neurofibrillary tangle formation. FEBS Lett. 2005;579:5704–5712. doi: 10.1016/j.febslet.2005.09.047. [DOI] [PubMed] [Google Scholar]
- Tomaselli S, Locatelli F, Gallo A. The RNA editing enzymes ADARs: Mechanism of action and human disease. Cell Tissue Res. 2014;356:527–532. doi: 10.1007/s00441-014-1863-3. [DOI] [PubMed] [Google Scholar]
- Wang Q, Khillan J, Gadue P, Nishikura K. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science. 2000;290:1765–1768. doi: 10.1126/science.290.5497.1765. [DOI] [PubMed] [Google Scholar]
- Wulff BE, Sakurai M, Nishikura K. Elucidating the inosinome: Global approaches to adenosine-to-inosine RNA editing. Nat Rev Genet. 2011;12:81–85. doi: 10.1038/nrg2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Tokunaga T, Furuta Y, Nada S, Yoshida M, Tsukada T, Saga Y, Takeda N, Ikawa Y, Aizawa S. A novel ES cell line, TT2, with high germline-differentiating potency. Anal Biochem. 1993;214:70–76. doi: 10.1006/abio.1993.1458. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.