

Abstract
There are three dominant contributors to the pathogenesis of dysfunctional adipose tissue (AT) in obesity: unresolved inflammation, inappropriate extracellular matrix (ECM) remodeling and insufficient angiogenic potential. The interactions of these processes during AT expansion reflect both a linear progression as well as feed-forward mechanisms. For example, both inflammation and inadequate angiogenic remodeling can drive fibrosis, which can in turn promote migration of immune cells into adipose depots and impede further angiogenesis. Therefore, the relationship between the members of this triad is complex but important for our understanding of the pathogenesis of obesity. Here we untangle some of these intricacies to highlight the contributions of inflammation, angiogenesis, and the ECM to both “healthy” and “unhealthy” AT expansion.
An old perspective on a new problem
Adipose tissue (AT) has evolved to be a highly dynamic organ, readily remodeling to meet the demands of an ever-changing metabolic landscape, as illustrated by the rapid changes in adipose depots of rodents undergoing various nutritional stresses. It takes as little as one week of high-fat feeding (HFF) in rodents for adipocytes to become enlarged, storing four-fold the amount of triglyceride per cell compared with baseline (1). The mouse visceral AT weight can double within one week of the initiation of a high-fat diet (HFD) (2) but can undergo a dramatic reduction within 24 hours of fasting. It is this plasticity that has made AT an essential organ for maintaining metabolic homeostasis during times when food is either abundant or scarce.
The ability of AT to rapidly remodel is the result of the coordinated response of resident AT cell types: adipocytes, immune cells, endothelial cells, and fibroblasts. When nutrient availability is in excess, adipocytes store various lipid species in lipid droplets, which quickly expand in size and, in the process, reach the diffusional limit of oxygen. The ensuing hypoxia is mild but induces a stress signal that drives angiogenesis and remodeling of the extracellular matrix (ECM) to facilitate further expansion of AT and reduce hypoxia (Figure 1). This acute effect can be thought of as “healthy” AT expansion. But in the evolutionary history of AT, the necessity to expand and store lipids was short lived, with periods of fasting or starvation guaranteeing reversal of weight gain. The current pervasive state of chronic overnutrition results in an increase in unresolved AT hypoxia. At some point during the progression of obesity, adipose depots expand beyond the tissue’s capacity for adequate angiogenesis, resulting in persistent hypoxia, fibrosis, cellular senescence, and necrotic adipocyte death (Figure 1), ultimately leading to unhealthy AT tissue expansion, a major contributor to the systemic metabolic disturbances that are characteristic of obesity and type 2 diabetes.
Figure 1. The complex contributions of angiogenesis, inflammation, and fibrosis in healthy and unhealthy AT expansion.

Hypoxia during early AT expansion induces stress signaling that facilitates angiogenesis via upregulation of an array of genes including VEGF, initiates inflammatory cell infiltration, and induces organized ECM remodeling to encourage appropriate AT expansion. However, persistent stress signaling translates into injury signaling, caused by long-term and severe hypoxia. This hypoxic condition fails to induce VEGF, resulting in impaired angiogenesis. Limited oxygen supply also triggers chronic low-grade inflammation predominantly characterized by proinflammatory macrophage infiltration. Upregulation of HIF-1α also increases the fibrotic program in AT. The integrated responses among those three factors promote significant hypotrophy in adipose tissue, and the consequence is an unhealthy AT expansion as well as obesity-associated metabolic consequences.
The adipocyte stress signal: mild hypoxia
Hypoxia is an important driver of the early AT response to increased dietary lipids. Within three days of HFF, rapid adipocyte hypertrophy along with mitochondrial uncoupling increase the demand for oxygen and reduce oxygen availability (3). Adipocytes respond by inducing glycolysis and angiogenesis in an attempt to alleviate the hypoxia (3). The major mediators of the hypoxia response are the hypoxia-inducible factor (HIF) transcription factors. HIFs contain an α subunit that, at normal tissue oxygenation, is hydroxylated by oxygen-sensing prolyl hydroxylase enzymes (PHD1, -2, and -3), resulting in ubiquitylation and proteosomal degradation of HIFα. In contrast, reduced oxygen tension inhibits PHDs, resulting in subsequent accumulation of HIFα. There are two major subunits of HIFα, HIF-1α and HIF-2α, which have opposing roles. HIF-1α activation is classically described as pro-angiogenic, but recent studies have shown it to also be pro-fibrotic and proinflammatory (4). In contrast, HIF-2α attenuates HFD-induced inflammation (3). Although HIF-1α and HIF-2α pathways can interact, HIF-1α signaling dominates in obesity, playing a causative role in early AT dysfunction (3). An important determinant of HIF-1α activity relative to HIF-2α activity is the abundance of specific PHD enzymes, which have differential specificities for prolyl hydroxylation sites between the HIFα isoforms (5). The importance and intricacy of PHD-specific regulation of HIFα can be illustrated by the phenotypes of mice deficient in either PHD1 or PHD2. PHD1 KO mice exhibit a greater fat pad mass and impaired glucose tolerance but do not show exacerbated diet-induced metabolic changes compared with WT mice (6). In contrast, adipocyte-specific deletion of PHD2 produces mice that are resistant to HFD-induced obesity and, despite elevated levels of both HIF-1α and HIF-2α, the mice display improved glucose tolerance and insulin sensitivity (7). Therefore, although further studies are required to understand the exact role for HIF-1α and HIF-2α in the regulation of AT expansion, it is clear that hypoxia signaling is paramount to the early response of AT to nutrient excess.
Acute inflammation.
In lean individuals, resident AT immune cells are indispensable for adipocyte function; however, AT inflammation is one of the most widely recognized contributors to metabolic dysfunction in obesity. Cells of both the innate and adaptive immune systems have been identified in AT (8, 9). Macrophages comprise the largest population of resident immune cells in visceral AT, constituting up to 10% of AT cells in mice (10). Macrophages are responsible for many housekeeping processes, such as removal of apoptotic and necrotic cells, modulation of angiogenesis, ECM remodeling, and differentiation of adipocyte precursors (8). Macrophages are often characterized by their “polarized” state, displaying more of a proinflammatory or antiinflammatory phenotype. Pathologic activation of both innate and adaptive immunity has been observed during obesity, even though the degree to which each component participates differs temporally. For instance, ApoE3-Leiden mice, which exhibit a more humanized lipid metabolism compared with WT mice, display increased acute-phase response genes within one week of HFF, whereas chronic inflammatory responses are increased after six weeks of HFF (2). This is not seen under all circumstances, as CD8+ T cell infiltration has been observed to precede and induce AT macrophage accumulation in other settings (11).
As an early event in AT expansion, hypoxia likely plays a fundamental role in the initiation of inflammation. Adipocytes release proinflammatory factors such as IL-6 and macrophage migration inhibitory factor (12). Immune cells, particularly monocytes, migrate to areas of hypoxia, where they differentiate into proinflammatory macrophages (13, 14). Macrophage infiltration into AT occurs within one day of HFF (15). Although few studies have directly addressed the effect of mild hypoxia during early AT expansion on AT inflammatory processes, there have been reports of both pro- and antiinflammatory responses during the first four days of HFF. Lee et al. demonstrated a mild proinflammatory response in the epididymal adipose depot of mice fed a HFD for three days (16), and the proinflammatory polarization of macrophages was of particular importance in this response. In contrast, others have reported that acute HFD stress leads to activation of antiinflammatory macrophages (17, 18). This effect is mediated though NKTs, which are activated by lipid antigens presented on CD1d molecules of antigen-presenting cells. The actions of NKT cells are likely beneficial for the adaptation of AT to acute HFF because mice deficient in CD1d do not activate AT NKT cells and display exacerbated insulin resistance (17). Other studies have corroborated these findings by demonstrating that acute HFF induces antiinflammatory immune cell actions, cell repair signaling, and angiogenesis (1, 17, 18). Thus, the early response to AT expansion is likely the result of a balanced action between pro-and antiinflammatory signals.
Healthy angiogenesis.
The basic function of blood vessels in regulating AT biology has been well studied. The vasculature supplies adipocytes with the necessary oxygen, nutrients, hormones, and growth factors for AT viability and growth and removes metabolic waste products. The vasculature may also make a direct contribution to the maintenance of AT. For example, circulating stem cells can contribute to adipogenesis (19). Interestingly, populations of white adipocyte progenitors have been identified in the mural compartment of vascular structures. Tang et al. demonstrated that these mural cells with high adipogenic potential can be identified by expression of PDGFR-β (20). Additionally, Gupta et al. used a genetic reporter mouse driven by the promoter of a transcription factor that is important for pre-adipocyte determination, ZFP423, to demonstrate a perivascular origin of pre-adipocytes for both white and brown ATs (21–23).
Local hypoxia is a potent stimulus for new blood vessel formation through the pro-angiogenic actions of HIF-1α and HIF-2α (24, 25). AT-derived angiogenic factors have been intensively studied, including VEGF-A (26, 27), VEGF-C (28), placental growth factor (29), HGF (30), IGF-1 (31), angiopoietin-1 and -2 (ANG-1 and ANG-2) (32, 33), and FGF2 (34). Among these factors, the VEGF-A/VEGFR2 signaling pathway is considered to play a particularly important role in angiogenesis and is one of the most potent activators of beiging in the subcutaneous depots of rodents. Additionally, new angiogenic modulators have recently been identified as part of the secretory repertoire of adipocytes. For example, the VEGF-B/VEGFR1 pathway also has a positive effect on AT vascularity and insulin sensitivity (35). In addition to authentic angiogenic factors, AT secretes several important adipokines and endogenous angiogenesis inhibitors to modulate the balance of new blood vessel formation, including adiponectin (36, 37), leptin (34), resistin (38), visfatin/thrombospondin 1 (39), and plasminogen activator inhibitor (40). Moreover, adipocytes can secrete pro-angiogenic lipid species such as monobutyrin (41).
Angiogenesis can be divided into two major subtypes: (a) sprouting angiogenesis, which is slow and the major developmental form of vessel growth and (b) looping/splitting angiogenesis, which is fast and occurs predominantly during adult wound healing. VEGF is a major regulator of sprouting angiogenesis (42). During sprouting, new vessels are headed by specialized endothelial cells (tip cells), which migrate in response to chemotactic and pro-angiogenic factors such as VEGF. In looping/splitting angiogenesis, existing vessels are split into two by the force of contracting myofibroblasts associated with a wound. There is no direct evidence for the existence of these types of angiogenesis in obese adult AT, but we could speculate that distinct angiogenic mechanisms participate during AT expansion and are dependent on the extent of hypoxia and the complex regulation in early or late stages of expansion. For example, VEGF is upregulated in AT of mice fed a HFD for three days, but there are reports of both enhanced and reduced VEGF expression in chronic obesity (3, 13, 43, 44), suggesting that sprouting angiogenesis is activated early and may or may not persist. In contrast, the combination of low VEGF signaling and increased myofibroblast activation suggests that splitting angiogenesis occurs more frequently during chronic obesity. Further studies are required to understand how these different types of angiogenesis contribute to obesity-associated vascular dysfunction. However, given the circumstantial evidence, it seems clear that, compared with any other tissue, a disproportionate level of vascular remodeling persists in AT, even at steady state.
Appropriate ECM remodeling.
The ECM of AT has a multi-faceted function in tissue homeostasis. It consists of various components including collagens, fibrillins, proteoglycans and non-proteoglycan polysaccharides. These elements act as a scaffold to maintain tissue structure, contribute to inflammatory signaling and sequester various growth factors for time- and context-dependent release. The ECM is maintained through secretion of ECM components by both resident adipocytes and stromal cells, particularly fibroblasts. Many ECM molecules, such as hyaluronic acid (HA), signal through direct binding of cell receptors (45). HA is a non-sulfated glycosaminoglycan that exists as a heterogeneous mixture of greatly varying molecular weights, allowing for modulation of multiple signaling pathways involved in angiogenesis, cell migration, inflammation and ECM remodeling. Other ECM molecules rely on the proteolytic release of biologically active peptides. For example collagen VI is cleaved at the C5 domain of the α3 subunit and the resulting peptide induces multiple processes including increasing collagen deposition, endothelial cell migration and macrophage infiltration (46, 47).
Bona fide fibrosis, defined as excessive ECM protein deposition, is a pathological consequence of long-term obesity, but acute ECM remodeling is essential for early healthy AT expansion. Genes involved in collagen breakdown, particularly MMP14, are significantly upregulated in mice fed a HFD for one week (48). Mice with heterozygous deletion of MMP-14 are not able to appropriately re-model the ECM, resulting in reduced capacity for adipose expansion (48). Conversely, mice deficient in collagen VI exhibit unimpeded tissue expansion (49). Thus, in early AT expansion, there is a strong ECM degradation signal to promote AT growth, which is important for maintaining whole body metabolic homeostasis (49).
The integrated response.
Hypoxia is a powerful trigger for ECM remodeling and inflammation as a means to facilitate angiogenesis. A potential mechanism for hypoxia-induced angiogenesis during early AT expansion is the production of pro-angiogenic, low-molecular-weight HA (LMW-HA). Gao et al. have demonstrated that LMW-HA production likely occurs in cells cultured in low oxygen through enhanced activity of the HA-degrading enzyme hyaluronidase (50). HA is also a potent inducer of the innate immune response through binding and activation of immune cell TLR2 and -4 (51). Finally, HA can participate in TGF-β1–dependent proliferation of fibroblasts to promote fibrosis. Immune cells can also promote angiogenesis in hypoxia. The hypoxic regions at the tip of the mouse epididymal fat pad have been shown to promote infiltration of a specific species of macrophage expressing the lymphatic endothelial marker LYVE-1. These LYVE-1+ macrophages promote angiogenesis through secretion of MMP-9, -12, and -7 to remodel the ECM and angiogenic factors like VEGF to stimulate the growth of a dense vascular network. This new vasculature provides the framework for adipogenesis and growth of the fat pad (52). Furthermore, the actions of MMP-9 both break down the fibrous collagen matrix to make space for new blood vessels and release matrix-bound VEGF (53).
There is a reciprocal interaction between immune cell function and angiogenesis. Blood vessels facilitate the physical infiltration of macrophages and leukocytes into tissues. These cells in turn promote endothelial cell proliferation and new blood vessel formation by secreting pro-angiogenic factors (54–56). Additionally, several cytokines produced by inflammatory cells have angiogenic effects, including TNF, IL-6, and IL-8 (57, 58). Adipocyte- and endothelial cell–derived ANG-2 exerts effects on both endothelial cells and immune cells. ANG-2 binds the endothelial cell RTK Tie2, activating endothelial cell migration and proliferation, but also initiating an NF-κB–dependent inflammatory signaling cascade (59). Thus, endothelial cells recruit more inflammatory cells. Interestingly, both ANG-2 and VEGF-A promote an antiinflammatory macrophage polarization (60, 61). The relationship between angiogenesis and immune cell signaling is complex, but at least in the context of a non-pathologic hypoxic stress signal, well-controlled inflammation and angiogenesis promote the healthy expansion of AT with a net beneficial effect on metabolic homeostasis.
The adipocyte injury signal: wound healing
There is a physiologic limit to adipocyte cell size. Adipocytes in the mouse inguinal and epididymal fat pads can increase in size three- and seven-fold, respectively, during 12 weeks of HFF (62). Cell size correlates strongly with the frequency of adipocyte death. The percentage of dead epididymal adipocytes increases progressively from 0.1% at one week to as much as 16% at 12 weeks of HFF (62). Independent of how adipocytes die — from necrosis or apoptosis — the reaction of AT to adipocyte death can be likened to the initiation of a wound healing response, triggering a considerable increase in immune cell infiltration. Monocyte recruitment and differentiation to proinflammatory macrophages are of particular importance. These macrophages surround the dead adipocytes, forming what is described histologically as “crown-like structures” (62–64). Concomitantly, activated myofibroblasts in the area secrete collagen to maintain the integrity of the damaged tissue (65). As macrophages and neutrophils clean the damaged area, they produce toxic products such as ROS and reactive nitrogen species (RNS) that further injure surrounding cells and promote fibrosis. Dermal white AT plays an important role in topical wound healing, with the adipocytes as critical mediators (66). Inflammation during wound healing is self-limiting, provided the force producing the injury signal is removed. In fact, early removal of macrophages is essential to proper wound healing and prevention of scarring (67). However, in obesity the injury signal persists, causing chronic activation of myofibroblasts and immune cells, resulting in further tissue damage, fibrosis, and ultimately AT dysfunction.
Unresolved inflammation.
The regulation of immune cell function and cytokine production in obese AT has been well studied (68), and it is now evident that the balance of pro- and antiinflammatory signals is critical for disease progression. Although a proinflammatory program is activated during early AT expansion, the immune response is dominated by antiinflammatory signals. During chronic obesity, both innate and adaptive immunity are activated and skewed to a proinflammatory response triggered by adipocyte death, hypoxia, and reduced fatty acid storage capacity in dysfunctional adipocytes.
One of the best studied consequences of proinflammatory signaling in AT is insulin resistance. TNF-α, produced by immune cells, was the first cytokine demonstrated to directly impede insulin action in the adipocyte (69). It downregulates the major insulin-responsive glucose transporter GLUT4 and inhibits insulin-dependent tyrosine phosphorylation of the insulin receptor and IRS-1 through ceramide production (70, 71). Other factors produced by inflammatory cells have since been shown to inhibit insulin signaling in the adipocyte including IL-6, IFN-γ and CCL2 (72, 73). Adaptive immune cells also produce factors that influence insulin signaling. B cells are divided into two main subfamilies, B1 and B2. B1 cells produce germline-encoded natural IgM and IgA antibodies. B2 cells are responsive to T cells upon antigen stimulation and produce a host of different adaptive antibodies as well as cytokines. In addition to cytokines, B2 cells also produce over a hundred different antibodies that have been associated with insulin resistance in human AT (8, 9).
It has been suggested that the production of ROS and RNS by immune cells contribute to the oxidative damage observed in obese AT (74, 75). Although this theory has yet to be directly tested, the degree of macrophage infiltration into obese AT and what we know about macrophage function in wound healing make this a plausible scenario. During the inflammatory phase of wound healing, activation of monocytes and macrophages induces a “respiratory burst,” in which large quantities of superoxide are generated by a membrane-bound NADPH oxidase. This respiratory burst precedes the production of multiple ROS including hydroxyl radicals and the longer-lived pro-oxidant hydrogen peroxide. This response makes macrophages lethal to pathogens, but it does not come without collateral damage. Macrophages need to be removed before surrounding tissues can be repaired (76). Increased oxidative stress and reduced antioxidant defense have been demonstrated in obese AT (74). Further work will be needed to determine whether this is a direct consequence of AT macrophage activation.
Interestingly, in humans, oxidative stress may precede AT inflammatory cell infiltration. Men fed a high-calorie diet for one week gained significant weight accompanied with insulin resistance (77). Oxidative damage was detected in AT biopsies without an increase in inflammatory cell markers. In mice, the chronology of oxidative stress and inflammation onset is not known; however, treatment of mice with antioxidants can attenuate diet-induced inflammation and insulin resistance, suggesting a strong influence of oxidative stress on obesity-associated disease (78–80). In addition, oxidative modification of cellular components such as glutathionylated 4-hydroxy-2-nonenal can directly stimulate a macrophage-mediated proinflammatory response (81).
Although the detrimental consequences of proinflammatory signaling in obese AT are evident, our group has shown that it is also required for appropriate expansion of AT and safe storage of potentially toxic lipid species (82). In fact, TNF-α plays an essential role in the adaptation of AT to HFF. Adipose-specific overexpression of a dominant-negative form of TNF-α causes severe glucose intolerance and a reduction in the insulin-sensitizing adipokine adiponectin in mice fed a HFD (82). Thus, AT inflammation in obese individuals cannot be assigned a strictly pathologic role.
Impaired angiogenesis.
Under the persistent metabolic challenge of chronic HFF, the demand for oxygen is great but the capacity to form new blood vessels is poor. The consequence is disorganized and pathologic angiogenesis; however, the underlying mechanisms are not fully understood. Long-term HFF results in abnormal regulation of VEGF-A expression; not only do the VEGF-A levels decrease in the AT of obese mice and humans (13), but there is also evidence that the presence of VEGF in obese mice can block the regulation of neovascularization and vessel normalization (83). Interestingly, unlike acute AT expansion, the hypoxia response of obese ob/ob mice fails to induce VEGF expression, and instead a decrease in vascular density is observed (4). Although the majority of studies have shown that obese AT is hypoxic, we do not know if the degree of hypoxia is sufficient to enforce persistent angiogenesis, particularly in human AT. Indeed, it has been argued that the oxygen tension in AT is not low enough to be defined as hypoxic (84). Regardless of the intensity of the hypoxia, our studies have shown that activating the classic hypoxia response is not sufficient to ameliorate angiogenic deficiencies in obese mice. Genetic introduction of dominant-active HIF-1α in mice was unable to induce an angiogenic response but instead increased the expression of several fibrotic genes (4). Conversely, either overexpression of a dominant-negative HIF-1α or pharmacologic inhibition of HIF-1α significantly reduced AT fibrosis and improved AT function in mice fed a HFD (85). Further efforts are required to understand how AT angiogenesis is disrupted in obesity and how this can be therapeutically overcome.
One potential determinant of vessel density and integrity is the type of angiogenesis that occurs during long-term obesity. We can speculate that, as a result of myofibroblast activation in obese AT, new vascular networks are formed frequently through intussusception, or splitting of existing vessels. This rapid form of angiogenesis can be beneficial in the special conditions of cutaneous wound healing but may contribute to vascular dysfunction in obese fibrotic AT. Mechanical forces that exist during the rapid growth of a tumor induce this kind of angiogenesis (86). As in obese AT, tumor vasculature is significantly more permeable than that of normal tissue (87), a characteristic that promotes immune cell extravasation and fibrosis. Thus, future therapeutic strategies will benefit from an understanding of the mechanisms behind the pathologic angiogenesis observed in obesity.
Given the importance of appropriate vascularization for AT expansion, angiogenesis has been proposed as a potential therapeutic target to treat obesity and its related metabolic problems. Chemical angiogenic inhibitors such as angiostatin, endostatin, and thalidomide as well as the VEGFR2-blocking antibody TNP-470, have been tested in diet-induced obese mice or genetically obese ob/ob mice and shown to significantly reduce body weight and fat pad mass (88, 89). Additionally, a recent study suggested treatment with docosahexaenoic acid might improve insulin resistance through attenuation of AT angiogenesis (90). Several natural products extracted from plants (91–93) have also effectively reduced body weight in ob/ob and HFD-induced obese mice by inhibiting angiogenesis. Despite these results, the inhibition of angiogenesis as a therapeutic strategy for obesity remains a controversial notion (94). Any metabolic improvements afforded by these systemically introduced compounds cannot be directly attributed to reduced angiogenesis in AT. Our group has reported that WAT-specific overexpression of VEGF-A resulted in differential metabolic effects in mice challenged with a HFD or mice that are genetically obese (22). In HFD-challenged mice, VEGF-A significantly increased WAT vascularization and beiging, which augmented energy expenditure and prevented unfavorable metabolic changes, while blockade of VEGF-A/VEGFR2 caused aggravated systemic insulin insensitivity. In contrast, in ob/ob mice with pre-existing obesity, inhibiting the VEGF-A/VEGFR2 signaling pathway resulted in improved insulin sensitivity and decreased body weight.
Several key questions about the role of angiogenesis in AT remain. First, what types of pro-angiogenic mechanisms are in effect either simultaneously or sequentially (and in which order) during the expansion of AT? Second, how do we properly define the stages of AT expansion and further identify when angiogenesis is appropriate and when it is pathologic? Third, can modulators of angiogenesis be activated locally in different fat pads and achieve distinct effects? And finally, would an angiogenic strategy be more effective if combined with other existing anti-obesity therapies?
Fibrosis and ECM dysfunction.
Fibrosis occurs during chronic obesity and has been accepted as a major contributor to obesity-associated AT dysfunction. Although deposition of fibrous collagen proteins is well known to promote metabolic dysfunction in obesity, many other ECM components are dysregulated in obese AT. Recent studies have shown an important role of microfibril-associated glycoprotein 1 (MAGP1) in obese AT. MAGP1 binds active TGF-β, sequestering it in the ECM (95). The actions of MAGP1 have been shown to protect mice from obesity and associated metabolic defects (95). Additionally, HA accumulates in insulin-resistant AT in obese mice and is thought to negatively regulate adipocyte insulin signaling (96). Fibrosis has been suggested to limit AT angiogenic capacity. Genetic knockout of the collagen-binding receptor integrin in the muscle of diet-induced obese mice increases vascularization (97).
Interestingly, the detrimental effects of fibrosis-mediated restriction of AT expansion during obesity on metabolic health may be dependent on the adipose depot affected. Mice genetically deficient in interferon regulatory factor 5 display enhanced expansion of subcutaneous AT but limited expansion of visceral AT on a HFD (98). Restricted epididymal AT expansion was associated with a large increase in antiinflammatory macrophage infiltration, collagen deposition, and improved insulin sensitivity. Thus, the ability of fibrosis to affect AT processes and signaling pathways in a depot-specific manner makes it a fertile topic for future research.
An integrated response: a role for cellular senescence in obesity.
Recent studies have identified an important role for cellular senescence in many diseases including those associated with obesity. Senescent cells can originate from most cell types and are potent modulators of inflammation, angiogenesis, and fibrosis. Cells undergo so-called replicative senescence during natural aging when they have reached the genetically determined limit of division. Division-competent cells can also be pushed to senescence though damaging stresses such has oncogene induction, oxidative stress, and double-strand DNA breaks (99). Although growth arrested, senescent cells are highly metabolic and exhibit a senescence-associated secretory phenotype (SASP), producing factors that have profound effects on neighboring cells such as proinflammatory cytokines (IL-6, IL-8, TNF-α, monocyte chemoattractant protein 1 [MCP-1]), VEGF, MMPs, and PDGF-AA (100). Targeting senescent cells or their products alleviates age-related dysfunction of adipocyte progenitors and metabolism, as elegantly demonstrated in mice expressing a p16INK4a promoter–driven inducible caspase-8 (INK4-ATTAC mice) (101). In this model, the loss of senescent cells blunted age-related fat loss and enhanced adipogenic transcription factor expression within three weeks.
Little is known about the role of cellular senescence in obesity-associated disease. However, several studies suggest that senescence may be a contributing factor (Figure 2). Increased AT DNA damage and resulting cellular senescence have been identified in mice fed a HFD for 20 weeks (102). This study established a strong link between cell senescence and obesity by demonstrating that inducing genomic instability through ablation of polymerase η increased the number of senescent AT cells and exacerbated AT dysfunction. The prevention of cellular senescence through inhibition of the major senescence regulator p53 attenuates metabolic abnormalities (102, 103). Obesity is thought to promote cell senescence through various means including oxidative stress, high glucose concentrations in the microenvironment, and increased IGF and ceramides (104). Surprisingly, preadipocytes and mature adipocytes as well as endothelial cells can become senescent during obesity (105–109). Reduced blood flow, as seen in obese AT, is sufficient to trigger endothelial cell senescence (105, 110). Systemically, the circulating endothelial cell precursor population undergoes premature senescence in obese individuals (107), contributing to the impairments in vascular function and repair observed in obesity. Thus, cell senescence of adipocytes, endothelial cells, or their precursors might have major effects on AT homeostasis.
Figure 2. The role of senescence in obesity-associated AT dysfunction.
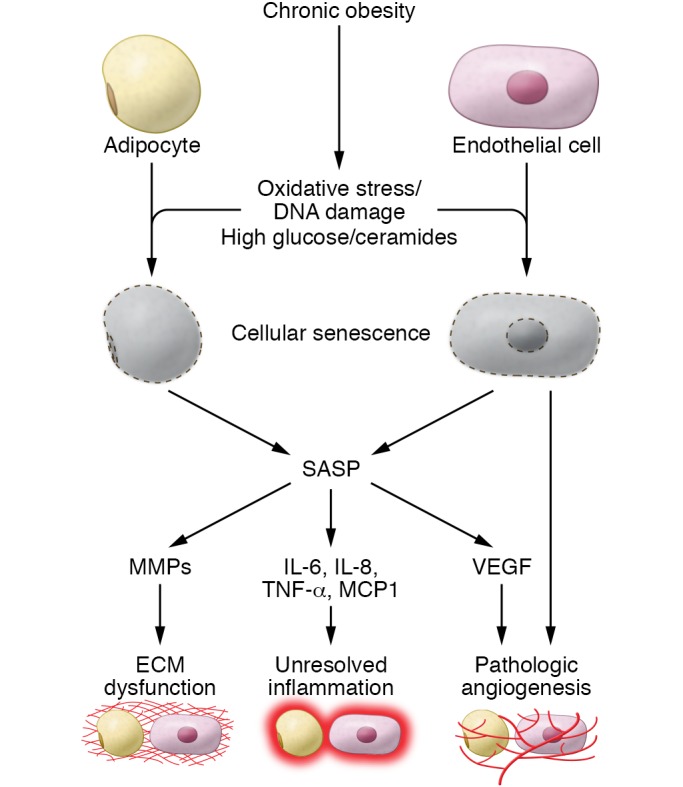
Chronic obesity can cause AT oxidative stress, DNA damage, and increased exposure to high glucose and ceramide concentrations. These deleterious factors can drive cellular senescence in many cells types. Adipocyte and endothelial cell senescence have been specifically studied in the context of obesity. Mature adipocytes and endothelial cells as well as their precursors can undergo senescence and take on the SASP. SASP factors can promote AT dysfunction through dysregulation of AT ECM remodeling, inflammation, and angiogenesis.
SASP factors are also likely to have considerable effects on AT immunity, angiogenesis, and fibrosis. SASP has recently been shown to have a beneficial effect on wound healing by recruiting immune cells to clear dead cells, promote angiogenesis, limit fibrosis, and stimulate wound closure (100, 111, 112). In contrast, the proinflammatory cytokines released from senescent cells, including MCP-1, TNF-α, and IL-1β, are among those considered most important for the progression to dysfunctional AT and chronic fibrotic disease (65, 113). Therefore, in chronic obesity the beneficial effects of time-limited cellular senescence in wound healing are replaced by the detrimental effects of chronic SASP.
AT inflammation, fibrosis, and angiogenesis in human obesity.
Although rodent models are crucial for our continued understanding of adipocyte biology in obesity, the translation of these findings to human disease is the ultimate goal. Briefly outlined below are findings from human studies on this subject.
Inflammation.
Weisberg et al. demonstrated that subcutaneous AT macrophage infiltration was increased in obese patients, similar to that observed in obese mice (10). Several detailed studies have also reported a higher macrophage count in visceral AT of insulin-resistant obese patients compared with lean or insulin-sensitive obese counterparts, an effect that was significantly mitigated following bariatric surgery (10, 114–116). However, conflicting findings concerning AT inflammation in humans have also been reported. Boden et al. showed that healthy men can become insulin insensitive during acute excessive caloric intake but do not display inflammatory changes in AT (77). Moreover, others have shown that 12 months after bariatric surgery, AT inflammation remained elevated in approximately 40% of patients (117). Therefore, whether inflammation is the trigger or result of obesity-associated metabolic defects remains an important area for human studies.
Angiogenesis.
Similar to findings in mice, human studies have reported that VEGF expression is reduced in obese humans (13). Consistent with mouse models of adipocyte-specific overexpression of VEGF-A, higher VEGF-A expression correlated with improved capillary density and insulin sensitivity in non-diabetic obese individuals (118). However, contradictory reports have demonstrated elevated levels of VEGF in obese subjects as well as a positive correlation between VEGF expression and insulin resistance (43). Thus, in both rodent models and human studies, VEGF-A–mediated AT angiogenesis exerts dichotomous effects in AT expansion and function.
Fibrosis.
The majority of human studies have reported fibrosis in obese AT (119–121). However, a recent human study compared obese patients with or without T2D and found significantly less fibrosis in the visceral AT of the diabetic subjects (122). Another independent study also reported less collagen content in visceral AT from metabolically unhealthy obese patients (123). Less fibrosis was associated with adipocyte hypertrophy, reduced pre-adipocyte hyperplasia, and AT dysfunction (122). Thus, the role of ECM remodeling and fibrosis in human AT dysfunction is stage dependent and likely fat depot specific. This debate is also a reflection of the fact that no consensus has emerged as to how we should define fibrosis. Is it the total tissue collagen content? Is it a reflection of how intensely the collagen bundles are cross-linked? Is it about interstitial pericellular collagen accumulation, or is it about the density of the septa that separate individual functional units within adipose depots (124)?
Concluding remarks
We are beginning to understand the importance of the complex interactions between inflammation, the ECM, and angiogenesis in the context of obesity. Therapeutic strategies are extremely challenging at all three levels. The limited successes for pharmacologic intervention at the level of inflammation (94, 125), the established unmet needs in the area of anti-fibrotic therapies, and obvious difficulties in promoting a healthy pro-angiogenic response versus the creation of a tumor-friendly angiogenic environment pose major obstacles to attempts to promote exogenously healthy expansion of WAT and will remain an important challenge for future research endeavors.
Acknowledgments
The authors are supported by National Institute of Health grants R01-DK55758, R01-DK099110, P01-DK088761 and P01-AG051459, as well as a grant from the Cancer Prevention and Research Institute of Texas (CPRIT RP140412).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2017;127(1):74–82. doi:10.1172/JCI88883.
Contributor Information
Clair Crewe, Email: clair.crewe@utsouthwestern.edu.
Yu Aaron An, Email: yu.an@UTSouthwestern.edu.
Philipp E. Scherer, Email: philipp.scherer@utsouthwestern.edu.
References
- 1.Li J, Yu X, Pan W, Unger RH. Gene expression profile of rat adipose tissue at the onset of high-fat-diet obesity. Am J Physiol Endocrinol Metab. 2002;282(6):E1334–E1341. doi: 10.1152/ajpendo.00516.2001. [DOI] [PubMed] [Google Scholar]
- 2.Kleemann R, et al. Time-resolved and tissue-specific systems analysis of the pathogenesis of insulin resistance. PLoS One. 2010;5(1):e8817. doi: 10.1371/journal.pone.0008817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee YS, et al. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell. 2014;157(6):1339–1352. doi: 10.1016/j.cell.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halberg N, et al. Hypoxia-inducible factor 1α induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. 2009;29(16):4467–4483. doi: 10.1128/MCB.00192-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Appelhoff RJ, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 6.Thomas A, et al. Hypoxia-inducible factor prolyl hydroxylase 1 (PHD1) deficiency promotes hepatic steatosis and liver-specific insulin resistance in mice. Sci Rep. 2016;6:24618. doi: 10.1038/srep24618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuura H, et al. Prolyl hydroxylase domain protein 2 plays a critical role in diet-induced obesity and glucose intolerance. Circulation. 2013;127(21):2078–2087. doi: 10.1161/CIRCULATIONAHA.113.001742. [DOI] [PubMed] [Google Scholar]
- 8.Schipper HS, Prakken B, Kalkhoven E, Boes M. Adipose tissue-resident immune cells: key players in immunometabolism. Trends Endocrinol Metab. 2012;23(8):407–415. doi: 10.1016/j.tem.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Winer DA, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17(5):610–617. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishimura S, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15(8):914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 12.Trayhurn P. Hypoxia and adipocyte physiology: implications for adipose tissue dysfunction in obesity. Annu Rev Nutr. 2014;34:207–236. doi: 10.1146/annurev-nutr-071812-161156. [DOI] [PubMed] [Google Scholar]
- 13.Pasarica M, et al. Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes. 2009;58(3):718–725. doi: 10.2337/db08-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujisaka S, et al. Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an HIF-1α-dependent and HIF-1α-independent manner in obese mice. Diabetologia. 2013;56(6):1403–1412. doi: 10.1007/s00125-013-2885-1. [DOI] [PubMed] [Google Scholar]
- 15.Yoshida H, et al. Naringenin suppresses macrophage infiltration into adipose tissue in an early phase of high-fat diet-induced obesity. Biochem Biophys Res Commun. 2014;454(1):95–101. doi: 10.1016/j.bbrc.2014.10.038. [DOI] [PubMed] [Google Scholar]
- 16.Lee YS, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes. 2011;60(10):2474–2483. doi: 10.2337/db11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ji Y, Sun S, Xia S, Yang L, Li X, Qi L. Short term high fat diet challenge promotes alternative macrophage polarization in adipose tissue via natural killer T cells and interleukin-4. J Biol Chem. 2012;287(29):24378–24386. doi: 10.1074/jbc.M112.371807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Camell C, Smith CW. Dietary oleic acid increases m2 macrophages in the mesenteric adipose tissue. PLoS One. 2013;8(9):e75147. doi: 10.1371/journal.pone.0075147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gavin KM, et al. De novo generation of adipocytes from circulating progenitor cells in mouse and human adipose tissue. FASEB J. 2016;30(3):1096–1108. doi: 10.1096/fj.15-278994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang W, et al. White fat progenitor cells reside in the adipose vasculature. Science. 2008;322(5901):583–586. doi: 10.1126/science.1156232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta RK, et al. Transcriptional control of preadipocyte determination by Zfp423. Nature. 2010;464(7288):619–623. doi: 10.1038/nature08816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta RK, et al. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab. 2012;15(2):230–239. doi: 10.1016/j.cmet.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vishvanath L, et al. Pdgfrβ+ mural preadipocytes contribute to adipocyte hyperplasia induced by high-fat-diet feeding and prolonged cold exposure in adult mice. Cell Metab. 2016;23(2):350–359. doi: 10.1016/j.cmet.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, et al. Adipose tissue-specific inhibition of hypoxia-inducible factor 1{alpha} induces obesity and glucose intolerance by impeding energy expenditure in mice. J Biol Chem. 2010;285(43):32869–32877. doi: 10.1074/jbc.M110.135509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.García-Martín R, et al. Adipocyte-specific hypoxia-inducible factor 2α deficiency exacerbates obesity-induced brown adipose tissue dysfunction and metabolic dysregulation. Mol Cell Biol. 2015;36(3):376–393. doi: 10.1128/MCB.00430-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun K, et al. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc Natl Acad Sci U S A. 2012;109(15):5874–5879. doi: 10.1073/pnas.1200447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michailidou Z, et al. Increased angiogenesis protects against adipose hypoxia and fibrosis in metabolic disease-resistant 11β-hydroxysteroid dehydrogenase type 1 (HSD1)-deficient mice. J Biol Chem. 2012;287(6):4188–4197. doi: 10.1074/jbc.M111.259325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gómez-Ambrosi J, et al. Involvement of serum vascular endothelial growth factor family members in the development of obesity in mice and humans. J Nutr Biochem. 2010;21(8):774–780. doi: 10.1016/j.jnutbio.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Lijnen HR, et al. Impaired adipose tissue development in mice with inactivation of placental growth factor function. Diabetes. 2006;55(10):2698–2704. doi: 10.2337/db06-0526. [DOI] [PubMed] [Google Scholar]
- 30.Bell LN, Cai L, Johnstone BH, Traktuev DO, March KL, Considine RV. A central role for hepatocyte growth factor in adipose tissue angiogenesis. Am J Physiol Endocrinol Metab. 2008;294(2):E336–E344. doi: 10.1152/ajpendo.00272.2007. [DOI] [PubMed] [Google Scholar]
- 31.Gealekman O, et al. Control of adipose tissue expandability in response to high fat diet by the insulin-like growth factor-binding protein-4. J Biol Chem. 2014;289(26):18327–18338. doi: 10.1074/jbc.M113.545798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Voros G, Maquoi E, Demeulemeester D, Clerx N, Collen D, Lijnen HR. Modulation of angiogenesis during adipose tissue development in murine models of obesity. Endocrinology. 2005;146(10):4545–4554. doi: 10.1210/en.2005-0532. [DOI] [PubMed] [Google Scholar]
- 33.Xue Y, et al. FOXC2 controls Ang-2 expression and modulates angiogenesis, vascular patterning, remodeling, and functions in adipose tissue. Proc Natl Acad Sci U S A. 2008;105(29):10167–10172. doi: 10.1073/pnas.0802486105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao R, Brakenhielm E, Wahlestedt C, Thyberg J, Cao Y. Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF. Proc Natl Acad Sci U S A. 2001;98(11):6390–6395. doi: 10.1073/pnas.101564798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robciuc MR, et al. VEGFB/VEGFR1-induced expansion of adipose vasculature counteracts obesity and related metabolic complications. Cell Metab. 2016;23(4):712–724. doi: 10.1016/j.cmet.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee HP, et al. Adiponectin promotes VEGF-A-dependent angiogenesis in human chondrosarcoma through PI3K, Akt, mTOR, and HIF-α pathway. Oncotarget. 2015;6(34):36746–36761. doi: 10.18632/oncotarget.5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shibata R, Ouchi N, Kihara S, Sato K, Funahashi T, Walsh K. Adiponectin stimulates angiogenesis in response to tissue ischemia through stimulation of amp-activated protein kinase signaling. J Biol Chem. 2004;279(27):28670–28674. doi: 10.1074/jbc.M402558200. [DOI] [PubMed] [Google Scholar]
- 38.Robertson SA, Rae CJ, Graham A. Induction of angiogenesis by murine resistin: putative role of PI3-kinase and NO-dependent pathways. Regul Pept. 2009;152(1–3):41–47. doi: 10.1016/j.regpep.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 39.Tan BK, et al. Metformin decreases angiogenesis via NF-kappaB and Erk1/2/Erk5 pathways by increasing the antiangiogenic thrombospondin-1. Cardiovasc Res. 2009;83(3):566–574. doi: 10.1093/cvr/cvp131. [DOI] [PubMed] [Google Scholar]
- 40.Scroyen I, Jacobs F, Cosemans L, De Geest B, Lijnen HR. Effect of plasminogen activator inhibitor-1 on adipogenesis in vivo. Thromb Haemost. 2009;101(2):388–393. [PubMed] [Google Scholar]
- 41.Wilkison WO, Choy L, Spiegelman BM. Biosynthetic regulation of monobutyrin, an adipocyte-secreted lipid with angiogenic activity. J Biol Chem. 1991;266(25):16886–16891. [PubMed] [Google Scholar]
- 42.Gerhardt H, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161(6):1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tinahones FJ, et al. Obesity-associated insulin resistance is correlated to adipose tissue vascular endothelial growth factors and metalloproteinase levels. BMC Physiol. 2012;12:4. doi: 10.1186/1472-6793-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miyazawa-Hoshimoto S, Takahashi K, Bujo H, Hashimoto N, Saito Y. Elevated serum vascular endothelial growth factor is associated with visceral fat accumulation in human obese subjects. Diabetologia. 2003;46(11):1483–1488. doi: 10.1007/s00125-003-1221-6. [DOI] [PubMed] [Google Scholar]
- 45.Clause KC, Barker TH. Extracellular matrix signaling in morphogenesis and repair. Curr Opin Biotechnol. 2013;24(5):830–833. doi: 10.1016/j.copbio.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park J, Scherer PE. Adipocyte-derived endotrophin promotes malignant tumor progression. J Clin Invest. 2012;122(11):4243–4256. doi: 10.1172/JCI63930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun K, et al. Endotrophin triggers adipose tissue fibrosis and metabolic dysfunction. Nat Commun. 2014;5:3485. doi: 10.1038/ncomms4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chun TH, et al. Genetic link between obesity and MMP14-dependent adipogenic collagen turnover. Diabetes. 2010;59(10):2484–2494. doi: 10.2337/db10-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan T, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29(6):1575–1591. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao F, et al. Hypoxia-induced alterations in hyaluronan and hyaluronidase. Adv Exp Med Biol. 2005;566:249–256. doi: 10.1007/0-387-26206-7_33. [DOI] [PubMed] [Google Scholar]
- 51.Jiang D, Liang J, Noble PW. Hyaluronan as an immune regulator in human diseases. Physiol Rev. 2011;91(1):221–264. doi: 10.1152/physrev.00052.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cho CH, et al. Angiogenic role of LYVE-1-positive macrophages in adipose tissue. Circ Res. 2007;100(4):e47–e57. doi: 10.1161/01.RES.0000259564.92792.93. [DOI] [PubMed] [Google Scholar]
- 53.Bergers G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2(10):737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gong R, Rifai A, Dworkin LD. Anti-inflammatory effect of hepatocyte growth factor in chronic kidney disease: targeting the inflamed vascular endothelium. J Am Soc Nephrol. 2006;17(9):2464–2473. doi: 10.1681/ASN.2006020185. [DOI] [PubMed] [Google Scholar]
- 55.Funa K, Uramoto H. Regulatory mechanisms for the expression and activity of platelet-derived growth factor receptor. Acta Biochim Pol. 2003;50(3):647–658. [PubMed] [Google Scholar]
- 56.Lee YC. The involvement of VEGF in endothelial permeability: a target for anti-inflammatory therapy. Curr Opin Investig Drugs. 2005;6(11):1124–1130. [PubMed] [Google Scholar]
- 57.Kwon YW, et al. Tumor necrosis factor-α-activated mesenchymal stem cells promote endothelial progenitor cell homing and angiogenesis. Biochim Biophys Acta. 2013;1832(12):2136–2144. doi: 10.1016/j.bbadis.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 58.Wagner M, et al. Inflamed tumor-associated adipose tissue is a depot for macrophages that stimulate tumor growth and angiogenesis. Angiogenesis. 2012;15(3):481–495. doi: 10.1007/s10456-012-9276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fiedler U, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med. 2006;12(2):235–239. doi: 10.1038/nm1351. [DOI] [PubMed] [Google Scholar]
- 60.Elias I, Franckhauser S, Bosch F. New insights into adipose tissue VEGF-A actions in the control of obesity and insulin resistance. Adipocyte. 2013;2(2):109–112. doi: 10.4161/adip.22880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scholz A, Plate KH, Reiss Y. Angiopoietin-2: a multifaceted cytokine that functions in both angiogenesis and inflammation. Ann N Y Acad Sci. 2015;1347:45–51. doi: 10.1111/nyas.12726. [DOI] [PubMed] [Google Scholar]
- 62.Strissel KJ, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56(12):2910–2918. doi: 10.2337/db07-0767. [DOI] [PubMed] [Google Scholar]
- 63.Alkhouri N, et al. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J Biol Chem. 2010;285(5):3428–3438. doi: 10.1074/jbc.M109.074252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cinti S, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46(11):2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 65.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kruglikov IL, Scherer PE. Dermal Adipocytes: From Irrelevance to Metabolic Targets? Trends Endocrinol Metab. 2016;27(1):1–10. doi: 10.1016/j.tem.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Duffield JS, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115(1):56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.DiSpirito JR, Mathis D. Immunological contributions to adipose tissue homeostasis. Semin Immunol. 2015;27(5):315–321. doi: 10.1016/j.smim.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 70.Lumeng CN, Deyoung SM, Saltiel AR. Macrophages block insulin action in adipocytes by altering expression of signaling and glucose transport proteins. Am J Physiol Endocrinol Metab. 2007;292(1):E166–E174. doi: 10.1152/ajpendo.00284.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peraldi P, Hotamisligil GS, Buurman WA, White MF, Spiegelman BM. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem. 1996;271(22):13018–13022. doi: 10.1074/jbc.271.22.13018. [DOI] [PubMed] [Google Scholar]
- 72.Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem. 2003;278(46):45777–45784. doi: 10.1074/jbc.M301977200. [DOI] [PubMed] [Google Scholar]
- 73.Weisberg SP, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116(1):115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Furukawa S, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114(12):1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marseglia L, et al. Oxidative stress in obesity: a critical component in human diseases. Int J Mol Sci. 2014;16(1):378–400. doi: 10.3390/ijms16010378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Darr D, Fridovich I. Free radicals in cutaneous biology. J Invest Dermatol. 1994;102(5):671–675. doi: 10.1111/1523-1747.ep12374036. [DOI] [PubMed] [Google Scholar]
- 77.Boden G, et al. Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci Transl Med. 2015;7(304):304re7. doi: 10.1126/scitranslmed.aac4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Priyanka A, Anusree SS, Nisha VM, Raghu KG. Curcumin improves hypoxia induced dysfunctions in 3T3-L1 adipocytes by protecting mitochondria and down regulating inflammation. Biofactors. 2014;40(5):513–523. doi: 10.1002/biof.1175. [DOI] [PubMed] [Google Scholar]
- 79.Xue P, et al. Adipose deficiency of Nrf2 in ob/ob mice results in severe metabolic syndrome. Diabetes. 2013;62(3):845–854. doi: 10.2337/db12-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alcalá M, et al. Vitamin E reduces adipose tissue fibrosis, inflammation, and oxidative stress and improves metabolic profile in obesity. Obesity (Silver Spring) 2015;23(8):1598–1606. doi: 10.1002/oby.21135. [DOI] [PubMed] [Google Scholar]
- 81.Frohnert BI, Long EK, Hahn WS, Bernlohr DA. Glutathionylated lipid aldehydes are products of adipocyte oxidative stress and activators of macrophage inflammation. Diabetes. 2014;63(1):89–100. doi: 10.2337/db13-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wernstedt Asterholm I, et al. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014;20(1):103–118. doi: 10.1016/j.cmet.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xiao L, et al. Anti-vascular endothelial growth factor treatment induces blood flow recovery through vascular remodeling in high-fat diet induced diabetic mice. Microvasc Res. 2016;105:70–76. doi: 10.1016/j.mvr.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 84.Goossens GH, et al. Increased adipose tissue oxygen tension in obese compared with lean men is accompanied by insulin resistance, impaired adipose tissue capillarization, and inflammation. Circulation. 2011;124(1):67–76. doi: 10.1161/CIRCULATIONAHA.111.027813. [DOI] [PubMed] [Google Scholar]
- 85.Sun K, Halberg N, Khan M, Magalang UJ, Scherer PE. Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol Cell Biol. 2013;33(5):904–917. doi: 10.1128/MCB.00951-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kreuger J, Phillipson M. Targeting vascular and leukocyte communication in angiogenesis, inflammation and fibrosis. Nat Rev Drug Discov. 2016;15(2):125–142. doi: 10.1038/nrd.2015.2. [DOI] [PubMed] [Google Scholar]
- 87.Nishimura S, et al. In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. J Clin Invest. 2008;118(2):710–721. doi: 10.1172/JCI33328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rupnick MA, et al. Adipose tissue mass can be regulated through the vasculature. Proc Natl Acad Sci U S A. 2002;99(16):10730–10735. doi: 10.1073/pnas.162349799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bråkenhielm E, et al. Angiogenesis inhibitor, TNP-470, prevents diet-induced and genetic obesity in mice. Circ Res. 2004;94(12):1579–1588. doi: 10.1161/01.RES.0000132745.76882.70. [DOI] [PubMed] [Google Scholar]
- 90.Luo X, et al. Docosahexaenoic acid attenuates adipose tissue angiogenesis and insulin resistance in high fat diet-fed middle-aged mice via a sirt1-dependent mechanism. Mol Nutr Food Res. 2016;60(4):871–885. doi: 10.1002/mnfr.201500714. [DOI] [PubMed] [Google Scholar]
- 91.Klaus S, Pültz S, Thöne-Reineke C, Wolfram S. Epigallocatechin gallate attenuates diet-induced obesity in mice by decreasing energy absorption and increasing fat oxidation. Int J Obes (Lond) 2005;29(6):615–623. doi: 10.1038/sj.ijo.0802926. [DOI] [PubMed] [Google Scholar]
- 92.Ejaz A, Wu D, Kwan P, Meydani M. Curcumin inhibits adipogenesis in 3T3-L1 adipocytes and angiogenesis and obesity in C57/BL mice. J Nutr. 2009;139(5):919–925. doi: 10.3945/jn.108.100966. [DOI] [PubMed] [Google Scholar]
- 93.Park BY, et al. Reduction of Adipose Tissue Mass by the Angiogenesis Inhibitor ALS-L1023 from Melissa officinalis. PLoS One. 2015;10(11):e0141612. doi: 10.1371/journal.pone.0141612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kusminski CM, Bickel PE, Scherer PE. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov. 2016;15(9):639–660. doi: 10.1038/nrd.2016.75. [DOI] [PubMed] [Google Scholar]
- 95.Craft CS, et al. The extracellular matrix protein MAGP1 supports thermogenesis and protects against obesity and diabetes through regulation of TGF-β. Diabetes. 2014;63(6):1920–1932. doi: 10.2337/db13-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhu Y, Crewe C, Scherer PE. Hyaluronan in adipose tissue: Beyond dermal filler and therapeutic carrier. Sci Transl Med. 2016;8(323):323ps4. doi: 10.1126/scitranslmed.aad6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kang L, et al. Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin alpha2beta1 in mice. Diabetes. 2011;60(2):416–426. doi: 10.2337/db10-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dalmas E, et al. Irf5 deficiency in macrophages promotes beneficial adipose tissue expansion and insulin sensitivity during obesity. Nat Med. 2015;21(6):610–618. doi: 10.1038/nm.3829. [DOI] [PubMed] [Google Scholar]
- 99.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192(4):547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Demaria M, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31(6):722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu M, et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife. 2015;4:e12997. doi: 10.7554/eLife.12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen YW, Harris RA, Hatahet Z, Chou KM. Ablation of XP-V gene causes adipose tissue senescence and metabolic abnormalities. Proc Natl Acad Sci U S A. 2015;112(33):E4556–E4564. doi: 10.1073/pnas.1506954112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Minamino T, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009;15(9):1082–1087. doi: 10.1038/nm.2014. [DOI] [PubMed] [Google Scholar]
- 104.Palmer AK, Tchkonia T, LeBrasseur NK, Chini EN, Xu M, Kirkland JL. Cellular senescence in type 2 diabetes: a therapeutic opportunity. Diabetes. 2015;64(7):2289–2298. doi: 10.2337/db14-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Warboys CM, et al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler Thromb Vasc Biol. 2014;34(5):985–995. doi: 10.1161/ATVBAHA.114.303415. [DOI] [PubMed] [Google Scholar]
- 106.Rouault C, et al. Roles of chemokine ligand-2 (CXCL2) and neutrophils in influencing endothelial cell function and inflammation of human adipose tissue. Endocrinology. 2013;154(3):1069–1079. doi: 10.1210/en.2012-1415. [DOI] [PubMed] [Google Scholar]
- 107.Tobler K, et al. Reduction of both number and proliferative activity of human endothelial progenitor cells in obesity. Int J Obes (Lond) 2010;34(4):687–700. doi: 10.1038/ijo.2009.280. [DOI] [PubMed] [Google Scholar]
- 108.Wang CY, et al. Obesity increases vascular senescence and susceptibility to ischemic injury through chronic activation of Akt and mTOR. Sci Signal. 2009;2(62):ra11. doi: 10.1126/scisignal.2000143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Villaret A, et al. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes. 2010;59(11):2755–2763. doi: 10.2337/db10-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Toda N, Okamura T. Obesity impairs vasodilatation and blood flow increase mediated by endothelial nitric oxide: an overview. J Clin Pharmacol. 2013;53(12):1228–1239. doi: 10.1002/jcph.179. [DOI] [PubMed] [Google Scholar]
- 111.Zhu F, et al. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS One. 2013;8(9):e74535. doi: 10.1371/journal.pone.0074535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Krizhanovsky V, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Martinez-Santibañez G, Lumeng CN. Macrophages and the regulation of adipose tissue remodeling. Annu Rev Nutr. 2014;34:57–76. doi: 10.1146/annurev-nutr-071812-161113. [DOI] [PubMed] [Google Scholar]
- 114.Cancello R, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes. 2005;54(8):2277–2286. doi: 10.2337/diabetes.54.8.2277. [DOI] [PubMed] [Google Scholar]
- 115.Harman-Boehm I, et al. Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesity. J Clin Endocrinol Metab. 2007;92(6):2240–2247. doi: 10.1210/jc.2006-1811. [DOI] [PubMed] [Google Scholar]
- 116.Kloting N, et al. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab. 2010;299(3):E506–E515. doi: 10.1152/ajpendo.00586.2009. [DOI] [PubMed] [Google Scholar]
- 117.Schmitz J, et al. Obesogenic memory can confer long-term increases in adipose tissue but not liver inflammation and insulin resistance after weight loss. Mol Metab. 2016;5(5):328–339. doi: 10.1016/j.molmet.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gealekman O, et al. Depot-specific differences and insufficient subcutaneous adipose tissue angiogenesis in human obesity. Circulation. 2011;123(2):186–194. doi: 10.1161/CIRCULATIONAHA.110.970145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dankel SN, et al. COL6A3 expression in adipocytes associates with insulin resistance and depends on PPARγ and adipocyte size. Obesity (Silver Spring) 2014;22(8):1807–1813. doi: 10.1002/oby.20758. [DOI] [PubMed] [Google Scholar]
- 120.Spencer M, et al. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. Am J Physiol Endocrinol Metab. 2010;299(6):E1016–E1027. doi: 10.1152/ajpendo.00329.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vila IK, et al. Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep. 2014;7(4):1116–1129. doi: 10.1016/j.celrep.2014.03.062. [DOI] [PubMed] [Google Scholar]
- 122.Muir LA, et al. Adipose tissue fibrosis, hypertrophy, and hyperplasia: Correlations with diabetes in human obesity. Obesity (Silver Spring) 2016;24(3):597–605. doi: 10.1002/oby.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lackey DE, et al. Contributions of adipose tissue architectural and tensile properties toward defining healthy and unhealthy obesity. Am J Physiol Endocrinol Metab. 2014;306(3):E233–E246. doi: 10.1152/ajpendo.00476.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wernstedt Asterholm I, Scherer PE. Fibrosis-streaks and splatters: Some things are not always what they seem to be. Obesity (Silver Spring) 2016;24(3):552–553. doi: 10.1002/oby.21430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Goldfine AB, et al. Salicylate (salsalate) in patients with type 2 diabetes: a randomized trial. Ann Intern Med. 2013;159(1):1–12. doi: 10.7326/0003-4819-159-1-201307020-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]