

Abstract
Over the last years, hypothalamic inflammation has been linked to the development and progression of obesity and its sequelae. There is accumulating evidence that this inflammation not only impairs energy balance but also contributes to obesity-associated insulin resistance. Elevated activation of key inflammatory mediators such as JNK and IκB kinase (IKK) occurs rapidly upon consumption of a high-fat diet, even prior to significant weight gain. This activation of hypothalamic inflammatory pathways results in the uncoupling of caloric intake and energy expenditure, fostering overeating and further weight gain. In addition, these inflammatory processes contribute to obesity-associated insulin resistance and deterioration of glucose metabolism via altered neurocircuit functions. An understanding of the contributions of different neuronal and non-neuronal cell types to hypothalamic inflammatory processes, and delineation of the differences and similarities between acute and chronic activation of these inflammatory pathways, will be critical for the development of novel therapeutic strategies for the treatment of obesity and metabolic syndrome.
Introduction
For many years obesity has been known to induce a state of systemic low-grade inflammation that contributes to the development of numerous comorbidities, such as type 2 diabetes, dyslipidemia, cardiovascular diseases, and neurodegenerative disorders (1–3). In addition to metabolic derangements in peripheral organs, obesity-related inflammatory changes perturb brain function, especially affecting brain areas that regulate energy homeostasis and systemic metabolism. The importance of the brain in controlling energy homeostasis is underscored by the fact that monogenetic defects underlying the development of obesity are almost exclusively caused by mutations in genes that act in the brain to regulate food intake (4). Furthermore, examination of genes at loci associated with BMI has provided strong support for a role of the CNS in obesity susceptibility (5, 6). In addition to an individual’s genetic predisposition to hyperphagia, the virtually unlimited availability of calorie-dense, high-fat– and high-carbohydrate–containing foods has contributed to the recent, ever-increasing obesity epidemic (7, 8). Over the last years, hypothalamic inflammation has been linked to the development and progression of obesity and its sequelae and has emerged not only as an important driver of impaired energy balance, but also as a contributor to obesity-associated insulin resistance via altered neurocircuit functions.
Hypothalamic regulation of energy homeostasis
The hypothalamus controls a number of neuroendocrine functions that integrate metabolic feedback and regulate energy homeostasis (9, 10). Early lesion experiments revealed that alterations in the hypothalamic area of the brain resulted in changes in feeding behavior and energy expenditure (11, 12). Key to this regulatory function is the melanocortin system, which consists of two functionally antagonistic neuronal populations — one subset of neurons expresses the orexigenic neuropeptides agouti-related peptide (AgRP) and neuropeptide Y (NPY), the second subset expresses the anorexigenic peptides proopiomelanocortin (POMC) and cocaine and amphetamine regulated transcript (CART). These neuronal subsets are located in the arcuate nucleus (ARC) of the mediobasal hypothalamus (MBH) (13, 14). Both of these neuronal populations express high levels of receptors that bind hormones and thus respond to metabolic cues to control food intake and energy expenditure, depending on the energy state of the body (15–17). Circulating levels of insulin and leptin, proportionate to nutritional status and adipose tissue stores, inhibit AgRP neurons and activate POMC neurons, leading to decreased energy intake and increased energy expenditure (18–20). Neuronal intracellular insulin signaling triggers phosphorylation of forkhead box protein O1 (FOXO1), resulting in nuclear exclusion of FOXO1 and inhibition of FOXO1-mediated repression of POMC gene expression (21–23). Leptin receptor signaling leads to the phosphorylation of STAT3, which dimerizes and translocates to the nucleus, inducing POMC gene expression (24). POMC is expressed as a precursor peptide, which is processed posttranslationally into several peptides, including melanocortin receptor ligands (25–27). These melanocyte-stimulating hormones (MSHs) are released from synaptic endings of POMC neurons and act on melanocortin receptors (MC3R/MC4R) expressed on second-order neurons that are located, among other target regions, in the paraventricular nucleus (PVN) of the hypothalamus to suppress food intake (16). These second-order neurons are part of the energy homeostasis circuit projecting to hindbrain autonomic centers, such as the nucleus of the solitary tract (NTS), which process the response to satiety signals and contribute to adaptive feeding behavior (ref. 28 and Figure 1).
Figure 1. Hypothalamic control of energy homeostasis.

The hypothalamus senses and integrates feedback from adipostatic hormones, which circulate in levels proportionate to nutritional status and adipose tissue stores. Insulin and leptin act directly on neuronal subsets in the ARC of the hypothalamus to control energy homeostasis. Through activation of POMC neurons and inhibition of AgRP neurons, adipostatic signals activate MC4R-expressing neurons in the PVN. During fasting conditions, the expression of AgRP increases, whereas POMC expression is reduced, resulting in decreased MC4R signaling. In the fed state, AgRP levels are diminished and POMC levels increase, which triggers MC4R signaling and culminates in satiety and stimulation of energy expenditure. Neuronal inflammation and the subsequent insulin and leptin resistance of ARC neurons disrupts this metabolic feedback loop, further promoting increased food intake and body weight gain.
Under fasting conditions AgRP-expressing neurons are activated to induce feeding, to inhibit energy expenditure, and to regulate glucose metabolism (13, 14, 29). More recently, we demonstrated that an obesity-associated increase in circulating uridine concentrations can increase hypothalamic UDP concentrations, which in turn activate AgRP neurons in a P2Y6 receptor–dependent manner (30). AgRP acts as an inverse agonist of MC3R/MC4R and counteracts the anorectic effect of α- and β-MSH, thereby positively regulating feeding behavior (31). AgRP neuronal activity also immediately affects peripheral glucose homeostasis (32, 33). Interestingly, signals that promote feeding also enhance POMC neuronal activity. Activation of the cannabinoid receptor CBR1, which is known to mediate cannabinoid-induced food intake, leads to the (paradoxical) activation of POMC neurons and the selective release of β-endorphin instead of α-MSH (34). This preferential production of β-endorphin is dependent on endocannabinoid receptor expression in mitochondria and modulation of mitochondrial uncoupling protein 2.
In this context it is remarkable how rapidly overconsumption of a fat-rich diet results in acute changes in the feedback to metabolic endocrine signals (such as leptin and insulin signaling) and in hypothalamic inflammatory responses (35). Inflammatory processes in the hypothalamus occur in two phases. A transient, early inflammatory phase and, with sustained exposure to high-fat diet (HFD), a secondary phase, in which prolonged inflammatory cascades lead to the activation of cellular stress mechanisms. Markers of hypothalamic inflammation increase significantly during the first days of HFD feeding, with reactive gliosis and neuronal injury manifesting during the first week, even prior to body weight gain (35).
Acute effects of HFD feeding
Regulation of energy homeostasis depends on input to the hypothalamus from metabolic feedback signals such as insulin and leptin (36). Diet-induced obesity causes activation of cytokines and inflammatory pathways in the hypothalamus (37). In parallel to the early occurrence of inflammation, three days of HFD feeding is sufficient to significantly reduce hypothalamic insulin sensitivity in rodents (38). Importantly, these processes precede inflammatory events in peripheral tissues, such as the liver (39).
Several studies have shown that specific lipid species are linked with resistance to the main adipostatic hormones insulin and leptin in peripheral tissues (40–43). Fatty acids, especially long-chain saturated fatty acids (SFAs), can acutely modulate neuronal control of energy homeostasis. Enteric gavage with SFAs induces hypothalamic inflammation within days, whereas administration of monounsaturated fatty acids did not result in compromised hypothalamic function (44). SFAs, such as palmitate and stearate, are able to cross the blood-brain barrier (BBB) and accumulate specifically in the hypothalamus, where they blunt anorexigenic signaling by insulin and leptin and thereby promote positive energy balance (44–46). SFAs, unlike unsaturated fatty acids, trigger the activation of inflammatory signaling cascades via TLR4 signaling and the adaptor molecule myeloid differentiation primary response gene 88 (MyD88) (refs. 47, 48, and Figure 2). Pharmacologic inhibition of neuronal TLR4 signaling inhibits fatty acid–induced insulin (45) and leptin resistance (49). Furthermore, mice with CNS-specific ablation of MyD88 are protected from HFD-induced weight gain and deterioration of glucose metabolism (48). Downstream signaling occurs via the IKK complex and NF-κB activation, leading to the expression of proinflammatory genes in the hypothalamus (45, 49, 50). Brain-specific activation of IKKβ results in increased food intake and body weight gain and interrupts central insulin and leptin signaling (50). Furthermore, activation of NF-κB induces expression of suppressor of cytokine signaling 3 (SOCS3), which subsequently inhibits neuronal insulin signaling (50). Upon acute exposure to HFD, SOCS3 is upregulated in AgRP neurons, leading to hyperphagia, energy imbalance, and development of insulin and leptin resistance. In contrast, intracerebroventricular (ICV) injections of unsaturated fatty acids, namely omega-3 fatty acids, restore leptin and insulin sensitivity (51–53).
Figure 2. Molecular pathways of metabolic inflammation in the hypothalamus.

Activation of the insulin signaling pathway leads to FOXO1 phosphorylation, resulting in nuclear exclusion and inhibition of FOXO1-mediated activation/repression of target genes. By removing FOXO1 repression, insulin increases the anorexigenic tone via induced POMC gene expression in POMC neurons with simultaneous suppressed AgRP/NPY expression in AgRP neurons. Binding of leptin to the leptin receptor activates and recruits STAT3. Tyrosine phosphorylation triggers dimerization of STAT3 and translocation to the nucleus, where the STAT3 complex induces POMC gene expression in POMC neurons and suppresses AgRP/NPY in AgRP neurons. TNF-α binding promotes the trimerization of the TNF receptor, which promotes the assembly of a signaling complex that activates JNK and inhibits cellular insulin signaling. In addition, TNF signaling triggers activation of NF-κB. In the TLR4 signaling pathway, MyD88 serves as a scaffold protein for downstream signaling molecules. NF-κB dimers are sequestered in the cytoplasm by the inhibitor of NF-κB α (IκBα). Phosphorylation by the IKK complex and subsequent proteolysis of IκBα leads to the release and nuclear translocations of NF-κB. NF-κB heterodimer RelA and p50 bind to motifs in the promoter of targets genes, such as SOCS3, a common inhibitor of insulin and leptin signaling, and proinflammatory genes. Induction of ceramides leads to increased phosphorylation and localization of PKC with caveolin-enriched lipid microdomains to inactivate Akt.
TLR4-mediated signaling pathways also activate the MAPK pathway, which triggers p38- and JNK-dependent signaling and activation of various activator protein-1 (AP-1) subunits. JNK mediates inhibitory phosphorylation of insulin receptor substrate (IRS) proteins at serine 307, thereby impairing insulin action (54). Constitutive JNK activation in AgRP neurons of the hypothalamus induces weight gain and adiposity in mice as a consequence of hyperphagia (55). In fact, conditional JNK1 knockout specifically in the brain, but not in other tissues, leads to protection against insulin resistance, hyperinsulinemia, and glucose intolerance (56, 57). Interestingly, activation of TLR4 signaling controls apoptotic activity of cells in the hypothalamus but subsequently activates proinflammatory pathways that ultimately lead to the development of central insulin and leptin resistance (58).
Elevations of sphingolipids such as ceramides, whose synthesis depends on SFAs, and alterations in downstream sphingolipid-mediated signaling pathways might provide an additional mechanism by which SFA-induced inflammatory pathways mediate the deterioration of insulin and leptin signaling (59, 60). Induction of ceramides during obesity has been shown to promote insulin resistance (61). Ceramide is a common mediator of cellular stress, and inhibition of ceramide biosynthesis blocks the ability of SFAs to induce insulin resistance in obese rodent models (62). Although hypothalamic metabolic inflammation affects both males and females, sphingolipids and palmitic acid show a sexually dimorphic accumulation pattern during high-fat feeding and increase more in the CNS of male mice than in female mice (63). Furthermore, SFAs are able to mediate central insulin resistance via activation of the PKC isoform PKC-θ. Palmitic acid induces translocation of PKC-θ to the cell membrane exclusively in AgRP neurons, resulting in inhibition of PI3K signaling (64). This finding is specific for palmitic acid, as CNS exposure to the monounsaturated fatty acid oleic acid did not induce insulin resistance. Conversely, ARC-specific knockdown of PKC-θ attenuated diet-induced obesity (64). These HFD-induced perturbations are further amplified by the ER system via activation of unfolded protein response (UPR) signaling pathways (52, 65, 66). In the hypothalamus, ER stress and activation of UPR signaling pathways lead to the development of insulin and leptin resistance. ER stress and IKK/NF-κB promote each other during HFD feeding and accelerate the energy imbalance underlying obesity (50). Central administration of an ER stress inducer inhibits the anorexigenic and weight-reducing effects of leptin and insulin (67). Conversely, mice with neuron-specific deletion of ER stress activator X-box binding protein 1 (Xbp1) exhibit increased leptin resistance and adiposity (65). Hypothalamic POMC neurons are critical mediators of hypothalamic ER stress. Constitutive expression of Xbp1s selectively in POMC neurons represses Socs3 and protein tyrosine phosphatase 1B (Ptp1B) expression and protects against HFD-induced obesity (68). ER stress and the UPR are potent regulators of POMC neurons and are therefore interesting targets for the amelioration of central insulin and leptin resistance and the regulation of metabolic disorders.
Collectively, multiple inflammatory and stress response pathways are rapidly activated during HFD-feeding and promote the development of neuronal insulin and leptin resistance, raising the possibility that nutrient excess itself is the primary driver of hypothalamic inflammation.
Chronic effects of HFD feeding
Inflammatory mediators released from non-neuronal cell types give rise to long-lasting impaired metabolic control of the hypothalamus. Proinflammatory cytokines, such as TNF-α, promote the onset of insulin and leptin resistance by further activation of JNK/AP-1 and NF-κB signal transduction pathways. CNS exposure to low-dose TNF-α promotes local insulin and leptin resistance (69), whereas hypothalamic administration of the anti–TNF-α antibody infliximab markedly reduces body weight gain during HFD feeding (70). Furthermore, TNF-α induces the expression of PTP1B, a major negative regulator of insulin and leptin signaling, via NF-κB activation (71). Elevated levels of PTP1B after long-term HFD feeding attenuate insulin signaling via direct dephosphorylation of the insulin receptor as wells as its downstream substrate, IRS-1 (72, 73). Neuronal-specific deletion of Ptp1b in mice has been shown to reduce body weight and adiposity, increase energy expenditure, and improve glucose homeostasis (72). POMC-specific ablation of PTP1B recapitulated these findings and identified POMC neurons as important targets of PTP1B action (74, 75).
HFD feeding also alters synaptic plasticity, a change in synaptic strength in response to stimuli, in key hypothalamic neuronal systems (76–78). When exposed to a HFD, both POMC and AgRP neurons present fewer synapses on their perikarya compared with mice fed a normal chow diet. The decrease in POMC synapses affects inhibitory contacts specifically, suggesting a decreased inhibitory tone (78). In addition, prolonged inflammation leads to apoptosis of hypothalamic neurons and a reduction of synaptic inputs in the ARC and lateral hypothalamus (58, 79, 80). POMC neurons in the hypothalamus are the main targets of inflammation-induced apoptosis, resulting in an imbalance of the hypothalamic control of energy homeostasis (35, 58, 80). Furthermore, long-term HFD feeding leads to functional impairment of the neurogenic capacity of hypothalamic neural stem cells (htNSCs), in which HFD-induced activation of IKK/NF-κB triggers apoptotic pathways, resulting in reduced proliferation and survival of htNSCs (80).
Hypothalamic microglia and metabolic inflammation
Prolonged HFD feeding leads to metabolic inflammation in peripheral organs such as white adipose tissue, liver, and skeletal muscle (81). In contrast, hypothalamic inflammation occurs before significant body weight gain, with rapid activation of a complex network of cells (Figure 3 and ref. 35). Activation of hypothalamic microglia in obese mice is influenced by dietary composition and by fat- and gut-derived hormones, rather than obesity or adiposity per se (82). Activated microglia accumulate within the hypothalamus (35, 49) and are associated with the buildup of SFAs (44). Interestingly, the depletion of microglia in the hypothalamus reduces SFA-induced inflammation and neuronal stress, which enhances leptin signaling and leads to decreased food intake (44). Conversely, ICV infusion of stearic acid leads to hypothalamic inflammation that mimics TNF-α action (83). Furthermore, neuropeptides such as α-MSH and NPY can immediately modulate the production of cytokines by microglial cells (84, 85). Activated hypothalamic microglia produce a variety of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 (86, 87); however, IL-6 was shown to be involved in exercise-induced suppression of IKKβ/NF-κB activity and ER stress in the brain, leading to a reduction in food intake (88). Apart from direct proinflammatory action on neurons, these microglia-derived cytokines induce the expression of fractalkine (CX3CL1) in neurons (89), thereby amplifying the inflammatory processes through recruitment of peripheral monocytes to the hypothalamus. Although considered to be immune privileged, recent findings show that the brain is tightly coupled to the immune system, as exemplified by the discovery of a classical lymphatic system in the CNS that allows lymphatic drainage and leukocyte trafficking (90). Indeed, reciprocal interactions between the CNS and the innate immune system may allow the brain to rapidly sense peripheral metabolic and inflammatory cues (91).
Figure 3. Cellular network of hypothalamic inflammation.
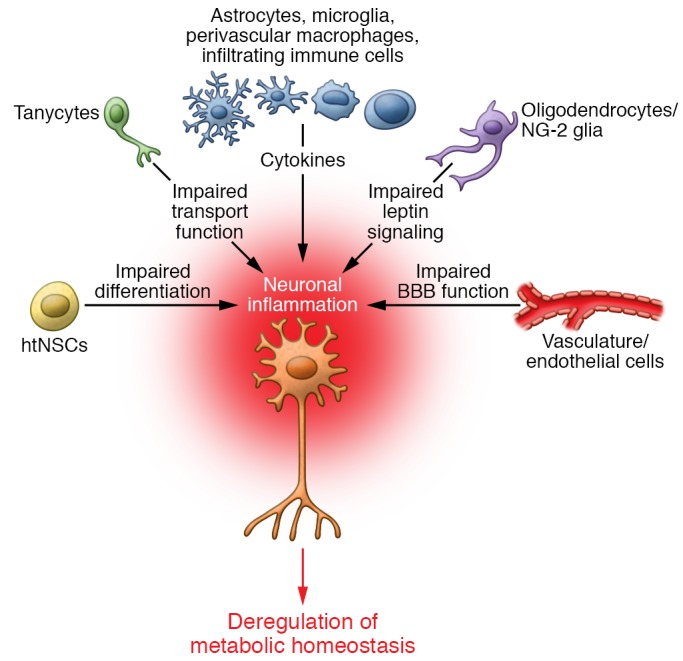
Neurons are embedded within populations of non-neuronal cells, and HFD-induced inflammation rapidly impairs a complex network of different cell types. HFDs impair BBB function at the level of endothelial cells and astrocytes and also impair the transport function of hypothalamic tanycytes. The release of proinflammatory cytokines from microglia, astrocytes, perivascular macrophages, and infiltrating immune cells impairs neuronal function and translates into altered feeding behavior and energy expenditure. This proinflammatory environment leads to functional impairment of supporting cells, such as oligodendrocytes, and of the neurogenic capacity of the hypothalamus.
Hypothalamic astrocytes
Metabolically, the brain critically depends on sustained energy supply and relies almost exclusively on glucose as its main energy source, although the brain is able to use other energy substrates such as ketone bodies during development and starvation (92). Glucose is primarily taken up by astrocytes located around blood vessels and metabolized to lactate, which is supplied to neurons (93). However, a recent study is challenging this view and shows that neurons, and not astrocytes, are the primary consumers of glucose (94). Apart from this shuttling function, astrocytes play a key role in maintaining synaptic plasticity and survival (95).
HFD is associated with the accumulation and activation of astrocytes in the hypothalamus (35, 96). Reactive astrocytosis in the hypothalamus occurs as early as 24 hours after HFD intake (97). In response to HFD feeding, hypothalamic astrocytes produce a variety of inflammatory factors. Again, SFAs activate inflammatory signaling pathways in primary astrocyte cultures and trigger the release of inflammatory cytokines (98). Astrocytes also express TLRs that respond to inflammatory triggers via activation of the NF-κB pathway, which in turn regulates the expression of proinflammatory cytokines (99). In addition, obesity-induced production of TGF-β by astrocytes induces a hypothalamic RNA stress response to accelerate Ikba mRNA decay, leading to atypical NF-κB activation in the hypothalamus (100). Thus, crosstalk of astrocytes and neurons in response to HFD intake plays a fundamental role in hypothalamic inflammation.
Hypothalamic vasculature and the BBB
Alterations of the BBB are fundamentally involved in the development of hypothalamic inflammation (101). The BBB acts as an interface between the CNS and peripheral tissues, regulating the exchange of substances between the circulation and the brain parenchyma. Pathologic changes in the hypothalamic vasculature have been observed in response to a HFD/high-sucrose diet in rodents as well as in patients with type 2 diabetes (102). Moreover, the unique, fenestrated capillary system in the median eminence exposes hypothalamic cell populations to stress-promoting signals emanating from peripheral circulation (35, 103, 104). Interestingly, obesity-associated astrogliosis does not occur uniformly across the hypothalamus, but is associated with microvessels, suggesting a possible response by astrocytes to changes at the BBB and/or the peripheral circulation (105). IL-1β is upregulated in the CNS during HFD feeding and contributes to BBB dysfunction via the transcriptional repression of claudin-5 (106), a tight junction protein critically involved in maintaining BBB integrity (107). Furthermore, long-term intake of a hypercaloric diet decreases expression of other tight junction proteins on brain endothelial cells and increases in microgliosis (108). This disruption of BBB integrity permits leukocyte infiltration into the brain (109). Critically, the function of the BBB is controlled by VEGF, which is the key regulator of neovascularization, blood vessel homeostasis, and hypervascularization (110). Recently, work from our lab has shown that acute HFD feeding leads to reduced brain glucose uptake. Subsequent activation of perivascular macrophages increases VEGF production, which is sufficient to reconstitute glucose transporter expression at the BBB (91). Long-term HFD feeding results in the expression of VEGF in astrocytes (111) as well as in tanycytes in the hypothalamus (112), which promotes microvessel permeability and disruption of BBB integrity. HFD-fed mice show increased accumulation of IgG in the ARC, whereas in chow-fed obese ob/ob mice, IgG deposition was comparable to control wild-type mice fed regular chow (113). These findings imply that increased exposure to dietary lipids, but not increased body weight, is the predominant driver of BBB impairment. Furthermore, mice that are fed a long-term HFD show increased hypothalamic angiopathy with increased blood vessel density and length (102).
Contributions of other cell types
Tanycytes are specialized glial cells that play a crucial role in the transport of and response to leptin (112, 114). These cells have long processes that bridge the cerebrospinal fluid to the portal capillaries. Recently another cell type, NG2-glia cells (also known as oligodendrocyte precursors) were determined to be critical for hypothalamic function, as the ARC leptin receptor neurons lose responsiveness to leptin after NG2-glia ablation (115). These cell types are part of a complex regulatory network involved in maintenance of metabolic homeostasis and underscore the importance of understanding the interactions between neuronal and non-neuronal cells in hypothalamic inflammation.
Hypothalamic deregulation in the human brain
There is growing evidence that consumption of a high-fat, high carbohydrate diet is associated with hypothalamic deregulation in the human brain, but so far only a few studies have addressed anomalous hypothalamic function in the human brain directly. A retrospective analysis of MRIs of non-obese and obese patients showed evidence of increased gliosis in the MBH of obese humans (35). A study using diffusion tensor imaging reported obesity-associated hypothalamic damage associated with inflammatory markers and worse cognitive performance (116). Furthermore, obesity-related systemic inflammation reduces the integrity of brain structures involved in reward and feeding behaviors (117). Mouse models of obesity provide unique insights into disease mechanisms but have their translational limitations, as exemplified by the lack of β-MSH in mice (27). In light of the limited knowledge of metabolic inflammation in the human brain, further neuroimaging studies on human patients are needed to validate findings from animal models and potential therapeutic strategies. Neuroimaging techniques such as functional MRI, PET, electroencephalography, and magnetoencephalography are powerful tools for directly investigating changes in brain function associated with human obesity and provide an important link between animal studies and clinical research in obese patients.
Hypothalamic inflammation and deregulation of peripheral tissues
Hypothalamic neurons project to autonomic sites such as the rostral ventrolateral medulla, NTS, and dorsal motor nucleus of the vagus nerve in the hindbrain (118, 119). The sympathetic nervous system stimulates thermogenesis and stabilizes body weight via activation of β-adrenergic receptors. HFD-induced hypothalamic inflammation disrupts this regulatory pathway of energy homeostasis. Overnutrition is associated with excessive sympathoexcitation, which increases weight gain as a consequence of reduced sensitivity of β-adrenergic receptors (120). In this context, NF-κB–mediated inflammation in POMC neurons, rather than AgRP neurons, is required for overnutrition-induced sympathoexcitation (121). Furthermore, a reduction in parasympathetic tone has been demonstrated to contribute to the activation of inflammatory responses in obesity (122, 123). Activation of α7 nicotinic acetylcholine receptors on immune cells could thus limit obesity-induced inflammation and subsequently improve systemic insulin resistance (124, 125). Hypothalamic inflammation exerts broad effects on peripheral tissues, such as modulation of insulin secretion by pancreatic β cells (83). Similar to β cells (126), glucose sensing in POMC neurons is lost during the development of metabolic syndrome, which subsequently leads to impaired control of glucose metabolism and energy homeostasis (127). In rodents, three days of HFD feeding were sufficient to impair the ability of hypothalamic insulin to suppress white adipose tissue lipolysis and hepatic glucose production (128). In addition, hypothalamic inflammation leads to deregulation of peripheral insulin action (129) and a reduction in adaptive thermogenesis (130, 131).
Maternal metabolic imprinting
Increasing evidence from human and animal studies suggests that maternal obesity and/or diabetes has long-term consequences for the future health of offspring (132–135). Maternal feeding of a HFD only during lactation impairs hypothalamic melanocortin circuitry, leading to malformation of neural projections as a consequence of abnormal insulin signaling (134). The maternal diet affects hypothalamic function and plasticity, resulting in alterations to energy homeostasis in the offspring. Furthermore, HFD exposure prior to and during pregnancy in rodents (136, 137) and non-human primates (138, 139) leads to activation of the maternal immune system and subsequent increases in brain inflammatory markers of the offspring. Therefore, inflammatory pathways in the brain may already be primed toward a proinflammatory response as a consequence of maternal diet.
Conclusion
Over the last years it has become evident that the brain plays a crucial role in the development of obesity and associated co-morbidities. Most of the evidence points to uncoupling of food intake and energy expenditure due to diet-induced inflammation and gliosis in the hypothalamus. Obesogenic diets elicit early inflammatory effects in the hypothalamus, which precede inflammatory events in peripheral tissues. Metabolic processes are not regulated via neuronal cells alone, but rather they are embedded in a complex regulatory system of different cell types. Prolonged over-nutrition leads to sustained hypothalamic inflammatory processes via interactions between neurons and non-neuronal cell populations, perpetuating these initially reversible processes and ultimately resulting in uncoupling between caloric intake and energy expenditure, fostering overeating and further weight gain. Understanding the contributions of different neuronal and non-neuronal cell types to hypothalamic inflammatory processes and delineating differences and similarities between acute and chronic activation of inflammatory pathways will be critical to the development of novel therapeutic strategies for the treatment of metabolic syndrome.
Acknowledgments
The authors apologize to all authors not cited who have made important contributions. This work was supported by a grant from the DFG (BR 1492/7-1) to JCB, by the DFG-funded TRR134 collaborative research center, and by the CMMC and the Excellence Initiative by German Federal and State Governments (CECAD). The authors were supported in part by the Helmholtz Alliance Imaging and Curing Environmental Metabolic Diseases (ICEMED), through the Initiative and Networking Fund of the Helmholtz Association. The authors also received funding from the European Union Seventh Framework Program (FP7/2007-2013) under grant 266408.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2017;127(1):24–32. doi:10.1172/JCI88878.
References
- 1.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 2.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 4.O’Rahilly S, Farooqi IS, Yeo GS, Challis BG. Minireview: human obesity-lessons from monogenic disorders. Endocrinology. 2003;144(9):3757–3764. doi: 10.1210/en.2003-0373. [DOI] [PubMed] [Google Scholar]
- 5.Locke AE, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Speliotes EK, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill JO, Wyatt HR, Reed GW, Peters JC. Obesity and the environment: where do we go from here? Science. 2003;299(5608):853–855. doi: 10.1126/science.1079857. [DOI] [PubMed] [Google Scholar]
- 8.Finucane MM, et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet. 2011;377(9765):557–567. doi: 10.1016/S0140-6736(10)62037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams KW, Elmquist JK. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci. 2012;15(10):1350–1355. doi: 10.1038/nn.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Könner AC, Klöckener T, Brüning JC. Control of energy homeostasis by insulin and leptin: targeting the arcuate nucleus and beyond. Physiol Behav. 2009;97(5):632–638. doi: 10.1016/j.physbeh.2009.03.027. [DOI] [PubMed] [Google Scholar]
- 11.Kennedy GC. Experimental hypothalamic obesity. Proc R Soc Med. 1951;44(10):899–902. [PMC free article] [PubMed] [Google Scholar]
- 12.Anand BK, Dua S, Shoenberg K. Hypothalamic control of food intake in cats and monkeys. J Physiol (Lond) 1955;127(1):143–152. doi: 10.1113/jphysiol.1955.sp005244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gropp E, et al. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8(10):1289–1291. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- 14.Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310(5748):683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 15.Belgardt BF, Brüning JC. CNS leptin and insulin action in the control of energy homeostasis. Ann N Y Acad Sci. 2010;1212:97–113. doi: 10.1111/j.1749-6632.2010.05799.x. [DOI] [PubMed] [Google Scholar]
- 16.Vogt MC, Brüning JC. CNS insulin signaling in the control of energy homeostasis and glucose metabolism - from embryo to old age. Trends Endocrinol Metab. 2013;24(2):76–84. doi: 10.1016/j.tem.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Yi CX, Tschöp MH. Brain-gut-adipose-tissue communication pathways at a glance. Dis Model Mech. 2012;5(5):583–587. doi: 10.1242/dmm.009902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benoit SC, et al. The catabolic action of insulin in the brain is mediated by melanocortins. J Neurosci. 2002;22(20):9048–9052. doi: 10.1523/JNEUROSCI.22-20-09048.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choudhury AI, et al. The role of insulin receptor substrate 2 in hypothalamic and beta cell function. J Clin Invest. 2005;115(4):940–950. doi: 10.1172/JCI24445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cowley MA, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411(6836):480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 21.Ernst MB, et al. Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. J Neurosci. 2009;29(37):11582–11593. doi: 10.1523/JNEUROSCI.5712-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitamura T, et al. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12(5):534–540. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- 23.Belgardt BF, et al. PDK1 deficiency in POMC-expressing cells reveals FOXO1-dependent and -independent pathways in control of energy homeostasis and stress response. Cell Metab. 2008;7(4):291–301. doi: 10.1016/j.cmet.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz MW, et al. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46(12):2119–2123. doi: 10.2337/diab.46.12.2119. [DOI] [PubMed] [Google Scholar]
- 25.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385(6612):165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 26.Biebermann H, et al. A role for beta-melanocyte-stimulating hormone in human body-weight regulation. Cell Metab. 2006;3(2):141–146. doi: 10.1016/j.cmet.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 27.Lee YS, et al. A POMC variant implicates beta-melanocyte-stimulating hormone in the control of human energy balance. Cell Metab. 2006;3(2):135–140. doi: 10.1016/j.cmet.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Morton GJ, et al. Leptin action in the forebrain regulates the hindbrain response to satiety signals. J Clin Invest. 2005;115(3):703–710. doi: 10.1172/JCI22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mesaros A, et al. Activation of Stat3 signaling in AgRP neurons promotes locomotor activity. Cell Metab. 2008;7(3):236–248. doi: 10.1016/j.cmet.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 30.Steculorum SM, et al. Hypothalamic UDP increases in obesity and promotes feeding via P2Y6-dependent activation of AgRP neurons. Cell. 2015;162(6):1404–1417. doi: 10.1016/j.cell.2015.08.032. [DOI] [PubMed] [Google Scholar]
- 31.Aponte Y, Atasoy D, Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci. 2011;14(3):351–355. doi: 10.1038/nn.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steculorum SM, et al. AgRP neurons control systemic insulin sensitivity via myostatin expression in brown adipose tissue. Cell. 2016;165(1):125–138. doi: 10.1016/j.cell.2016.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Könner AC, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5(6):438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Koch M, et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature. 2015;519(7541):45–50. doi: 10.1038/nature14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thaler JP, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122(1):153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Könner AC, Brüning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 2012;16(2):144–152. doi: 10.1016/j.cmet.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 37.De Souza CT, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146(10):4192–4199. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- 38.Clegg DJ, et al. Consumption of a high-fat diet induces central insulin resistance independent of adiposity. Physiol Behav. 2011;103(1):10–16. doi: 10.1016/j.physbeh.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prada PO, et al. Western diet modulates insulin signaling, c-Jun N-terminal kinase activity, and insulin receptor substrate-1ser307 phosphorylation in a tissue-specific fashion. Endocrinology. 2005;146(3):1576–1587. doi: 10.1210/en.2004-0767. [DOI] [PubMed] [Google Scholar]
- 40.Boden G. Interaction between free fatty acids and glucose metabolism. Curr Opin Clin Nutr Metab Care. 2002;5(5):545–549. doi: 10.1097/00075197-200209000-00014. [DOI] [PubMed] [Google Scholar]
- 41.Boden G, Shulman GI. Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur J Clin Invest. 2002;32(suppl 3):14–23. doi: 10.1046/j.1365-2362.32.s3.3.x. [DOI] [PubMed] [Google Scholar]
- 42.Dube JJ, Bhatt BA, Dedousis N, Bonen A, O’Doherty RM. Leptin, skeletal muscle lipids, and lipid-induced insulin resistance. Am J Physiol Regul Integr Comp Physiol. 2007;293(2):R642–R650. doi: 10.1152/ajpregu.00133.2007. [DOI] [PubMed] [Google Scholar]
- 43.Delarue J, Magnan C. Free fatty acids and insulin resistance. Curr Opin Clin Nutr Metab Care. 2007;10(2):142–148. doi: 10.1097/MCO.0b013e328042ba90. [DOI] [PubMed] [Google Scholar]
- 44.Valdearcos M, Robblee MM, Benjamin DI, Nomura DK, Xu AW, Koliwad SK. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014;9(6):2124–2138. doi: 10.1016/j.celrep.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Posey KA, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. 2009;296(5):E1003–E1012. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borg ML, Omran SF, Weir J, Meikle PJ, Watt MJ. Consumption of a high-fat diet, but not regular endurance exercise training, regulates hypothalamic lipid accumulation in mice. J Physiol (Lond) 2012;590(17):4377–4389. doi: 10.1113/jphysiol.2012.233288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276(20):16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- 48.Kleinridders A, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009;10(4):249–259. doi: 10.1016/j.cmet.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Milanski M, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci. 2009;29(2):359–370. doi: 10.1523/JNEUROSCI.2760-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKβ/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135(1):61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oh DY, Olefsky JM. Omega 3 fatty acids and GPR120. Cell Metab. 2012;15(5):564–565. doi: 10.1016/j.cmet.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cintra DE, et al. Unsaturated fatty acids revert diet-induced hypothalamic inflammation in obesity. PLoS One. 2012;7(1):e30571. doi: 10.1371/journal.pone.0030571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olofsson LE, Unger EK, Cheung CC, Xu AW. Modulation of AgRP-neuronal function by SOCS3 as an initiating event in diet-induced hypothalamic leptin resistance. Proc Natl Acad Sci U S A. 2013;110(8):E697–E706. doi: 10.1073/pnas.1218284110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275(12):9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 55.Tsaousidou E, et al. Distinct roles for JNK and IKK activation in agouti-related peptide neurons in the development of obesity and insulin resistance. Cell Rep. 2014;9(4):1495–1506. doi: 10.1016/j.celrep.2014.10.045. [DOI] [PubMed] [Google Scholar]
- 56.Sabio G, et al. Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes Dev. 2010;24(3):256–264. doi: 10.1101/gad.1878510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Belgardt BF, et al. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc Natl Acad Sci U S A. 2010;107(13):6028–6033. doi: 10.1073/pnas.1001796107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moraes JC, et al. High-fat diet induces apoptosis of hypothalamic neurons. PLoS One. 2009;4(4):e5045. doi: 10.1371/journal.pone.0005045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holland WL, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 2011;121(5):1858–1870. doi: 10.1172/JCI43378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Summers SA, Nelson DH. A role for sphingolipids in producing the common features of type 2 diabetes, metabolic syndrome X, and Cushing’s syndrome. Diabetes. 2005;54(3):591–602. doi: 10.2337/diabetes.54.3.591. [DOI] [PubMed] [Google Scholar]
- 61.Turpin SM, et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014;20(4):678–686. doi: 10.1016/j.cmet.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 62.Holland WL, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5(3):167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 63.Morselli E, et al. Hypothalamic PGC-1α protects against high-fat diet exposure by regulating ERα. Cell Rep. 2014;9(2):633–645. doi: 10.1016/j.celrep.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benoit SC, et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-θ subcellular localization in rodents. J Clin Invest. 2009;119(9):2577–2589. doi: 10.1172/JCI36714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ozcan L, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9(1):35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 66.Ozcan U, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 67.Won JC, et al. Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity (Silver Spring) 2009;17(10):1861–1865. doi: 10.1038/oby.2009.194. [DOI] [PubMed] [Google Scholar]
- 68.Williams KW, et al. Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab. 2014;20(3):471–482. doi: 10.1016/j.cmet.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Romanatto T, et al. TNF-alpha acts in the hypothalamus inhibiting food intake and increasing the respiratory quotient — effects on leptin and insulin signaling pathways. Peptides. 2007;28(5):1050–1058. doi: 10.1016/j.peptides.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 70.Romanatto T, et al. Deletion of tumor necrosis factor-alpha receptor 1 (TNFR1) protects against diet-induced obesity by means of increased thermogenesis. J Biol Chem. 2009;284(52):36213–36222. doi: 10.1074/jbc.M109.030874. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 71.Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem. 2008;283(21):14230–14241. doi: 10.1074/jbc.M800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bence KK, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12(8):917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 73.Dubé N, Tremblay ML. Involvement of the small protein tyrosine phosphatases TC-PTP and PTP1B in signal transduction and diseases: from diabetes, obesity to cell cycle, and cancer. Biochim Biophys Acta. 2005;1754(1-2):108–117. doi: 10.1016/j.bbapap.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 74.Banno R, et al. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J Clin Invest. 2010;120(3):720–734. doi: 10.1172/JCI39620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Jonghe BC, et al. Deficiency of PTP1B in POMC neurons leads to alterations in energy balance and homeostatic response to cold exposure. Am J Physiol Endocrinol Metab. 2011;300(6):E1002–E1011. doi: 10.1152/ajpendo.00639.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chun SK, Jo YH. Loss of leptin receptors on hypothalamic POMC neurons alters synaptic inhibition. J Neurophysiol. 2010;104(5):2321–2328. doi: 10.1152/jn.00371.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Horvath TL. Synaptic plasticity in energy balance regulation. Obesity (Silver Spring) 2006;14(suppl 5):228S–233S. doi: 10.1038/oby.2006.314. [DOI] [PubMed] [Google Scholar]
- 78.Horvath TL, et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc Natl Acad Sci U S A. 2010;107(33):14875–14880. doi: 10.1073/pnas.1004282107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McNay DE, Briançon N, Kokoeva MV, Maratos-Flier E, Flier JS. Remodeling of the arcuate nucleus energy-balance circuit is inhibited in obese mice. J Clin Invest. 2012;122(1):142–152. doi: 10.1172/JCI43134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li J, Tang Y, Cai D. IKKβ/NF-κB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat Cell Biol. 2012;14(10):999–1012. doi: 10.1038/ncb2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121(6):2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gao Y, et al. Hormones and diet, but not body weight, control hypothalamic microglial activity. Glia. 2014;62(1):17–25. doi: 10.1002/glia.22580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Calegari VC, et al. Inflammation of the hypothalamus leads to defective pancreatic islet function. J Biol Chem. 2011;286(15):12870–12880. doi: 10.1074/jbc.M110.173021. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 84.Delgado R, et al. Melanocortin peptides inhibit production of proinflammatory cytokines and nitric oxide by activated microglia. J Leukoc Biol. 1998;63(6):740–745. doi: 10.1002/jlb.63.6.740. [DOI] [PubMed] [Google Scholar]
- 85.Ferreira R, et al. Neuropeptide Y inhibits interleukin-1β-induced phagocytosis by microglial cells. J Neuroinflammation. 2011;8:169. doi: 10.1186/1742-2094-8-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nakanishi M, Niidome T, Matsuda S, Akaike A, Kihara T, Sugimoto H. Microglia-derived interleukin-6 and leukaemia inhibitory factor promote astrocytic differentiation of neural stem/progenitor cells. Eur J Neurosci. 2007;25(3):649–658. doi: 10.1111/j.1460-9568.2007.05309.x. [DOI] [PubMed] [Google Scholar]
- 87.Lambertsen KL, et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci. 2009;29(5):1319–1330. doi: 10.1523/JNEUROSCI.5505-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ropelle ER, et al. IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and ER stress inhibition. PLoS Biol. 2010;8(8):e1000465. doi: 10.1371/journal.pbio.1000465. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 89.Morari J, et al. Fractalkine (CX3CL1) is involved in the early activation of hypothalamic inflammation in experimental obesity. Diabetes. 2014;63(11):3770–3784. doi: 10.2337/db13-1495. [DOI] [PubMed] [Google Scholar]
- 90.Louveau A, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jais A, et al. Myeloid-cell-derived VEGF maintains brain glucose uptake and limits cognitive impairment in obesity. Cell. 2016;165(4):882–895. doi: 10.1016/j.cell.2016.03.033. [DOI] [PubMed] [Google Scholar]
- 92.Nehlig A. Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot Essent Fatty Acids. 2004;70(3):265–275. doi: 10.1016/j.plefa.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 93.Figley CR. Lactate transport and metabolism in the human brain: implications for the astrocyte-neuron lactate shuttle hypothesis. J Neurosci. 2011;31(13):4768–4770. doi: 10.1523/JNEUROSCI.6612-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lundgaard I, et al. Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat Commun. 2015;6:6807. doi: 10.1038/ncomms7807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Allen NJ. Astrocyte regulation of synaptic behavior. Annu Rev Cell Dev Biol. 2014;30:439–463. doi: 10.1146/annurev-cellbio-100913-013053. [DOI] [PubMed] [Google Scholar]
- 96.Marino JS, Xu Y, Hill JW. Central insulin and leptin-mediated autonomic control of glucose homeostasis. Trends Endocrinol Metab. 2011;22(7):275–285. doi: 10.1016/j.tem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Buckman LB, Thompson MM, Lippert RN, Blackwell TS, Yull FE, Ellacott KL. Evidence for a novel functional role of astrocytes in the acute homeostatic response to high-fat diet intake in mice. Mol Metab. 2015;4(1):58–63. doi: 10.1016/j.molmet.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gupta S, Knight AG, Gupta S, Keller JN, Bruce-Keller AJ. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J Neurochem. 2012;120(6):1060–1071. doi: 10.1111/j.1471-4159.2012.07660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59(2):242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- 100.Yan J, et al. Obesity- and aging-induced excess of central transforming growth factor-β potentiates diabetic development via an RNA stress response. Nat Med. 2014;20(9):1001–1008. doi: 10.1038/nm.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Thaler JP, Guyenet SJ, Dorfman MD, Wisse BE, Schwartz MW. Hypothalamic inflammation: marker or mechanism of obesity pathogenesis? Diabetes. 2013;62(8):2629–2634. doi: 10.2337/db12-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yi CX, et al. High calorie diet triggers hypothalamic angiopathy. Mol Metab. 2012;1(1–2):95–100. doi: 10.1016/j.molmet.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yi CX, et al. Exercise protects against high-fat diet-induced hypothalamic inflammation. Physiol Behav. 2012;106(4):485–490. doi: 10.1016/j.physbeh.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 104.Peruzzo B, et al. A second look at the barriers of the medial basal hypothalamus. Exp Brain Res. 2000;132(1):10–26. doi: 10.1007/s002219900289. [DOI] [PubMed] [Google Scholar]
- 105.Buckman LB, Thompson MM, Moreno HN, Ellacott KL. Regional astrogliosis in the mouse hypothalamus in response to obesity. J Comp Neurol. 2013;521(6):1322–1333. doi: 10.1002/cne.23233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Beard RS, et al. Non-muscle Mlck is required for β-catenin- and FoxO1-dependent downregulation of Cldn5 in IL-1β-mediated barrier dysfunction in brain endothelial cells. J Cell Sci. 2014;127(pt 8):1840–1853. doi: 10.1242/jcs.144550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nitta T, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161(3):653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Freeman LR, Granholm AC. Vascular changes in rat hippocampus following a high saturated fat and cholesterol diet. J Cereb Blood Flow Metab. 2012;32(4):643–653. doi: 10.1038/jcbfm.2011.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Varvel NH, et al. Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc Natl Acad Sci U S A. 2012;109(44):18150–18155. doi: 10.1073/pnas.1210150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee S, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130(4):691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Argaw AT, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122(7):2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Langlet F, et al. Tanycytic VEGF-A boosts blood-hypothalamus barrier plasticity and access of metabolic signals to the arcuate nucleus in response to fasting. Cell Metab. 2013;17(4):607–617. doi: 10.1016/j.cmet.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yi CX, Tschöp MH, Woods SC, Hofmann SM. High-fat-diet exposure induces IgG accumulation in hypothalamic microglia. Dis Model Mech. 2012;5(5):686–690. doi: 10.1242/dmm.009464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Balland E, et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab. 2014;19(2):293–301. doi: 10.1016/j.cmet.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Djogo T, et al. Adult NG2-glia are required for median eminence-mediated leptin sensing and body weight control. Cell Metab. 2016;23(5):797–810. doi: 10.1016/j.cmet.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 116.Puig J, et al. Hypothalamic damage is associated with inflammatory markers and worse cognitive performance in obese subjects. J Clin Endocrinol Metab. 2015;100(2):E276–E281. doi: 10.1210/jc.2014-2682. [DOI] [PubMed] [Google Scholar]
- 117.Cazettes F, Cohen JI, Yau PL, Talbot H, Convit A. Obesity-mediated inflammation may damage the brain circuit that regulates food intake. Brain Res. 2011;1373:101–109. doi: 10.1016/j.brainres.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443(7109):289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 119.Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- 120.Esler M, Straznicky N, Eikelis N, Masuo K, Lambert G, Lambert E. Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension. 2006;48(5):787–796. doi: 10.1161/01.HYP.0000242642.42177.49. [DOI] [PubMed] [Google Scholar]
- 121.Purkayastha S, Zhang G, Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-β and NF-κB. Nat Med. 2011;17(7):883–887. doi: 10.1038/nm.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Andersson U, Tracey KJ. Reflex principles of immunological homeostasis. Annu Rev Immunol. 2012;30:313–335. doi: 10.1146/annurev-immunol-020711-075015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rosas-Ballina M, Tracey KJ. The neurology of the immune system: neural reflexes regulate immunity. Neuron. 2009;64(1):28–32. doi: 10.1016/j.neuron.2009.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang X, Yang Z, Xue B, Shi H. Activation of the cholinergic antiinflammatory pathway ameliorates obesity-induced inflammation and insulin resistance. Endocrinology. 2011;152(3):836–846. doi: 10.1210/en.2010-0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bencherif M, Lippiello PM, Lucas R, Marrero MB. Alpha7 nicotinic receptors as novel therapeutic targets for inflammation-based diseases. Cell Mol Life Sci. 2011;68(6):931–949. doi: 10.1007/s00018-010-0525-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307(5708):384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 127.Parton LE, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449(7159):228–232. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- 128.Scherer T, Lindtner C, Zielinski E, O’Hare J, Filatova N, Buettner C. Short term voluntary overfeeding disrupts brain insulin control of adipose tissue lipolysis. J Biol Chem. 2012;287(39):33061–33069. doi: 10.1074/jbc.M111.307348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci U S A. 2011;108(7):2939–2944. doi: 10.1073/pnas.1006875108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Arruda AP, et al. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology. 2011;152(4):1314–1326. doi: 10.1210/en.2010-0659. [DOI] [PubMed] [Google Scholar]
- 131.Arruda AP, et al. Hypothalamic actions of tumor necrosis factor alpha provide the thermogenic core for the wastage syndrome in cachexia. Endocrinology. 2010;151(2):683–694. doi: 10.1210/en.2009-0865. [DOI] [PubMed] [Google Scholar]
- 132.Steculorum SM, Vogt MC, Brüning JC. Perinatal programming of metabolic diseases: role of insulin in the development of hypothalamic neurocircuits. Endocrinol Metab Clin North Am. 2013;42(1):149–164. doi: 10.1016/j.ecl.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 133.Sullivan EL, Grove KL. Metabolic imprinting in obesity. Forum Nutr. 2010;63:186–194. doi: 10.1159/000264406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vogt MC, et al. Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell. 2014;156(3):495–509. doi: 10.1016/j.cell.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Howie GJ, Sloboda DM, Kamal T, Vickers MH. Maternal nutritional history predicts obesity in adult offspring independent of postnatal diet. J Physiol. 2009;587(pt 4):905–915. doi: 10.1113/jphysiol.2008.163477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bilbo SD, Tsang V. Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J. 2010;24(6):2104–2115. doi: 10.1096/fj.09-144014. [DOI] [PubMed] [Google Scholar]
- 137.Rother E, et al. Hypothalamic JNK1 and IKKβ activation and impaired early postnatal glucose metabolism after maternal perinatal high-fat feeding. Endocrinology. 2012;153(2):770–781. doi: 10.1210/en.2011-1589. [DOI] [PubMed] [Google Scholar]
- 138.Grayson BE, Levasseur PR, Williams SM, Smith MS, Marks DL, Grove KL. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology. 2010;151(4):1622–1632. doi: 10.1210/en.2009-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sullivan EL, et al. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J Neurosci. 2010;30(10):3826–3830. doi: 10.1523/JNEUROSCI.5560-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]