

Abstract
Key points
The mechanisms of action of anaesthetics on the living brain are still poorly understood. In this respect, the analysis of the differential effects of anaesthetics on spontaneous and sensory‐evoked cortical activity might provide important and novel cues.
Here we show that the anaesthetic sevoflurane strongly silences the brain but potentiates in a dose‐ and frequency‐dependent manner the cortical visual response.
Such enhancement arises from a linear scaling by sevoflurane of the power‐law relation between light intensity and the cortical response.
The fingerprint of sevoflurane action suggests that circuit silencing can boost linearly synaptic responsiveness presumably by scaling the number of responding units and/or their correlation following a sensory stimulation.
Abstract
General anaesthetics, which are expected to silence brain activity, often spare sensory responses. To evaluate differential effects of anaesthetics on spontaneous and sensory‐evoked cortical activity, we characterized their modulation by sevoflurane and propofol. Power spectra and the bust‐suppression ratio from EEG data were used to evaluate anaesthesia depth. ON and OFF cortical responses were elicited by light pulses of variable intensity, duration and frequency, during light and deep states of anaesthesia. Both anaesthetics reduced spontaneous cortical activity but sevoflurane greatly enhanced while propofol diminished the ON visual response. Interestingly, the large potentiation of the ON visual response by sevoflurane was found to represent a linear scaling of the encoding mechanism for light intensity. To the contrary, the OFF cortical visual response was depressed by both anaesthetics. The selective depression of the OFF component by sevoflurane could be converted into a robust potentiation by the pharmacological blockade of the ON pathway, suggesting that the temporal order of ON and OFF responses leads to a depression of the latter. This hypothesis agrees with the finding that the enhancement of the ON response was converted into a depression by increasing the frequency of light‐pulse stimulation from 0.1 to 1 Hz. Overall, our results support the view that inactivity‐dependent modulation of cortical circuits produces an increase in their responsiveness. Among the implications of our findings, the silencing of cortical circuits can boost linearly the cortical responsiveness but with negative impact on their frequency transfer and with a loss of the information content of the sensory signal.
Keywords: anaesthesia, cortical silencing, gain modulation, inactivity‐dependent modulation, intensity sensitivity, visual cortex, visual evoked potential
Key points
The mechanisms of action of anaesthetics on the living brain are still poorly understood. In this respect, the analysis of the differential effects of anaesthetics on spontaneous and sensory‐evoked cortical activity might provide important and novel cues.
Here we show that the anaesthetic sevoflurane strongly silences the brain but potentiates in a dose‐ and frequency‐dependent manner the cortical visual response.
Such enhancement arises from a linear scaling by sevoflurane of the power‐law relation between light intensity and the cortical response.
The fingerprint of sevoflurane action suggests that circuit silencing can boost linearly synaptic responsiveness presumably by scaling the number of responding units and/or their correlation following a sensory stimulation.
Abbreviations
- BSR
burst‐suppression ratio
- CCF
cross‐correlation function
- EEG
electroencephalography
- l‐AP4
l‐2‐amino‐4‐phosphonobutyric acid
- MAP
mean arterial pressure
- mGluR6
metabotropic glutamate receptor 6
- N
number of readily releasable synaptic vesicles
- p
release probability of synaptic vesicles
- P1
first positive peak of the visual evoked potential
- PSD
power spectral density
- RMS
root mean square
- V1
primary visual cortex
- VEP
visual evoked potential
Introduction
In the past, the silencing effect of general anaesthetics has been studied and interpreted as the result of interactions with molecular targets that control neuronal excitability (Patel et al. 1999; Ouyang et al. 2003; Bieda & MacIver, 2004) or the strength of excitatory pathways. The latter scenario might relate to a direct inhibition of excitatory synaptic transmission (Yamakura et al. 1995; Nishikawa & MacIver, 2000; Westphalen & Hemmings, 2003; Wu et al. 2004; Solt et al. 2006; Ishizeki et al. 2008; Haseneder et al. 2009) or might follow the enhancement of inhibitory transmission (Mihic et al. 1997; de Sousa et al. 2000; Asahi et al. 2006; MacIver, 2014).
Because of the complexity of cortical circuits, the final in vivo outcome of anaesthetic treatment might be counterintuitive, for example resulting in a reduction of the strong synaptic inhibitory tone observed in wakefulness, which generates a more balanced ratio between excitation and inhibition (Haider et al. 2013). Furthermore, during general anaesthesia neuronal activity is not simply suppressed, but the brain seems to shift into a distinct electrical and functional mode (Brown et al. 2010). In this state, many sensory stimuli can still effectively reach the cerebral cortex, where a variable degree of cortical activation can be detected (see for review Kumar et al. 2000). These sensory responses are not simply altered in their amplitude but anaesthetics change their shape and duration compared to wakefulness (Imas et al. 2005, 2006; Saxena et al. 2013). While a depression of the amplitude of sensory responses would be expected (Saxena et al. 2013), because of the well‐known depressant effect of anaesthetics on thalamo‐cortical and cortico‐cortical firing rates, their maintenance and/or amplitude enhancement (Imas et al. 2005, 2006) is more difficult to reconcile with the currently available information.
Here, we explored the effects of the volatile anaesthetic sevoflurane on the visual processing of dark‐to‐light and light‐to‐dark transitions from rat primary visual cortex (V1) and compared its actions with those of another general anaesthetic molecule, propofol, which belongs to a different class of compounds. Our findings reveal an inactivity‐dependent modulation of cortical circuits by sevoflurane underlying a previously unrecognized multiplicative action of this compound on light‐intensity coding mechanisms.
Methods
Ethical approval and animal care procedures
Research and animal care procedures were approved by our Institutional Animal Care and Use Committee for Good Animal Experimentation in accordance with the Italian MIUR code of practice for the care and use of animals for scientific purposes (IACUC number: 541). Experiments were carried out on adult male Sprague–Dawley rats (350–540 g; n = 33). As indicated below, every effort was made to minimize the animals’ distress, pain and suffering during the entire course of the experimentation. All animals were individually caged with free access to food and water ad libitum and were exposed to 12 h light–dark cycles at 23°C constant room temperature.
Surgical and electrophysiological protocols
Cortical activity was recorded by three metal electrodes (stainless steel screws, 1.4 mm diameter) chronically implanted on the skull, positioned at the level of left–right V1 and right motor cortex. The coordinates for implantation were as follows: for V1, mediolateral ±4.6 mm from the sagittal axis and rostrocaudal 7.00 mm below bregma; for the motor cortex, mediolateral –1.82 mm and rostrocaudal 2.4 mm above bregma. For electrode implantation, rats were placed in an anaesthetic induction chamber (sevoflurane 5%; Abbvie, North Chicago, IL, USA) for a few minutes until unconscious. Rats were then removed from the chamber, the loss of the righting reflex was tested, and then they were quickly connected through a nose cone to a mechanical ventilator (sevoflurane 3.75%; SERVO900C, Siemens, München, Germany; gas vaporizer, Vapor 2000, Drägerwerk, Lübeck, Germany). Rats then received intraperitoneal gentamicin (1.5 mg kg−1, Italfarmaco, Milano, Italy), subcutaneous carprofen (5 mg kg−1, Pfizer, Latina, Italy) and dexamethasone (0.2 mg kg−1, Hospira, Napoli, Italy). Body temperature during the procedure was maintained at 36–37°C using a heating pad (ATC1000, WPI, Sarasota, FL, USA). Electrodes were secured by methylmethacrylate cement (Salmoiraghi Produzione Dentaria, Mulazzano, Italy). For the following 3 days, animals received intraperitoneal gentamicin (1.5 mg kg−1) twice a day.
After 1 week of recovery from electrode implantation, rats underwent two successive recording trials run under full anaesthesia, interleaved with a resting period of at least 3 days (3–6 days), during which their recovery and health conditions were carefully monitored. As described below, during these two experimental trials, animals were neither physically restrained nor exposed to any further surgical procedure or noxious stimuli. At the end of the second trial, before regaining consciousness, animals were killed by a lethal dose of thiopental (100 mg kg−1; i.v.), which induced a rapid flattening of the EEG (see below). Animals were disposed of only after death was confirmed by the clear onset of rigor mortis.
For the two recording trials, rats were anaesthetized as described above, quickly intubated (endotracheal polyethylene 16 GA cannula; the cannula was moistened using a 2.5% lidocaine chlorhydrate gel; Luan, Molteni, Italy) and the endotracheal cannula connected to the mechanical ventilator (sevoflurane 2.5%). Volume‐controlled air ventilation (tidal volume 6 ml; respiratory rate 70–80 breaths min−1) was set and the end‐expiratory sevoflurane and CO2 concentration continuously monitored by a gas analyser (Vamos, Drägerwerk, Lübeck, Germany). Rats were placed on a custom‐built apparatus for electrophysiological recordings, where body temperature was maintained at 36–37°C using a heating pad. The tail vein was then cannulated (26 GA cannula) and from this point on, the anaesthesia was maintained either with sevoflurane (2.5–5%) or with propofol (intravenous 1–2 mg kg−1 min−1; AstraZeneca, London, UK), using as starting dosage for both compounds the lowest one (sevoflurane 2.5%; propofol 1 mg kg−1 min−1). For propofol anaesthesia, the intravenous injection of an initial bolus (10 mg kg−1) was followed by continuous intravenous infusion of the anaesthetic at maintenance dosage (Pump11‐elite, Harvard Apparatus, Holliston, MA, USA). Following the induction of anaesthesia, electrodes were connected to the recording system. During the initial preparation phase (∼20 min), the stability of anaesthesia was evaluated by on‐line monitoring of the EEG and its spectrum as well as by evaluating the loss of the righting reflex. The lowest anaesthetic dosages used in these experiments were found to abolish the righting reflex in 100% of cases (sevoflurane 2.5%; propofol 1 mg kg−1 min−1). Following this preparation phase, atracurium (GlaxoSmithKline, Brentford, UK) was administered as an intravenous bolus (5 mg kg−1 every 18 min) in order to suppress involuntary eye bulb movements, which are preserved during anaesthesia (Nair et al. 2011). This treatment was used to maximize visual responses reproducibility. EEG monitoring was maintained throughout the experiment to evaluate the stability and depth of anaesthesia. Regarding the anaesthetic concentrations, in all experiments, the sequence used always went from the lowest to the highest concentration (one anaesthetic compound, three sequential anaesthetic concentrations for each trial). For intravitreal injection of the metabotropic glutamate receptor 6 (mGluR6) agonist l‐2‐amino‐4‐phosphonobutyric acid (l‐AP4; Tocris Bioscience, Bristol, UK), during the initial preparation phase (sevoflurane 2.5%), 5 μl of a standard Tyrode solution containing 20 mm l‐AP4 was injected into the eye bulb (estimated final concentration of l‐AP4 2 mm). In some experiments the mean arterial pressure (MAP) was recorded by a catheter (26 GA) inserted into the femoral artery and connected to a pressure transducer. Electrical activity was recorded by a custom‐built amplifier featuring a total gain of 5000 and band‐pass filtering of 0.1–3000 Hz. The signal was digitalized at 20 kHz (16 bit) using the ITC‐18 data acquisition interface (HEKA Elektronik, Lambrecht, Germany) controlled by a custom software developed in the LabVIEW environment (National Instruments, Austin, TX, USA). A ground electrode was positioned on the ear. The inter‐hemispheric occipito‐occipital derivation was used to record visual evoked potentials (VEPs) and the inter‐hemispheric occipito‐frontal derivation was used for spontaneous activity.
Experimental design and visual stimuli
To reduce animal‐to‐animal variability, the same animals were exposed to both anaesthetics in sequential experimental sessions. In these two separate recording trials, the administration sequence for the two anaesthetic compounds was randomly assigned to animals. Spontaneous activity recordings were performed in complete darkness without light stimulation. For light stimulation protocols, rats were maintained in complete darkness with their left eye covered with a black tape. The light source was a white, high‐brightness light‐emitting diode placed 2 cm away from the right eye. Light stimuli were applied at a fixed rate (0.1 Hz). The irradiance and the pulse duration were randomly varied by the software during the course of experiments (irradiance range, 1–330 μW cm−2; pulse duration range, 20–800 ms). In some experiments the rate of light stimuli was also varied (frequency range, 0.1–1 Hz) with fixed light pulse intensity and duration (150 μW cm−2 and 300 ms, respectively).
EEG and VEP analysis
Power spectral density (PSD) of raw electroencephalographic (EEG) signal in the resting condition was computed as follows: in each condition 110 s of spontaneous brain activity was subdivided into 10 s sweeps, band‐pass filtered (0.5–90 Hz) and band‐block filtered (49–51 Hz) for removal of line noise. Each sweep was Hamming‐windowed followed by fast Fourier transform (FFT). For normalization the square magnitude of FFT was divided by the number of samples N and by the sampling frequency F s. A set of successive power spectra (n = 11) were averaged. For the delta (0.5–4 Hz), theta (4–8 Hz), alpha (8–12 Hz), sigma or spindle (12–15 Hz), beta (15–25 Hz) and gamma (25–80 Hz) bands, the average PSD and overall sum for all densities was calculated. For burst detection, 10 s sweeps were down‐sampled to 5 kHz, band‐pass filtered (12–15 Hz) and rectified; burst events were then detected based on their amplitude (mean voltage and its SD) and duration (minimal duration ≥ 500 ms; when inter‐event interval was ≤ 300 ms, the events were considered as parts of the same burst. Burst peak‐amplitude (rectified signal), duration and PSD (same procedure as above) were then extracted for analysis. Short‐time Fourier transform (STFT) was performed on raw signals with 120 s Hamming windows (99% overlapping). The state of burst suppression was quantified by the burst‐suppression ratio (BSR), which specifies the fractional time spent in suppression (100%, all suppression; 0%, no suppression; Vijn & Sneyd, 1998).
Quantitative estimates of VEPs were obtained from ensemble averages as well as from single traces. VEP latency was expressed as the latency of 50% of the first positive peak from the beginning of the light pulse (50% P1 latency). VEP amplitude was obtained as the root mean square (RMS) of the first 150 ms on the ensemble average starting from stimulus onset:
Where x is the voltage sample and n is the sample number. For the OFF response, the end of the stimulus was used as reference starting point.
The weighted least‐square method was used to fit linear functions. A non‐linear fitting process was performed using the Levenberg–Marquardt algorithm. Error bars were used as weights in fitting procedures. Light‐intensity and pulse‐rate response curves were fitted with the following power‐function:
where y is the RMS amplitude, b is the power exponent, a is the multiplicative coefficient and x either the light intensity or stimulation frequency. Power exponents b were computed by linear fitting of log–log plots and then averaged for every condition. For the effects of light‐pulse stimulation frequency on VEP amplitude, the amplitudes of single VEPs that were uncorrupted by spontaneous cortical noise (selection threshold equal to the mean RMS + 1 SD of the first 20 ms from stimulus onset) were measured, averaged and fitted by the following exponential decay function:
where y is the RMS amplitude, x is the stimulus number, τ is the time constant, y∞ is the plateau value, and y 0 is set to the amplitude of the first VEP in the series.
Cross‐correlation functions (CCFs) were computed using a 150 ms window starting from stimulus onset (same temporal window used for RMS analysis). The lag of the first positive peak of the cross‐correlation function (between pairs of VEP traces) was detected by searching for the first local maximum of the CCF using a moving search window of ±3 neighbouring samples.
Statistics
Results are expressed as the mean ± standard error of the mean. The normality of distribution of 50% P1 latency and of VEP RMS of ON and OFF responses in the experimental population was assessed by applying the Shapiro–Wilk test. In a repeated measures design, principal and interaction effects were tested with one‐way or two‐way repeated measures ANOVA (rANOVA). Greenhouse–Geisser correction was applied when sphericity could not be assumed. Group comparisons in repeated measures design were tested with Student's paired‐samples t test and the Bonferroni–Holm correction was applied to multiple hypothesis testing. The two‐samples t test was chosen for non‐repeated measures design and the Welch correction for unequal variance was applied. To evaluate the goodness of fit, adjusted R 2 was computed and the F test was performed to establish a P value. For the analysis of the effects of light‐pulse stimulation frequency on VEP amplitude, the Spearman correlation coefficient (ρ) was calculated and the t test used to compute a P value. Non‐parametric statistics were applied to test comparisons between ratio estimates and standard deviations. In the repeated measures design, principal effects were tested with Friedman's test, while group comparisons in repeated measures design were tested with Wilcoxon's signed‐rank test. The criterion for significance was set at P < 0.05. All statistical tests were two‐tailed. Symbols used to indicate P‐values are as follows: P > 0.05, ns; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Results
Initial evaluation of anaesthesia depth
To evaluate the depth of anaesthesia we recorded spontaneous cortical activity and tested the effects of increasing dosages of sevoflurane and propofol. Concentrations used in these experiments ranged for sevoflurane from 2.5 to 5% and for propofol from 1 to 2 mg kg−1 min−1. At the lowest dosages, spontaneous cortical activity showed the characteristic high‐voltage low‐frequency oscillations consistent with non‐REM sleep and anaesthesia (Fig. 1 A; Brown et al. 2010). When the concentrations of these agents were increased (sevoflurane, 3.75 and 5%; propofol, 1.5 and 2 mg kg−1 min−1), a clear burst‐suppression pattern was induced (Fig. 1 A and B), reflecting a strong brain inactivation which is characteristic of deep general anaesthesia and coma (Kroeger & Amzica, 2007; Ching et al. 2012). The burst‐suppression ratio (BSR; Vijn & Sneyd, 1998) confirmed that at low dosages this behaviour remains close to zero for both anaesthetics (Fig. 1 B; n = 7 rats; BSR: sevoflurane 2.5%, 3.44 ± 2.01%; propofol 1 mg kg−1 min−1, 1.39 ± 0.34%; P > 0.05, Wilcoxon's signed‐rank test) while a large increase of BSR was found when drug concentration was increased (Fig. 1 B; n = 7 rats; P < 0.001 for both sevoflurane and propofol, Friedman's test). At the highest dosages, no significant differences in BSR were found between the two anaesthetics, suggesting a similar state of cortical inactivation (Fig. 1 B; n = 7 rats; BSR: sevoflurane 5%, 95.35 ± 0.78%; propofol 2 mg kg−1 min−1, 94.53 ± 0.81%; P > 0.05, Wilcoxon's signed‐rank test). This bursting behaviour emerged as a dose‐dependent reduction of EEG power spectral density (Fig. 1 C–F; n = 7 rats, 0.5–80 Hz window; P sevoflurane < 0.01; P propofol < 0.001; one‐way rANOVA) even at the level of individual frequency bands with the sole exception of the gamma band (25–80 Hz) under sevoflurane anaesthesia (Fig. 1 E and F; gamma band P sevoflurane > 0.05; for all other bands and conditions in all cases P < 0.05; one‐way rANOVA).
Figure 1. Changes in spontaneous cortical activity induced by sevoflurane and propofol.

A, spontaneous cortical voltage oscillations observed in an exemplar experiment where the animal was exposed to three different concentrations of sevoflurane (top; 2.5, 3.75 and 5%) and propofol (bottom; 1, 1.5 and 2 mg kg−1 min−1; traces from the same rat kept in the dark and exposed to the two different anaesthetics in consecutive experimental sessions, 3 days apart). Notice the dose‐dependent change in spontaneous activity with a burst‐suppression pattern for the intermediate and highest anaesthetics doses. B, BSR for both anaesthetics at all dosages (n = 7 rats exposed to both agents). C and D, average periodograms from three representative unfiltered EEG recordings (10 s sweeps, total trace length 110 s) obtained as above for sevoflurane (2.5, 3.75 and 5%; C) and propofol (1, 1.5 and 2 mg kg−1 min−1; D). The sharp peak at 50 Hz in D is from power‐line noise. E and F, mean PSD from quantitative spectral analysis for sevoflurane (E) and propofol (F), on the whole frequency band (0.5–80 Hz; left in both panels E and F) and for single frequency bands (right in both panels E and F) as a function of anaesthetic dosage (n = 7, animals exposed to both anaesthetics). Asterisks indicate significance values (panel B, principal effect by Friedman test; panels E and F, principal effect by one‐way rANOVA; P > 0.05, ns; * P < 0.05; ** P < 0.01; *** P < 0.001).
The analysis of individual bursts also showed that while burst occurrence and duration were significantly decreased at high dosages with anaesthetics, burst amplitude and spectral power were differentially affected (Fig. 2).
Figure 2. Analysis of spontaneous bursts induced by sevoflurane and propofol.
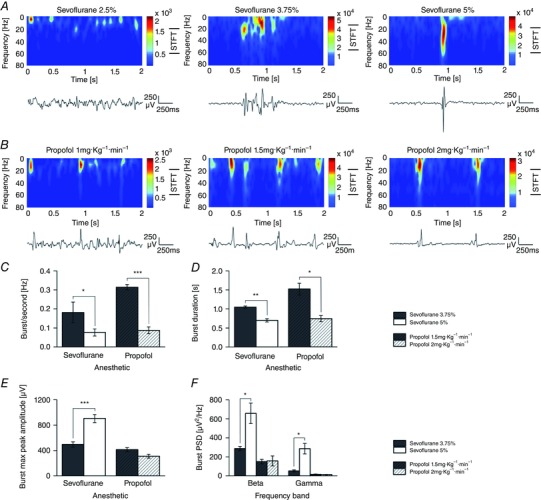
A and B, spontaneous cortical oscillations and their time–frequency analysis at increasing concentrations of sevoflurane (A; 2.5, 3.75 and 5%) and propofol (B; 1, 1.5 and 2 mg kg−1 min−1; same animal as in A). Notice that when the anaesthetic concentration is increased a clear burst‐suppression pattern is induced (middle and right in A and B). C–F, quantitative burst analysis obtained on identified burst episodes during burst suppression to illustrate the effects of these two anaesthetics on burst frequency (C; n = 5 rats; sevoflurane 3.75%, 0.18 ± 0.05 Hz; sevoflurane 5%, 0.08 ± 0.02 Hz; P < 0.05; propofol 1.5 mg kg−1 min−1, 0.31 ± 0.01 Hz; propofol 2 mg kg−1 min−1, 0.09 ± 0.02 Hz; P < 0.001; paired‐samples t test), burst duration (D), burst maximum peak amplitude (E), and their beta and gamma power (F). A parallel decrement of burst duration (D) was detected with both anaesthetics and this was dose‐dependent (sevoflurane 3.75%, 1.04 ± 0.03 s; 5%, 0.70 ± 0.04 s; P < 0.01, paired‐samples t test; propofol 1.5 mg kg−1 min−1, 1.52 ± 0.15 s; 2 mg kg−1 min−1, 0.74 ± 0.08 s; P < 0.05, paired‐samples t test). Notice the opposite effects of sevoflurane and propofol on burst maximum peak amplitude (E; sevoflurane 3.75%, 495.39 ± 42.05 μV; 5%, 903.80 ± 63.77 μV; n = 5 rats; P < 0.001, paired‐samples t test). Notice that this potentiation of burst peak amplitude was not seen with propofol (propofol 1.5 mg kg−1 min−1, 414.18 ± 31.33 μV; 2 mg kg−1 min−1, 310.41 ± 30.21 μV; n = 5 rats; P > 0.05, paired‐samples t test). F, analysis of the beta and gamma power of identified bursts during the burst suppression phase. Notice the clear dose increase in gamma and beta power for sevoflurane but not propofol (beta power: sevoflurane 3.75%, 287.36 ± 22.8 μV2 Hz−1; sevoflurane 5%, 657.43 ± 109.02 μV2 Hz−1; P < 0.05; gamma power: sevoflurane 3.75%, 50.71 ± 10.92 μV2 Hz−1; sevoflurane 5%, 286.30 ± 55.41 μV2 Hz−1; P < 0.05;). (Panels C–F, paired‐samples t test; * P < 0.05; ** P < 0.01; *** P < 0.001.)
Selective potentiation of visual cortical responses by sevoflurane
We began by studying the action of sevoflurane on evoked cortical responses induced by light stimuli (VEPs) and compared its effects with those of propofol. These sensory responses displayed the standard set of positive and negative deflections (Creel et al. 1974) and remained stable both in amplitude and waveform up to the end of recordings for both pharmacological agents (pulse rate 0.1 Hz; stable up to 2 h from the beginning of anaesthesia). As depicted in Fig. 3, a dose‐dependent potentiation of VEP amplitude by sevoflurane was found (light‐pulse duration 20 ms; pulse irradiance 25 μW cm−2; pulse rate 0.1 Hz) while this was not seen with propofol, which produced the opposite effect (Fig. 3 A and B; n = 6 rats; RMS amplitude estimated from the average VEP waveform; principal effect of anaesthetic concentration, P < 0.05 for both sevoflurane and propofol, one‐way rANOVA; significant multiple comparisons, P sevoflurane (2.5 vs. 3.75%) < 0.05, P sevoflurane (2.5 vs. 5%) < 0.05, P propofol (1 vs. 2 mg kg–1 min–1) < 0.05, paired‐samples t test). In these experiments a clear dose‐dependent incremental delay of the onset of the P1 wave was found with both anaesthetics (Fig. 3 C; 50% P1 latency; P sevoflurane < 0.01; P propofol < 0.05; principal effect of anaesthetic concentration, one‐way rANOVA). Since sevoflurane and propofol are known to reduce arterial blood pressure (Claeys et al. 1988; Ebert et al. 1995), we wondered if the differential effects of these two molecules on visual responses could be explained by an uneven action on the cardiovascular system. We found that stepwise increments in sevoflurane and propofol concentrations produced comparable drops in the mean arterial pressure (Fig. 4), suggesting that their specific mode of action could not be attributed to differential effects on the cardiovascular system.
Figure 3. Opposite changes induced by sevoflurane and propofol on visual response.
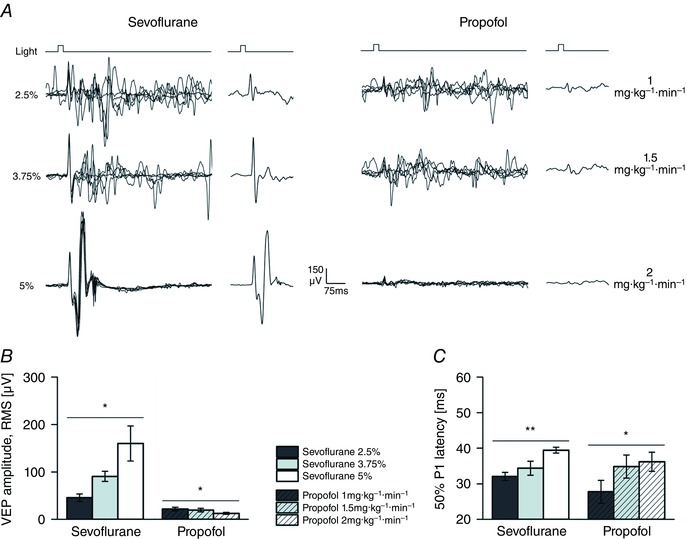
A, single VEP traces and ensemble averages from an individual animal exposed to brief light pulses (pulse duration 20 ms; pulse irradiance 25 μW cm−2; pulse rate 0.1 Hz) at incremental concentrations of sevoflurane (2.5, 3.75 and 5%; left) and propofol (1, 1.5 and 2 mg kg−1 min−1; right). B and C, bars graphs of VEP amplitude (B; RMS amplitude, first 150 ms from stimulus onset, estimated from the averaged waveform) and the onset of the P1 deflection (C; mean latency of the 50% of P1 amplitude). These estimates were obtained from the same animals, each exposed to all dosages of both anaesthetics in two separate experimental sessions, run a few days apart (n = 6 rats). Notice how, sevoflurane increased the VEP amplitude in a concentration‐dependent fashion, while propofol did the opposite. Also notice that the VEP latency was incremented by both drugs, indicating a common reduction in neuronal excitability. Asterisks indicate significance values (B and C, principal effect by one‐way rANOVA; P > 0.05, ns; * P < 0.05; ** P < 0.01; *** P < 0.001).
Figure 4. Comparable changes of mean arterial pressure induced by sevoflurane and propofol.
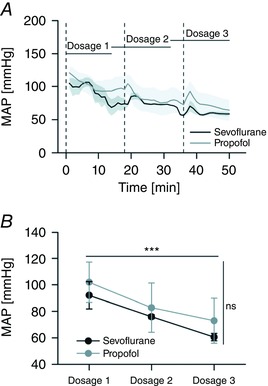
A, time courses of the mean arterial pressure (MAP) during stepwise increments of sevoflurane and propofol concentration (n = 4 rats; Dosage 1: sevoflurane 2.5%, propofol 1 mg kg−1 min−1; Dosage 2: sevoflurane 3.75%, propofol 1.5 mg kg−1 min−1; Dosage 3: sevoflurane 5%, propofol 2 mg kg−1 min−1). Dashed lines indicate the time points for atracurium injections. The grey envelopes plot the standard errors of the mean. MAP was recorded by the cannulation of the femoral artery. B, averages of MAP estimates (15 min epochs) for all dosages and anaesthetics. The increment of the concentration of both anaesthetics induced a comparable drop in MAP (n = 4 rats; P < 0.001; principal effect of anaesthetic dosage, two‐way rANOVA) but for each step in anaesthetic concentration the effects of sevoflurane and propofol were not significantly different (P > 0.05 for all anaesthetics pairs, paired‐samples t test; P > 0.05, principal effect of anaesthetic agent, two‐way rANOVA). (A and B, principal effects by two‐way rANOVA; P > ns; *** P < 0.001.)
To evaluate the consistency of the sevoflurane‐induced potentiation across individual trials and clarify the temporal reproducibility, we analysed in greater detail the behaviour of the sequential VEPs (Fig. 5 A; plotting the same experiment presented in Fig. 3 A). Individual responses obtained at all sevoflurane concentrations seemed highly reproducible despite a clear dose‐dependent reduction in pre‐stimulus and late cortical background activity. Similarly to estimates from average responses, a significant sevoflurane concentration‐dependent effect was found for both VEP amplitude (Fig. 5 B; P < 0.05; compare with Fig. 3 B) and background cortical activity (Fig. 5 C; P < 0.001; compare with Fig. 1 E) when using individual responses (n = 6 experiments; principal effect by one‐way rANOVA). The reliability of individual visual responses was confirmed by a cross‐correlation analysis (Fig. 5 D–F). At all sevoflurane concentrations, single trials showed a similar degree of lag 0 cross‐correlation with the corresponding VEP ensemble averages (n = 6; P > 0.05, Friedman's test), suggesting a similar representativeness of the latter descriptors. Furthermore when the cross‐correlation function (CCF) was computed in the same experiments among all pairs of individual VEPs, the distributions of the lag of the first CCF positive peak were found to be very similar at all sevoflurane concentrations (Fig. 5 F), without any significant difference in their variability (Fig. 5 E; n = 6; P > 0.05, Friedman's test). These results indicate that the potentiation of VEP responses by sevoflurane does not arise from an increased trial‐to‐trial synchronicity but represents a real modification of VEP amplitude.
Figure 5. Analysis of the effects of sevoflurane on single VEPs.
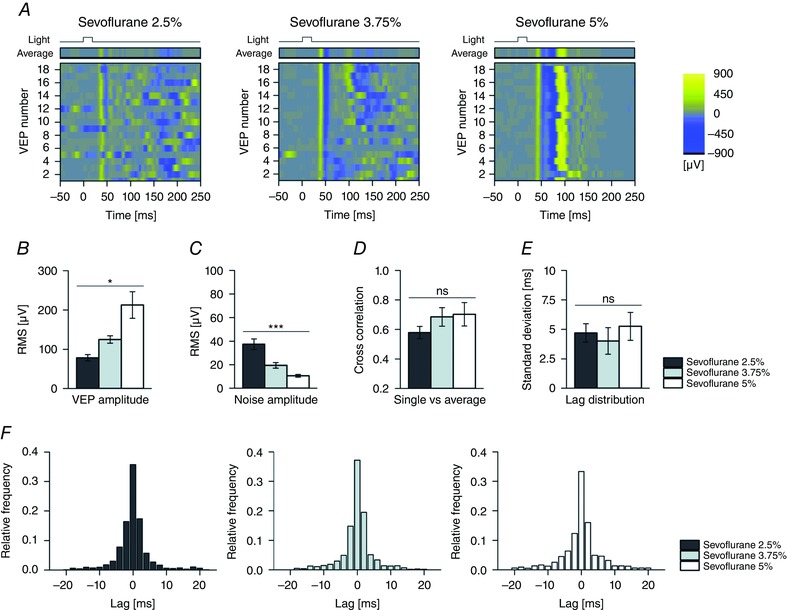
A, raster plots of individual and sequential visual responses and their ensemble averages (top) at three different sevoflurane concentrations (same experiment presented in Fig. 3 A; pulse duration, 20 ms; pulse irradiance, 25 μW cm−2; pulse rate, 0.1 Hz; sevoflurane, 2.5, 3.75 and 5%). Notice how in different trials, the VEP onset is highly reproducible while pre‐stimulus and late activity show large inter‐trial variability; B and C, bar graph of RMS amplitude estimated from individual VEPs and their pre‐stimulus spontaneous activity or cortical noise (B; first 150 ms from stimulus onset; C; first 50 ms before stimulus onset; n = 6 rats exposed to all dosages of sevoflurane). The RMS estimates from single VEPs agree with average waveform analysis (Fig. 3 B). The amplitude of cortical noise decreased with a concentration trend similar to that found with PSD (Fig. 1 E). D, bar graph of cross‐correlation at lag 0 between single VEP traces and their ensemble average (n = 6 rats). This result indicates that in our conditions the representativeness of the ensemble average (n = 19 individual VEPs) is not significantly affected by the dosage of sevoflurane. E and F, lag of the first positive cross‐correlation peak between all pairs of individual VEPs (E, standard deviation; F, lag distributions; n = 6 rats, each exposed to all dosages of sevoflurane). This analysis indicates that the phase jittering of individual visual evoked responses displays a high temporal reliability at all sevoflurane concentrations. Asterisks are significance values (B and C, principal effect by one‐way rANOVA; D and E, principal effect by Friedman's test; P > 0.05, ns; * P < 0.05; ** P < 0.01; *** P < 0.001).
Effects of sevoflurane and propofol on light‐intensity response curves
As for other types of sensory signalling, the V1 response to a light pulse is sensitive to its strength across a wide range of stimulus intensities (Stevens, 1970). The cortical visual response grew as a power function of irradiance (pulse irradiance 1–330 μW cm−2; pulse duration 20 ms; pulse rate 0.1 Hz). This was found to be true for both sevoflurane and propofol Fig. 6 B; at the lowest dosages the power exponent was ∼0.1). Interestingly, when the dose of the two anaesthetics was varied, a clear discrepancy between these two agents emerged (Fig. 6 C–E). With increasing concentration of sevoflurane, the entire light‐intensity response curve was found to be shifted upward in log–log coordinates (Fig. 6 C; n = 6 rats; P < 0.01; principal effect of sevoflurane concentration, two‐way rANOVA; P sevoflurane (2.5 vs. 3.75%) < 0.001, P sevoflurane (2.5 vs. 5%) < 0.001, P sevoflurane (3.75 vs. 5%) < 0.01; multiple comparisons, paired‐samples t test) and an interaction between the concentration of sevoflurane and the irradiance could be excluded (P > 0.05, two‐way rANOVA). These results contrasted with the effects of propofol. On the one hand, increasing propofol concentrations produced a dose‐dependent decrement of the VEP amplitude with significantly different light‐intensity response curves (Fig. 6 D; n = 6 rats; P < 0.001; principal effect of propofol concentration, two‐way rANOVA; P propofol (1 vs. 1.5 mg kg–1 min–1) < 0.05, P propofol (1 vs. 2 mg kg–1 min–1) < 0.001, P propofol (1.5 vs. 2 mg kg–1 min–1) < 0.001; multiple comparisons, paired‐samples t test). On the other hand at high stimulus intensities, these curves converged to a comparable response amplitude, causing a significant interaction between light‐intensity and propofol dosage (P < 0.01, two‐way rANOVA). This suggests some form of relief of propofol‐induced cortical inhibition at high stimulation intensities, which was not seen with sevoflurane.
Figure 6. Light‐intensity response curves reveal an interaction between light signalling and propofol but not sevoflurane.
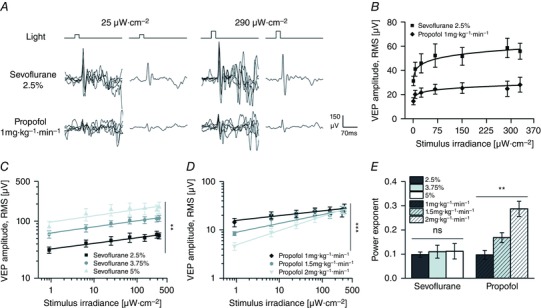
A, single VEPs and ensemble averages from a representative animal exposed to light pulses of different intensities (same rat of Fig. 3 A; irradiance values: 25 μW cm−2, left; 290 μW cm−2, right; duration, 20 ms; rate, 0.1 Hz) in the presence of sevoflurane (2.5%; top) and propofol (1 mg kg−1 min−1; bottom). B, superimposition of light‐intensity response curves obtained in the presence of sevoflurane (2.5%) and propofol (1 mg kg−1 min−1) (n = 6 rats exposed to both anaesthetics). The continuous lines are power fitting of these curves (sevoflurane: adjusted R² = 0.97, power exponent = 0.098, P < 0.001; propofol: adjusted R² = 0.93, power exponent = 0.099, P < 0.001; F test). Notice the curve scaling up with sevoflurane with similar power exponent. C and D, light‐intensity response curves in log–log plots obtained with sevoflurane (C) and propofol (D) at different anaesthetic dosages. The continuous lines are power fitting of these curves (sevoflurane 2.5%: adjusted R 2 = 0.97, P < 0.001; sevoflurane 3.75%: adjusted R 2 = 0.97, P < 0.001; sevoflurane 5%: adjusted R 2 = 0.72, P < 0.01; propofol 1 mg kg−1 min−1: adjusted R 2 = 0.92, P < 0.001; propofol 1.5 mg kg−1 min−1: adjusted R 2 = 0.95, P < 0.001; propofol 2 mg kg−1 min−1: adjusted R 2 = 0.99, P < 0.01; F test). The data used for the lowest dosage of both anaesthetics are the same as used in B. Notice how with increasing concentration of sevoflurane the stimulus–response curve fully translates to higher response values indicating a multiplicative action between the mechanisms of light stimulus processing and the neural effects of sevoflurane. Notice the inhibiting effect of propofol at low light intensities and the graded relief of this inhibition when the light intensity is increased. E, estimates of the power exponents obtained by fittings the light‐intensity response curves from individual experiments (n = 6 rats). A significant change in the exponent is seen with increasing concentration of propofol but not sevoflurane. Asterisks are significance values (panels C and D, principal effect by two‐way rANOVA; E, principal effect by one‐way rANOVA; P > 0.05, ns; * P < 0.05; ** P < 0.01; *** P < 0.001).
Differential effects of sevoflurane and propofol on ON and OFF visual responses
Because of the composite ON and OFF nature of the visual cortical response (Reid & Alonso, 1995; Martinez et al. 2005; Lien & Scanziani, 2013), we evaluated the effects of sevoflurane and propofol on the individual ON and OFF components (VEPOn and VEPOff, respectively). With low concentration of both anaesthetics, the prolongation of the light pulse produced two clear and well separated ON and OFF transients, seen at the beginning and at the end of the stimulus, respectively (Fig. 7 A; sevoflurane, 2.5%; light‐pulse duration, 300–800 ms; pulse irradiance, 150 μW cm−2; pulse rate, 0.1 Hz). When the light‐pulse duration was incremented, the OFF response remained time‐locked to the end of the stimulus, indeed suggesting that it represents a bona fide OFF response (Fig. 7 A). Similarly to the ON response, the VEPOff was sensitive to the light intensity, but it was smaller in amplitude than the VEPOn (Fig. 7 B; sevoflurane, 2.5%; light‐pulse irradiance, 6.5–290 μW cm−2; pulse duration, 300 ms; pulse rate, 0.1 Hz). Therefore both VEPOn and VEPOff amplitudes grew as a power function of irradiance with a similar exponent (Fig. 7 B; n = 7 rats; power exponent of VEPOn, 0.24 ± 0.04; power exponent of VEPOff, 0.19 ± 0.04; P > 0.05, paired‐samples t test), but the entire light‐intensity response curve of the VEPOff was scaled toward smaller amplitude values compared with ON responses (Fig. 7 B; n = 7 rats; P < 0.001, paired‐samples t test).
Figure 7. Effects of sevoflurane and propofol on VEPOn and VEPOff .
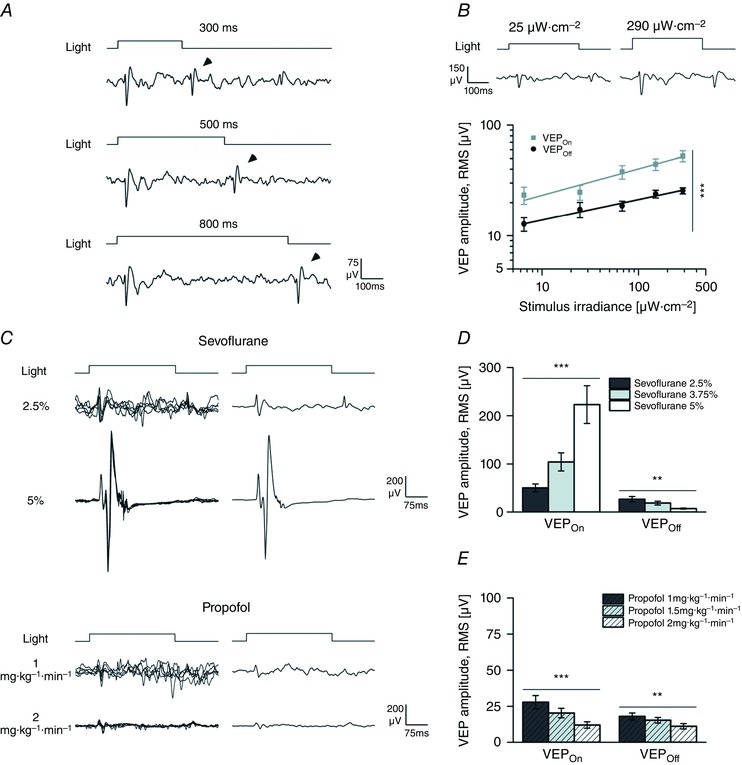
A, ensemble VEPs from a representative animal exposed to randomized light pulses of different duration (pulse duration range 300–800 ms; pulse irradiance 150 μW cm−2; pulse rate 0.1 Hz) recorded in the presence of sevoflurane (2.5%). Notice the presence of clear ON and OFF responses. By increasing stimulus duration the OFF response (arrow tips) was shifted in time. B, stimulus irradiance curves for the ON and OFF responses in log‐log plots (pulse irradiance range, 6.5–290 μW cm−2; pulse duration, 300 ms; pulse rate, 0.1 Hz; n = 7 rats) in the presence of sevoflurane (2.5%). Two exemplar responses (ensemble VEPs) are plotted at the top. In most experiments, with a 300 ms pulse duration the OFF response was smaller than the ON signal. Notice how both ON and OFF amplitudes similarly increased with increasing irradiances. The continuous lines are power fitting of these curves (VEPOn adjusted R 2 = 0.93; P < 0.01; VEPOff adjusted R 2 = 0.96; P < 0.01, F test). C, single traces (left) and ensemble VEP averages (right) obtained in the presence of two different sevoflurane (2.5 and 5%; top) and propofol (1 and 2 mg kg−1 min−1; bottom) concentrations from representative recordings (pulse duration, 300 ms; pulse irradiance, 150 μW cm−2; pulse rate, 0.1 Hz). Notice how high concentration of sevoflurane enhanced the ON and suppressed the OFF responses. Also notice that the effect of propofol was a more generalized reduction of both ON and OFF components. D and E, quantitative analysis of the effects of sevoflurane (D) and propofol (E) on VEPOn and VEPOff waveform amplitude. Asterisks are significance values (B, paired‐samples t test; D and E, principal effect by one‐way rANOVA; P > 0.05, ns; * P < 0.05; ** P < 0.01; *** P < 0.001).
When rats were exposed to prolonged light pulses at all anaesthetic dosages (Fig. 7 C–E; sevoflurane, 2.5, 3.75 and 5%; propofol, 1, 1.5 and 2 mg kg−1 min−1; light‐pulse duration, 300 ms; pulse irradiance, 150 μW cm−2; pulse rate, 0.1 Hz), the simultaneous analysis of the ON and OFF responses revealed that the potentiation of the amplitude of the VEPOn by sevoflurane contrasted with the dose‐dependent reduction of the amplitude of the VEPOff. The dose‐dependent decrement of the OFF response was often so large that in some experiments, at high sevoflurane concentrations, it was difficult to identify consistently an OFF wave (Fig. 7 C). In contrast to the effects on the VEPOn, the reduction of the VEPOff was seen with both anaesthetic compounds (Fig. 7 C–E; n = 6 rats; sevoflurane P VEPOn < 0.001, P VEPOff < 0.01; propofol P VEPOn < 0.001, P VEPOff < 0.01; one‐way rANOVA).
To evaluate if the contrasting effect of sevoflurane on the amplitude of ON and OFF waves related to some form of interaction between these two pathways, we abolished selectively the ON channel response by intravitreal injection of the mGluR6 agonist l‐AP4 (Nakajima et al. 1993). In these conditions, we recorded visual responses before and a few minutes (1 and 10 min) after the intravitreal administration of l‐AP4 (2–8 mm). In all experiments (n = 10 rats), l‐AP4 produced the expected clear and almost complete suppression of the ON response (Fig. 8 A and B; sevoflurane, 2.5%; light‐pulse duration, 300 ms; pulse irradiance, 150 μW cm−2; pulse rate, 0.1 Hz; n = 5 experiments with a precise 10 min waiting time from l‐AP4 injection and the beginning of light stimulation; VEPOn before, 40.79 ± 9.18 μV; VEPOn l‐AP4, 6.27 ± 0.76 μV; P < 0.05, paired‐samples t test). To evaluate if in the presence of l‐AP4, the effect of sevoflurane was conserved, we varied the concentration of this anaesthetic agent immediately after the administration l‐AP4 (Fig. 8 C and D; l‐AP4, 2–8 mm; sevoflurane, 2.5, 3.75 and 5%; light‐pulse duration, 300 ms; pulse irradiance, 150 μW cm−2; pulse rate, 0.1 Hz). Interestingly, when the ON response was absent because of the l‐AP4 administration, sevoflurane induced a paradoxical dose‐dependent potentiation on the VEPOff amplitude, thus reverting the inhibitory action into a potentiation (Fig. 8 C and D; n = 5 rats; P < 0.05; one‐way rANOVA; compare with Fig. 7 D). Strikingly, the dose‐dependent augmentation of the ON response by sevoflurane in the absence of l‐AP4 matched almost perfectly the potentiation of the OFF response seen in the presence of l‐AP4 (Fig. 8 E; P > 0.05, two‐sample t test for each sevoflurane concentration). The visualization of individual VEP traces (Fig. 8 F) highlights that as for ON responses, OFF responses occurred with precise timing, with a consistent potentiation across trials.
Figure 8. l‐AP4 reverts the sevoflurane‐induced inhibition of the VEPOff .
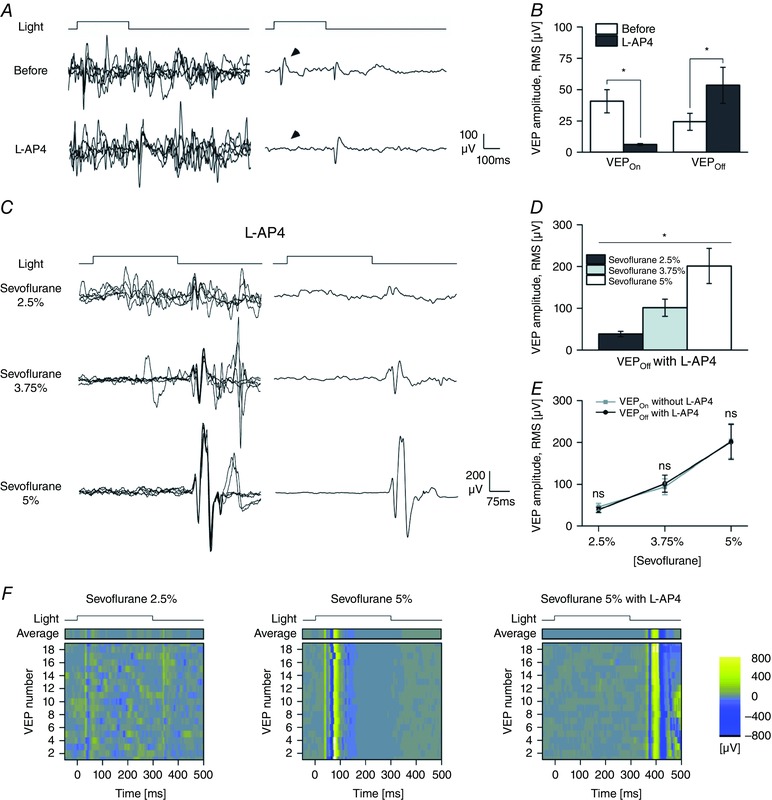
A, traces on the left are single sweeps and traces on the right are the ensemble averages of light responses (averages of n = 59 traces; pulse irradiance 150 μW cm−2; pulse duration 300 ms; pulse rate 0.1 Hz) obtained before (upper traces) and after the vitreal injection of the ON pathway suppressor l‐AP4 (bottom traces; final concentration of l‐AP4 2 mm; sevoflurane 2.5%) in a representative experiment. The arrowheads indicate the ON response. B, bars plot the amplitude of the VEPOn and VEPOff before and after the application of l‐AP4 (n = 5; sevoflurane, 2.5%). Notice how the intravitreal injection of l‐AP4 fully suppresses the VEPOn waveform while the VEPOff response is spared and potentiated (VEPOff before, 24.44 ± 6.77 μV; VEPOff l‐AP4, 53.44 ± 14.39 μV; P > 0.05, paired‐samples t test). C, single light responses (left) and ensemble VEP averages (right; averages of n = 24 traces) recorded in a representative animal exposed to the three different sevoflurane concentrations (2.5, 3.75 and 5%; light‐pulse duration, 300 ms; pulse irradiance, 150 μW cm−2; pulse rate, 0.1 Hz) in the presence of intravitreal l‐AP4 (2 mm). A clear dose‐dependent enhancement of the VEPOff waveform, reminiscent of the potentiation of the VEPOn response seen in control conditions is manifest. D, bar graph of the amplitude of the OFF response at increasing concentrations of sevoflurane (n = 5 rats). Notice how the increment of the VEPOff amplitude is dose‐dependent. E, the same VEPOff data plotted in D superimposed on the effects of sevoflurane on the VEPOn response seen in the absence of l‐AP4 (same data as plotted in Fig. 7 D) to illustrate the superimposable dose‐dependent behaviour of the potentiation of the VEPOn (control conditions) and VEPOff (in the presence of l‐AP4). F, raster plots of individual and sequential VEPs and their ensemble averages (top) from a control animal (same experiment presented in Fig. 7 C; 2.5 and 5% sevoflurane) and from an animal treated with l‐AP4 (same experiment as presented in C; 5% sevoflurane, 2 mm l‐AP4; pulse duration, 300 ms; pulse irradiance, 150 μW cm−2; pulse rate, 0.1 Hz). Notice how in different trials, the VEPOn and VEPOff responses can be easily recognized and occur with a precise timing. Also notice how the disappearance of the OFF response in 5% sevoflurane and conversely the suppression of the ON and the potentiation of the OFF responses in the presence of l‐AP4 (sevoflurane, 5%: l‐AP4, 2 mm) are consistent findings across trials. Asterisks are significance values (B, paired‐samples t test; E, two‐sample t test; D, principal effect by one‐way rANOVA; P > 0.05, ns; * P < 0.05; ** P < 0.01; *** P < 0.001).
An increase in the frequency of stimulation abolishes the sevoflurane‐induced potentiation and unmasks a depression
Since the ON and OFF pathways are known to converge onto single cortical neurons in layer IV (Reid & Alonso, 1995; Martinez et al. 2005; Lien & Scanziani, 2013), we tested for the induction of a use‐dependent depressive state triggered by the initial ON response. To evaluate the presence of an activity‐dependent change, rats were exposed to trains of brief light stimuli at different frequencies (0.1–1 Hz) in the presence of increasing dosages of sevoflurane (sevoflurane, 2.5, 3.75 and 5%; light‐pulse duration, 20 ms; pulse irradiance, 150 μW cm−2; pulse rate range, 1–0.1 Hz; n = 4 rats). At low frequency of light stimulation (0.1 Hz), no significant change in VEPOn amplitude could be detected along the sequence of stimuli for all sevoflurane concentrations (Fig. 9 A; n = 4 rats; sevoflurane 2.5%: ρ = –0.01, P > 0.05; sevoflurane 5%: ρ = –0.12, P > 0.05; Spearman correlation, t test). Conversely, when rats were stimulated by trains of light pulses at higher frequency (1 Hz), a quick decrement of VEP amplitude could be observed, which was much more evident at the higher anaesthetic dosages (Fig. 9 B; n = 4 rats; sevoflurane 2.5%: ρ = –0.47, P < 0.05; sevoflurane 5%: ρ = –0.62, P < 0.01; Spearman correlation, t test). In the latter condition, the amplitude time course could be well fitted by an exponential decay function, whose steady state value unmasked a depression (Fig. 9 B). The level of this activity‐dependent depression increased as a function of the anaesthetic dosage (Fig. 9 D; n = 4 rats; P < 0.05; one‐way rANOVA). When these steady state values were plotted as a function of stimulation frequency, the slope of the resulting curves in log–log plots decreased in a dose‐dependent fashion (Fig. 9 G; n = 4 rats; P < 0.001, one‐way rANOVA). These results confirm a clear interaction between the concentration of sevoflurane and the frequency of stimulation (Fig. 9 F; P < 0.05, two‐way rANOVA) and indicate that by increasing the frequency of sequential stimulation, a gradual conversion of the dose‐dependent potentiation into a dose‐dependent depression could be elicited.
Figure 9. The dose‐dependent potentiation induced by sevoflurane is gradually converted into a dose‐dependent depression by increasing the frequency of stimulation.
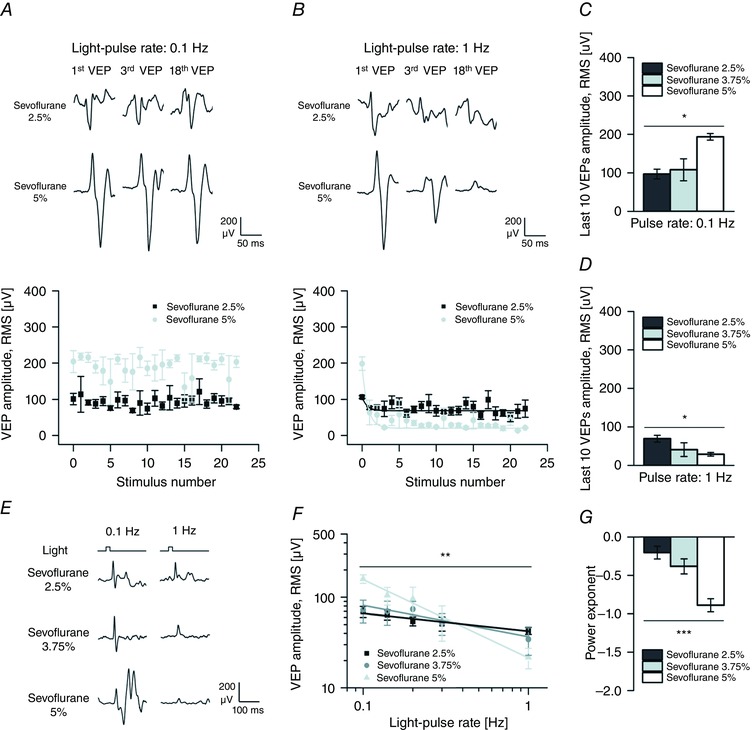
A and B, variation in time of the amplitude of single VEPs from rats (n = 4) exposed to trains of light pulses (light‐pulse duration 20 ms; pulse irradiance 150 μW cm−2) presented at a lower rate and a higher rate (0.1 Hz, A; 1 Hz, B) in the presence of low and high dosages of sevoflurane (2.5 and 5%). Top panels show 3 single responses from a representative rat, while the bottom panels show the mean amplitude of single responses plotted against the stimulation sequence. When stimuli were presented at low rate (0.1 Hz; A), the VEP amplitude in the presence of sevoflurane 2.5% was smaller than the VEP amplitude in the presence of sevoflurane 5% and no significant correlation between the VEP amplitude and the stimulation sequence could be detected. Conversely, when stimuli were presented at a higher rate (1 Hz; B), VEP amplitude exponentially decreased with both dosages of anaesthetic. Continuous lines represent exponential fitting of these relations (sevoflurane 2.5%: y ∞ = 68.62, τ = 0.66, adjusted R 2 = 0.79, P < 0.001; sevoflurane 5%: y ∞ = 20.04, τ = 0.74, adjusted R 2 = 0.48, P < 0.001; F test). With sevoflurane 5% the amplitude decay stabilized at lower values than with sevoflurane 2.5% as indicated by the respective y ∞ values. C, bar graph showing the significant dose‐dependent increase of the mean amplitude of the last 10 responses to the lower rate of stimulation (0.1 Hz; P < 0.05; one‐way rANOVA) during the exposure to increasing concentrations of sevoflurane (2.5, 3.75 and 5%). D, bar graph showing the significant dose‐dependent decrease of the mean amplitude of the last 10 responses to the higher rate of stimulation (1 Hz; P < 0.05; one‐way rANOVA) during the exposure to increasing concentrations of sevoflurane. E, ensemble averages of the last 24 VEPs from a representative animal exposed to light pulses at two different rates (0.1 Hz, left; 1 Hz, right) in the presence of increasing dosages of sevoflurane (2.5, 3.75 and 5%). F, superimposition of light‐pulse rate response curves obtained from rats exposed to all dosages of sevoflurane (2.5, 3.75 and 5%) and to all stimulation frequencies (light‐pulse rate range 0.1–1 Hz; pulse duration 20 ms; pulse irradiance 150 μW cm−2). Continuous lines represent the power‐function fitting of these curves in log–log plots (sevoflurane 2.5%: adjusted R 2 = 0.94, P < 0.01; sevoflurane 3.75%: adjusted R 2 = 0.77, P < 0.05; sevoflurane 5%: adjusted R 2 = 0.99, P < 0.001; F test). Notice how the VEP amplitude is significantly decreased as a power function of the light‐pulse rate (principal effect of the frequency of stimulation P < 0.01; two‐way rANOVA) and how the potentiating effect of sevoflurane observed at lower light‐pulse rates is gradually converted into a depression when the frequency of stimulation is increased. G, estimates of the power exponents obtained by fitting the light‐pulse rate response curves from individual experiments (n = 4 rats). A significant decrease in the power exponent is seen by increasing the concentration of sevoflurane. Asterisks are significance values (C, D and G, principal effect by one‐way rANOVA; F, principal effect by two‐way rANOVA; P > 0.05, ns; * P < 0.05; ** P < 0.01; *** P < 0.001).
Discussion
Silencing induced potentiation
Here we report that two general anaesthetic compounds, sevoflurane and propofol, display opposite actions on the sensory visual response recorded on the V1 cortex (Fig. 3). The attenuation by propofol, which agrees with a previous report (Saxena et al. 2013), is clearly consistent with the general reduction in neuronal excitability and firing (see for review Franks, 2008), here transpiring as a decrease in spectral power of spontaneous activity (Fig. 1) and increase in VEP latencies (Fig. 3). To the contrary, the large amplitude enhancement of cortical visual responses by sevoflurane, which is consistent with previous reports on the effects of other volatile anaesthetics (Imas et al. 2005, 2006), is more difficult to understand. One possible explanation is that cortical silencing and the enhancement of visual sensory responses are not independent phenomena. Indeed, in wakefulness, the primary visual cortex processes a large amount of spontaneous circuital activity, which is induced and sustained to a large extent by two‐way thalamo‐cortical interactions (Reinhold et al. 2015). These cortical dynamics are likely to induce various forms of activity‐dependent short‐term plasticity whose balance contributes to the input–output properties of the cortex (Markram et al. 1997; Zucker & Regehr, 2002). This might result in a persistent activity‐dependent synaptic depression in the awake state (Castro‐Alamancos & Oldford, 2002; Swadlow et al. 2002; Reinhold et al. 2015), more evident at those terminals which are prone to depress as the thalamo‐cortical synapses (Gil et al. 1997).
The expectation is that agents that reduce the thalamo‐cortical firing rate, such as anaesthetics, would counteract this synaptic depression. Indeed, our results show a clear inverse correlation between the dose‐dependent reduction in cortical spontaneous activity by sevoflurane (Figs 1, 2, 3) and the dose‐dependent potentiation of visual evoked responses (Fig. 3). Another prediction is that a cortex silenced by anaesthetics, when challenged repeatedly with externally controlled stimuli, should re‐enter the depressed state. Our results also confirmed this second prediction. After silencing of cortical activity, a series of visual stimuli presented at frequencies above 0.1 Hz were found to produce a decremental cortical response (Fig. 9). Interestingly, the increase in activation frequency not only abolished the dose‐dependent facilitation of the visual responses but revealed an underlying dose‐dependent depression by sevoflurane (Fig. 9). The reduced ability of brain tissue anaesthetized by sevoflurane to follow a series of close visual stimuli agrees with the recent findings by Reinhold et al. (2015), where another volatile anaesthetic, isoflurane, was found to disrupt high‐frequency thalamo‐cortical transmission.
Our interpretation is reinforced by the here described negative interaction between ON and OFF responses (Figs 7 and 8), whose elicitations were separated by a short interval (300 ms). Indeed, ON and OFF information are known to converge onto subfields of individual simple cells in the primary visual cortex (Reid & Alonso, 1995; Martinez et al. 2005; Lien & Scanziani, 2013). When the dosage of sevoflurane was increased, the clear enhancement of the ON response was accompanied by a progressive reduction in the OFF response (Fig. 7). As depicted in Fig. 8, the abolition of the ON response by l‐AP4 was found to convert the depression of the OFF component into a sevoflurane dose‐dependent potentiation, with a trend analogous to the potentiation of the ON response seen in control conditions. This result suggests that both ON and OFF modalities are shaped by the same use‐dependent synaptic phenomena.
A mechanistic synaptic hypothesis for the sevoflurane‐induced potentiation of visual responses
Short‐term use‐dependent changes in synaptic efficacy are in most cases expressed at the level of the presynaptic compartment (Zucker & Regehr, 2002). Central synapses, which are characterized by a low release probability p, display a small number of readily releasable synaptic vesicles N (Abenavoli et al. 2002; Lamanna et al. 2015), a figure which is the final outcome of a series of docking, priming and super‐priming steps (Lee et al. 2013; Jackman et al. 2016). These features, on the one hand render their output very unreliable, especially in vivo (Borst, 2010), and on the other hand allow large degrees of modulation. If sevoflurane were to scale either p or N, this change would translate into a large enhancement of visual responses, as the one seen here. The increment in p is unlikely, because of the many in vitro studies showing that release probability is reduced by volatile anaesthetics (Westphalen & Hemmings, 2003; Wu et al. 2004; Hemmings et al. 2005; Xie et al. 2013; Baumgart et al. 2015). For example, Baumgart et al. (2015) have recently reported that in hippocampal cultures, the volatile anaesthetic isoflurane produces a clear decrement of synaptic vesicle fusion probability. An alternative and more likely hypothesis relates to an increase in N by sevoflurane. This vesicular pool is not stable but undergoes continuous activity‐dependent changes (Zucker & Regehr, 2002). The partial depression of cortical synaptic circuits found in the awake state (Castro‐Alamancos & Oldford, 2002; Swadlow et al. 2002; Reinhold et al. 2015) might then simply represent the use‐dependent depletion of the vesicular pool N by exuberant spontaneous activity. Conversely, neural inactivity by sevoflurane would counteract the awake low‐N state generating in some conditions a higher Np binomial product, resulting in a larger visual evoked response.
One important caveat of this hypothesis relates to the fact that the compound propofol, found to silence cortical circuits to a similar extent (Figs 1, 2, 3), does not potentiate visual evoked responses (Figs 3 and 6). One possible interpretation is that propofol might antagonize the inactivity‐dependent replenishment of the vesicular pool N. In support of this interpretation, propofol has been recently found to increase spontaneous vesicle exocytosis at the neuromuscular junction (Leite et al. 2011). An increase in spontaneous vesicle exocytosis would clearly compete with the increment of the vesicular pool N induced by inactivity postulated here.
The linear scaling of stimulus–response curve by sevoflurane
The visual response requires the activation of several axonal fibres, neurons and synapses whose activity in the end generates the ensemble field potentials seen at the cortical surface. The amplitude of these evoked responses essentially relates to (i) the number and average firing of thalamo‐cortical axons; (ii) the number and average strength of thalamo‐cortical synapses; (iii) the balance between excitatory and inhibitory cortical circuits (Murphy & Miller, 2003) and the contribution of feedback mechanisms such as the cortico‐thalamic one (Lien & Scanziani, 2013); and (iv) the synchronicity of the responding neurons as well as the fine spatial distribution of cortical activation (Buzsáki et al. 2012). As for other sensory systems, the natural expectation is that an increase in the intensity of light stimulation should produce an increase in cortical response by an increase in the firing rate of the sensory fibres (Maguire & Baizer, 1982; Derrington & Lennie, 1984). Across a large range of stimulation intensities the standard psychophysics power function should then be detected (Stevens, 1970). In our experiments, the cortical response grew as a power function of light intensity (Fig. 6). The response curve matched closely a straight line in log–log coordinates with a slope close to 0.1, in conditions where the stimulus intensity values covered a range of ∼2.5 log units. When the concentration of sevoflurane was increased, this log–log relation was shifted to higher response values across the entire range of intensities, while its slope was left unchanged. This pure linear effect contrasts with the action of propofol which seemed more complex, including linear and non‐linear changes in the response curve (Fig. 6).
Is our mechanistic interpretation for the effect of sevoflurane compatible with this linear scaling of light‐intensity response curves? At least in theory, some factors among those listed above (i–iv) could produce a linear change of the stimulus–response curve, for example the balance between excitation and inhibition (Murphy & Miller, 2003; Ayaz & Chance, 2009; Atallah et al. 2012), feedback circuits (Olsen et al. 2012) and modulation by background synaptic noise (Chance et al. 2002; Murphy & Miller, 2003; Ayaz & Chance, 2009). Despite this, it is difficult to reconcile the above mechanisms with the frequency‐dependent abolition of the sevoflurane‐induced potentiation (Fig. 9), which suggests, also in light of its temporal dynamics, an activity‐dependent change in synaptic strength (Zucker & Regehr, 2002). The advantage of our mechanistic hypothesis is that it would explain both the linear scaling of the stimulus–response curve and the use‐dependency of potentiation. Indeed an increase in the number of releasable vesicles N by the sevoflurane‐induced circuit silencing would render the activated synapses more reliable, with an increased average output. Since this change would be independent from the intensity of sensory activation, a linear scaling of stimulus–response curves would be expected. In addition, a series of closely repeated stimuli would deplete again the vesicular pool N, restoring the depressed synaptic state (Castro‐Alamancos & Oldford, 2002; Swadlow et al. 2002; Borst, 2010; Reinhold et al. 2015). It is conceivable that with a higher synaptic reliability, postsynaptic targets would become more prone to respond synchronously, an idea which is consistent with earlier suggestions about the effects of some other anaesthetic compounds (Greenberg et al. 2008; Hudetz et al. 2009).
Conclusions
A formal testing of the here proposed model would require an independent quantification of the two N and p quantal parameters at in vivo brain synapses. In this respect, the analysis of synaptic vesicle dynamics would provide the most definitive answer. This information would be valuable not only to understand the effects of these drugs, but more importantly to make realistic models for subtractive and divisive gain modulation of cortical circuits for both tuning and intensity curves (Salinas & Thier, 2000; Murphy & Miller, 2003; Ayaz & Chance, 2009) that should also integrate presynaptic dynamics. Unfortunately no technique is today available to approach quantitatively vesicle dynamics in the living brain. Despite this limitation, some experimentally testable predictions can still be made. For example it would be very valuable to combine VEPs and single intracellular recordings from the V1 cortex during sevoflurane anaesthesia. Besides providing a more detailed analysis of the relationships between evoked and spontaneous activity, this combined approach might represent an indirect testing of our presynaptic depression hypothesis. If the stimulation of the thalamo‐cortical path could be restricted to a single action potential, for example by using modern optogenetic tools (Fenno et al. 2011), it should be possible to evaluate the interactions between anaesthetics and some forms of short‐term plasticity known to be expressed presynaptically (Smith & Augustine, 1988; Zucker & Regehr, 2002; Jackman et al. 2016). Conversely, by using the same optogenetic approach under deep anaesthesia, the generation of variable degrees and dynamics of visual stimulus‐uncorrelated synaptic activity should restore the depressed level seen at low sevoflurane concentration or in the awake state, where a maximal level of depression should be reached. To conclude, our results might be used in the future to strengthen our understanding of the synaptic mechanisms underlying gain modulation in the cortex (Olsen et al. 2012). This would be especially important when it is relevant to address the contribution of background noise or stimulus‐uncorrelated activity (Chance et al. 2002; Montesano et al. 2015) to the modulation of visual and other sensory modalities perception by attention and wakefulness.
Additional information
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author contributions
The experiments were performed at the Scientific Institute San Raffaele, Division of Neuroscience, Neurobiology of Learning Unit, Via Olgettina 58, 20132 Milan, Italy. A.A., J.L. and A.M. were responsible for study design; A.A. performed the experiments; A.A. and J.L. analysed the electrophysiological data; A.A., J.L., A.M., M.G., M.R., G.R., V.Z., A.D.V. and L.B. helped in data acquisition and interpretation of results. A.A., J.L. and A.M. wrote the manuscript draft. All authors were then involved in revising the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This research was supported by the Italian Ministry of Education, Universities and Research, Rome, Italy (PRIN 2012), Cariplo Foundation, Milan, Italy (Ricerca Scientifica 2011) and Regione Lombardia, Milan, Italy.
References
- Abenavoli A, Forti L, Bossi M, Bergamaschi A, Villa A & Malgaroli A (2002). Multimodal quantal release at individual hippocampal synapses: evidence for no lateral inhibition. J Neurosci 22, 6336–6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahi T, Hirota K, Sasaki R, Mitsuaki Y & Roth SH (2006). Intravenous anesthetics are more effective than volatile anesthetics on inhibitory pathways in rat hippocampal CA1. Anesth Analg 102, 772–778. [DOI] [PubMed] [Google Scholar]
- Atallah BV, Bruns W, Carandini M & Scanziani M (2012). Parvalbumin‐expressing interneurons linearly transform cortical responses to visual stimuli. Neuron 73, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayaz A & Chance FS (2009). Gain modulation of neuronal responses by subtractive and divisive mechanisms of inhibition. J Neurophysiol 101, 958–968. [DOI] [PubMed] [Google Scholar]
- Baumgart JP, Zhou Z, Hara M, Cook DC, Hoppa MB, Ryan TA & Hemmings HC (2015). Isoflurane inhibits synaptic vesicle exocytosis through reduced Ca2+ influx , not Ca2+‐exocytosis coupling. Proc Natl Acad Sci USA 112, 11959–11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieda MC & MacIver MB (2004). Major role for tonic GABAA conductances in anesthetic suppression of intrinsic neuronal excitability. J Neurophysiol 92, 1658–1667. [DOI] [PubMed] [Google Scholar]
- Borst JGG (2010). The low synaptic release probability in vivo. Trends Neurosci 33, 259–266. [DOI] [PubMed] [Google Scholar]
- Brown EN, Lydic R & Schiff ND (2010). General anesthesia, sleep, and coma. N Engl J Med 363, 2638–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Anastassiou CA & Koch C (2012). The origin of extracellular fields and currents – EEG, ECoG, LFP and spikes. Nat Rev Neurosci 13, 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro‐Alamancos MA & Oldford E (2002). Cortical sensory suppression during arousal is due to the activity‐dependent depression of thalamocortical synapses. J Physiol 541, 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance FS, Abbott LF & Reyes AD (2002). Gain modulation from background synaptic input. Neuron 35, 773–782. [DOI] [PubMed] [Google Scholar]
- Ching S, Purdon PL, Vijayan S, Kopell NJ & Brown EN (2012). A neurophysiological‐metabolic model for burst suppression. Proc Natl Acad Sci USA 109, 3095–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claeys MA, Gepts E & Camu F (1988). Haemodynamic changes during anaesthesia induced and maintained with propofol. Br J Anaesth 60, 3–9. [DOI] [PubMed] [Google Scholar]
- Creel D, Dustman RE & Beck EC (1974). Intensity of flash illumination and the visually evoked potential of rats, guinea pigs and cats. Vision Res 14, 725–729. [DOI] [PubMed] [Google Scholar]
- Derrington AM & Lennie P (1984). Spatial and temporal contrast sensitivities of neurones in lateral geniculate nucleus of macaque. J Physiol 357, 219–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa SL, Dickinson R, Lieb WR & Franks NP (2000). Contrasting synaptic actions of the inhalational general anesthetics isoflurane and xenon. Anesthesiology 92, 1055–1066. [DOI] [PubMed] [Google Scholar]
- Ebert TJ, Harkin CP & Muzi M (1995). Cardiovascular responses to sevoflurane: a review. Anesth Analg 81, 11S–22S. [DOI] [PubMed] [Google Scholar]
- Fenno L, Yizhar O & Deisseroth K (2011). The development and application of optogenetics. Annu Rev Neurosci 34, 389–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP (2008). General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci 9, 370–386. [DOI] [PubMed] [Google Scholar]
- Gil Z, Connors BW & Amitai Y (1997). Differential regulation of neocortical synapses by neuromodulators and activity. Neuron 19, 679–686. [DOI] [PubMed] [Google Scholar]
- Greenberg DS, Houweling AR & Kerr JND (2008). Population imaging of ongoing neuronal activity in the visual cortex of awake rats. Nat Neurosci 11, 749–751. [DOI] [PubMed] [Google Scholar]
- Haider B, Häusser M & Carandini M (2013). Inhibition dominates sensory responses in the awake cortex. Nature 493, 97–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseneder R, Kratzer S, von Meyer L, Eder M, Kochs E & Rammes G (2009). Isoflurane and sevoflurane dose‐dependently impair hippocampal long‐term potentiation. Eur J Pharmacol 623, 47–51. [DOI] [PubMed] [Google Scholar]
- Hemmings HC, Yan W, Westphalen RI & Ryan TA (2005). The general anesthetic isoflurane depresses synaptic vesicle exocytosis. Mol Pharmacol 67, 1591–1599. [DOI] [PubMed] [Google Scholar]
- Hudetz AG, Vizuete JA & Imas OA (2009). Desflurane selectively suppresses long‐latency cortical neuronal response to flash in the rat. Anesthesiology 111, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imas OA, Ropella KM, Ward BD, Wood JD & Hudetz AG (2005). Volatile anesthetics enhance flash‐induced gamma oscillations in rat visual cortex. Anesthesiology 102, 937–947. [DOI] [PubMed] [Google Scholar]
- Imas OA, Ropella KM, Wood JD & Hudetz AG (2006). Isoflurane disrupts anterio‐posterior phase synchronization of flash‐induced field potentials in the rat. Neurosci Lett 402, 216–221. [DOI] [PubMed] [Google Scholar]
- Ishizeki J, Nishikawa K, Kubo K, Saito S & Goto F (2008). Amnestic concentrations of sevoflurane inhibit synaptic plasticity of hippocampal CA1 neurons through γ‐aminobutyric acid‐mediated mechanisms. Anesthesiology 108, 447–456. [DOI] [PubMed] [Google Scholar]
- Jackman SL, Turecek J, Belinsky JE & Regehr WG (2016). The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature 529, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeger D & Amzica F (2007). Hypersensitivity of the anesthesia‐induced comatose brain. J Neurosci 27, 10597–10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Bhattacharya A & Makhija N (2000). Evoked potential monitoring in anaesthesia and analgesia. Anaesthesia 55, 225–241. [DOI] [PubMed] [Google Scholar]
- Lamanna J, Signorini MG, Cerutti S & Malgaroli A (2015). A pre‐docking source for the power‐law behavior of spontaneous quantal release: application to the analysis of LTP. Front Cell Neurosci 9, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Ho W‐K, Neher E & Lee S‐H (2013). Superpriming of synaptic vesicles after their recruitment to the readily releasable pool. Proc Natl Acad Sci USA 110, 15079–15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite LF, Gomez RS, Fonseca MDC, Gomez MV & Guatimosim C (2011). Effect of intravenous anesthetic propofol on synaptic vesicle exocytosis at the frog neuromuscular junction. Acta Pharmacol Sin 32, 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien AD & Scanziani M (2013). Tuned thalamic excitation is amplified by visual cortical circuits. Nat Neurosci 16, 1315–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver MB (2014). Anesthetic agent‐specific effects on synaptic inhibition. Anesth Analg 119, 558–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire WM & Baizer JS (1982). Luminance coding of briefly presented stimuli in area 17 of the rhesus monkey. J Neurophysiol 47, 128–137. [DOI] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M & Sakmann B (1997). Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275, 213–215. [DOI] [PubMed] [Google Scholar]
- Martinez LM, Wang Q, Reid RC, Pillai C, Alonso J‐M, Sommer FT & Hirsch JA (2005). Receptive field structure varies with layer in the primary visual cortex. Nat Neurosci 8, 372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, Harris RA & Harrison NL (1997). Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature 389, 385–389. [DOI] [PubMed] [Google Scholar]
- Montesano G, Belfiore M, Ripamonti M, Arena A, Lamanna J, Ferro M, Zimarino V, Ambrosi A & Malgaroli A (2015). Effects of the concomitant activation of ON and OFF retinal ganglion cells on the visual thalamus: evidence for an enhanced recruitment of GABAergic cells. Front Neural Circuits 9, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BK & Miller KD (2003). Multiplicative gain changes are induced by excitation or inhibition alone. J Neurosci 23, 10040–10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair G, Kim M, Nagaoka T, Olson DE, Thulé PM, Pardue MT & Duong TQ (2011). Effects of common anesthetics on eye movement and electroretinogram. Doc Ophthalmol 122, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y, Iwakabe H, Akazawa C, Nawa H, Shigemoto R, Mizuno N & Nakanishi S (1993). Molecular characterization of a novel retinal metabotropic glutamate receptor mGluR6 with a high agonist selectivity for L‐2‐amino‐4‐phosphonobutyrate. J Biol Chem 268, 11868–11873. [PubMed] [Google Scholar]
- Nishikawa K & MacIver MB (2000). Excitatory synaptic transmission mediated by NMDA receptors is more sensitive to isoflurane than are non‐NMDA receptor‐mediated responses. Anesthesiology 92, 228–236. [DOI] [PubMed] [Google Scholar]
- Olsen SR, Bortone DS, Adesnik H & Scanziani M (2012). Gain control by layer six in cortical circuits of vision. Nature 483, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Wang G & Hemmings HC (2003). Isoflurane and propofol inhibit voltage‐gated sodium channels in isolated rat neurohypophysial nerve terminals. Mol Pharmacol 64, 373–381. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E, Lesage F, Fink M, Romey G & Lazdunski M (1999). Inhalational anesthetics activate two‐pore‐domain background K+ channels. Nat Neurosci 2, 422–426. [DOI] [PubMed] [Google Scholar]
- Reid CR & Alonso J (1995). Specificity of monosynaptic connections from thalamus to visual cortex. Nature 378, 281–284. [DOI] [PubMed] [Google Scholar]
- Reinhold K, Lien AD & Scanziani M (2015). Distinct recurrent versus afferent dynamics in cortical visual processing. Nat Neurosci 18, 1789–1796. [DOI] [PubMed] [Google Scholar]
- Salinas E & Thier P (2000). Gain modulation: a major computational principle of the central nervous system. Neuron 27, 15–21. [DOI] [PubMed] [Google Scholar]
- Saxena N, Muthukumaraswamy SD, Diukova A, Singh K, Hall J & Wise R (2013). Enhanced stimulus‐induced gamma activity in humans during propofol‐induced sedation. PLoS One 8, e57685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SJ & Augustine GJ (1988). Calcium ions, active zones and synaptic transmitter release. Trends Neurosci 11, 458–464. [DOI] [PubMed] [Google Scholar]
- Solt K, Eger EI & Raines DE (2006). Differential modulation of human N‐methyl‐D‐aspartate receptors by structurally diverse general anesthetics. Anesth Analg 102, 1407–1411. [DOI] [PubMed] [Google Scholar]
- Stevens SS (1970). Neural events and the psychophysical law. Science 170, 1043–1050. [DOI] [PubMed] [Google Scholar]
- Swadlow HA, Gusev AG & Bezdudnaya T (2002). Activation of a cortical column by a thalamocortical impulse. J Neurosci 22, 7766–7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijn PC & Sneyd JR (1998). I.v. anaesthesia and EEG burst suppression in rats: bolus injections and closed‐loop infusions. Br J Anaesth 81, 415–421. [DOI] [PubMed] [Google Scholar]
- Westphalen RI & Hemmings HC (2003). Selective depression by general anesthetics of glutamate versus GABA release from isolated cortical nerve terminals. J Pharmacol Exp Ther 304, 1188–1196. [DOI] [PubMed] [Google Scholar]
- Wu X‐S, Sun J‐Y, Evers AS, Crowder M & Wu L‐G (2004). Isoflurane inhibits transmitter release and the presynaptic action potential. Anesthesiology 100, 663–670. [DOI] [PubMed] [Google Scholar]
- Xie Z, McMillan K, Pike CM, Cahill AL, Herring BE, Wang Q & Fox AP (2013). Interaction of anesthetics with neurotransmitter release machinery proteins. J Neurophysiol 109, 758–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakura T, Sakimura K, Shimoji K & Mishina M (1995). Effects of propofol on various AMPA‐, kainate‐ and NMDA‐selective glutamate receptor channels expressed in Xenopus oocytes. Neurosci Lett 188, 187–190. [DOI] [PubMed] [Google Scholar]
- Zucker RS & Regehr WG (2002). Short‐term synaptic plasticity. Annu Rev Physiol 64, 355–405. [DOI] [PubMed] [Google Scholar]