

Abstract
Key points
The effects of short‐term (ST; 10 days) and long‐term (LT; 30 days) intermittent hypoxia (IH) on blood pressure (BP), breathing and carotid body (CB) chemosensory reflex were examined in adult rats.
ST‐ and LT‐IH treated rats exhibited hypertension, irregular breathing with apnoea and augmented the CB chemosensory reflex, with all these responses becoming normalized during recovery from ST‐ but not from LT‐IH.
The persistent cardiorespiratory responses to LT‐IH were associated with elevated reactive oxygen species (ROS) levels in the CB and adrenal medulla, which were a result of DNA methylation‐dependent suppression of genes encoding anti‐oxidant enzymes (AOEs).
Treating rats with decitabine either during LT‐IH or during recovery from LT‐IH prevented DNA methylation of AOE genes, normalized the expression of AOE genes and ROS levels, reversed the heightened CB chemosensory reflex and hypertension, and also stabilized breathing.
Abstract
Rodents exposed to chronic intermittent hypoxia (IH), simulating blood O2 saturation profiles during obstructive sleep apnoea (OSA), have been shown to exhibit a heightened carotid body (CB) chemosensory reflex and hypertension. CB chemosensory reflex activation also results in unstable breathing with apnoeas. However, the effect of chronic IH on breathing is not known. In the present study, we examined the effects of chronic IH on breathing along with blood pressure (BP) and assessed whether the autonomic responses are normalized after recovery from chronic IH. Studies were performed on adult, male, Sprague–Dawley rats exposed to either short‐term (ST; 10 days) or long‐term (LT, 30 days) IH. Rats exposed to either ST‐ or LT‐IH exhibited hypertension, irregular breathing with apnoeas, an augmented CB chemosensory reflex as indicated by elevated CB neural activity and plasma catecholamine levels, and elevated reactive oxygen species (ROS) levels in the CB and adrenal medulla (AM). All these effects were normalized after recovery from ST‐IH but not from LT‐IH. Analysis of the molecular mechanisms underlying the persistent effects of LT‐IH revealed increased DNA methylation of genes encoding anti‐oxidant enzymes (AOEs). Treatment with decitabine, a DNA methylation inhibitor, either during LT‐IH or during recovery from LT‐IH, prevented DNA methylation, normalized the expression of AOE genes, ROS levels, CB chemosensory reflex and BP, and also stabilized breathing. These results suggest that persistent cardiorespiratory abnormalities caused by LT‐IH are mediated by epigenetic re‐programming of the redox state in the CB chemosensory reflex pathway.
Keywords: adrenal medulla, anti‐oxidant enzymes, blood pressures, carotid body, catecholamine, epigenetic changes
Key points
The effects of short‐term (ST; 10 days) and long‐term (LT; 30 days) intermittent hypoxia (IH) on blood pressure (BP), breathing and carotid body (CB) chemosensory reflex were examined in adult rats.
ST‐ and LT‐IH treated rats exhibited hypertension, irregular breathing with apnoea and augmented the CB chemosensory reflex, with all these responses becoming normalized during recovery from ST‐ but not from LT‐IH.
The persistent cardiorespiratory responses to LT‐IH were associated with elevated reactive oxygen species (ROS) levels in the CB and adrenal medulla, which were a result of DNA methylation‐dependent suppression of genes encoding anti‐oxidant enzymes (AOEs).
Treating rats with decitabine either during LT‐IH or during recovery from LT‐IH prevented DNA methylation of AOE genes, normalized the expression of AOE genes and ROS levels, reversed the heightened CB chemosensory reflex and hypertension, and also stabilized breathing.
Abbreviations
- AM
adrenal medulla
- AOE
anti‐oxidant enzymes
- BP
blood pressure
- BB
breath‐to‐breath
- BBi
breath‐to‐breath interval
- BBi+1
breath‐to‐breath subsequent interval
- Cat
gene for catalase
- CB
carotid body
- CPAP
continuous positive airway pressure
- Dnmt
DNA methyltransferase
- Gpx2
gene for glutathione peroxidase 2
- IH
intermittent hypoxia
- LT‐IH
long term‐intermittent hypoxia
- MDA
malondialdehyde
- NE
norepinephrine
- OSA
obstructive sleep apnoea
- qPCR
quantitative real‐time PCR
- gene for Prdx4
peroxiredoxin 4
- ROS
reactive oxygen species
- RT
reverse transcription
- Sod
gene for superoxide dismutase
- ST‐IH
short term‐intermittent hypoxia
- gene for Txnrd2
thioredoxin reductase 2
Introduction
Obstructive sleep apnoea (OSA) is a prevalent respiratory disease affecting an estimated 10% of the population (Punjabi, 2008; Peppard et al. 2013). OSA is characterized by periodic obstruction of the upper airway during sleep, resulting in a cessation of air flow leading to intermittent hypoxia (IH). Population‐based studies report an increased incidence of hypertension in OSA patients with strong correlation between the severity of apnoea and hypertension (Young et al. 1997; Lavie et al. 2000; Nieto et al. 2000; Peppard et al. 2000). Rodents exposed to IH, simulating the blood O2 saturation profiles during OSA, exhibit hypertension (Fletcher, 1995; Kumar et al. 2006; Peng et al. 2006; Troncoso Brindeiro et al. 2007). Studies in humans and rodents suggest that an exaggerated carotid body (CB) chemosensory reflex contributes to IH‐induced hypertension (Prabhakar et al. 2015). Disrupting the chemosensory reflex, either by ablating the CB (afferent pathway) or by sectioning the sympathetic nerves innervating the adrenal medulla (AM; efferent pathway), prevents IH‐induced hypertension in adult rats (Fletcher et al. 1992; Peng et al. 2014). Emerging evidence suggests that reactive oxygen species (ROS) signalling is a major cellular mechanism contributing to IH‐induced activation of the CB chemosensory reflex (Prabhakar et al. 2015).
In addition to causing hypertension, activation of the CB chemosensory reflex by IH has been proposed to contribute to progression of OSA by inducing unstable breathing with greater incidence of apnoeas (Prabhakar, 2001). Such a possibility was partly supported by a finding that adult rats exposed to IH in the neonatal period exhibit increased incidence of apnoeas, and this effect was attributed to an enhanced CB chemosensory reflex (Nanduri et al. 2012). It remains unknown whether IH also leads to irregular breathing with apnoea in adult rodents. Therefore, one objective of the present study is to determine the effects of IH on breathing.
An unresolved question is whether IH‐induced changes in BP and breathing triggered by the CB chemosensory reflex are reversed during recovery from IH. Understanding post‐IH recovery is important because continuous positive airway pressure (CPAP), which is a major treatment option that improves oxygenation and thereby prevents IH, is not effective with respect to normalizing BP and breathing in a subset of OSA patients (Thomas et al. 2004; Mulgrew et al. 2010; Dudenbostel & Calhoun, 2012; Eckert et al. 2013). It is possible that untreated OSA of long‐term duration may lead to CPAP‐resistant hypertension and residual apnoeas. Consequently, the second objective of the present study was to test the hypothesis that prolonged IH exposure leads to an irreversible CB chemosensory reflex resulting in hypertension, as well as breathing instability that persist during the recovery from long‐term IH. We tested this possibility by examining the recovery of BP, breathing and CB chemosensory reflex responses in adult rats exposed to short‐term (10 days) and long‐term (30 days) IH.
Methods
Experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Chicago. Experiments were performed on adult, male Sprague–Dawley rats weighing between 200 and 300 g. Rats were killed with an overdose of anaesthesia (urethane 3 g/kg; i.p.) at the termination of the experiment.
Exposure to IH
Adult male Sprague–Dawley rats were exposed to IH between 09.00 h and 17.00 h, either for 10 days (ST‐IH) or 30 days (LT‐IH), as described previously (Peng et al. 2003; Peng et al. 2014). Following either ST‐IH or LT‐IH, rats were allowed to recover in room air for either 10 or 30 days, respectively. Control experiments were performed on age‐matched rats exposed to alternating cycles of room air instead of hypoxia. Experiments were performed on freely mobile rats fed ad libitum. All measurements were performed within 1 day after IH exposures and after completion of the recovery period. Decitabine was administered by i.p. injection at a dose of 1 mg kg–1 every other day either during the last 20 days of LT‐IH or during the room air recovery from LT‐IH.
Measurement of BP and breathing
BP was measured in conscious rats between 09.00 h and 11.00 h by tail‐cuff method using a non‐invasive BP system (IITC Life Science Inc., Woodland Hills, CA, USA) as described previously (Peng et al. 2006). Ventilation was monitored by whole‐body plethysmography in unsedated rats (Peng et al. 2011; Nanduri et al. 2012). Baseline ventilation was recorded for 3 h when the rats were breathing room air. All recordings were made at a mean ± SD ambient temperature of 25 ± 1°C.
Measurement of plasma norepinephrine
Arterial blood samples were collected from urethane anaesthetized rats (1.2 g/kg; i.p.) in heparinized vials (heparin, 30 IU ml–1; n = 8). Plasma was separated and norepinephrine (NE) was extracted with cis‐diol‐specific affinity gel, acetylated and quantitated by competitive enzyme‐linked immunoassay kit (Labor‐Diagnostika, Nord Gmbh & Co. KG, Nordhorn, Germany).
Measurement of CB sensory nerve activity
Sensory nerve activity of the CB was recorded in anaesthetized rats as described previously (Peng et al. 2003). Briefly, the carotid sinus nerve was transected where it joins the glossopharyngeal nerve, treated with collagenase, and several nerve bundles were isolated. Action potentials from one of the nerve bundles were recorded using a monopolar platinum–iridium wire electrode with a reference electrode placed in a nearby neck muscle. In general, two or three action potentials of varying size and amplitude were seen in a given nerve bundle. CB activity was identified by the prompt increase in sensory discharge in response to 30 s of asphyxia and prompt decrease in response to 100% O2. Reducing the pressure in the carotid sinus by occluding the common carotid artery for 10 s caused either no change or an increase in sinus nerve activity, but never a decrease, indicating that the sensory nerve activity originated from the CB but not from baroreceptors. For analysis of sensory nerve activity, action potentials of the same height, duration and shape (i.e. single unit) were selected using Spike Histogram software (Labchart 7 Pro; ADInstruments, Sydney, Australia). Arterial blood samples were collected for measurement of O2 and CO2 partial pressures, as well as pH (ABL‐5; Radiometer, Copenhagen, Denmark).
Reverse transcription (RT) and quantitative real‐time PCR (qPCR) assay
Anti‐oxidant enzyme (AOE) gene expression in the CB and AM was analysed by qPCR assay using SYBR GreenER (Invitrogen, Carlsbad, CA, USA) as described previously (Nanduri et al. 2012). Briefly, RNA was extracted from the CB (two CBs from a single rat) and AM (one AM from a single rat) using Trizol (Thermo Scientific, Waltham, MA, USA) and reverse‐transcribed using SuperScript III (Thermo Fisher). Relative mRNA quantification, expressed as fold change (F), was calculated using the formula F = 2−ΔΔCT where ΔC T is the difference between the threshold cycles of the given target cDNA and 18S rRNA, and Δ(ΔC T) is the difference between the ΔC T values under normoxia and IH. PCR specificity was confirmed by omitting the template and by performing a standard melting curve analysis. The nucleotide sequences of primers used for qPCR are given in Table 1.
Table 1.
Sequence of primers
| Gene | Sequence | Gene Bank number |
|---|---|---|
| 18S |
|
NR_046237.1 |
|
||
| Sod‐1 |
|
NM_017050.1 |
|
||
| Sod‐2 |
|
NM_017051.2 |
|
||
| Catalase |
|
NM_012520.2 |
|
||
| Txnrd2 |
|
NM_022584.2 |
|
||
| Prdx4 |
|
NM_053512.2 |
|
||
| Gpx2 |
|
NM_183403.2 |
|
||
| Dnmt1 |
|
NM_053354.3 |
|
||
| Dnmt3a |
|
NM_001003958.1 |
|
||
| Dnmt3b |
|
NM_001003959.1 |
|
DNA methylation assay
Genomic DNA was isolated from the AM and DNA methylation status of AOE genes were analysed using Epitect Methyl II custom PCR array (Qiagen Inc., Valencia, CA, USA) as described previously (Nanduri et al. 2012). Briefly, methylation‐sensitive and insensitive restriction enzymes were used to selectively digest unmethylated or methylated DNA, respectively. The relative amount of DNA remaining after each digestion was quantified by real‐time qPCR using primers that flanked CpG islands near the target promoter region. Gene methylation status expressed as percentage of cytosine methylated represents the fraction of input genomic DNA containing two or more methylated CpG sites in the targeted region of a gene. Methylation status of the Sod2 gene (encoding superoxide dismutase 2) was analysed in the AM by bisulphite sequencing. Genomic DNA was isolated and incubated with 40% sodium bisulphite in 10 mm hydroquinone for 18 h at 55°C, which converted non‐methylated cytosine to uracil. The primers chosen based on the region of interest (–2 to +1 kb from the transcription start site) were used to amplify the bisulphite‐treated DNA segments, which were purified and sequenced. The ratio of signal C/(C + T) was used for quantification of methylation at individual CpG sites.
Measurement of malondialdehyde (MDA) levels, aconitase, Sod2 and DNA methyltransferase (Dnmt) activities
The CB and AM were homogenized in 10 volumes of 20 mm phosphate buffer (pH 7.4) at 4°C and centrifuged at 500 g for 10 min at 4°C. MDA levels were analysed in the supernatant as described previously (Peng et al. 2006) and are reported as nmol MDA (mg protein)–1. Mitochondrial and cytosolic fractions were isolated from the AM by differential centrifugation. Aconitase enzyme activity was measured by monitoring the increase in absorbance at 340 nm associated with the formation of NADPH during the conversion of isocitrate to α‐ketoglutarate. The rate of NADPH production is proportional to aconitase activity and is expressed as nmol isocitrate min–1 (mg protein)–1 (Khan et al. 2011). Sod2 activity in the mitochondrial fractions was measured by SOD assay kit‐WST (Dojondo Molecular Technologies Inc., Rockville, MD, USA). Protein concentrations were determined and the data were normalized to mg protein. Dnmt activity was analysed using a commercially available kit (Active Motif, Carlsbad, CA, USA).
Immunoblot assay
Tissue lysates were fractionated by 6% polyacrylamide‐SDS gel electrophoresis and immunoblot assays were performed with antibodies against Sod2 (dilution 1:3000; Millipore, Billerica, MA, USA), Dnmt1 (dilution 1:2000; Novus Biologicals, Littleton, CO, USA), Dnmt3a and Dnmt3b (dilution 1:2000; Cell Signaling Technology, Beverly, MA, USA), TATA binding protein (dilution 1:2000; Abcam, Cambridge, MA, USA), and tubulin (dilution 1:3000; Sigma‐Aldrich, St Louis, MO, USA).
Statistical analysis
Data are expressed as the mean ± SEM. Statistical analysis was performed by ANOVA and, for the analysis of normalized data, the Wilcoxon–Mann–Whitney test was used. P < 0.05 was considered statistically significant.
Results
Effect of ST‐IH on BP, breathing and CB chemosensory reflex normalize during room air recovery
Adult rats were exposed to alternating cycles of 5% O2 for 15 s and 5 min of room air, 9 episodes h–1 and 8 h day–1 for 10 days (ST‐IH). This IH paradigm was shown to reduce arterial blood O2 saturation from 97% to ∼80% with each episode of hypoxia (Peng et al. 2014), which is similar to the O2 desaturation observed in OSA patients (Young et al. 1993).
Rats exposed to ST‐IH exhibited elevated mean BP and displayed irregular breathing with apnoeas (cessation of breathing for ∼3 breaths duration; Fig. 1 A and B). Irregular breathing was quantified by analysing the breath‐to‐breath (BB) interval (BBi) vs. the subsequent interval (BBi+1) for 500 breaths and presented as Poincarè plots (Fig. 1 C, top). Analysis of the SD of BB intervals showed that SD1 (representing the y‐axis) and SD2 (representing the x‐axis) were significantly greater after ST‐IH compared to pre‐IH controls (Fig. 1 C, bottom). ST‐IH‐exposed rats showed increased incidence of apnoeas compared to pre‐IH controls (Fig. 1 D). Recovery in room air for 10 days completely normalized BP and restored stable breathing (Fig. 1 A–D).
Figure 1. Reversal of BP, breathing, CB sensory nerve activity and plasma NE responses to ST‐IH.
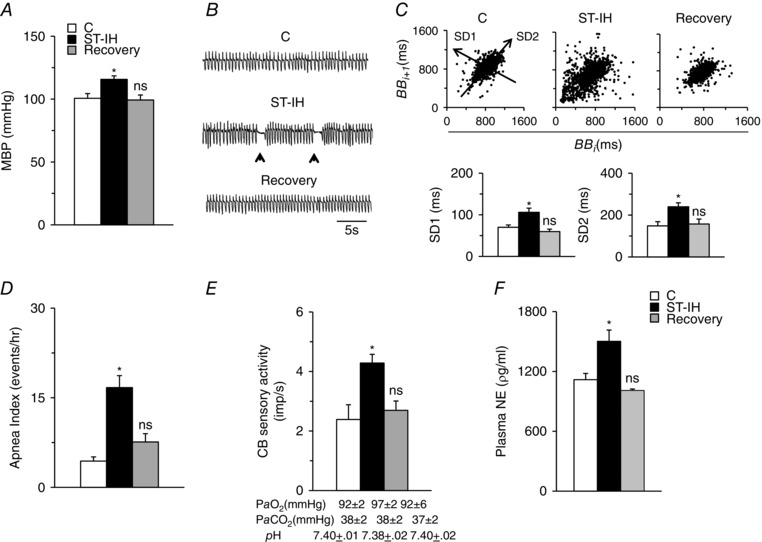
A and B, mean BP (MBP) (A) and breathing (B) in control (C), ST‐IH exposed (ST‐IH) and ST‐IH exposed rats after room air recovery for 10 days (Recovery). Arrowheads in (B) represent the incidence of spontaneous apnoeas. C, top: analysis of irregular breathing by Poincarè plots of BBi (x‐axis) vs. BBi+1 (y‐axis) for 500 breaths analysed in control (C), ST‐IH exposed and ST‐IH rats after room air recovery for 10 days (Recovery). Bottom: quantitation of SD1, representing the SD of data points from the ascending 45º line, and SD2, representing the SD of data points from the line orthogonal to SD1 shown at the top. D–F, number of apnoeas per hour (D), CB sensory nerve activity (impulses per second, imp s–1) (E) and plasma norepinephrine (NE) levels (F) in control (C), ST‐IH exposed and ST‐IH rats after room air recovery for 10 days (Recovery). Arterial , and pH values during room air breathing are shown for each group of rats in (E). Data are the mean ± SEM from eight rats for each treatment. * P < 0.01. ns, not significant compared to normoxic control rats (C).
To assess the effects of ST‐IH on the CB chemosensory reflex, baseline CB sensory nerve activity (afferent limb of the reflex) and plasma NE levels, an index of sympathetic activation (efferent limb of the reflex), were monitored. During room air breathing, arterial blood PO2, PCO2 and pH of ST‐IH‐exposed rats being comparable with control rats (Fig. 1 E). CB sensory nerve activity and plasma NE levels were elevated in rats exposed to ST‐IH and these effects returned to baseline values following room air recovery (Fig. 1 E and F).
Effects of LT‐IH on BP, breathing and CB chemosensory reflex persist during room air recovery
To examine the effects of LT‐IH, rats were exposed to IH for 30 days followed by 30 days of recovery in room air. LT‐IH resulted in elevated BP and irregular breathing with apnoeas (Fig. 2 A–D). CB sensory nerve activity and plasma NE levels were elevated in LT‐IH treated rats despite arterial blood PO2, PCO2 and pH being comparable with control rats (Fig. 2 E and F). The effects of LT‐IH on BP, breathing, CB sensory activity and plasma NE levels persisted during 30 days of recovery in room air (Fig. 2).
Figure 2. LT‐IH induces persistent changes in BP, breathing, CB sensory nerve activity and plasma NE levels.
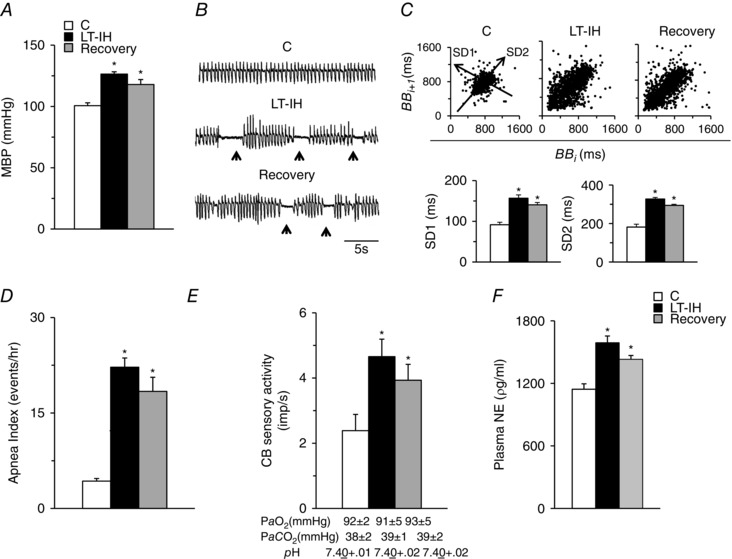
A and B, Mean BP (MBP) (A) and breathing (B) in control (C), LT‐IH exposed and LT‐IH exposed rats after room air recovery for 30 days (Recovery). Arrowheads in (B) represent spontaneous apnoeas. C, top: analysis of irregular breathing by Poincarè plots of BBi (x‐axis) vs. BBi+1 (y‐axis) for 500 breaths analysed in control (C), LT‐IH exposed and LT‐IH rats after recovery in room air for 30 days (Recovery). Bottom: quantitation of SD1, representing the SD of data points from the ascending 45º line, and SD2, representing the SD of data points from the line orthogonal to SD1 shown at the top. D–F, number of apnoeas per hour (D), CB sensory nerve activity (impulses/second; imp s–1) (E) and plasma norepinephrine (NE) levels (F) in control (C), LT‐IH exposed and LT‐IH rats after recovery in room air for 30 days. Arterial , and pH values during room air breathing are shown for each group of rats in (E). Data are the mean ± SEM from eight rats for each treatment. * P < 0.01 compared to normoxic control rats (C).
These results demonstrate that, unlike ST‐IH, LT‐IH leads to irreversible changes in BP and breathing and these effects are associated with persistent activation of the CB chemosensory reflex.
LT‐IH leads to persistent elevation of ROS levels in the CB chemosensory reflex pathway
Previous studies suggest that ROS signalling is central to IH‐induced activation of the CB chemosensory reflex (Prabhakar et al. 2006). We examined the effects of ST‐ and LT‐IH on ROS levels in the CB chemosensory reflex pathway. ROS levels were monitored in the CB (afferent limb of the chemosensory reflex) and AM, a major end‐organ of the sympathetic nervous system (efferent limb of the chemosensory reflex). Two approaches were employed to determine ROS levels: one by monitoring MDA levels, which represent the oxidized lipids (Ramanathan et al. 2005) and the other by measuring the aconitase enzyme activity, a robust in vivo biochemical marker of ROS in the cytosolic and mitochondrial compartments (Gardner et al. 1995). MDA levels were determined in the CB and AM, whereas aconitase activity was determined in in the AM only, because isolating the cytosolic and mitochondrial fractions was not feasible in the CB as a result of the limited availability of the tissue (wet weight of CB is ∼60 μg).
MDA levels were elevated the CB and AM, and aconitase activity decreased in the cytosolic and mitochondrial fractions in the AM of ST‐IH exposed rats. These effects were reversed following room air recovery (Fig. 3 A and B). LT‐IH also increased ROS levels, as measured by MDA levels in the CB and AM and aconitase activity in the AM (Fig. 3 C and D), although ROS levels remained elevated after room air recovery for 30 days (Fig. 3 C and D).
Figure 3. ROS levels in the CB and AM of ST‐IH and LT‐IH exposed rats.
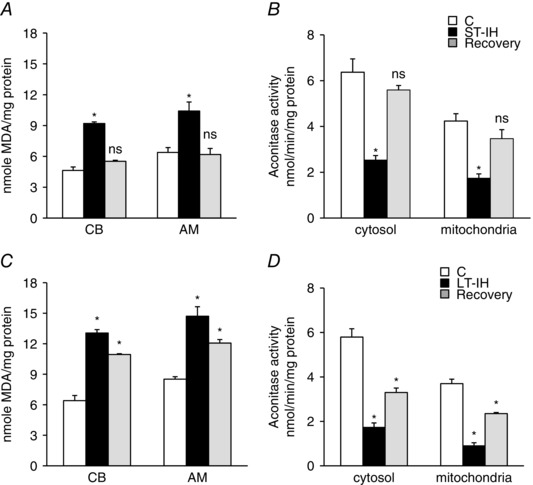
MDA levels and aconitase enzyme activity were monitored as indices of ROS generation. A and B, MDA levels in CB and AM (A) and aconitase activity in mitochondrial and cytosolic fractions of AM (B) from control (C), ST‐IH and ST‐IH exposed rats after recovery in room air for 10 days (Recovery). C and D, MDA levels in CB and AM (C) and aconitase activity in mitochondrial and cytosolic fractions of AM (D) from control (C), LT‐IH and LT‐IH exposed rats after recovery in room air for 30 days (Recovery). Data represent the mean ± SEM from five independent experiments for each treatment (CB and AM tissues were pooled from two rats for each experiment). * P < 0.01. ns, not significant compared to normoxic control rats (C).
LT‐IH leads to persistent down‐regulation of AOE gene expression
The mechanism(s) underlying the persistent elevation of ROS levels in the CB and AM during recovery from LT‐IH were examined. We previously reported that reduced expression of AOEs contributes in part to the IH‐induced increase in ROS levels in the CB and AM (Nanduri et al. 2009). Therefore, we determined AOE gene expression in the CB and AM of ST‐IH and LT‐IH exposed rats after 10 and 30 days of recovery in room air, respectively. The AOE genes analysed were: superoxide dismutase 1 and 2 (Sod1, Sod2), catalase (Cat), thioredoxin reductase 2 (Txnrd2), peroxiredoxin 4 (Prdx4) and glutathione peroxidase 2 (Gpx2). The AOE mRNA levels in both CB and AM of ST‐IH exposed rats after 10 days of recovery were almost the same as controls (Fig. 4 A and B). By contrast, all six AOE genes were significantly reduced in CBs of LT‐IH exposed rats after 30 days of recovery, whereas AM showed reduced expression of Sod1, Sod 2, Txnrd2 and Prdx4 compared to controls (Fig. 4 C and D).
Figure 4. AOE gene expression in ST‐IH and LT‐IH treated rats after recovery in room air.
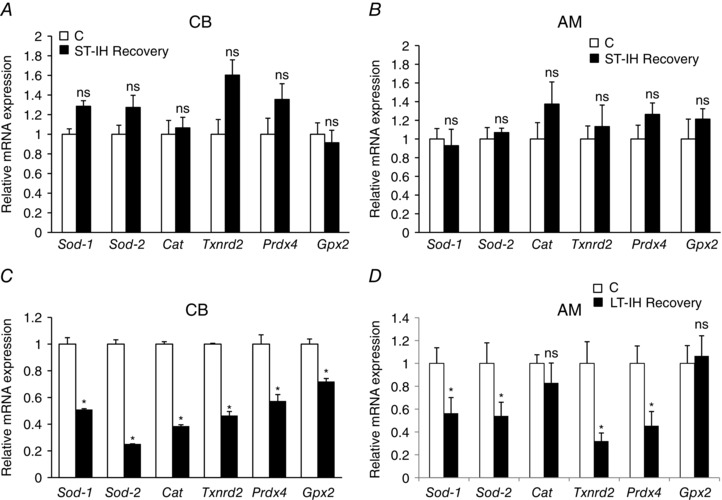
A and B, qPCR analysis of mRNAs encoding superoxide dismutase 1 and 2 (Sod1, Sod2), catalase (Cat), thioredoxin reductase (Txnrd2), peroxiredoxin 4 (Prdx4) and glutathione peroxidase 2 (Gpx2) in the CB (A) and AM (B) of control (C) and ST‐IH exposed rats after recovery in room air for 10 days (ST‐IH Recovery). C and D, qPCR analysis of Sod1, Sod2, Cat, Txnrd2, Prdx4 and Gpx2 mRNAs in the CB (C) and AM (D) of control (C) and LT‐IH exposed rats after recovery in room air for 30 days (LT‐IH Recovery). AOE gene expressions was normalized to 18S rRNA and expressed as the relative change from normoxic controls (C). Data are the mean ± SEM from five independent experiments for each treatment. * P < 0.01. ns, not significant compared to controls (C).
LT‐IH induces increased DNA methylation of AOE genes
Epigenetic regulation by DNA methylation leads to a long‐lasting suppression of gene expression (Miranda & Jones, 2007). We investigated whether increased DNA methylation accounts for persistent down‐regulation of AOE gene expression by LT‐IH. DNA methylation was analysed in AM tissues of ST‐IH and LT‐IH exposed rats after 10 and 30 days of room air recovery, respectively. ST‐IH exposed rats exhibited no changes in DNA methylation of AOE genes after 10 days of recovery (Fig. 5 A). By contrast, LT‐IH rats showed increased DNA methylation of Sod1, Sod2, Txnrd2 and Prdx4 (but not of Cat and Gpx2) after 30 days of room air recovery (Fig. 5 B). These results suggest that LT‐IH but not ST‐IH leads to increased DNA methylation of AOE genes.
Figure 5. LT‐IH induces DNA methylation of AOE genes.
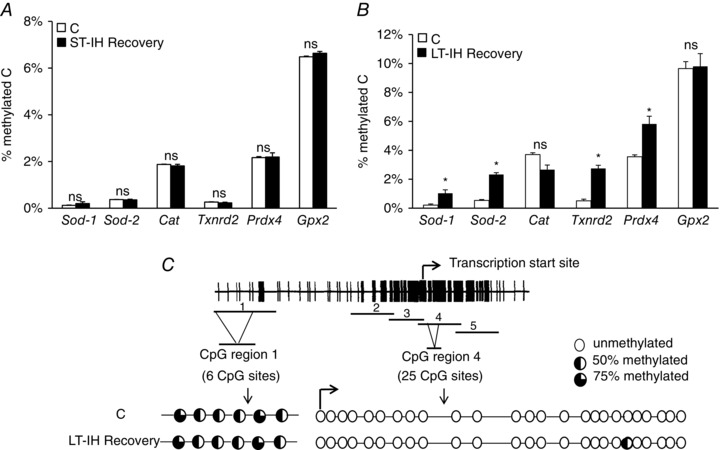
A and B, DNA methylation status of AOE genes in the AM of control rats (C) and ST‐IH rats after recovery in room air for 10 days (ST‐IH Recovery) and LT‐IH rats after recovery in room air for 30 days (LT‐IH Recovery). Data are expressed as percentage of methylated cytosines (methylated C). C, top: schematic presentation of the Sod2 locus showing CpG sites and PCR amplicons (1–5) used for the bisulphite analysis. Bent arrow indicates transcription start site. The percentage of each cytosine methylation was calculated from the formula C/C + T, where C and T represent the peak heights of cytidine and thymidine, respectively. Bottom: methylation status of CpG sites in regions 1 and 4 are shown in the AM of control (C) and LT‐IH rats after recovery in room air for 30 days (LT‐IH Recovery). Data are shown as the mean ± SEM from three independent experiments in each group. * P < 0.01. ns, not significant compared to normoxic control rats (C).
DNA methylation occurs at cytosine residues located immediately 5′ to a guanine residue, which are known as CpG dinucleotides and when found as clusters are referred to as CpG islands (Illingworth & Bird, 2009). We aimed to identify specific CpG dinucleotides that were methylated in response to LT‐IH using the Sod2 as a representative AOE gene. Bisulphite sequencing analysis was performed using primers that spanned from –2 to +1 kb relative to the transcription start site of the Sod2 gene (Fig. 5 C, top). Only a single CpG dinucleotide in the CpG region 4 at +157 bp, out of the 25 CpG sites, was found to be methylated in AM samples of LT‐IH treated rats after 30 days of room air recovery (Fig. 5 C, bottom). Six CpG sites in CpG region 1 were constitutively methylated and LT‐IH had no further effect (Fig. 5 C, bottom).
LT‐IH increases Dnmt activity
DNA methylation is catalysed by Dnmt, which include Dnmt1, Dnmt3a and Dnmt3b (Bird, 2002). To further establish the effects of LT‐IH on DNA methylation, Dnmt enzyme activity, mRNA and protein levels of Dnmt 1, 3a and 3b were determined in the AM samples of rats after 30 days of recovery from LT‐IH. Dnmt enzyme activity (but not Dnmt mRNA levels) was elevated after recovery from LT‐IH (Fig. 6 A and B ). The increased enzyme activity was associated with increased Dnmt1 and Dnmt3b (but not Dnmt3a) protein levels (Fig. 6 C and D).
Figure 6. Dnmt activity and expression of Dnmt mRNA and protein in the AM of LT‐IH rats after recovery in room air.
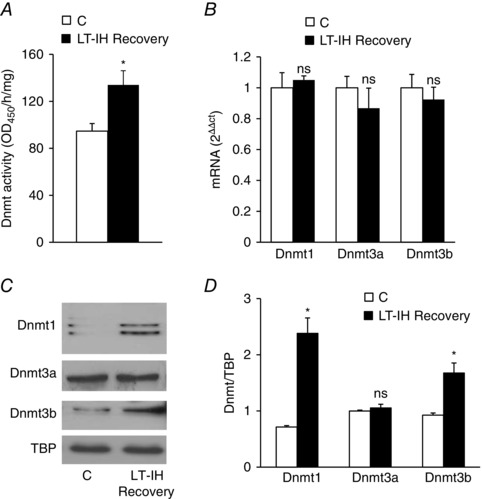
Analysis of Dnmt activity (A) and mRNA levels (B) of Dnmt1, Dnmt3a and Dnmt3b relative to 18S rRNA and normalized to normoxic controls (C) (mean ± SEM from four independent experiments). C, representative immunoblots of Dnmt1, Dnmt3a, Dnmt3b and TATA binding protein (TBP) as a loading control in nuclear extracts prepared from the AM of control (C) and LT‐IH rats after recovery in room air for 30 days (LT‐IH recovery). D, quantitation by densitometric analysis presented as the mean ± SEM from three independent experiments. * P < 0.01. ns, not significant compared to normoxic control rats (C).
Decitabine treatment prevents LT‐IH‐induced DNA methylation
We next investigated whether treating rats with decitabine, an inhibitor of DNA methylation (Yoo & Jones, 2006), either during LT‐IH or during recovery from LT‐IH, prevents DNA methylation of AOE genes. Rats were treated with decitabine (1 mg kg–1) via an i.p. route every alternate day during 10–30 days of IH exposure, as well as during 30 days of recovery from LT‐IH (Fig. 7, top). The choice of decitabine dose and frequency was based on preliminary studies, which showed that doses higher than 1 mg kg–1 or the administration of 1 mg kg decitabine–1 every day caused hair loss and impaired mobility of the rats. Decitabine treatment, either during LT‐IH or during LT‐IH recovery, prevented DNA methylation of AOE genes (Fig. 7, bottom).
Figure 7. Blockade of DNA methylation of AOE genes with decitabine.
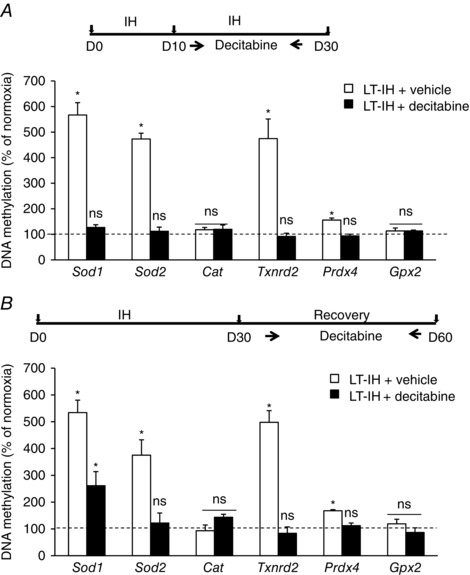
A and B, rats were treated with either vehicle or decitabine during 10–30 days of exposure to LT‐IH (A) or from days 30–60 during the period of recovery from LT‐IH in room air (B). DNA methylations of Sod1, Sod2, Cat, Txnrd2, Prdx4 and Gpx2 (AOE) genes were analysed in the AM and presented as percentage of normoxic control rats treated with decitabine (dotted line). Data are the mean ± SEM from three independent experiments in each group. * P < 0.01. ns, not significant compared to normoxic control rats (C).
Decitabine treatment during LT‐IH recovery restores AOE gene expression, ROS levels and normalizes cardiorespiratory functions
We next examined the effects of decitabine treatment during LT‐IH recovery on AOE gene expression in the CB and AM. Decitabine treatment prevented the down‐regulation of AOE gene expression in the CB and AM, and restored Sod2 protein, enzyme activity and normalized ROS levels in the AM (Fig. 8). Remarkably, decitabine treatment also normalized the CB sensory activity, plasma NE levels, BP and breathing to pre‐IH levels (Fig. 9).
Figure 8. Decitabine treatment during recovery from LT‐IH normalizes AOE gene expression, Sod2 and ROS levels.
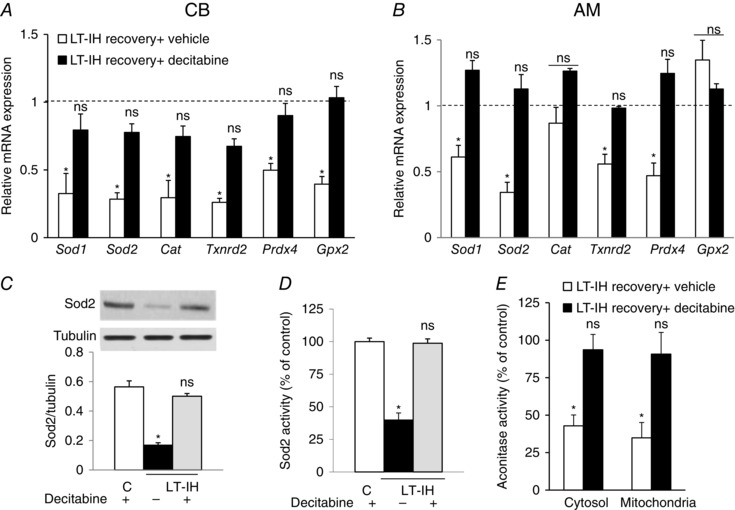
A and B, LT‐IH exposed rats were treated with either vehicle (LT‐IH recovery + vehicle) or decitabine (LT‐IH recovery + decitabine) during 30 days of room air recovery. Sod1, Sod2, Cat, Txnrd2, Prdx4 and Gpx2 mRNA abundances were determined in the CB and AM. The data were normalized with 18S rRNA abundance and expressed as mRNA expression relative to normoxic controls (dotted line). Data are shown as the mean ± SEM from three independent experiments in each group. C–E, Sod2 protein (C, top: representative immunoblot; bottom: densitometric analysis), Sod2 enzyme activity (D) and aconitase activity in cytosol and mitochondrial fractions (E) in the AM of control rats treated with decitabine (C) and LT‐IH exposed rats treated during recovery with either vehicle (LT‐IH recovery + vehicle) or decitabine (LT‐IH recovery + decitabine) in room air for 30 days. Data are shown as the mean ± SEM from three independent experiments in each group. * P < 0.01. ns, not significant.
Figure 9. Decitabine treatment during recovery from LT‐IH normalizes CB sensory nerve activity, plasma NE levels, BP and breathing.
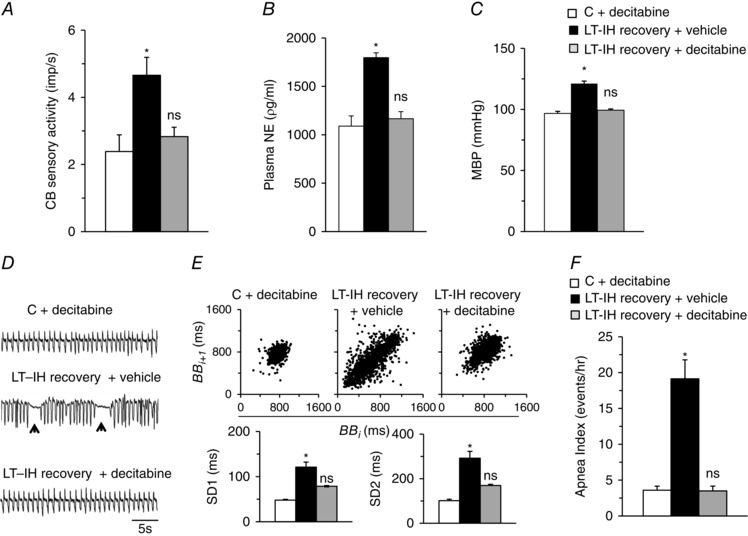
Three groups of rats were studied: (a) control rats treated with decitabine (C+ decitabine), (b) LT‐IH exposed rats treated with either vehicle (LT‐IH recovery + vehicle) or (c) decitabine (LT‐IH recovery + decitabine) during 30 days of recovery in room air. A–C, CB sensory activity (impulses/sec; imp s–1) (A), plasma NE levels (B) and mean BP (MBP) (C). D, representative examples of breathing (arrowheads represent spontaneous apnoeas). E, top: analysis of irregular breathing by Poincarè plots of BBi (x‐axis) vs. BBi+1 (y‐axis) for 500 breaths analysed in rats from each group. Bottom: quantitation of SD1, representing the SD of data points from the ascending 45º line, and SD2, representing the SD of data points from the line orthogonal to SD1 shown at the top. F, number of apnoeas per hour. Data are the mean ± SEM from eight rats in each group. * P < 0.01. ns, not significant.
Discussion
The major findings of the present study are: (i) IH, in addition to causing hypertension, as reported previously (Nieto et al. 2000; Peng et al. 2006; Peng et al. 2014), also leads to irregular breathing with a high incidence of spontaneous apnoeas, and these effects were associated with a heightened CB chemosensory reflex; (ii) The adverse effects of ST‐IH on BP, breathing and CB chemosensory reflex are reversible, whereas those caused by LT‐IH are irreversible upon room air recovery; and (iii) the persistent effects of LT‐IH on BP, breathing and CB chemosensory reflex are partly the result of a long‐lasting suppression of AOE genes by DNA methylation, resulting in persistent elevation of ROS levels in the CB chemosensory reflex pathway (Fig. 10).
Figure 10. Schematic representation of molecular mechanisms underlying the cardiorespiratory responses triggered by CB chemosensory reflex in ST‐ and LT‐IH exposed rats.
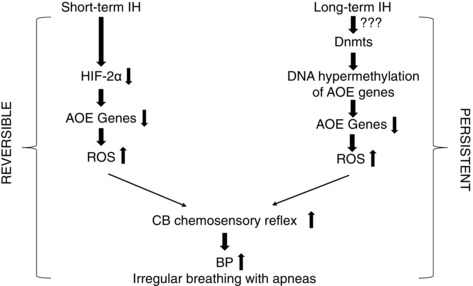
Cardiorespiratory responses to ST‐IH or LT‐IH require ROS‐dependent activation of the CB chemosensory reflex. ST‐IH increases ROS levels partly as a result of insufficient transcriptional activation of AOE genes by hypoxia‐inducible factor‐2 (HIF‐2). Activation of Dnmts and the resulting suppression of AOE genes by DNA hypermethylation contributes to increased ROS generation by LT‐IH. The effects of ST‐IH are reversible, whereas those induced by LT‐IH persist during room air recovery.
Two earlier studies that examined the effects of IH on breathing did not identify any abnormalities in anaesthetized preparations (Peng & Prabhakar, 2004; Rey et al. 2004), suggesting that anaesthesia masked the effects of IH on breathing stability. The increased incidence of apnoeas was associated with elevated CB sensory nerve activity and plasma NE levels, indicating an enhanced CB chemosensory reflex. We attribute the IH‐induced abnormal breathing to an augmented CB chemosensory reflex, which is known to cause breathing instability with apnoea (Cherniack & Longobardo, 2006; Dempsey et al. 2012; Marcus et al. 2014). Apnoea can arise either as a result of a defective respiratory rhythm generation by the central nervous system (central apnoea) or obstruction of the upper airway (obstructive apnoea). Whether the IH‐induced apnoeas are of central origin or of an obstructive phenotype remains to be determined. In the present study, we employed an IH paradigm that produces blood O2 saturation profiles similar to those encountered in sleep apnoea patients. Various paradigms of IH have been employed in other studies (Prabhakar et al. 2015). It remains to be investigated whether paradigms of IH, other than that employed in the present study, also lead to irregular breathing with apnoea. Nonetheless, these results demonstrate that activation of the CB chemosensory reflex by IH, in addition to causing hypertension, also results in irregular breathing with apnoea.
An important finding of the present study concerns the reversibility of cardiorespiratory responses during recovery from IH. Although the cardiorespiratory responses to ST‐IH were completely reversible, those evoked by LT‐IH persisted during the 30 day recovery period. The CB chemosensory reflex, which is a major contributor to autonomic responses to IH (Prabhakar et al. 2015), completely reversed after terminating ST‐IH but persisted after recovery from LT‐IH. These findings suggest that persistent augmentation of the CB chemosensory reflex contributes to the irreversible cardiorespiratory changes caused by LT‐IH.
How might LT‐IH lead to irreversible increase in the CB chemosensory reflex? Previous studies suggest that ROS signalling is central to IH‐induced activation of the CB chemosensory reflex (Peng & Prabhakar, 2003; Prabhakar et al. 2006). IH increases ROS levels in glomus cells, the primary O2 sensing cells of the CB (Makarenko et al. 2016), nucleus tractus solitarious and rostral ventrolateral medulla, the two major brainstem areas associated with processing of sensory information from the CB, and, in AM, a major end organ of the sympathetic nervous system (Kumar et al. 2006; Souvannakitti et al. 2009; Peng et al. 2014). ROS in turn activates the CB and AM chromaffin cells via activating neurotransmitter mechanisms, as well as ion channels (Prabhakar et al. 2015). We previously reported that insufficient transcriptional activation of AOEs by the hypoxia‐inducible factor‐2 contributes in part to IH‐induced increase in ROS levels (Nanduri et al. 2009). Normalization of the CB chemosensory reflex during recovery from ST‐IH was reflected by a return of ROS levels and also AOE gene expression to control levels. By contrast, failure of the recovery of CB chemosensory reflex from LT‐IH was associated with a persistent increase in ROS levels and down‐regulation of AOE gene expression in the CB and AM. These findings indicate that mechanism(s) other than transcriptional factor‐mediated regulation contribute to the persistent suppression of AOE genes and elevated ROS levels by LT‐IH, which, in turn, lead to long‐lasting activation of the CB chemosensory reflex.
What mechanism(s) might contribute to persistent suppression of AOE genes by LT‐IH? Epigenetic changes involving DNA methylation lead to long‐term suppression of gene expression (Bird & Wolffe, 1999; Miranda & Jones, 2007). DNA methylation is a time‐dependent process. Previous studies have shown that de novo DNA methylation does not contribute to the silencing of gene expression during the initial stages of a given stimulus (Gautsch & Wilson, 1983; Pannell et al. 2000); instead, it is initiated during prolonged perturbations leading to long‐lasting suppression of gene expression (Niwa et al. 1983). Consistent with these studies, ST‐IH‐induced down‐regulation of AOE genes was reversible, with no increase in DNA methylation during the room air recovery. On the other hand, there is evidence to demonstrate that DNA methylation contributes to persistent suppression of AOE genes by LT‐IH. First, down‐regulation of AOE genes was associated with increased DNA methylation. Second, using the Sod2 as a representative AOE gene, we identified a single CpG dinucleotide near the transcription initiation site as a target of LT‐IH‐induced DNA methylation. Third, LT‐IH increased Dnmt enzyme activity, which is an essential prerequisite for DNA methylation, and this effect was associated with increased Dnmt1 and Dnmt3b protein levels. Fourth, decitabine treatment either during LT‐IH or during recovery from LT‐IH prevented DNA methylation of AOE genes. Fifth, decitabine treatment during recovery from LT‐IH normalized AOE gene expression and ROS levels, which were associated with normalization of the CB chemosensory reflex and cardiorespiratory functions. Although DNA methylation was assessed only in the AM, similar changes probably also occur in the CB because both these organs exhibit similar responses to IH. However, further studies are needed to determine whether genes other than AOEs are also targets of LT‐IH‐induced DNA methylation and whether LT‐IH induces DNA methylation in tissues other than CB and AM.
How might LT‐IH activate Dnmts and the ensuing DNA methylation? Previous studies indicate that ROS can either directly activate DNA methylation (Wu & Ni, 2015) or indirectly through affecting the protein stability of Dnmts (Lin & Wang, 2014). It may be that ROS generated during LT‐IH trigger de novo methylation by activating Dnmts, resulting in persistent down‐regulation of AOE genes, which in turn leads to irreversible increase in ROS levels (i.e. a positive feedforward mechanism). However, detailed investigations are needed to assess such a possibility, and these are beyond the scope of the present study.
Emerging evidence suggests that DNA methylation mediates adult‐onset diseases as a result of perturbations in the neonatal period (Santos & Dean, 2004; Anway et al. 2005; Ho et al. 2006; Dolinoy et al. 2007; Feinberg, 2007). A previous study showed that rats exposed to IH in the neonatal period, simulating apnoea of prematurity, exhibit DNA hypermethylation of the Sod2 gene, and the resulting persistent oxidative stress contributes to hypertension in adulthood (Nanduri et al. 2012). The present study demonstrates that de novo DNA methylation can also occur in adult life in response to prolonged IH. These findings demonstrate a hitherto uncharacterized role for DNA hypermethylation in evoking the cardiorespiratory abnormalities elicited by long‐term environmental perturbations in adults.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
JN and NRP conceived the study. JN and NRP designed the research. JN, Y‐JP, NW, SAK and GKK performed the experiments. JN, Y‐JP, NW, SAK and GKK analysed the data. JN, GLS, GKK and NRP wrote the paper. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by National Institutes of Health grants P01‐HL‐90554 and UH2‐HL‐123610.
Translational perspective
Although CPAP, the current treatment of choice for OSA, improves oxygenation, it is not effective with respect to normalizing BP and restoring stable breathing in a subset of OSA patients (Mulgrew et al. 2010; Dudenbostel & Calhoun, 2012). The mechanisms underlying the ineffectiveness of CPAP in these patients are not known. Because the paradigm of IH employed in the present study produces blood O2 saturation profiles similar to that seen during OSA, our findings might be of relevance to our understanding of CPAP‐resistant cardiorespiratory abnormalities in OSA patients. We speculate that CPAP resistance may be a consequence of LT‐IH associated with chronically undiagnosed and untreated OSA, leading to persistent CB chemosensory reflex activation through epigenetic programing of the redox state. In this scenario, the administration of DNA hypomethylating drugs, which are currently used for cancer therapy, might correct the altered redox state in the CB chemosensory reflex pathway and thereby correct cardiorespiratory abnormalities in CPAP‐resistant OSA patients.
References
- Anway MD, Cupp AS, Uzumcu M & Skinner MK (2005). Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308, 1466–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A (2002). DNA methylation patterns and epigenetic memory. Genes Dev 16, 6–21. [DOI] [PubMed] [Google Scholar]
- Bird AP & Wolffe AP (1999). Methylation‐induced repression – belts, braces, and chromatin. Cell 99, 451–454. [DOI] [PubMed] [Google Scholar]
- Cherniack NS & Longobardo GS (2006). Mathematical models of periodic breathing and their usefulness in understanding cardiovascular and respiratory disorders. Exp Physiol 91, 295–305. [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Smith CA, Blain GM, Xie A, Gong Y & Teodorescu M (2012). Role of central/peripheral chemoreceptors and their interdependence in the pathophysiology of sleep apnea. Adv Exp Med Biol 758, 343–349. [DOI] [PubMed] [Google Scholar]
- Dolinoy DC, Weidman JR & Jirtle RL (2007). Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod Toxicol 23, 297–307. [DOI] [PubMed] [Google Scholar]
- Dudenbostel T & Calhoun DA (2012). Resistant hypertension, obstructive sleep apnoea and aldosterone. J Hum Hypertens 26, 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert DJ, White DP, Jordan AS, Malhotra A & Wellman A (2013). Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Respir Crit Care Med 188, 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP (2007). Phenotypic plasticity and the epigenetics of human disease. Nature 447, 433–440. [DOI] [PubMed] [Google Scholar]
- Fletcher EC (1995). An animal model of the relationship between systemic hypertension and repetitive episodic hypoxia as seen in sleep apnoea. J Sleep Res 4, 71–77. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Culman J, Miller CC & Unger T (1992). Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension 20, 612–619. [DOI] [PubMed] [Google Scholar]
- Gardner PR, Raineri I, Epstein LB & White CW (1995). Superoxide radical and iron modulate aconitase activity in mammalian cells. J Biol Chem 270, 13399–13405. [DOI] [PubMed] [Google Scholar]
- Gautsch JW & Wilson MC (1983). Delayed de novo methylation in teratocarcinoma suggests additional tissue‐specific mechanisms for controlling gene expression. Nature 301, 32–37. [DOI] [PubMed] [Google Scholar]
- Ho SM, Tang WY, Belmonte de Frausto J & Prins GS (2006). Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res 66, 5624–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth RS & Bird AP (2009). CpG islands – ‘a rough guide’. FEBS Lett 583, 1713–1720. [DOI] [PubMed] [Google Scholar]
- Khan SA, Nanduri J, Yuan G, Kinsman B, Kumar GK, Joseph J, Kalyanaraman B & Prabhakar NR (2011). NADPH oxidase 2 mediates intermittent hypoxia‐induced mitochondrial complex I inhibition: relevance to blood pressure changes in rats. Antioxid Redox Signal 14, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar GK, Rai V, Sharma SD, Ramakrishnan DP, Peng YJ, Souvannakitti D & Prabhakar NR (2006). Chronic intermittent hypoxia induces hypoxia‐evoked catecholamine efflux in adult rat adrenal medulla via oxidative stress. J Physiol 575, 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavie P, Herer P & Hoffstein V (2000). Obstructive sleep apnoea syndrome as a risk factor for hypertension: population study. BMJ 320, 479–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RK & Wang YC (2014). Dysregulated transcriptional and post‐translational control of DNA methyltransferases in cancer. Cell Biosci 4, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarenko VV, Ahmmed GU, Peng YJ, Khan SA, Nanduri J, Kumar GK, Fox AP & Prabhakar NR (2016). CaV3.2 T‐type Ca2+ channels mediate the augmented calcium influx in carotid body glomus cells by chronic intermittent hypoxia. J Neurophysiol 115, 345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus NJ, Del Rio R, Schultz EP, Xia XH & Schultz HD (2014). Carotid body denervation improves autonomic and cardiac function and attenuates disordered breathing in congestive heart failure. J Physiol 592, 391–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda TB & Jones PA (2007). DNA methylation: the nuts and bolts of repression. J Cell Physiol 213, 384–390. [DOI] [PubMed] [Google Scholar]
- Mulgrew AT, Lawati NA, Ayas NT, Fox N, Hamilton P, Cortes L & Ryan CF (2010). Residual sleep apnea on polysomnography after 3 months of CPAP therapy: clinical implications, predictors and patterns. Sleep Med 11, 119–125. [DOI] [PubMed] [Google Scholar]
- Nanduri J, Makarenko V, Reddy VD, Yuan G, Pawar A, Wang N, Khan SA, Zhang X, Kinsman B, Peng YJ, Kumar GK, Fox AP, Godley LA, Semenza GL & Prabhakar NR (2012). Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis. Proc Natl Acad Sci USA 109, 2515–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA & Prabhakar NR (2009). Intermittent hypoxia degrades HIF‐2alpha via calpains resulting in oxidative stress: implications for recurrent apnea‐induced morbidities. Proc Natl Acad Sci USA 106, 1199–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D'Agostino RB, Newman AB, Lebowitz MD & Pickering TG (2000). Association of sleep‐disordered breathing, sleep apnea, and hypertension in a large community‐based study. Sleep Heart Health Study. JAMA 283, 1829–1836. [DOI] [PubMed] [Google Scholar]
- Niwa O, Yokota Y, Ishida H & Sugahara T (1983). Independent mechanisms involved in suppression of the Moloney leukemia virus genome during differentiation of murine teratocarcinoma cells. Cell 32, 1105–1113. [DOI] [PubMed] [Google Scholar]
- Pannell D, Osborne CS, Yao S, Sukonnik T, Pasceri P, Karaiskakis A, Okano M, Li E, Lipshitz HD & Ellis J (2000). Retrovirus vector silencing is de novo methylase independent and marked by a repressive histone code. EMBO J 19, 5884–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Khan SA, Yuan G, Wang N, Kinsman B, Vaddi DR, Kumar GK, Garcia JA, Semenza GL & Prabhakar NR (2011). Hypoxia‐inducible factor 2α (HIF‐2α) heterozygous‐null mice exhibit exaggerated carotid body sensitivity to hypoxia, breathing instability, and hypertension. Proc Natl Acad Sci USA 108, 3065–3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK & Prabhakar NR (2003). Induction of sensory long‐term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci USA 100, 10073–10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ & Prabhakar NR (2003). Reactive oxygen species in the plasticity of respiratory behavior elicited by chronic intermittent hypoxia. J Appl Physiol 94, 2342–2349. [DOI] [PubMed] [Google Scholar]
- Peng YJ & Prabhakar NR (2004). Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol 96, 1236–1242; discussion 1196. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Khan S, Nanduri J, Makarenko VV, Reddy VD, Vasavda C, Kumar GK, Semenza GL & Prabhakar NR (2014). Regulation of hypoxia‐inducible factor‐α isoforms and redox state by carotid body neural activity in rats. J Physiol 592, 3841–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch‐Marce M, Kumar GK, Semenza GL & Prabhakar NR (2006). Heterozygous HIF‐1alpha deficiency impairs carotid body‐mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol 577, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW & Hla KM (2013). Increased prevalence of sleep‐disordered breathing in adults. Am J Epidemiol 177, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppard PE, Young T, Palta M & Skatrud J (2000). Prospective study of the association between sleep‐disordered breathing and hypertension. N Engl J Med 342, 1378–1384. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR (2001). Oxygen sensing during intermittent hypoxia: cellular and molecular mechanisms. J Appl Physiol 90, 1986–1994. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Kumar GK & Nanduri J (2015). Peripheral chemoreception and arterial pressure responses to intermittent hypoxia. Compr Physiol 5, 561–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Yuan G & Kumar GK (2006). Reactive oxygen species facilitate oxygen sensing. Novartis Found Symp 272, 95–99. [PubMed] [Google Scholar]
- Punjabi NM (2008). The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc 5, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan L, Gozal D & Siegel JM (2005). Antioxidant responses to chronic hypoxia in the rat cerebellum and pons. J Neurochem 93, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Alcayaga J & Iturriaga R (2004). Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol 560, 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos F & Dean W (2004). Epigenetic reprogramming during early development in mammals. Reproduction 127, 643–651. [DOI] [PubMed] [Google Scholar]
- Souvannakitti D, Kumar GK, Fox A & Prabhakar NR (2009). Neonatal intermittent hypoxia leads to long‐lasting facilitation of acute hypoxia‐evoked catecholamine secretion from rat chromaffin cells. J Neurophysiol 101, 2837–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RJ, Terzano MG, Parrino L & Weiss JW (2004). Obstructive sleep‐disordered breathing with a dominant cyclic alternating pattern – a recognizable polysomnographic variant with practical clinical implications. Sleep 27, 229–234. [DOI] [PubMed] [Google Scholar]
- Troncoso Brindeiro CM, da Silva AQ, Allahdadi KJ, Youngblood V & Kanagy NL (2007). Reactive oxygen species contribute to sleep apnea‐induced hypertension in rats. Am J Physiol Heart Circ Physiol 293, H2971–H2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q & Ni X (2015). ROS‐mediated DNA methylation pattern alterations in carcinogenesis. Curr Drug Targets 16, 13–19. [DOI] [PubMed] [Google Scholar]
- Yoo CB & Jones PA (2006). Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 5, 37–50. [DOI] [PubMed] [Google Scholar]
- Young T, Palta M, Dempsey J, Skatrud J, Weber S & Badr S (1993). The occurrence of sleep‐disordered breathing among middle‐aged adults. N Engl J Med 328, 1230–1235. [DOI] [PubMed] [Google Scholar]
- Young T, Peppard P, Palta M, Hla KM, Finn L, Morgan B & Skatrud J (1997). Population‐based study of sleep‐disordered breathing as a risk factor for hypertension. Arch Intern Med 157, 1746–1752. [PubMed] [Google Scholar]