

Abstract
Metabolic diseases affect millions of individuals across the world and represent a group of chronic diseases of very high prevalence and relatively low therapeutic success, making them suitable candidates for pathophysiological studies. The sympathetic nervous system (SNS) contributes to the regulation of energy balance and energy expenditure both in physiological and pathological states. For instance, drugs that stimulate sympathetic activity decrease food intake, increase resting metabolic rate and increase the thermogenic response to food, while pharmacological blockade of the SNS has opposite effects. Likewise, dysmetabolic features such as insulin resistance, dyslipidaemia and obesity are characterized by a basal overactivation of the SNS. Recently, a new line of research linking the SNS to metabolic diseases has emerged with the report that the carotid bodies (CBs) are involved in the development of insulin resistance. The CBs are arterial chemoreceptors that classically sense changes in arterial blood O2, CO2 and pH levels and whose activity is known to be increased in rodent models of insulin resistance. We have shown that selective bilateral resection of the nerve of the CB, the carotid sinus nerve (CSN), totally prevents diet‐induced insulin resistance, hyperglycaemia, dyslipidaemia, hypertension and sympathoadrenal overactivity. These results imply that the beneficial effects of CSN resection on insulin action and glucoregulation are modulated by target‐related efferent sympathetic nerves through a reflex that is initiated in the CBs. It also highlights modulation of CB activity as a putative future therapeutic intervention for metabolic diseases.
Abbreviations
- CB
carotid body
- CSN
carotid sinus nerve
- NEFAs
non‐esterified fatty acids
- OSA
obstructive sleep apnoea
- SNS
sympathetic nervous system
Epidemiology of metabolic diseases
In the last decades we have witnessed a dramatic increase in the prevalence of arterial hypertension, insulin resistance, obesity and dyslipidaemia, central features of metabolic diseases such as the metabolic syndrome, type 2 diabetes mellitus and obstructive sleep apnoea (OSA). Recent data estimated the prevalence of metabolic syndrome in nearly 35% of all adults and of 50% in those aged 60 years or older in the United States (Ford, 2005; Aguilar et al. 2015). Also, type 2 diabetes affects over 8.8% of the world population and it is expected that, in 2040, 642 million people will be diabetic worldwide (International Diabetes Federation, 2015). This represents an alarming health problem, with severe economic and social repercussions, therefore it is imperative to elucidate the biological mechanisms underlying it as well as to identify prevention and treatment strategies.
Sympathetic nervous system contribution to metabolic diseases
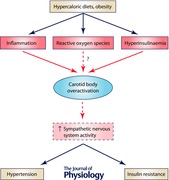
The pathophysiological mechanisms underlying the generation of insulin resistance and insulin resistance‐related illnesses are complex and extend beyond sedentary lifestyle, hypercaloric diets and genetic predisposition. In the last decades, visceral obesity has been considered the common pathophysiological pathway responsible for metabolic changes present in the metabolic syndrome, type 2 diabetes and OSA (Ferranini et al. 2007; Katagiri et al. 2007; Lambert et al. 2010). Indeed, several epidemiological studies have shown that visceral obesity has a strong association with insulin resistance and hypertension (Ferranini et al. 1997; Katagiri et al. 2007), being the mechanism of disease based on the release of non‐esterified fatty acids (NEFAs) and bioactive mediators, known as adipokines, by adipocytes (Katagiri et al. 2007; Yanai et al. 2008). The augmentation in circulating NEFAs leads to the decrease in glucose uptake by the skeletal muscle (Boden & Chen, 1995) and also contributes to a decrease in insulin production (Kashyap et al. 2003). In liver, NEFAs cause an increase in glucose production (Boden et al. 2001), elevated hepatic very low density lipoprotein–triacylglycerol output (Byrne et al. 1991) and decreased insulin clearance by the liver (Wiesenthal et al. 1999). Together these metabolic changes lead to insulin resistance and compensatory hyperinsulinaemia, glucose intolerance and hyperlipidaemia. The adipokines expressed by the adipocytes, which include cytokines, growth factors, leptin, resistin, adiponectin, adipsin and components of the renin–angiotensin system, may act in organs of metabolic relevance such as brain, liver, muscle and the immune system, contributing to the modulation of haemostasis, blood pressure, inflammation, atherosclerosis, glucose and lipid metabolism (Kwon & Pessin, 2013). However, the picture is not as clear as it initially seemed: according to some studies insulin resistance is associated with sleep apnoea and with increased cardiovascular risk, independently of obesity (West et al. 2006). Also, hypertension appears to be independent of the amount of fat mass in OSA patients (Pepper et al. 2000). In fact, obesity has been challenged as a major player in the pathophysiology of disrupted glucose homeostasis by the peripheral nervous system. Increasing evidence points towards the sympathetic nerves as having a pivotal role in the generation of organ‐specific insulin resistance, insulin resistance‐related illnesses and also obesity (see Fig. 1) (Lambert GW et al. 2010; Lambert EA et al. 2015; Thorp & Schlaich, 2015). The sympathetic nervous system (SNS) is key in both circulatory and metabolic control. In high energy expenditure situations, sympathetic activation leads to the release of noradrenaline in the nerve endings and stimulation of adrenergic receptors. The responses are organ specific and depend on the adrenoreceptor isoforms expressed in the tissues (for a review, see Lambert et al. 2010). Acute sympathoexcitation (Fig. 2) leads to activation of hepatic sympathetic nerves which stimulate glycogenolysis in the fed state and gluconeogenesis in fasting conditions. In the pancreas, sympathetic stimulation leads to increased glucagon release into the portal vein and to a moderate inhibition of insulin secretion. Activation of sympathetic fibres that innervate adipose tissue leads to lipolysis and release of NEFAs into the circulation. In response to sympathetic stimulation of the kidney, renin is released and, at higher firing rates, sodium retention and local vasoconstriction also occur. In the adrenal glands, sympathetic stimulation causes release of adrenaline into the bloodstream. These effects, if sustained in the long term, may contribute to the development of insulin resistance since they adversely affect metabolic control. Prolonged deregulation of hepatic glucose output and increased glucagon secretion by the pancreas contribute to increase plasma glucose levels. Increased lipolysis and NEFA release into the circulation affect insulin signalling transduction pathways and contribute to decrease insulin action (Boden, 2011). Enduring sympathetic discharges cause pronounced neural‐mediated vasoconstriction and rarefaction in peripheral arterioles, associated with a marked decrease in blood flow and impaired nutrient uptake in skeletal muscle (Lambadiari et al. 2015) and adipose tissue (Ardilouze et al. 2012).
Figure 1. Insulin resistance.
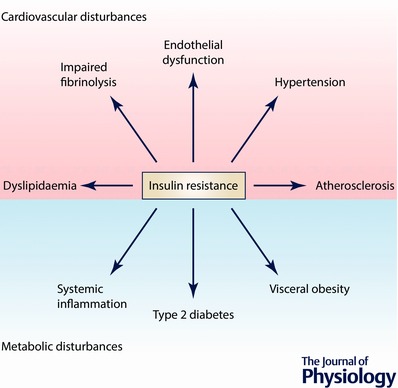
Insulin resistance is a core pathological feature of several metabolic and cardiovascular disturbances, being a principal characteristic of type 2 diabetes and also a risk factor for the development of cardiovascular diseases such as hypertension and atherosclerosis.
Figure 2. Acute activation of the sympathetic nervous system results in the release of noradrenaline (norepinephrine) and the subsequent stimulation of regionally specific adrenergic receptors, causing significant changes in glucose disposal by several organs.
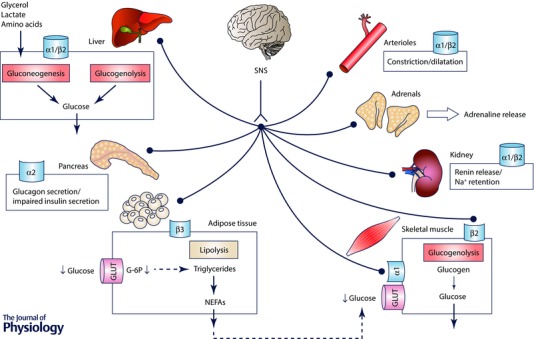
Acute sympathoexcitation leads to increased gluconeogenesis (mediated by α1 adrenergic receptors) and glycogenolysis (mediated by β2 adrenergic receptors) by the liver to provide energetic substrate for the brain. In the pancreas, acute sympathetic activation promotes glucagon release and impairs insulin secretion. In adipose tissue, sympathetic activation triggers β3‐mediated lipolysis and elevates non‐esterified fatty acids (NEFAs) in the circulation. Also, constriction of adipose arterioles causes decreased glucose uptake and decreased triglyceride synthesis. In the skeletal muscle, sympathetic activation triggers glycogenolysis mediated by β2 receptors and glucose uptake is directly related to arteriole tone: in case α1 receptors are activated arteriole constriction decreases glucose uptake mediated by glucose transporters (GLUT). In the kidney, sympathetic activation causes renin release and, at higher neuronal firing rates, sodium (Na+) retention. Sympathetic stimulation of the adrenal glands leads to the release of adrenaline (epinephrine) into circulation mediated by muscarinic receptors. Acute increase in sympathetic nervous activity causes α1 receptor‐mediated vasoconstriction and arteriole rarefaction.
Despite increasing knowledge in this area of research, the precise mechanism and the evolutive pathochrony linking sympathetic overactivation, increased insulin secretion and peripheral insulin resistance is complex. Several theories have been postulated to link features of the metabolic syndrome with changes in sympathetic activation. Landsberg and Reaven's work supports the idea that overeating and obesity lead first to peripheral insulin resistance followed by compensatory hyperinsulinaemia and subsequent sympathetic activation (Landsberg & Young, 1978; Reaven, 2004). Alternatively, other groups have postulated that sympathetic overactivation is the trigger that initiates insulin resistance by compromising glucose disposal and lipid kinetics (Laakso et al. 1990; Jamerson et al. 1993). The latter paradigm postulates that hyperinsulinaemia is a compensatory mechanism for decreased glucose uptake at the skeletal muscle caused by sympathetic overactivation (Julius et al. 1992) and is supported by evidence derived from prospective trials, demonstrating that increased sympathetic activation precedes and predicts obesity and insulin resistance development (Masuo et al. 1997; Flaa et al. 2008).
Data in the literature also agree that insulin resistance states are characterized by sympathetic predominance in a resting/basal state and reduced sympathetic responsiveness after physiological sympathetic stimuli. In fact, sympathetic nervous system responses to carbohydrate ingestion are blunted in insulin‐resistant states (Straznicky et al. 2015), and β‐adrenoreceptor‐mediated lipolysis and lipid oxidation in adipose tissue are severely impaired in obesity (Guo et al. 2014). Decreased responsiveness to the sympathetic nervous system could be caused by polymorphisms in genes that are involved in catecholamine signal transduction and have effects on fat cell lipolysis (Arner, 2001).
Interestingly, sympathetic activation has also been associated with triggering of the hypothalamic–pituitary axis and to increased inflammatory cytokine production (Björntorp, 1995). Cortisol is associated with glucose intolerance and may be one of the pathophysiological mechanisms involved in insulin resistance modulated by sympathetic overdrive, although the presence of hypercortisolism in insulin‐resistant individuals is not ubiquitous. Chronic overactivation of the sympathetic nervous system also induces a proinflammatory state mediated by IL‐6 production by adipose tissue, which results in an acute phase response by the liver, indicating that the increased levels of inflammatory markers seen in insulin‐resistant states may also, at least in part, be mediated by the sympathetic nervous system. Pro‐inflammatory cytokines also cause insulin resistance in adipose tissue, skeletal muscle and liver by inhibiting insulin signal transduction (de Luca & Olefsky, 2008). Noticeably, maintenance of all or part of the aforementioned adaptor responses induced by chronic activation of the sympathetic nervous system culminates in impaired insulin action.
Hyperinsulinaemia contributes to sympathetic overactivation
Among the several factors that have been proposed to be responsible for the increased sympathetic nerve activity in metabolic abnormalities is hyperinsulinaemia (Reaven, 1988; Landsberg, 2005; Lambert GW et al. 2010; Lambert EA et al. 2015). Increased insulin levels contribute to aggravate pathological features of metabolic disturbances by enhancing atherogenesis, increasing blood pressure and endothelial dysfunction, increasing adipose tissue mass and systemic inflammation and contributing to obesity and the development of type 2 diabetes (Reaven, 1988; Arcaro et al. 2002; Landsberg, 2005; Pedersen et al. 2015).
Several lines of evidence suggest that the excitatory effects of insulin on the SNS are mediated by the central nervous system (for a review see Dampney, 2011). In fact, animal studies have shown that the administration of insulin in the arcuate nucleus and paraventricular nucleus produced an increase in spinal sympathetic outflow, mediated by dorsal hypothalamus and rostral ventrolateral medulla (Cassaglia et al. 2011, Dampney, 2011). In agreement with these results, the injection of anti‐insulin affibody at the arcuate nucleus prevents the sympathetic excitation induced by insulin (Luckett et al. 2013). These data suggested that sympatho‐excitation induced by insulin is mediated by the arcuate nucleus, and therefore by the central nervous system. However, in euglycaemic conditions, intracarotid administration of insulin in anaesthetized dogs produced an increase in blood pressure and sympathetic activity higher than systemic insulin administration strongly suggesting the presence of a peripheral insulin sensor that mediates sympathetic activity (Pereda et al. 1962). Additionally, combined with higher sympatho‐excitation observed in insulin‐resistant states, the fact that insulin transport through the blood–brain barrier is decreased (Kaiyala et al. 2000) or unchanged (Israel et al. 1993), both in animal models of diet‐induced obesity and in insulin‐resistant patients (Kern et al. 2006; Heni et al. 2014), corroborates the hypothesis of the existence of an insulin‐sensitive sympatho‐modulator in the periphery.
The carotid body: a sympatho‐modulator in the peripheral nervous system
The CBs are peripheral chemoreceptors that classically sense changes in arterial blood O2, CO2 and pH levels. Hypoxia (O2 deprivation), hypercapnia (CO2 retention) and acidosis (pH drop) activate the CB. The response is an increase in the action potential frequency of the CB sensory nerve, the carotid sinus nerve (CSN). CSN activity is integrated in the brain stem to induce a set of respiratory reflexes aimed, primarily, to normalize the altered blood gases via hyperventilation (Gonzalez et al. 1994) and to regulate blood pressure and cardiac performance via sympathetic nervous system activation (Marshall, 1994).
In the last decades, several reports have linked sympathetic overactivation present in essential hypertension, OSA and chronic heart failure with an increase in CB activity. The first work that demonstrated that CB chemoreceptors are involved in the progression of chronic intermittent hypoxia‐induced hypertension dates from 1992 (Fletcher et al. 1992). In that pioneer animal study, the authors showed that bilateral CB denervation prevented the development of hypertension in rats exposed to chronic intermittent hypoxia for 35 days (Fletcher et al. 1992), which mimics OSA in humans. Moreover, subsequent work from other authors demonstrated that chronic intermittent hypoxia leads to an overactivation of the CB, manifested by its increased hypoxic sensory response (Peng & Prabhakar, 2004; Rey et al. 2004). Furthermore, an enhanced chemoreceptor reflex was described in both hypertensive animals and humans that may contribute to the excess sympathetic activity present in essential hypertension (Przybylski et al. 1982; Trzebski et al. 1982; Fukuda et al. 1987; Somers et al. 1988). More recently, a role for the CB in the pathogenesis of essential hypertension was observed in spontaneously hypertensive rats since animals submitted to bilateral CSN denervation exhibited a delay in the development and maintenance of hypertension, a reduction in sympathetic vasomotor tone and a decreased renal sympathetic activity (Abdala et al. 2012; McBryde et al. 2013). Also, the authors demonstrated that unilateral CSN resection was ineffective in decreasing arterial pressure and that bilateral CSN resection was more effective in reducing arterial pressure than the renal denervation (McBryde et al. 2013). These preclinical results were confirmed in humans, where it was shown that the functional abolishment of CB activity with 100% O2 induced a reduction in both arterial pressure and sympathetic activity in human hypertensive patients (Siński et al. 2012).
In contrast with the data obtained in spontaneously hypertensive rats, recent data from Fudim et al. (2015) showed that the unilateral resection of CB tumours in patients with hypertension was effective in decreasing blood pressure (Fudim et al. 2015). However, over the long term, the effect on pulse pressure and systolic blood pressure was small and without statistical significance (Fudim et al. 2015). The latter results are in agreement with the work of Paton et al. (Paton, 2015) where he showed that CB unilateral ablation decreases short‐term arterial pressure although the effect was attenuated 12 months after ablation suggesting a compensation of the remaining carotid body.
The CB chemoreceptors have also been described to be involved in chronic heart failure in humans and animal models, being associated with the sympathetic activation observed in this syndrome (Sun et al. 1999; Ponikowski et al. 2001). In both rat and rabbit models of chronic heart failure CB ablation, performed by cryogenic destruction, has been shown to reduce the hyperventilation and oscillatory breathing, as well as the tonic sympathetic outflow, resulting in an improvement in cardiac function and prolonged survival (Del Rio et al. 2013; Marcus et al. 2014). These preclinical results are supported by recent data obtained in patients (Niewiński et al. 2013, 2014) showing that unilateral CB removal in a patient with chronic heart failure resulted in a decrease in peripheral chemosensitivity, which was accompanied by improvements in autonomic function, cardiac function, exercise capacity and reduced resting ventilation (Niewiński et al. 2013). Also, in 2014, another study with six chronic heart failure patients showed that bilateral CB removal produced a reduction in ventilatory and blood pressure responses to hypoxia, suggesting a decrease in sympathetic tone (Niewiński et al. 2014). Moreover, CB removal in heart failure patients did not modify the heart chronotropic response, suggesting other peripheral chemoreceptors, may be involved in this response to hypoxia (Niewiński et al. 2014). As a whole, data reflect that CB dysfunction is implicated in the pathophysiology of various human cardiovascular diseases through the modulation of the SNS. Additionally, in the last couple of years, a new line of research has emerged from our laboratory, linking the CB‐mediated sympathetic nerve activation to metabolic diseases (Ribeiro et al. 2013; Conde et al. 2014).
What causes carotid body deregulation in metabolic diseases?
The fact that peripheral insulin administration elicited a higher increase in sympathetic activity than systemic administration (Pereda et al. 1962), together with the evidence that CB overactivation characterizes essential hypertension, OSA, chronic heart failure as well other sympathetically mediated diseases lead us to hypothesize that the CB is a peripheral insulin sensor. According to this new paradigm, CB overstimulation by inadequate insulin levels contributes to the genesis of peripheral insulin resistance and hypertension present in metabolic diseases via SNS activation (Fig. 3).
Figure 3. Schematic representation of the stimuli that activate the carotid body to induce an increase in sympathetic activity that promotes insulin resistance and glucose deregulation and hypertension.
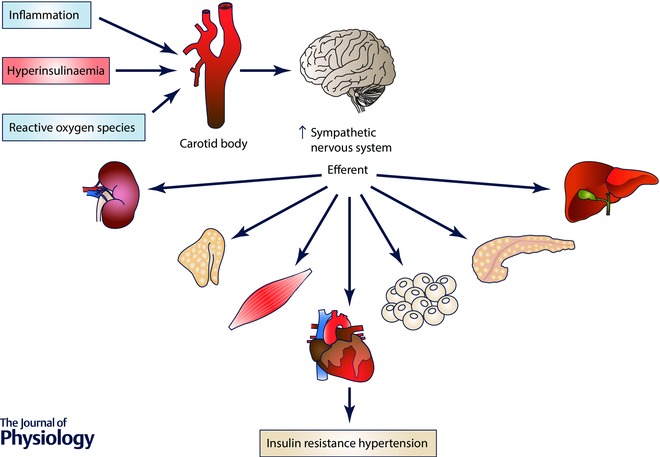
Hyperinsulinaemia, inflammation and reactive oxygen species induce carotid body overactivation leading to an increase in sympathetic nervous system activity that promotes insulin resistance and hypertension.
We have recently described that insulin receptors are present in the whole CB and that they become phosphorylated in response to insulin (Ribeiro et al. 2013). These results are in agreement with the findings of Gallego‐Martin et al. (2014) where it was observed that whole CBs incubated with insulin accumulate more 2‐deoxyglucose than the diaphragm muscle. Also, we have shown that insulin, in physiological concentrations, is capable of eliciting a neurosecretory response in chemoreceptor cells, measured by the increase in intracellular Ca2+ concentrations and by the release of dopamine and ATP from whole CBs (Ribeiro et al. 2013). These results obtained in vitro were confirmed by testing the in vivo effect of insulin on ventilation. In euglycaemic conditions, insulin increased ventilation in anaesthetized animals in a dose‐dependent manner, an effect fully mediated by the CB since it was absent in animals that had their CSN resected (Ribeiro et al. 2013). This effect of insulin on ventilation was not new, as Bin‐Jaliah et al. (2004) observed that insulin infusion in hypoglycaemic conditions increased minute ventilation and the rate of O2 consumption (), an effect that was also totally mediated by the CB since CSN denervation blunted it. Comparing both experimental protocols, and since we have tested the effect of insulin on ventilation in euglycaemic conditions instead of hypoglycaemic conditions as in Bin‐Jaliah et al. (2004), we trust that the effect of insulin on ventilation is mediated by insulin per se and not by low plasma glucose. Therefore, insulin has the ability to activate the CBs and this organ may be the peripheral insulin sensor that mediates sympatho‐excitation.
Apart from insulin, other humoral and local factors have been described as activating the CB, such as leptin, inflammatory cytokines and reactive oxygen species (ROS) (see Fig. 3). It has been suggested that leptin may contribute to peripheral ventilatory control, as the administration of the hormone can reverse hypoxia and hypercapnia in animal models with no functional leptin gene (Tankersley et al. 1998; O'Donnel et al. 1999). The results suggest that the ventilatory effects of leptin are mediated by the CB chemoreceptors and, in fact, the CBs express the leptin‐B receptor (Porzionato et al. 2011). Yet, we have recently shown that the acute stimulatory effect of leptin on ventilation is not CB controlled (Olea et al. 2015).
Other local mediators that are known to activate the CB are ROS (Peng et al. 2009; Del Rio et al. 2010). It has been described that ROS production and regional oxidative stress play a role in the CB chemosensory potentiation and in the progression of hypertension in rats exposed to chronic intermittent hypoxia (Peng et al. 2009; Del Rio et al. 2010); however, we are not aware of any effect of oxidative stress mediators in CB‐dependent glucose metabolism. Additionally, it is well established that the CB senses inflammatory mediators. The expression of receptors for interleukins IL‐1, IL‐6 and IL‐10, as well as for tumour necrosis factor receptor (TNFα), has been shown in the human CB (Mkrtchian et al. 2012). In the cat, Fernandez et al. (2008) demonstrated the co‐localization of TNFα receptors and tyrosine hydroxylase in CB chemoreceptor cells and its functionality. When the authors administered TNFα, this pro‐inflammatory cytokine was incapable of modifying basal CSN chemosensory discharge ex vivo, but reduced the hypoxia‐induced enhanced frequency of chemosensory discharge in a dose‐dependent manner (Fernandez et al. 2008). This inhibitory effect of TNFα observed in the cat is in contrast with the findings of Lam et al. (2008, 2012) in the rat, where the authors showed in dissociated CB chemoreceptor cells that TNFα enhances the [Ca2+]i response to acute hypoxia, this increase being significantly larger in cells from the CB of rats exposed to chronic hypoxia or to chronic intermittent hypoxia. Yet, TNFα is not the only cytokine that acts on the CB. Rat CB chemoreceptor cells showed a strong expression of interleukin‐1 (IL‐1) receptor type I (Wang et al. 2002) and interleukin‐6 (IL‐6) receptor α (Wang et al. 2006). In rat CB chemoreceptor cells IL‐1β significantly decreased the outward potassium current and triggered a transient rise in [Ca2+]i (Shu et al. 2007). Moreover, IL‐1β stimulated CSN discharges. In the same way application of exogenous IL‐6 induced an increase in [Ca2+]i and the release of catecholamines from rat CB chemoreceptor cells (Fan et al. 2009). Knowing that both subclinical inflammation and oxidative stress are correlated with insulin resistance (de Rooij et al. 2009) and both mediators stimulate the CB, it is possible that these molecules also play a role in the modulation of CB‐mediated insulin resistance.
The carotid bodies control whole body glucose homeostasis
Animals submitted to hypercaloric diets exhibit CB overactivation: they present an increase in spontaneous ventilation, an increase in the respiratory responses to ischaemic hypoxia, an increase in hypoxia‐evoked release of dopamine from the CB and an increase in the CB expression of tyrosine hydroxylase (Ribeiro et al. 2013). This chronic overactivation of the carotid bodies is tied to enhanced sympatho‐excitation, acknowledged by increased circulating and adrenal medulla catecholamines, that culminates in the development of insulin resistance through the mechanisms mentioned above (Ribeiro et al. 2013; Conde et al. 2014). Moreover, we have shown that bilateral CSN resection prevents the development of these features (Ribeiro et al. 2013) confirming the CB as a key player in controlling peripheral insulin sensitivity (Fig. 3). Our hypothesis of involvement of the CB in the genesis of metabolic disturbances was also supported by the findings of Shin et al. (2014). They observed that mice exposed for 4‐6 weeks to chronic intermittent hypoxia exhibited increased fasting blood glucose, increased hepatic glucose output and insulin resistance. The authors have shown that CSN denervation prevented the chronic intermittent hypoxia‐induced hyperglycaemia and the increase in baseline glucose hepatic output, an effect that was associated with the abolishment of sympathetic overactivation induced by the CB (Shin et al. 2014). The latter results in chronic intermittent hypoxia animals (Shin et al. 2014), as well as our data in hypercaloric animal models (Ribeiro et al. 2013; Conde et al. 2014), are in accordance with the findings by Limberg et al. (2014) where hyperoxic silencing of carotid chemoreceptors reduced muscle sympathetic nerve activity in hyperinsulinaemic conditions, suggesting that the CB mediates insulin‐dependent sympatho‐excitation in humans. Confirming this role, as well as the involvement of the CB in metabolic disease pathogenesis, we have recently shown that the suppression of CB activity with hyperbaric oxygen therapy (100% O2 at 2.5 absolute atmospheres, 70 min, 20 sessions) ameliorates fasting glycaemia and post‐prandial glucose tolerance in type 2 diabetes patients (Vera‐Cruz et al. 2015). Additionally, other works performed in healthy humans, exposed to hyperoxia, have also shown the involvement of CB in the counterregulatory response to hyperinsulinaemia‐induced hypoglycaemia, since a decrease in the release of counterregulatory hormones was observed (Wehrwein et al. 2010). Also, blood pressure responses to hyperinsulinaemia‐induced hypoglycaemia are reduced in hyperoxic conditions in healthy humans, suggesting that the sympathetic control of blood pressure is attenuated (Wehrwein et al. 2012). Recently, the same authors provided corroborative results that show that the effect of hyperoxia on the hypoglycaemia counterregulatory response is mediated by the CBs (Wehrwein et al. 2015). However, in patients who had had bilateral CB resection due to glomus cell tumours, the counterregulatory response to insulin‐induced hypoglycaemia was not modified, suggesting that physiological adaptations may occur over time and/or that the response to hypoglycaemic conditions in humans do not rely specifically on CB glucose sensing (Wehrwein et al. 2015). The long‐term adaptation that may occur after CB resection may explain why these results contrast with the data obtained by Koyama et al. (2000) in dogs, where 16 days after CB resection a decrease in the counterregulatory response during insulin‐induced hypoglycaemia was observed. Nonetheless, all these findings highlight a role for the CB in metabolic control, not only in pathological, but also in physiological conditions.
In conclusion, we propose that insulin‐triggered CB activation is a key step in the development of the excessive sympatho‐excitation that characterizes metabolic diseases, creating a vicious cycle that originates insulin resistance and hypertension. Therefore, the modulation of CB activity emerges as a possible therapeutic strategy for the treatment of metabolic diseases.
Additional information
Competing interests
None declared.
Funding
J.F.S. and M.J.R. are supported by PhD Grants from the Portuguese Foundation for Science and Technology (FCT), PD/BD/105890/2014 and SFR/BD/88983/2012, respectively.
Biography
Silvia Conde’s research focuses on understanding the physiology of the autonomic nervous system, in particular the carotid body, and in the application of its findings to pathological states allowing the identification of molecular targets for therapy. Silvia received her degree in biochemistry from the Faculty of Sciences, University of Lisbon. She pursued her PhD in 2007 from both NOVA University of Lisbon and from the University of Valladolid. She is Principal Investigator at CEDOC – Chronic Disease Research Center and tenured assistant professor at NOVA Medical School. In 2009 she was awarded a L'Oréal Medal of Honour for Women in Science.

This review was presented at the symposium “Targeting autonomic imbalance in pathophysiology: is the carotid body the new nirvana?”, which took place at the meeting of International Society for Autonomic Neuroscience in Stresa, Italy, 26–29 September 2015.
References
- Abdala AP, McBryde FD, Marina N, Hendy EB, Engelman ZJ, Fudim M, Sobotka PA, Gourine AV & Paton JFR (2012). Hypertension is critically dependent on the carotid body input in the spontaneously hypertensive rat. J Physiol 590, 4269–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar M, Bhuket T, Torres S, Liu B & Wong RJ (2015). Prevalence of the metabolic syndrome in the United States, 2003–2012. JAMA 313, 1973–1974. [DOI] [PubMed] [Google Scholar]
- Arcaro G, Cretti A, Balzano S, Lechi A, Muggeo M, Bonora E & Bonadonna RC (2002). Insulin causes endothelial dysfunction in humans: sites and mechanisms. Circulation 105, 576–582. [DOI] [PubMed] [Google Scholar]
- Ardilouze JL, Sotorník R, Dennis LA, Fielding BA, Frayn KN & Karpe F (2012). Failure to increase postprandial blood flow in subcutaneous adipose tissue is associated with tissue resistance to adrenergic stimulation. Diabetes Metab 38, 27–33. [DOI] [PubMed] [Google Scholar]
- Arner P (2001). Genetic variance and lipolysis regulation: implications for obesity. Ann Med 33, 542–546. [DOI] [PubMed] [Google Scholar]
- Bin‐Jaliah I, Maskell PD & Kumar P (2004). Indirect sensing of insulin‐induced hypoglycaemia by the carotid body in the rat. J Physiol 556, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björntorp P (1995). Neuroendocrine abnormalities in human obesity. Metabolism 44, 38–41. [DOI] [PubMed] [Google Scholar]
- Boden G & Chen X (1995). Effects of fat on glucose uptake and utilization in patients with non‐insulin‐dependent diabetes. J Clin Invest 96, 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden G, Chen X, Capulong E & Mozzoli M (2001). Effects of free fatty acids on gluconeogenesis and autoregulation of glucose production in type 2 diabetes. Diabetes 50, 810–816. [DOI] [PubMed] [Google Scholar]
- Boden G (2011). Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes 18, 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne CD, Brindle NP, Wang TW & Hales CN (1991). Interaction of non‐esterified fatty acid and insulin in control of triacylglycerol secretion by Hep G2 cells. Biochem J 280, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassaglia PA, Hermes SM, Aicher SA & Brooks VL (2011). Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. J Physiol 589, 1643–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde SV, Sacramento JF, Diogo L, Gonzalez C, Guarino MP, Monteiro EC, Obeso A & Ribeiro MJ (2014). Carotid body, insulin and metabolic diseases: unraveling the links. Front Physiol 5, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampney RAL (2011). Arcuate nucleus – a gateway for insulin's action on sympathetic activity. J Physiol 589, 2109–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio R, Marcus NJ & Schultz HD (2013). Carotid chemoreceptor ablation improves survival in heart failure: rescuing autonomic control of cardiorespiratory function. J Am Coll Cardiol 62, 2422–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio R, Moya EA & Iturriaga R (2010). Carotid body and cardiorespiratory alterations in intermittent hypoxia: the oxidative link. Eur Respir J 36, 143–150. [DOI] [PubMed] [Google Scholar]
- de Luca C & Olefsky JM (2008). Inflammation and insulin resistance. FEBS Lett 582, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij SR, Nijpels G, Nilsson PM, Nolan JJ, Gabriel R, Bobbioni‐Harsch E, Mingrone G & Dekker JM; Relationship Between Insulin Sensitivity and Cardiovascular Disease (RISC) Investigators (2009). Low‐grade chronic inflammation in the relationship between insulin sensitivity and cardiovascular disease (RISC) population: associations with insulin resistance and cardiometabolic risk profile. Diabetes Care 32, 1295–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Zhang B, Shu HF, Zhang XY, Wang X, Kuang F, Liu L, Peng ZW, Wu R, Zhou Z & Wang BR (2009). Interleukin‐6 increases intracellular Ca2+ concentration and induces catecholamine secretion in rat carotid body glomus cells. J Neurosci Res 87, 2757–2762. [DOI] [PubMed] [Google Scholar]
- Ferranini E, Balkau B, Coppack SW, Dekker JM, Mari A, Nolan J, Walker M, Natali A, Beck‐Nielsen H & RISC Investigators (2007). Insulin resistance, insulin response, and obesity as indicators of metabolic risk. J Clin Endocrinol Metab 92, 2885–2892. [DOI] [PubMed] [Google Scholar]
- Ferranini E, Natali A, Bell P, Cavallo‐Perin P, Lalic N & Mingrone G; European Group for the Study of Insulin Resistance (EGIR) (1997). Insulin resistance and hypersecretion in obesity. J Clin Invest 100, 1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández R, González S, Rey S, Cortés PP, Maisey KR, Reyes EP, Larraín C & Zapata P (2008). Lipopolysaccharide‐induced carotid body inflammation in cats: functional manifestations, histopathology and involvement of tumour necrosis factor‐alpha. Exp Physiol 93, 892–907. [DOI] [PubMed] [Google Scholar]
- Flaa A, Aksnes TA, Kjeldsen SE, Eide I & Rostrup M (2008). Increased sympathetic reactivity may predict insulin resistance: an 18‐year follow‐up study. Metabolism 57, 1422–1427. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Behm R, Miller CC, Stauss H & Unger T (1992). Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol (1985) 72, 1978–1984. [DOI] [PubMed] [Google Scholar]
- Ford ES (2005). Prevalence of the metabolic syndrome defined by the International Diabetes Federation among adults in the U.S. Diabetes Care 28, 2745–2749. [DOI] [PubMed] [Google Scholar]
- Fudim M, Groom KL, Laffer CL, Netterville JL, Robertson D & Elijovich F (2015). Effects of carotid body tumor resection on the blood pressure of essential hypertensive patients. J Am Soc Hypertens 9, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y, Sato A & Trzebski A (1987). Carotid chemoreceptor discharge responses to hypoxia and hypercapnia in normotensive and spontaneously hypertensive rats. J Auton Nerv Syst 19, 1–11. [DOI] [PubMed] [Google Scholar]
- Gallego Martin T, Olea E, Gonzalez C & Yubero S (2014). Interaction between intermittent hypoxia and high fat diet to generate oxidative stress, sympathetic hyperactivity, insulin resistance, and systemic hypertension. Proc Physiol Soc 31, SA097. [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A & Rigual R (1994). Carotid body chemoreceptors: From natural stimuli to sensory discharges. Physiol Rev 74, 829–898. [DOI] [PubMed] [Google Scholar]
- Guo T, Marmol P, Moliner A, Björnholm M, Zhang C, Shokat KM & Ibanez CF (2014). Adipocyte ALK7 links nutrient overload to catecholamine resistance in obesity. Elife 25, e03245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heni M, Schöpfer P, Peter A, Sartorius T, Fritsche A, Synofzik M, Häring HU, Maetzler W & Hennige AM (2014). Evidence for altered transport of insulin across the blood–brain barrier in insulin‐resistant humans. Acta Diabetol 51, 679–681. [DOI] [PubMed] [Google Scholar]
- International Diabetes Federation (2015). IDF Diabetes Atlas, 7th edn. IDF, Brussels. http://www.diabetesatlas.org/
- Israel PA, Park CR, Schwartz MW, Green PK, Sipols AJ, Woods SC, Porte D Jr & Figlewicz DP (1993). Effect of diet‐induced obesity and experimental hyperinsulinemia on insulin uptake into CSF of the rat. Brain Res Bull 30, 571–575. [DOI] [PubMed] [Google Scholar]
- Jamerson KA, Julius S, Gudbrandsson T, Andersson O & Brant DO(1993). Reflex sympathetic activation induces acute insulin resistance in the human forearm. Hypertension 21, 618–623. [DOI] [PubMed] [Google Scholar]
- Julius S, Gudbrandsson T, Jamerson K & Andersson O (1992). The interconnection between sympathetics, microcirculation, and insulin resistance in hypertension. Blood Press 1, 9–19. [DOI] [PubMed] [Google Scholar]
- Kaiyala KJ, Prigeon RL, Kahn SE, Woods SC & Schwartz MW (2000). Obesity induced by a high‐fat diet is associated with reduced brain insulin transport in dogs. Diabetes 49, 1525–1533. [DOI] [PubMed] [Google Scholar]
- Kashyap S, Belfort R, Gastaldelli A, Pratipanawatr T, Berria R, Pratipanawatr W, Bajaj M, Mandarino L, DeFronzo R & Cusi K (2003). A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes 52, 2461–2474. [DOI] [PubMed] [Google Scholar]
- Katagiri H, Yamada T & Oka Y (2007). Adiposity and cardiovascular disorders: disturbance of the regulatory system consisting of humoral and neuronal signals. Circ Res 101, 27–39. [DOI] [PubMed] [Google Scholar]
- Kern W, Benedict C, Schultes B, Plohr F, Moser A, Born J, Fehm HL & Hallschmid M (2006). Low cerebrospinal fluid insulin levels in obese humans. Diabetologia 49, 2790–2792. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Coker RH, Stone EE, Lacy DB, Jabbour K, Williams PE & Wasserman DH (2000). Evidence that carotid bodies play an important role in glucoregulation in vivo. Diabetes 49, 1434–1442. [DOI] [PubMed] [Google Scholar]
- Kwon H & Pessin JE (2013). Adipokines mediate inflammation and insulin resistance. Front Endocrinol (Lausanne) 4, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakso M, Edelman SV, Olefsky JM, Brechtel G, Wallace P & Baron AD (1990). Kinetics of in vivo muscle insulin‐mediated glucose uptake in human obesity. Diabetes 39, 965–974. [DOI] [PubMed] [Google Scholar]
- Lam SY, Liu Y, Ng KM, Lau CF, Liong EC, Tipoe GL & Fung ML (2012). Chronic intermittent hypoxia induces local inflammation of the rat carotid body via functional upregulation of proinflammatory cytokine pathways. Histochem Cell Biol 137, 303–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SY, Tipoe GL, Liong EC & Fung ML (2008). Chronic hypoxia upregulates the expression and function of proinflammatory cytokines in the rat carotid body. Histochem Cell Biol 130, 549–559. [DOI] [PubMed] [Google Scholar]
- Lambadiari V, Triantafyllou K & Dimitriadis GD (2015). Insulin action in muscle and adipose tissue in type 2 diabetes: The significance of blood flow. World J Diabetes 6, 626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert EA, Straznicky NE, Dixon JB & Lambert GW (2015). Should the sympathetic nervous system be a target to improve cardiometabolic risk in obesity? Am J Physiol Heart Circ Physiol 309, H244–H258. [DOI] [PubMed] [Google Scholar]
- Lambert GW, Straznicky NE, Lambert EA, Dixon JB & Schlaich MP (2010). Sympathetic nervous activation in obesity and the metabolic syndrome – causes, consequences and therapeutic implications. Pharmacol Ther 162, 159–172. [DOI] [PubMed] [Google Scholar]
- Landsberg L (2005). Insulin resistance and the metabolic syndrome. Diabetologia 48, 1244–1246. [DOI] [PubMed] [Google Scholar]
- Landsberg L & Young JB (1978). Fasting, feeding and regulation of the sympathetic nervous system. N Engl J Med 298, 1295–1301. [DOI] [PubMed] [Google Scholar]
- Limberg J, Mozer M, Joyner M & Curry T (2014). Role of the carotid chemoreceptors in insulin‐mediated sympathoexcitation in humans. FASEB J 28 (1 Suppl.), 873.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckett BS, Frielle JL, Wolfgang L & Stocker SD (2013). Arcuate nucleus injection of an anti‐insulin affibody prevents the sympathetic response to insulin. Am J Physiol Heart Circ Physiol 304, H1538–H1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus NJ, Del Rio R, Schultz EP, Xia XH & Schultz HD (2014). Carotid body denervation improves autonomic and cardiac function and attenuates disordered breathing in congestive heart failure. J Physiol 592, 391–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JM (1994). Peripheral chemoreceptors and cardiovascular regulation. Physiol Rev 74, 543–594. [DOI] [PubMed] [Google Scholar]
- Masuo K, Mikami H, Ogihara T & Tuck ML (1997). Sympathetic nerve hyperactivity precedes hyperinsulinemia and blood pressure elevation in a young, nonobese Japanese population. Am J Hypertens 10, 77–83. [DOI] [PubMed] [Google Scholar]
- McBryde FD, Abdala AP, Hendy EB, Pijacka W, Marvar P, Moraes DJ, Sobotka PA & Paton JF (2013). The carotid body as a putative therapeutic target for the treatment of neurogenic hypertension. Nat Commun 4, 2395. [DOI] [PubMed] [Google Scholar]
- Mkrtchian S, Kåhlin J, Ebberyd A, Gonzalez C, Sanchez D, Balbir A, Kostuk EW, Shirahata M, Fagerlund MJ & Eriksson LI (2012). The human carotid body transcriptome with focus on oxygen sensing and inflammation—a comparative analysis. J Physiol 590, 3807–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiński P, Janczak D, Rucinski A, Jazwiec P, Sobotka PA, Engelman ZJ, Fudim M, Tubek S, Jankowska EA, Banasiak W, Hart EC, Paton JF & Ponikowski P (2013). Carotid body removal for treatment of chronic systolic heart failure. Int J Cardiol 168, 2506–2509. [DOI] [PubMed] [Google Scholar]
- Niewiński P, Janczak D, Rucinski A, Tubek S, Engelman ZJ, Jazwiec P, Banasiak W, Sobotka PA, Hart EC, Paton JF & Ponikowski P (2014). Dissociation between blood pressure and heart rate response to hypoxia after bilateral carotid body removal in men with systolic heart failure. Exp Physiol 99, 552–561. [DOI] [PubMed] [Google Scholar]
- O'Donnel CP, Schaub CD, Haines AS, Berkowitz DE, Tankersley CG, Schwartz AR & Smith PL (1999). Leptin prevents respiratory depression in obesity. Am J Respir Crit Care Med 159, 1477–1484. [DOI] [PubMed] [Google Scholar]
- Olea E, Ribeiro MJ, Gallego‐Martin T, Yubero S, Rigual R, Masa JF, Obeso A, Conde SV & Gonzalez C (2015). The carotid body does not mediate the acute ventilatory effects of leptin. Adv Exp Med Biol 860, 379–385. [DOI] [PubMed] [Google Scholar]
- Paton JFR (2015). Hypertension – a visceral afferent problem. Auton Neurosci 192, 16 DOI: https://doi.org/10.1016/j.autneu.2015.07.297. [Google Scholar]
- Pedersen DJ, Guilherme A, Danai LV, Heyda L, Matevossian A, Cohen J, Nicoloro SM, Straubhaar J, Noh HL, Jung D, Kim JK & Czech MP (2015). A major role of insulin in promoting obesity‐associated adipose tissue inflammation. Mol Metab 4, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Yuan G, Wang N, Deneris E, Pendyala S, Natarajan V, Kumar GK & Prabhakar NR (2009). NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J Neurosci 29, 4903–4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ & Prabhakar NR (2004). Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol (1985) 96, 1236–1242. [DOI] [PubMed] [Google Scholar]
- Peppard PE, Young T, Palta M & Skatrud J (2000). Prospective study of the association between sleep‐disordered breathing and hypertension. N Engl J Med 342, 1378–1384. [DOI] [PubMed] [Google Scholar]
- Pereda SA, Eckstein JW & Abboud FM (1962). Cardiovascular responses to insulin in the absence of hypoglycemia. Am J Physiol 202, 249–252.14485087 [Google Scholar]
- Ponikowski P, Chua TP, Anker SD, Francis DP, Doehner Banasiak W, Poole‐Wilson PA, Piepoli MF & Coats AJ (2001). Peripheral chemoreceptor hypersensitivity: an ominous sign in patients with chronic heart failure. Circulation 104, 544–549. [DOI] [PubMed] [Google Scholar]
- Porzionato A, Rucinski M, Macchi V, Stecco C, Castagliuolo I, Malendowicz LK & De Caro R (2011). Expression of leptin and leptin receptor isoforms in the rat and human carotid body. Brain Res 1385, 56–67. [DOI] [PubMed] [Google Scholar]
- Przybylski J, Trzebski A, Czyzewski T & Jodkowski J (1982). Responses to hyperoxia, hypoxia, hypercapnia and almitrine in spontaneously hypertensive rats. Bulletin Europeen de Physiopathologie Respiratoire – Clinical Respiratory Physiology 18, 145–154.6797493 [Google Scholar]
- Reaven G (2004). The metabolic syndrome or the insulin resistance syndrome? Different names, different concepts, and different goals. Endocrinol Metab Clin North Am 33, 283–303. [DOI] [PubMed] [Google Scholar]
- Reaven GM (1988). Banting Lecture 1988. Role of insulin resistance in human disease. Diabetes 37, 1595–1607. [DOI] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Alcayaga J & Iturriaga R (2004). Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol 560, 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro MJ, Sacramento JF, Gonzalez C, Guarino MP, Monteiro EC & Conde SV (2013). Carotid body denervation prevents the development of insulin resistance and hypertension induced by hypercaloric diets. Diabetes 62, 2905–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin MK, Yao Q, Jun JC, Bevans‐Fonti S, Yoo DY, Han W, Mesarwi O, Richardson R, Fu Y, Pasricha PJ, Schwartz AR, Shirahata M & Polotsky VW (2014). Carotid body denervation prevents fasting hyperglycemia during chronic intermittent hypoxia. J Appl Physiol (1985) 117, 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu HF, Wang BR, Wang SR, Yao W, Huang HP, Zhou Z, Wang X, Fan J & Wang T & Ju G (2007). IL‐1β inhibits I K and increases [Ca2+]i in the carotid body glomus cells and increases carotid sinus nerve firings in the rat. Eur J Neurosci 25, 3638–3647. [DOI] [PubMed] [Google Scholar]
- Siński M, Lewandowski J, Przybylski J, Bidiuk J, Abramczyk P, Ciarka A & Gaciong Z (2012). Tonic activity of carotid body chemoreceptors contributes to the increased sympathetic drive in essential hypertension. Hypertens Res 35, 487–491. [DOI] [PubMed] [Google Scholar]
- Somers VK, Mark AL & Abboud FM (1988). Potentiation of sympathetic nerve responses to hypoxia in borderline hypertensive subjects. Hypertension 11, 608–612. [DOI] [PubMed] [Google Scholar]
- Straznicky NE, Grima MT, Sari CI, Eikelis N, Lambert GW, Nestel PJ, Richards K, Dixon JB, Schlaich MP & Lambert EA (2015). Pioglitazone treatment enhances the sympathetic nervous system response to oral carbohydrate load in obese individuals with metabolic syndrome. Metabolism 64, 797–803. [DOI] [PubMed] [Google Scholar]
- Sun SY, Wang W, Zucker IH & Schultz HD (1999). Enhanced peripheral chemoreflex function in conscious rabbits with pacing‐induced heart failure. J Appl Physiol (1985) 86, 1264–1272. [DOI] [PubMed] [Google Scholar]
- Tankersley CG, O'Donnell C, Daood MJ, Watchko JF, Mitzner W, Schwartz A & Smith P (1998). Leptin attenuates respiratory complications associated with the obese phenotype. J Appl Physiol (1985) 85, 2261–2269. [DOI] [PubMed] [Google Scholar]
- Thorp AA & Schlaich MP (2015). Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J Diabetes Res 341583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzebski A, Tafil M, Zoltowski M & Przybylski J (1982). Increased sensitivity of the arterial chemoreceptor drive in young men with mild hypertension. Cardiovasc Res 16, 163–172. [DOI] [PubMed] [Google Scholar]
- Vera‐Cruz P, Guerreiro F, Ribeiro MJ, Guarino MP & Conde SV (2015). Hyperbaric oxygen therapy improves glucose homeostasis in type 2 diabetes patients: a likely involvement of the carotid bodies. Adv Exp Med Biol 860, 221–225. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang BR, Duan XL, Zhang P, Ding YQ, Jia Y, XY Jiao & Ju G (2002). Strong expression of interleukin‐1 receptor type I in the rat carotid body. J Histochem Cytochem 50, 1677–1684. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang XJ, Xu Z, Li X, Li GL, Ju G & Wang BR (2006). Morphological evidence for existence of IL‐6 receptor alpha in the glomus cells of rat carotid body. Anat Rec A Discov Mol Cell Evol Biol 288, 292–296. [DOI] [PubMed] [Google Scholar]
- Wehrwein EA, Basu R, Basu A, Curry TB, Rizza RA & Joyner MJ (2010). Hyperoxia blunts counterregulation during hypoglycaemia in humans: possible role for the carotid bodies? J Physiol 588, 4593–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrwein EA, Curry TB, Basu A, Rizza RA, Basu R & Joyner M (2012). Do the carotid bodies modulate hypoglycemic counterregulation and baroreflex control of blood pressure in humans? Adv Exp Med Biol 758, 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrwein EA, Limberg JK, Taylor JL, Dube S, Basu A, Basu R, Rizza RA, Curry TB & Joyner MJ (2015). Effect of bilateral carotid body resection on the counterregulatory response to hypoglycaemia in humans. Exp Physiol 100, 69–78. [DOI] [PubMed] [Google Scholar]
- West SD, Nicoll DJ & Stradling JR (2006). Prevalence of obstructive sleep apnoea in men with type 2 diabetes. Thorax 61, 945–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesenthal SR, Sandhu H, McCall RH, Tchipashvili V, Yoshii H, Polonsky K, Shi ZQ, Lewis GF, Mari A & Giacca A (1999). Free fatty acids impair hepatic insulin extraction in vivo. Diabetes 48, 766–774. [DOI] [PubMed] [Google Scholar]
- Yanai H, Tomono Y, Ito K, Furutani N, Yoshida H & Tada N (2008). The underlying mechanisms for development of hypertension in the metabolic syndrome. Nutr J 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]