

Abstract
Key points
The midbrain periaqueductal grey (PAG) forms part of an endogenous analgesic system which is tightly regulated by the neurotransmitter GABA.
The role of endocannabinoids in regulating GABAergic control of this system was examined in rat PAG slices.
Under basal conditions GABAergic neurotransmission onto PAG output neurons was multivesicular.
Activation of the endocannabinoid system reduced GABAergic inhibition by reducing the probability of release and by shifting release to a univesicular mode.
Blockade of endocannabinoid system unmasked a tonic control over the probability and mode of GABA release.
These findings provides a mechanistic foundation for the control of the PAG analgesic system by disinhibition.
Abstract
The midbrain periaqueductal grey (PAG) has a crucial role in coordinating endogenous analgesic responses to physiological and psychological stressors. Endocannabinoids are thought to mediate a form of stress‐induced analgesia within the PAG by relieving GABAergic inhibition of output neurons, a process known as disinhibition. This disinhibition is thought to be achieved by a presynaptic reduction in GABA release probability. We examined whether other mechanisms have a role in endocannabinoid modulation of GABAergic synaptic transmission within the rat PAG. The group I mGluR agonist DHPG ((R,S)‐3,5‐dihydroxyphenylglycine) inhibited evoked IPSCs and increased their paired pulse ratio in normal external Ca2+, and when release probability was reduced by lowering Ca2+. However, the effect of DHPG on the coefficient of variation and kinetics of evoked IPSCs differed between normal and low Ca2+. Lowering external Ca2+ had a similar effect on evoked IPSCs to that observed for DHPG in normal external Ca2+. The low affinity GABAA receptor antagonist TPMPA ((1,2,5,6‐tetrahydropyridin‐4‐yl)methylphosphinic acid) inhibited evoked IPSCs to a greater extent in low than in normal Ca2+. Together these findings indicate that the normal mode of GABA release is multivesicular within the PAG, and that DHPG and lowering external Ca2+ switch this to a univesicular mode. The effects of DHPG were mediated by mGlu5 receptor engagement of the retrograde endocannabinoid system. Blockade of endocannabinoid breakdown produced a similar shift in the mode of release. We conclude that endocannabinoids control both the mode and the probability of GABA release within the PAG.
Keywords: endocannabinoid, multivesicular, synaptic transmission, gaba, pain
Key points
The midbrain periaqueductal grey (PAG) forms part of an endogenous analgesic system which is tightly regulated by the neurotransmitter GABA.
The role of endocannabinoids in regulating GABAergic control of this system was examined in rat PAG slices.
Under basal conditions GABAergic neurotransmission onto PAG output neurons was multivesicular.
Activation of the endocannabinoid system reduced GABAergic inhibition by reducing the probability of release and by shifting release to a univesicular mode.
Blockade of endocannabinoid system unmasked a tonic control over the probability and mode of GABA release.
These findings provides a mechanistic foundation for the control of the PAG analgesic system by disinhibition.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- AM251
1‐(2,4‐dichlorophenyl)‐5‐(4‐iodophenyl)‐4‐methyl‐N‐1‐piperidinyl‐1H‐pyrazole‐3‐carboxamide
- CGP55845
(2S)‐3‐[[(1S)‐1‐(3,4‐dichlorophenyl)ethyl]amino‐2‐hydroxypropyl](phenylmethyl)phosphinic acid hydrochloride
- CNQX
6‐cyano‐2,3‐dihdroxy‐7‐nitro‐quinoxaline
- CPCCOEt
(E)‐ethyl 1,1a,7,7a‐tetrahydro‐7‐(hydroxyimino)cyclopropa [b]chromene‐1a‐carboxylate
- DHPG
(R,S)‐3,5‐dihydroxyphenylglycine
- FAAH
fatty acid amide hydrolase
- gabazine
2‐(3‐Carboxypropyl)‐3‐amino‐6‐(4 methoxyphenyl)pyridazinium bromide
- JZL195
4‐nitrophenyl 4‐(3‐phenoxybenzyl)piperazine‐1‐carboxylate
- MAGL
monacylglycerol lipase
- MPEP
2‐methyl‐6‐(phenylethynyl)pyridine hydrochloride
- NO711
1,2,5,6‐tetrahydro‐1‐[2‐[[(diphenylmethylene)amino]oxy]ethyl]‐3‐pyridinecarboxylic acid hydrochloride
- PAG
periaqueductal grey
- PPR
paired pulse ratio
- RVM
rostroventral medial medulla
- SNAP5114
(S)‐1‐[2‐[Tris(4‐methoxyphenyl)methoxy]ethyl]‐3‐piperidinecarboxylic acid
- TPMPA
(1,2,5,6‐tetrahydropyridin‐4‐yl)methylphosphinic acid (±)WIN55212 mesylate [2,3‐dihydro‐5‐methyl‐3‐(4‐morpholinylmethyl)pyrrolo[1,2,3‐de]‐1,4‐benzoxazin‐6‐yl]‐1‐naphthalenyl‐methanone, methanesulfonate
Introduction
The midbrain periaqueductal grey (PAG) plays a pivotal role in integrating analgesic, behavioural and autonomic responses to physiological and psychological stressors, including pain (Keay & Bandler, 2001). Analgesia from within the PAG is mediated by a descending pathway which projects via the medulla to the spinal dorsal horn (Fields et al. 2006). This descending analgesic pathway is regulated so that it is quiescent under basal conditions (Helmstetter et al. 1998; Oliveira & Prado, 2001). In addition to stressors, exogenously applied cannabinoid receptor agonists activate this descending analgesic system from within the PAG (Martin et al. 1995). Furthermore, natural stressors and pain induce the release of endocannabinoids with the PAG, and these mediate a form of stress‐induced analgesia (Walker et al. 1999; Hohmann et al. 2005).
A number of PAG functions, such as analgesia, are tightly regulated by the neurotransmitter GABA. Under basal conditions, tonically active GABAergic synaptic inputs inhibit PAG descending output neurons (Lau & Vaughan, 2014). Cannabinoids activate PAG outputs by relieving this inhibitory GABAergic influence through a process called disinhibition. Thus, exogenously applied cannabinoid receptor agonists and endogenously generated cannabinoids (endocannabinoids) act via presynaptic cannabinoid CB1 receptors to decrease the probability of release of GABA within the PAG (Vaughan et al. 2000; Drew et al. 2008, 2009; Mitchell et al. 2009, 2011; Ho et al. 2011). Presynaptic signalling can, however, be controlled by mechanisms other than release probability. At some central excitatory and inhibitory synapses, multivesicular release occurs when more than one synaptic vesicle is released from a terminal in response to an action potential (Rudolph et al. 2015). In addition, presynaptic metabotropic receptor activation modulates multivesicular release within a number of brain regions (Takahashi et al. 1995; Gordon & Bains, 2005; Higley et al. 2009; Chalifoux & Carter, 2010; Rudolph et al. 2011). As a result, multivesicular signalling and its modulation by presynaptic receptors can have a major impact upon the strength and reliability of synaptic transmission (Foster et al. 2005; Bender et al. 2009; Chamberland et al. 2014).
We examined whether modulation of vesicle release mode has a role within the PAG by exploring the influence of release probability on endocannabinoid modulation of inhibitory GABAergic transmission. We show that under basal conditions multivesicular release is the preferred synaptic vesicle release mode, and this acts to strengthen GABAergic inhibition. In addition to reducing release probability, presynaptic cannabinoid CB1 receptor activation reversibly shifts the vesicle release mode from multi‐ to univesicular. The combined modulation of release mode and probability provides an enhanced mechanism for presynaptic disinhibition and allows rapid switching between quiescence and activation of the descending PAG output.
Methods
Ethical approval
Experiments were carried out on male and female Sprague Dawley rats (22–30 days old) obtained from Animal Resources Centre (Canning Vale, Australia), following the guidelines of the ‘NH&MRC Code of Practice for the Care and Use of Animals in Research in Australia’ and with the approval of the Royal North Shore Hospital Animal Ethics Committee. Animals were housed in groups of three in individually ventilated cages under a 12:12 h light/dark cycle, with environmental enrichment and free access to water and standard rat chow.
Retrograde tracer injections
In some experiments, fluorescent microspheres (Molecular Probes, Eugene, OR, USA) were microinjected into the rostroventral medial medulla (RVM) of 19–24 day old rats (n = 10), as described previously (Drew et al. 2009). The animals were anaesthetized (1–2% isoflurane in O2), placed in a stereotaxic frame and the dura exposed by trephination. A glass micropipette (tip diameter 30 μm) was advanced into the RVM under stereotaxic guidance, and microsphere injections were made over a 2–3 min period (100–120 nl total in 10 nl steps; Drummond Nanoject, Broomall, PA, USA). The micropipette was left in place for 3 min before being slowly withdrawn. The craniotomy was covered with bone wax, a topical antibiotic applied and the incision was closed. Animals received an analgesic (buprenorphine, 0.05 mg kg−1), and were then allowed to recover from anaesthesia in a warmed box before being returned to holding cages. Animals were used for in vitro experiments ≥ 3 days after tracer injection.
Brain slice preparation
Animals were deeply anaesthetized with isoflurane, decapitated and transverse midbrain slices (300 μm) containing PAG (and RVM in tracer injection experiments) were cut using a vibratome (VT1200S, Leica Microsystems AG, Wetzlar, Germany) in ice‐cold artificial cerebrospinal fluid (ACSF), as described previously (Drew et al. 2008). PAG slices were maintained at 34°C in a submerged chamber containing ACSF equilibrated with 95% O2 and 5% CO2. The slices were then individually transferred to a chamber and superfused continuously (2.5 ml min−1) with ACSF (34°C) of composition (in mm): 126 NaCl, 2.5 KCl, 1.4 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose and 25 NaHCO3; plus 0.05 % biocytin in tracer injection experiments.
Electrophysiology
PAG neurons were visualized using Dodt‐tube contrast gradient optics on an upright microscope (BX51; Olympus, Tokyo, Japan). Whole cell voltage‐clamp recordings (Axopatch 200B, Molecular Devices, Gainseville, FL, USA) of synaptic currents (holding potential, –65 mV) were made from lateral and ventrolateral PAG neurons using a CsCl‐based internal solution containing (in mm): 140 CsCl, 0.2 EGTA, 10 Hepes, 1 MgCl2, 2 MgATP and 0.3 NaGTP (pH 7.3; osmolarity, 280–285 mosmol l−1). Series resistance (< 20 MΩ) was compensated by 80% and continuously monitored during experiments; neurons were not used for analysis if series resistance varied by more than 25%. Liquid junction potentials of –4 mV were corrected. Recordings of IPSCs were obtained in the presence of the non‐NMDA glutamate receptor antagonist 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX) (5 μm) and the glycine receptor antagonist strychnine (3 μm). Recordings of EPSCs were obtained in the presence of picrotoxin (100 μm) and strychnine (3 μm). Evoked IPSCs and EPSCs were elicited every 6 s using a monopolar glass stimulating electrode (tip width 10–20 μm) placed 50–200 μm lateral to the recording electrode. In some experiments paired evoked IPSCs were elicited at an inter‐stimulus interval of 70 ms. Spontaneous miniature IPSCs were recorded in the additional presence of TTX (0.3 μm).
Data analysis
IPSCs and EPSCs were filtered (5–10 kHz low‐pass filter) and sampled (10–20 kHz) for on‐line and later off‐line analysis (Axograph X; Axograph Scientific Software, Berkeley, CA, USA). Miniature IPSCs above a pre‐set threshold (3–4 standard deviations above baseline noise) were automatically detected by a sliding template algorithm, and then manually checked off‐line. Multi‐peak IPSCs and EPSCs were excluded from analysis of kinetics. The following IPSC and EPSC characteristics were measured: amplitude relative to a 2 ms baseline preceding the stimulus artefact, rise time as the time from 20 to 80 % of the peak, time‐to‐peak as the time from the onset (5 % of peak) to the peak and width of the IPSC duration at 1/e (τ∼ 36.8 %) of the peak amplitude. IPSC and EPSC decay phases were best‐fit by one or two exponentials to obtain estimates of the decay amplitude and time constant, A and τ. In the case of the latter, the weighted decay time was calculated as τw = [A fast × τfast + A slow × τslow]/[A fast + A slow], where A and τ are the amplitudes and time constants for the fast and slow components of decay phases. The coefficient of variation (CV) of IPSC amplitudes was measured as the standard deviation divided by the mean (σ/M) over each epoch (minimum of 15 data points); CV2 as the variance divided by the square of the mean amplitude (σ 2/M 2). The ratio of CV−2 versus the mean of evoked IPSCs and EPSCs was compared for treatment groups by using a coefficient of variation analysis (Faber & Korn, 1991). Concentration response curves for varying external Ca2+ were constructed by fitting a sigmoidal curve with variable slope (Prism, GraphPad Software Inc., La Jolla, CA, USA).
Individual drug effects were calculated as the relative value in drug compared to the average of that before drug application and after drug washout for agonists, or as the relative value in drug compared to that before drug application for antagonists/blockers. Statistical comparisons of individual drug treatments were made using paired Student's t test (Prism). Comparisons between two different drug treatment groups were made using unpaired Student's t test, and for multiple drug treatment groups, comparisons were made using one‐way ANOVA with Dunnett's correction for post hoc comparisons to the control group. Differences were considered significant at P < 0.05.
Drug solutions
CNQX, picrotoxin and strychnine hydrochloride were obtained from Sigma (Sydney, Australia); CGP55845 ((2S)‐3‐[[(1S)‐1‐(3,4‐dichlorophenyl)ethyl]amino‐2‐hydroxypropyl](phenylmethyl)phosphinic acid hydrochloride), CPCCOEt ((E)‐ethyl 1,1a,7,7a‐tetrahydro‐7‐(hydroxyimino)cyclopropa [b]chromene‐1a‐carboxylate), DHPG ((R,S)‐3,5‐dihydroxyphenylglycine), gabazine, MPEP (2‐methyl‐6‐(phenylethynyl)pyridine) hydrochloride, NO711 (1,2,5,6‐tetrahydro‐1‐[2‐[[(diphenylmethylene)amino]oxy]ethyl]‐3‐pyridinecarboxylic acid hydrochloride) and TTX from Abcam Biochemicals (Cambridge, UK); AM251 (1‐(2,4‐dichlorophenyl)‐5‐(4‐iodophenyl)‐4‐methyl‐N‐1‐piperidinyl‐1H‐pyrazole‐3‐carboxamide), JZL195 (4‐nitrophenyl 4‐(3‐phenoxybenzyl)piperazine‐1‐carboxylate) and WIN55212 ([2,3‐dihydro‐5‐methyl‐3‐(4‐morpholinylmethyl)pyrrolo[1,2,3‐de]‐1,4‐benzoxazin‐6‐yl]‐1‐naphthalenyl‐methanone, methanesulfonate) from Cayman Chemicals (Ann Arbor, MI, USA); SNAP5114 ((S)‐1‐[2‐[Tris(4‐methoxyphenyl)methoxy]ethyl]‐3‐piperidinecarboxylic acid) and TPMPA ((1,2,5,6‐tetrahydropyridin‐4‐yl)methylphosphinic acid) from Tocris Bioscience (Bristol, UK). Stock solutions of drugs were made in distilled water, or DMSO, then diluted to working concentrations in ACSF (≤ 1:3000 solvent) immediately before use and applied by superfusion.
Immunohistochemistry
Immediately after recording from tracer injected animals, PAG and RVM slices were fixed overnight in a phosphate buffered paraformaldehyde solution. The slices were then washed 6–8 times and stored in a phosphate buffer. Biocytin‐filled PAG cells were visualized by incubation with Cy5‐labelled streptavidin (Jackson ImmunoResearch, West Grove, PA, USA). Slices were mounted and cover‐slipped and digital images of filled cells were collected using conventional fluorescence microscopy (Zeiss AX10). The RVM was examined to determine the extent of tracer injection sites.
Results
The mechanisms underlying Group I mGluR‐induced inhibition of evoked IPSCs are calcium dependent
We have previously shown that the group 1 mGlu receptor agonist DHPG presynaptically inhibits GABAergic synaptic transmission within the PAG (Drew & Vaughan, 2004). In this study we explored the synaptic mechanisms underlying this inhibition by varying external Ca2+ to determine the influence of release probability. A near‐maximal concentration of DHPG (10 μm) produced a reversible reduction in the amplitude of evoked GABAA receptor‐mediated IPSCs in normal (2.4 mm) and low (1.2 mm) external Ca2+ (Fig. 1 A, B, D, P < 0.0001, n = 19 and 9). This was associated with an increase in the paired pulse ratio (PPR) of evoked IPSCs under both conditions (Fig. 1 C, D, P = 0.009 and 0.03). The DHPG‐induced inhibition and paired pulse facilitation of evoked IPSCs were not significantly different in normal and low external Ca2+ (P = 0.7 and 0.1). These results indicate that the DHPG‐induced inhibition of GABAergic synaptic transmission involves a presynaptic reduction in release probability in normal and low external Ca2+.
Figure 1. DHPG inhibits evoked IPSCs by different mechanisms in normal and reduced external Ca2+ .
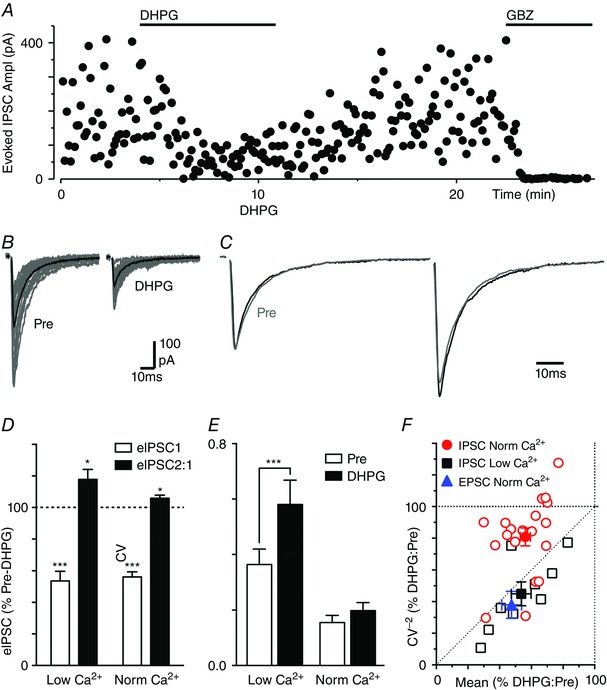
A, time plot of the amplitude of evoked IPSCs during superfusion of DHPG (10 μm) and gabazine (GBZ, 10 μm). B, traces of evoked IPSCs before (Pre) and during DHPG superfusion (15 raw IPSCs in grey and average trace in black). C, averaged traces of paired evoked IPSCs before (Pre) and during DHPG with the first IPSC normalized. D, bar chart of the effect of DHPG on evoked IPSC amplitude and paired pulse ratio (eIPSC2:1) in normal and low external Ca2+, shown as a percentage of the pre‐DHPG value. E, bar chart of the coefficient of variation (CV) of evoked IPSC amplitude before (Pre) and during DHPG superfusion, in normal and low external Ca2+. In D and E, asterisks denote * P < 0.05, ***0.0001 for DHPG versus pre‐DHPG. F, plot of the effect of DHPG on the CV−2 versus mean of evoked IPSCs in normal (red) and low external Ca2+ (black), and also for evoked EPSCs in normal Ca2+ (blue). In F, data are shown for individual neurons (open symbols) and the average for all neurons tested under each condition (filled symbols). [Colour figure can be viewed at wileyonlinelibrary.com]
We next examined these data using the CV method for analysing synaptic modulation (Faber & Korn, 1991). In normal external Ca2+, DHPG reduced the variance in evoked IPSC amplitude (Fig. 1 B). This equated to a lack of change in the CV (σ/M) of evoked IPSCs (Fig. 1 E, 113 ± 11 % of pre‐DHPG, P > 0.05). Under these conditions, the DHPG‐induced reduction in the CV−2 of evoked IPSCs was usually less than the change in their mean amplitude (Fig. 1 F). The average CV−2/mean ratio for DHPG consequently mapped between the lines of identity and unity (Fig. 1 F). In low external Ca2+, DHPG produced an increase in the CV of evoked IPSCs (Fig. 1 E, 171 ± 20% of pre‐DHPG, P < 0.0001). Under these conditions, DHPG usually produced an equal or greater reduction in the CV−2 of evoked IPSCs compared to the change in their mean amplitude (Fig. 1 F). The average CV−2/mean ratio for DHPG in low Ca2+ mapped onto the line of identity (Fig. 1 F). This analysis confirms that the DHPG inhibition of evoked IPSCs in low Ca2+ is mediated by a presynaptic reduction in release probability, but suggests that additional mechanisms are recruited in normal Ca2+.
In normal external Ca2+, DHPG also produced a reduction in the amplitude of evoked non‐NMDA‐mediated EPSCs, and an increase in their PPR and CV (mean amplitude = 47 ± 6%, PPR = 122 ± 6% and CV = 201 ± 35% of pre‐DHPG, P < 0.0001, P = 0.01 and 0.02, n = 8). Under these conditions, the average CV−2/mean ratio for DHPG mapped onto the line of identity (Fig. 1 F). Thus, the effects of DHPG on evoked EPSCs were similar to those observed for evoked IPSCs in low but not normal external Ca2+.
DHPG produces a calcium‐dependent shortening of evoked IPSC kinetics
DHPG also had a differential effect on the kinetics of evoked IPSCs in normal and low external Ca2+. In normal external Ca2+, DHPG produced a reduction in the width of evoked IPSCs (Figs 1 C, 2 Ai–ii, C, P < 0.0001). Under basal conditions, the decay phase of evoked IPSCs was best fit by a double exponential (τfast = 6.8 ± 0.7 ms, τslow = 41.4 ± 5.6 ms, A fast/A total = 76 ± 3%) and had a weighted decay time constant, τdecay, of 14.6 ± 2.3 ms. DHPG reduced the τdecay of evoked IPSCs to 12.6 ± 2.4 ms (Fig. 2 C, P < 0.0001). Furthermore, DHPG reduced the time‐to‐peak of evoked IPSCs from 2.0 ± 0.1 to 1.7 ± 0.1 ms (Fig. 2 Aiii, C, P = 0.0007). The reduction in the time‐to‐peak of evoked IPSCs was due to an increase in the latency to onset of the evoked IPSC, plus a reduction in rise time (Fig. 2 Aiii, C). In low external Ca2+, DHPG did not significantly alter the width of evoked IPSCs (Fig. 2 Bi–ii, C, P > 0.05). The decay phase of evoked IPSCs was best fit by a single exponential that had a τdecay of 9.1 ± 1.9 and 9.3 ± 2.0 ms before and after addition of DHPG (Fig. 2 C, P > 0.05). Under these conditions, DHPG had no significant effect on the rise time, or time‐to‐peak of evoked IPSCs (Fig. 2 Biii, C, P > 0.05).
Figure 2. DHPG shortens the decay phase of evoked IPSCs in normal but not low external Ca2+ .
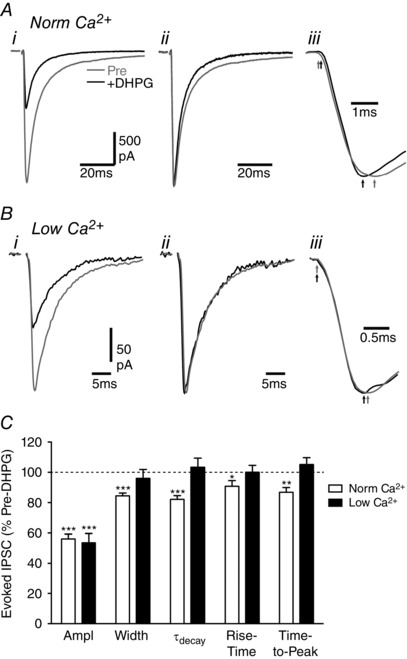
A and B, averaged traces of evoked IPSCs in (A) normal and (B) low external Ca2+ in two PAG neurons before (Pre) and during superfusion of DHPG (10 μm). Traces are shown for (i) raw averaged evoked IPSCs, (ii) normalized evoked IPSCs and (iii) normalized evoked IPSCs on an expanded time scale showing the onset and time of the IPSC peak (arrows in A). C, bar chart of the effect of DHPG on evoked IPSC amplitude, width, weighted decay phase time constant (τdecay), rise time and the time to peak, shown as a percentage of the pre‐DHPG value. Asterisks denote * P < 0.05, **0.01 and ***0.0001 for DHPG versus pre‐DHPG.
We examined whether the modulation of IPSC kinetics in normal external Ca2+ was specific to evoked release by assessing the effect of DHPG on quantal IPSCs in the presence of TTX (300 nm). In normal external Ca2+, DHPG (10 μm) produced a reduction in the rate but not amplitude of spontaneous miniature IPSCs (Fig. 3 A–C, E, P < 0.0001 and P = 0.4, n = 8). In addition, DHPG had no effect on the rise time or width of miniature IPSCs (Fig. 3 D, E, P = 0.8, 0.3). This confirms that DHPG acts presynaptically to reduce GABA release probability, but demonstrates that it has no effect on the kinetics of quantal IPSCs in normal external Ca2+. Thus, the modulation of evoked IPSC kinetics in normal Ca2+ is specific to evoked vesicular release and unlikely to be caused by a DHPG interaction with postsynaptic GABAA receptors.
Figure 3. DHPG has no effect on miniature IPSC kinetics.
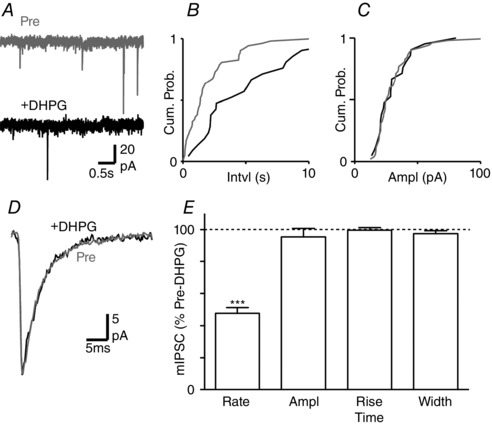
A, raw traces of miniature IPSCs in a PAG neuron, before (Pre) and during superfusion of DHPG (10 μm). B and C, cumulative distribution plots of miniature IPSC (B) inter‐event interval and (C) amplitude, before (Pre) and during DHPG. D, averaged traces of miniature IPSCs, before (Pre) and during DHPG. E, bar chart of the effect of DHPG on miniature IPSC rate, amplitude, rise time and width, shown as a percentage of the pre‐DHPG value. *** P < 0.0001 for DHPG versus pre‐DHPG.
Changes in evoked IPSC kinetics are not mediated by spill‐over
The differential effects of DHPG on the kinetics of evoked IPSCs in normal and low Ca2+ may be related to GABA transporter saturation and spill‐over, which only occurs when presynaptic release probability is elevated (Wadiche & Jahr, 2001). We assessed this by examining the effect of GABA transporter blockers on evoked IPSCs (Bagley et al. 2011). In normal external Ca2+, addition of the GAT‐1 and ‐3 blockers NO711 (10 μm) and SNAP5114 (40 μm) produced a reduction in the amplitude of evoked IPSCs, which was due to auto‐inhibition via presynaptic GABAB receptors as it was abolished by addition of the GABAB antagonist CGP55845 (1 μm, n = 3, data not shown). In the presence of CGP55845, application of NO711 (10 μm) and SNAP5114 (40 μm) produced an increase in the width but not the amplitude of evoked IPSCs (Fig. 4 A, B, 131 ± 9 and 109 ± 6% of pre‐NO711/SNAP, P = 0.04, 0.02, n = 6). Under these conditions, subsequent addition of DHPG still produced a reduction in the amplitude and width of evoked IPSCs, and an increase in their PPR (Fig. 4 A–C, P < 0.0001 and P = 0.03, 0.02, n = 6). Furthermore, the DHPG‐induced reduction in the CV−2 of evoked IPSCs was always less than the change in their mean amplitude (Fig. 4 D). The average CV−2/mean ratio for DHPG consequently mapped between the lines of identity and unity (Fig. 4 D). This was similar to our data in the absence of GAT blockers and suggests that the effect of DHPG on evoked IPSC width and CV in normal external Ca2+ was independent of GABA transporter function.
Figure 4. GAT blockade does not alter the effect of DHPG on evoked IPSC kinetics and CV.
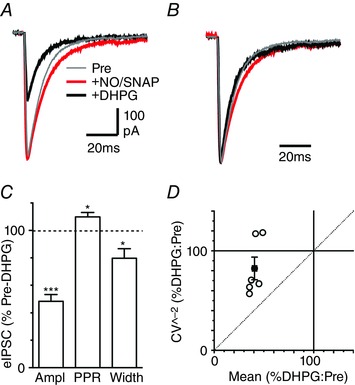
A, average traces of evoked IPSCs in a PAG neuron in the presence of the GABAB antagonist CGP55845 (1 μm), before (Pre) then after addition of NO711 (10 μm) plus SNAP5114 (40 μm) (NO/SNAP), then after further addition of DHPG (10 μm). B, normalized traces of averaged evoked EPSCs from A. C, bar chart of the effect of DHPG on the evoked IPSC amplitude (Ampl), paired pulse ratio and width, in the presence of CGP55845, NO711 and SNAP5114, shown as a percentage of the pre‐DHPG value. * P < 0.05 and ***0.0001 for DHPG versus pre‐DHPG. D, plot of the effect of DHPG on the CV−2 versus mean of evoked IPSCs in the presence of CGP55845 and GAT blockers; data are shown for individual neurons (open circles) and the average of neurons tested (filled circles). [Colour figure can be viewed at wileyonlinelibrary.com]
External calcium concentration influences presynaptic probability and release mode
In some brain regions basal synaptic transmission is multivesicular, and reducing release probability can shift this to a univesicular mode (Rudolph et al. 2015). The observed effects of DHPG on the CV and kinetics of evoked IPSCs in normal external Ca2+ were consistent with a similar shift in release mode. We therefore examined whether changing release probability by altering external Ca2+ produced similar changes in evoked IPSCs. The amplitude, PPR, CV and width of evoked IPSCs varied between low, normal and high (4.0 mm) external Ca2+ (P = 0.0001–0.002, F 2,10 = 18.4, 11.9, 15.4, 24.4, n = 6). Reducing external Ca2+ from 2.4 to 1.2 mm produced a decrease in the amplitude and width of evoked IPSCs (Fig. 5 A–C, P < 0.01, 0.05). It also produced an increase in the PPR and CV of evoked IPSCs (Fig. 5 B, D, P < 0.05). Thus, lowering external Ca2+ has a similar effect to DHPG on the amplitude, PPR and kinetics of evoked IPSCs. Lowering external Ca2+ reduced the CV−2 of evoked IPSCs to a lesser extent than their mean amplitude (Fig. 5 E). The average CV−2/mean ratio for lowering external Ca2+ mapped above the line of identity (Fig. 5 E), which was qualitatively similar to that observed for DHPG in normal Ca2+ (Fig. 1 F).
Figure 5. Altering external Ca2+ modulates evoked IPSC CV and width.
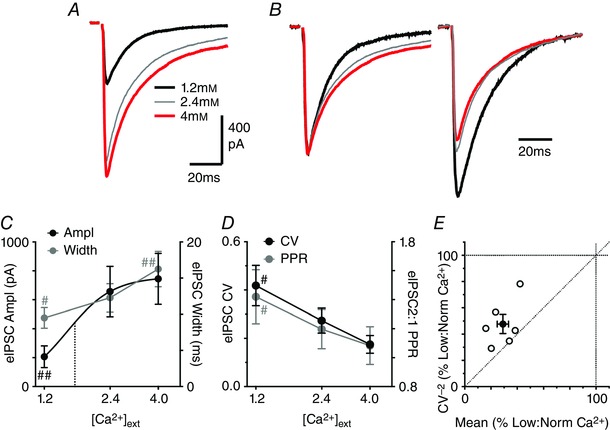
A, averaged traces of evoked IPSCs in a PAG neuron, in normal (2.4 mm), low (1.2 mm) and then high (4.0 mm) external Ca2+. B, averaged traces of paired evoked EPSCs for the neuron in A, normalized for the first evoked IPSC (inter‐event stimulus 100 ms). C, concentration response curve of the effect of external Ca2+ on evoked IPSC amplitude (Ampl) and width. The data for evoked IPSC amplitude were fit by a logistic function, with the EC50 (1.7 mm) shown by the dotted line. D, concentration response curve of the effect of external Ca2+ on the paired pulse ratio (PPR) and coefficient of variation (CV) of evoked IPSCs. In C and D, has marks denote # P < 0.05 and ##0.01 versus the value in 2.4 mm Ca2+. E, plot of the effect of low external Ca2+ on the CV−2 versus mean of evoked IPSCs, expressed as a percentage of the value in normal external Ca2+. Data are shown for individual neurons (open symbols) and the average for all neurons tested (filled symbols). [Colour figure can be viewed at wileyonlinelibrary.com]
In these neurons, increasing external Ca2+ from 2.4 to 4.0 mm produced an increase in the width, but had no significant effect on the amplitude of evoked IPSCs (Fig. 5 A–C, P < 0.001, > 0.05). The amplitude of evoked IPSCs displayed a sigmoidal Ca2+ dependence, with an EC50 of 1.5 ± 0.3 mm and a Hill slope of 4.1 ± 1.2 (Fig. 5 C). Increasing external Ca2+ did not produce a significant change in the PPR and CV of evoked IPSCs (Fig. 5 D, P > 0.05).
If lowering external Ca2+ shifts release from a multi‐ to univesicular mode then there should be a reduction in the concentration of synaptically released GABA. We tested this by examining the effect of the low affinity GABAA receptor antagonist TPMPA (Foster et al. 2005; Christie & Jahr, 2006; Balakrishnan et al. 2009). TPMPA (200 μm) produced a reduction in the amplitude of evoked IPSCs which varied with the concentration of external Ca2+ (Fig. 6 A, C, E, P = 0.003, F 2,12 = 7.5, n = 5 each concentration). While the TPMPA‐induced inhibition of evoked IPSCs was greater in low compared to normal external Ca2+ (P < 0.05), it was not significantly different between normal and high external Ca2+ (Fig. 6 D, P > 0.05). The effect of TPMPA on the PPR of evoked IPSC amplitudes also varied with external Ca2+ (Fig. 6 B, D, E, P = 0.002, F 2,12 = 11.1). TPMPA produced a similar reduction in the amplitude of the first and second IPSCs in normal and high external Ca2+, and consequently did not have a significant effect on the PPR of evoked IPSCs under these conditions (Fig. 6 B, E, P = 0.6, 0.7). In low external Ca2+, TPMPA produced a greater reduction in the first evoked IPSC which was observed as an increase in the PPR (Fig. 6 D, E, P = 0.01). This suggests that GABA release is multivesicular in normal and high external Ca2+, but univesicular in low external lowering Ca2+. It also indicates that paired pulse facilitation also involves a shift from univesicular to multivesicular release in low external Ca2+.
Figure 6. Reducing external Ca2+ affects TPMPA sensitivity.
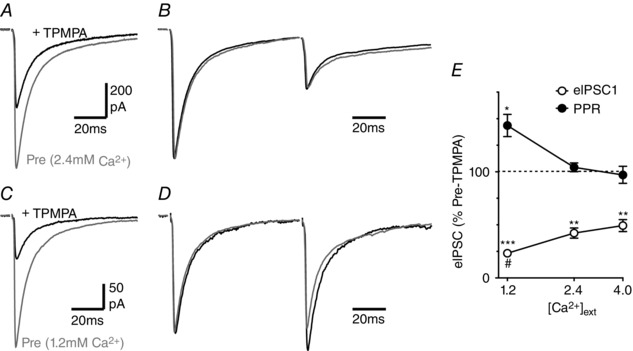
A and C, averaged traces of evoked IPSCs in a PAG neuron in (A) normal (2.4 mm) and (C) low (1.2 mm) external Ca2+, before (Pre) and after application of TPMPA (200 μm). B and D, averaged traces of paired evoked IPSCs for the neuron depicted in A and C, respectively, with the first IPSC normalized. E, bar chart of the effect of TPMPA on the evoked IPSC amplitude (eIPSC1) and paired pulse ratio (PPR) in normal (2.4 mm), low (1.2 mm) and high (4.0 mm) external Ca2+; data shown as a percentage of the pre‐TPMPA value. Asterisks denote * P < 0.05, **0.01 and ***0.0001 for TPMPA versus pre‐TPMPA; # P < 0.05 versus the values in 2.4 mm Ca2+.
The mGluR modulation of evoked IPSC kinetics is mediated by endocannabinoids
The DHPG‐induced inhibition of evoked IPSCs within the PAG is mediated by mGlu5 receptor‐induced retrograde endocannabinoid signalling (Drew et al. 2008). We examined whether the DHPG modulation of evoked IPSC kinetics was mediated by a similar mechanism. The DHPG‐induced reduction in evoked IPSC width was abolished in slices pre‐incubated in the mGlu5 receptor antagonist MPEP (10 μm), but not the mGluR1 antagonist CPCCOEt (100 μm) (Fig. 7 A, C, P = 0.9 and 0.02, n = 7, 5). The DHPG‐induced reduction in evoked IPSC width was also abolished by pre‐treatment with the CB1 receptor antagonist AM251 (1 μm), and by replacement of GTP with GDP‐βS in the internal postsynaptic recording solution (Fig. 7 B, C, P = 0.9 and 0.3, n = 8, 5). Furthermore, the actions of DHPG were mimicked by the pan‐cannabinoid agonist WIN55212 (1 μm), which produced a reduction in both the amplitude and the width of evoked IPSCs (46 ± 6 and 86 ± 2% of pre‐WIN55212, P < 0.0001, n = 12).
Figure 7. The DHPG‐induced reduction in evoked IPSC width is endocannabinoid mediated.
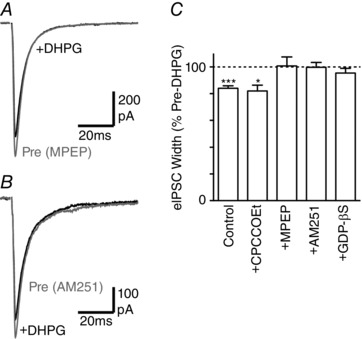
A and B, averaged traces of evoked IPSCs in PAG neurons before (Pre) and during superfusion of DHPG (10 μm), in slices pre‐incubated in (A) MPEP (10 μm) and (B) AM251 (1 μm). C, bar chart of the effect of DHPG on evoked IPSC width in untreated slices (control) and in slices pre‐incubated in CPCCOEt (100 μm), MPEP (10 μm) or AM251 (1 μm), or in neurons in which internal GTP was replaced with GDP‐βS. * P < 0.05, ***0.0001 for DHPG versus pre‐DHPG.
Within the PAG there is a basal endocannabinoid modulation of synaptic transmission (Kawahara et al. 2011; Lau et al. 2014). We therefore examined the effect of blockade of this endocannabinoid tone on evoked IPSCs. Superfusion of AM251 produced an increase in the width but not the amplitude of evoked IPSCs (Fig. 8 A, B, E, P = 0.02, 0.9, n = 7). Likewise, MPEP (10 μm) produced an increase in the width but not the amplitude of evoked IPSCs (126 ± 9 and 91 ± 8% of pre‐MPEP, P = 0.03, 0.3, n = 7). In addition, MPEP had no effect on the amplitude or width of evoked IPSCs in slices pre‐incubated in AM251 (107 ± 16 and 98 ± 5% of pre‐MPEP, P = 0.7, 0.6, n = 5). The effect of MPEP and AM251 on evoked IPSC kinetics may have been an artefact of the whole cell recording conditions as we only compared the IPSC parameters before and during antagonist application in these experiments. The width of evoked IPSCs, however, did not vary over a similar recording period when no antagonist was applied (IPSC width = 99 ± 1% of first 5 min at 25 min, F 4,48 = 0.7, P = 0.52, n = 13).
Figure 8. Basal endocannabinoid modulation of evoked IPSC kinetics.
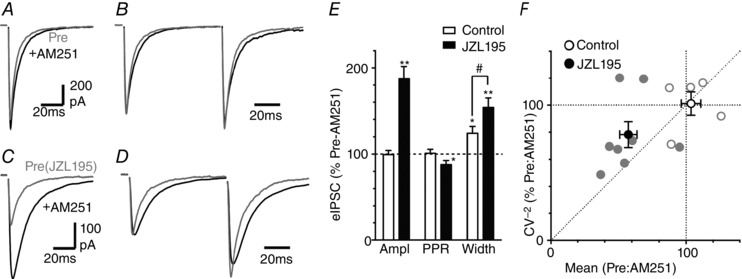
A and C, averaged traces of evoked IPSCs before and after addition of AM251 (1 μm), in (A) untreated control slices and (C) in slices pre‐incubated in JZL195 (1 μm). B and D, averaged traces of paired evoked IPSCs for the neuron depicted in A and C, respectively, with the first IPSC normalized. E, bar chart of the effect of AM251 on evoked IPSC amplitude, paired pulse ratio (PPR) and width, shown as a percentage of the pre‐AM251 value, in untreated control slices and slices pre‐incubated in JZL195; * P < 0.05 and **0.01 for AM251 versus pre‐ AM251; # P < 0.05 for control versus JZL194 treatment. F, plot of the effect of AM251 on the CV−2 versus mean of evoked IPSCs, expressed as a percentage of the value in normal external Ca2+.
Basal endocannabinoid modulation of GABAergic synaptic transmission within the PAG is regulated by both of the degradation enzymes fatty acid amide hydrolase (FAAH) and monacylglycerol lipase (MAGL) (Lau et al. 2014). When slices were pre‐incubated in the dual FAAH/MAGL inhibitor JZL195 (1 μm), AM251 produced an increase in both the amplitude and the width of evoked IPSCs (Fig. 8 C–E, P = 0.008, 0.003, n = 7). The effect of AM251 on the amplitude and width of evoked IPSCs was greater in the presence of JZL195 than in its absence (Fig. 8 E, P = 0.001, 0.04). We quantified the inverse of the effect of AM251 (Pre:AM251) for the CV analysis to assess the level of endocannabinoid tone. In the presence of JZL195, the CV−2 versus mean of evoked IPSCs prior to addition of AM251 mapped between the lines of identity and unity (Fig. 8 F). In the absence of JZL195, the CV−2 versus mean of evoked IPSCs was unaffected by AM251 (Fig. 8 F). Together, these observations indicate that basal release of endogenous cannabinoids modulates IPSC release mode and probability within the PAG, and reducing their degradation via both FAAH and MAGL enhances this basal tone.
Descending projection neurons
We finally examined whether the effects of DHPG on evoked IPSCs described above were also present in PAG output neurons identified as projecting along the descending analgesic pathway. At 3–6 days following injection of fluorescent microspheres into the RVM, PAG neurons which contain fluorescent microspheres were identified, as described previously (Drew et al. 2009) (Fig. 9 A). In the retrogradely labelled PAG neurons, DHPG produced a reduction in the amplitude and width of evoked IPSCs (Fig. 9 B–E, P < 0.0001 and = 0.003, n = 8). DHPG also produced an increase in the PPR of evoked IPSCs (Fig. 9 D, E, P < 0.05). In these neurons, the DHPG‐induced reduction in the CV−2 of evoked IPSCs was less than the change in their mean amplitude (Fig. 9 D). The average CV−2/mean ratio for DHPG consequently mapped onto the line of unity (Fig. 4 D).
Figure 9. DHPG shortens the evoked IPSCs decay phase in PAG descending projection neurons.
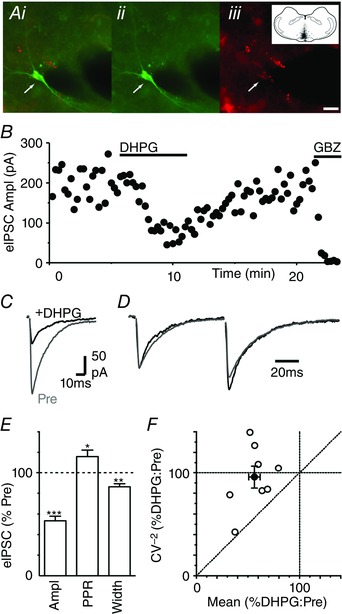
A, photomicrographs showing (i) merged and (ii, iii) individual images of biocytin and fluorescent microsphere labelling in a PAG neuron, following RVM injection (inset shows RVM injection site in black). Scale bar = 20 μm. B, time plots of evoked IPSC amplitude during superfusion of DHPG (10 μm) and gabazine (10 μm). C and D, averaged (C) raw and (D) normalized traces of evoked IPSCs in the same PAG neuron before (Pre) and during DHPG. A–D are from the same neuron. E, bar chart of the effect of DHPG on evoked IPSC amplitude, paired pulse ratio (PPR) and width, shown as a percentage of the pre‐DHPG value. * P < 0.05, **0.01, ***0.0001 for DHPG versus pre‐DHPG. F, plot of the effect of DHPG on the CV−2 versus mean of evoked IPSCs, expressed as a percentage of the pre‐DHPG value. Data are shown for individual neurons (open symbols) and the average for all neurons tested (filled symbols). [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
In the present study we have demonstrated that endocannabinoids modulate inhibitory GABAergic synaptic transmission onto PAG descending projection neurons by altering the mode and not just the probability of vesicle release. Under basal conditions, GABA release was multivesicular. This release mode would act to stabilize the unreliable, stochastic nature of synaptic transmission, thereby ensuring inhibition of the descending analgesic pathway. Activation of the endocannabinoid system shifted this to a lower probability, univesicular mode of GABA release. This switch to univesicular release would facilitate activation of the descending analgesic pathway by reducing GABAergic inhibition of PAG output neurons. This multifaceted regulation of GABAergic synaptic transmission provides a mechanistic foundation for the control of PAG output projection neurons by disinhibition.
Group I mGlu receptor activation reduces GABAergic synaptic transmission via multiple mechanisms
In the present study, DHPG reduced the amplitude of evoked IPSCs in normal conditions and when release probability was reduced by lowering the concentration of external Ca2+. This inhibition was associated with an increase in the PPR of evoked IPSCs under both conditions. In addition, DHPG produced a reduction in the rate but not the amplitude of spontaneous miniature IPSCs. These observations were consistent with DHPG producing a presynaptic reduction in the probability of GABA release (Drew & Vaughan, 2004).
DHPG produced a similar reduction in the CV−2 and the mean amplitude of evoked IPSCs in low external Ca2+. This was also consistent with a presynaptic reduction in release probability (Faber & Korn, 1991). This was not the case in normal external Ca2+ where DHPG produced a lesser reduction in CV−2 compared to the mean amplitude of evoked IPSCs. In addition, DHPG produced a reduction in the decay and rise times of evoked IPSCs in normal but not low external Ca2+. Together, these observations suggest that additional mechanisms become involved in the DHPG modulation of GABA release in normal external Ca2+. The effect of DHPG in normal external Ca2+ was not due to a direct action on GABAA or other postsynaptic ion channels because it had no effect on the kinetics of quantal miniature IPSCs. In addition, the effect of DHPG was not due to a reduction in GABA spill‐over because the modulation of evoked IPSC CV−2 and kinetics was unaffected by GABA transporter blockers (Wadiche & Jahr, 2001). An alternative explanation for the observed effects of DHPG was a shift in the mode of vesicular release. In hippocampal CA3 neurons, a disparate change in the CV−2 and mean amplitude of synaptic currents during train stimulation has been attributed to a shift in release mode (Chamberland et al. 2014). In addition, changes in the width of synaptic currents have been attributed to a shift in release mode, possibly involving desynchronized multivesicular release given the observed changes in rise time (Rudolph et al. 2011; Satake et al. 2012).
Release probability and multivesicular release
At some central inhibitory and excitatory synapses, release is multivesicular and can be shifted to a univesicular mode by reducing release probability. Indeed, lowering external Ca2+ produced a reduction in the amplitude of evoked IPSCs and an increase in their PPR, similar to that observed for DHPG. This was consistent with a reduction in release probability. Lowering external Ca2+ also led to a shortening of evoked IPSC kinetics and a lesser decrease in the CV−2 of evoked IPSCs compared to their mean amplitude, similar to that observed for DHPG in normal Ca2+. A shift from a multivesicular to a univesicular release mode leads to a reduction in the concentration of released transmitter which be detected with low affinity antagonists (Wadiche & Jahr, 2001; Foster et al. 2005; Christie & Jahr, 2006; Balakrishnan et al. 2009). In the present study, the low affinity GABAA receptor antagonist TPMPA produced a greater reduction in the amplitude of evoked IPSCs when release probability was reduced by lowering external Ca2+. Furthermore, TPMPA also produced paired pulse facilitation of evoked IPSCs in low but not in normal external Ca2+. This indicates that lowering external Ca2+ reduces the concentration of synaptically released GABA, and is consistent with a shift from multi‐ to univesicular release. It might also be noted that there was paired pulse facilitation of the width of evoked IPSCs in low but not normal external Ca2+. This suggests that the short‐term plasticity associated with paired pulse facilitation in low Ca2+ is due to a shift from univesicular to multivesicular release and not just an increase in release probability, as observed in the cerebellum and hippocampus (Satake et al. 2012; Chamberland et al. 2014; Satake & Imoto, 2014).
A consequence of multivesicular release is that the higher concentrations of synaptically released GABA can lead to saturation of postsynaptic GABAA receptors (Rudolph et al. 2015). We explored the state of GABAergic synapses within PAG by examining the effect of increasing external Ca2+. While increasing external Ca2+ produced a lengthening of the decay phase of evoked IPSCs, it did not alter their amplitude or the TPMPA‐induced reduction in evoked IPSC amplitude. Furthermore, increasing external Ca2+ reduced the CV of evoked IPSCs, and did not alter the PPR of the amplitude of evoked IPSCs. Indeed, the concentration dependence of evoked IPSC amplitude on external Ca2+ indicated that synaptic activation of GABAA receptors was at near saturation levels under normal conditions.
A role for endocannabinoids
We have previously shown that DHPG inhibits GABAergic synaptic transmission via postsynaptic mGlu 5 receptor coupling to the retrograde endocannabinoid signalling system (Drew et al. 2008). In the present study the effect of DHPG on evoked IPSC kinetics was shown to be mediated via the same retrograde signalling cascade. Furthermore, direct agonism at presynaptic cannabinoid receptors produced a change in the kinetics and CV of evoked IPSCs which was similar to that observed for DHPG. This suggests that GABA release within the PAG is multivesicular under basal conditions and that endocannabinoids can dynamically regulate the release mode. It is well known that endocannabinoids control the probability of neurotransmitter release throughout the brain. While endocannabinoid control of release mode has not been reported previously, it has been noted that retrograde endocannabinoid inhibition of glutamate release from climbing fibres to cerebellar Purkinje cells is associated with a reduction in evoked EPSC decay time (Maejima et al. 2001), and that multivesicular release is the dominant release mode at this synapse (Wadiche & Jahr, 2001). In addition, presynaptic control of vesicle release mode has previously been observed for muscarinic acetylcholine receptors in the striatum (Higley et al. 2009), GABAB receptors in the cortex (Chalifoux & Carter, 2010) and group II mGlu receptors in the cerebellum (Rudolph et al. 2011). It is therefore possible that endocannabinoids and other neurotransmitters act via metabotropic receptors to modulate release mode at a wide range of central synapses (Rudolph et al. 2015).
Under basal conditions, tonically released endocannabinoids influence release probability within the PAG (Kawahara et al. 2011; Lau et al. 2014). In the present study, the cannabinoid CB1 receptor antagonist AM251 produced an increase in the decay time but not the amplitude of evoked IPSCs. This suggests that AM251 unmasks a tonic endocannabinoid‐mediated suppression in the level of multivesicular release. This observation is qualitatively similar to inhibitory inputs to CA3 pyramidal neurons where multivesicular release is observed only in the presence of AM251 (Biro et al. 2006). In the current study, the effect of AM251 on IPSC kinetics was greatly enhanced by the dual FAAH and MAGL inhibitor JZL195. This suggests that the actions of basally released endocannabinoids are rapidly terminated by enzymatic degradation within PAG, and that blockade of endocannabinoid degradation greatly enhances the endogenous suppression of higher probability, multivesicular release. Thus, endocannabinoids have the potential to tonically modulate the vesicle release mode, thereby providing a powerful form of presynaptic plasticity. This action could account for the efficacious analgesic actions of JZL195 (Long et al. 2009; Anderson et al. 2014).
Functional significance of switching release modes within the PAG
The PAG descending analgesic pathway is quiescent under basal conditions. This state of quiescence is thought to be maintained by a dense network of GABAergic interneurons which inhibit neuronal outputs from within the PAG. In the present study we have demonstrated that GABA release onto PAG output neurons is multivesicular under basal conditions. Multivesicular release enhances synaptic potency by increasing the concentration of GABA within the synaptic cleft, thereby providing profound suppression of this endogenous analgesic system. The descending PAG analgesic pathway is activated under emergency situations, such as when an organism is subjected to painful, stressful or threatening stimuli, or following treatment with analgesic agents. These stressors induce the rapid release of endocannabinoids within the PAG which, in turn, indirectly activates the descending analgesic pathway (Walker et al. 1999; Hohmann et al. 2005). We have previously shown that endocannabinoids activate PAG neuronal outputs by a presynaptic cannabinoid CB1 receptor‐mediated reduction in GABA release probability (Vaughan et al. 2000; Drew et al. 2008, 2009). In the present study, we have extended this to show that endocannabinoids shift the mode of GABA release from a primarily multi‐ to a univesicular mode. The presynaptic control of both the mode and the probability of release provides a rationale for disinhibition as a means of ensuring rapid and reliable activation of the normally quiescent descending PAG analgesic pathway.
Additional information
Competing interests
The authors declare that they have no competing interests associated with this manuscript.
Author contributions
All the experiments were carried out in the Pain Management Research Institute Research Laboratories, Kolling Institute of Medical Research, Northern Clinical School, The University of Sydney and Royal North Shore Hospital, St. Leonards, New South Wales, Australia. The study was conceived and designed by CWV and GMD, with later contributions to study design by KRA and BKL. Experimental data acquisition, analysis and interpretation of the data was by KRA, GMD, HJJ, BKL and CWV. Drafting and revision of the manuscript was by KRA, GMD, HJJ, BKL and CWV. All authors have approved the final version of the manuscript. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by Australian National Health and Medical Research Council Project Grant 1002680 (CWV) and CJ Martin Biomedical Fellowship 402868 (GMD).
Significance statement
The midbrain periaqueductal grey plays an important role in modulating endogenous analgesic responses to environmental stressors. Normally the outputs from this brain region are maintained in a quiescent state by tonically active GABAergic inputs. Endocannabinoids activate, or disinhibit, these output neurons by presynaptically reducing GABA release. In this study we show that GABAergic synaptic transmission is normally multivesicular and that endocannabinoids shift this to a univesicular release mode. This dual modulation of release mode and probability provides a mechanistic basis for disinhibition as a means for the tight regulation of outputs from this brain structure.
References
- Anderson WB, Gould MJ, Torres RD, Mitchell VA & Vaughan CW (2014). Actions of the dual FAAH/MAGL inhibitor JZL195 in a murine inflammatory pain model. Neuropharmacology 81, 224–230. [DOI] [PubMed] [Google Scholar]
- Bagley EE, Hacker J, Chefer VI, Mallet C, McNally GP, Chieng BC, Perroud J, Shippenberg TS & Christie MJ (2011). Drug‐induced GABA transporter currents enhance GABA release to induce opioid withdrawal behaviours. Nat Neurosci 14, 1548–1554. [DOI] [PubMed] [Google Scholar]
- Balakrishnan V, Kuo SP, Roberts PD & Trussell LO (2009). Slow glycinergic transmission mediated by transmitter pooling. Nat Neurosci 12, 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender VA, Pugh JR & Jahr CE (2009). Presynaptically expressed long‐term potentiation increases multivesicular release at parallel fibre synapses. J Neurosci 29, 10974–10978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biro AA, Holderith NB & Nusser Z (2006). Release probability‐dependent scaling of the postsynaptic responses at single hippocampal GABAergic synapses. J Neurosci 26, 12487–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifoux JR & Carter AG (2010). GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron 66, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberland S, Evstratova A & Toth K (2014). Interplay between synchronization of multivesicular release and recruitment of additional release sites support short‐term facilitation at hippocampal mossy fibre to CA3 pyramidal cells synapses. J Neurosci 34, 11032–11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM & Jahr CE (2006). Multivesicular release at Schaffer collateral‐CA1 hippocampal synapses. J Neurosci 26, 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew GM, Lau BK & Vaughan CW (2009). Substance P drives endocannabinoid‐mediated disinhibition in a midbrain descending analgesic pathway. J Neurosci 29, 7220–7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew GM, Mitchell VA & Vaughan CW (2008). Glutamate spillover modulates GABAergic synaptic transmission in the rat midbrain periaqueductal grey via metabotropic glutamate receptors and endocannabinoid signalling. J Neurosci 28, 808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew GM & Vaughan CW (2004). Multiple metabotropic glutamate receptor subtypes modulate GABAergic neurotransmission in rat periaqueductal grey neurons in vitro . Neuropharmacology 46, 927–934. [DOI] [PubMed] [Google Scholar]
- Faber DS & Korn H (1991). Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J 60, 1288–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Basbaum AI & Heinricher MM (2006). Central nervous systems mechanisms of pain modulation In Textbook of Pain, 5th edn, ed. McMahon SB. & Koltzenburg M, pp. 125–142. Elsevier, Churchill Livingston, Philadelphia. [Google Scholar]
- Foster KA, Crowley JJ & Regehr WG (2005). The influence of multivesicular release and postsynaptic receptor saturation on transmission at granule cell to Purkinje cell synapses. J Neurosci 25, 11655–11665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR & Bains JS (2005). Noradrenaline triggers multivesicular release at glutamatergic synapses in the hypothalamus. J Neurosci 25, 11385–11395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstetter FJ, Tershner SA, Poore LH & Bellgowan PS (1998). Antinociception following opioid stimulation of the basolateral amygdala is expressed through the periaqueductal grey and rostral ventromedial medulla. Brain Res 779, 104–118. [DOI] [PubMed] [Google Scholar]
- Higley MJ, Soler‐Llavina GJ & Sabatini BL (2009). Cholinergic modulation of multivesicular release regulates striatal synaptic potency and integration. Nat Neurosci 12, 1121–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YC, Lee HJ, Tung LW, Liao YY, Fu SY, Teng SF, Liao HT, Mackie K & Chiou LC (2011). Activation of orexin 1 receptors in the periaqueductal grey of male rats leads to antinociception via retrograde endocannabinoid (2‐arachidonoylglycerol)‐induced disinhibition. J Neurosci 31, 14600–14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G & Piomelli D (2005). An endocannabinoid mechanism for stress‐induced analgesia. Nature 435, 1108–1112. [DOI] [PubMed] [Google Scholar]
- Kawahara H, Drew GM, Christie MJ & Vaughan CW (2011). Inhibition of fatty acid amide hydrolase unmasks CB1 receptor and TRPV1 channel‐mediated modulation of glutamatergic synaptic transmission in midbrain periaqueductal grey. Brit J Pharmacol 163, 1214–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keay KA & Bandler R (2001). Parallel circuits mediating distinct emotional coping reactions to different types of stress. Neurosci Biobehav Rev 25, 669–678. [DOI] [PubMed] [Google Scholar]
- Lau BK, Drew GM, Mitchell VA & Vaughan CW (2014). Endocannabinoid modulation by FAAH and monoacylglycerol lipase within the analgesic circuitry of the periaqueductal grey. Brit J Pharmacol 171, 5225–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau BK & Vaughan CW (2014). Descending modulation of pain: the GABA disinhibition hypothesis of analgesia. Curr Opin Neurobiol 29, 159–164. [DOI] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim‐Selley LJ, Lichtman AH, Wiley JL & Cravatt BF (2009). Dual blockade of FAAH and MAGL identifies behavioural processes regulated by endocannabinoid crosstalk in vivo . Proc Natl Acad Sci USA 106, 20270–20275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Aiba A & Kano M (2001). Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31, 463–475. [DOI] [PubMed] [Google Scholar]
- Martin WJ, Patrick SL, Coffin PO, Tsou K & Walker JM (1995). An examination of the central sites of action of cannabinoid‐induced antinociception in the rat. Life Sci 56, 2103–2109. [DOI] [PubMed] [Google Scholar]
- Mitchell VA, Jeong HJ, Drew GM & Vaughan CW (2011). Cholecystokinin exerts an effect via the endocannabinoid system to inhibit GABAergic transmission in midbrain periaqueductal grey. Neuropsychopharmacology 36, 1801–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell VA, Kawahara H & Vaughan CW (2009). Neurotensin inhibition of GABAergic transmission via mGluR‐induced endocannabinoid signalling in rat periaqueductal grey. J Physiol 587, 2511–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira MA & Prado WA (2001). Role of PAG in the antinociception evoked from the medial or central amygdala in rats. Brain Res Bull 54, 55–63. [DOI] [PubMed] [Google Scholar]
- Rudolph S, Overstreet‐Wadiche L & Wadiche JI (2011). Desynchronization of multivesicular release enhances Purkinje cell output. Neuron 70, 991–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph S, Tsai MC, von Gersdorff H & Wadiche JI (2015). The ubiquitous nature of multivesicular release. Trends Neurosci 38, 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake S & Imoto K (2014). Cav2.1 channels control multivesicular release by relying on their distance from exocytotic Ca2+ sensors at rat cerebellar granule cells. J Neurosci 34, 1462–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake S, Inoue T & Imoto K (2012). Paired‐pulse facilitation of multivesicular release and intersynaptic spillover of glutamate at rat cerebellar granule cell‐interneurone synapses. J Physiol 590, 5653–5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Kovalchuk Y & Attwell D (1995). Pre‐ and postsynaptic determinants of EPSC waveform at cerebellar climbing fibre and parallel fibre to Purkinje cell synapses. J Neurosci 15, 5693–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Connor M, Bagley EE & Christie MJ (2000). Actions of cannabinoids on membrane properties and synaptic transmission in rat periaqueductal grey neurons in vitro. Mol Pharmacol 57, 288–295. [PubMed] [Google Scholar]
- Wadiche JI & Jahr CE (2001). Multivesicular release at climbing fibre‐Purkinje cell synapses. Neuron 32, 301–313. [DOI] [PubMed] [Google Scholar]
- Walker JM, Huang SM, Strangman NM, Tsou K & Sanudo‐Pena MC (1999). Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci USA 96, 12198–12203. [DOI] [PMC free article] [PubMed] [Google Scholar]