

Abstract
Post-translational modifications (PTMs) are critical molecular events which alter protein conformation after their synthesis and diversity protein properties by modulating their stability, localization, interacting partners or the activity of their substrates, consequently exerting pivotal roles in regulating the functions of many important eukaryotic proteins. It has been well acknowledged that PTMs are of great importance in a broad range of biological processes such as gene regulation, cell proliferation, differentiation and apoptosis, tissue development, diseases, tumor progression and drug resistance. As the core and contributing catalytic subunit of Polycomb repressive complex 2(PRC2), Enhancer of zeste homolog 2 (EZH2) is a master epigenetic regulator, often serving as a highly conserved histone methyltransferase (HMTase) to induce histone H3 lysine 27 trimethylation (H3K27me3) and repress gene transcription and expression. Dysregulated EZH2 expression is frequently associated with cancer development and poor prognosis in a wide variety of cancers. Considered its essential role in carcinogenesis, EZH2 is a potential candidate for cancer targeted therapy. Remarkably, mounting evidence highlights that EZH2 expression, activity and stability can be regulated by PTMs including phosphorylation, acetylation, ubiquitination, sumoylation and GlcNAcylation aside from its well-validated modifications in transcriptional and post-transcriptional levels. However, the precise regulatory mechanisms underlying EZH2 PTMs and whether other types of PTMs orchestrate in EZH2 remain largely unclear. In this review, we summarize current advances in the understanding of EZH2 regulation by PTMs and their associated biological functions during tumorigenesis.
Keywords: Post-translational modifications (PTMs), polycomb group (PcG), polycomb repressive complex 2(PRC2), enhancer of zeste homolog 2 (EZH2), histone H3 lysine 27 trimethylation (H3K27me3)
Introduction
To the best of our knowledge, there are two major mechanisms responsible for the diversity of proteome, resulting in the number of proteins far exceeds that is estimated by DNA coding capacities: one is mRNA splicing, the other is PTMs [1]. PTMs, which occur after the completion of protein translation, regulate protein structures and functions by covalent addition of functional groups, peptides or other complex molecules reversibly or irreversibly, leading to extend and diversify protein properties and enhance the ability of modulating protein stability, localization and activity themselves or their interacting proteins [2,3]. To date, more than 300 types of PTMs are identified to occur physiologically, of which the major and common ones are methylation, acetylation, phosphorylation, ubiquitination, glycosylation and sumoylation [4,5]. PTMs are closely associated with many important biological processes involved in cellular proliferation and differentiation [6], organismal development [7] and human disease disorders including neurodegeneration [8], cardiovascular diseases [9] and cancer [10]. In recent years, PTMs have gradually emerged as one of available and diverse strategies for the fine-tuned modification of some essential regulators in the development of cancer, such as EZH2. Therefore, it is pivotal to explore the diversity of PTMs and clarify their complex mechanisms functioning in cellular regulation, homeostasis maintenance and carcinogenesis.
Epigenetic abnormalities represent key players in the initiation and progression of cancer [11]. PcG proteins are important epigenetic regulators that have been recognized as transcriptional repressors and key regulators of cell fate in cancer development [12]. Consisting of two complexes Polycomb Repressive Complex 1 and 2 (PRC1 and PRC2) [13], PcG proteins were originally identified as essential factors in genetic screens for homeotic transformations to regulate body segmentation during Drosophila melanogaster embryogenesis [14,15]. Among the subunits of PRC2, EZH2 is a highly conserved HMTase, functioning to trigger H3K27me3 and silence transcription and expression of target genes associated with numerous crucial biological processes including cell cycle regulation, cell fate decision, senescence, cell proliferation, differentiation, apoptosis and tumorigenesis [12,16]. Being a core epigenetic regulator, aberrant EZH2 expression is widely implicated in a broad array of aggressive and metastatic malignancies with poor prognosis [17-22]. It has been widely accepted that EZH2 can be regulated in a wide variety of human cancers at both transcriptional and post-transcriptional levels [23]. For example, E2Fs can bind to the promoter of EZH2 and transactivate its expression at transcriptional level [24]. At post-transcriptional level, EZH2 transcript can be regulated by a large number of micro-RNAs, such as miR-26a, miR-32, miR-101, miR-137, miR-138 and miR-506 [25-30]. Besides, accumulating evidence currently supports that EZH2 can also be modulated by PTMs in the development of cancer. Recent studies have focused on the functional importance of some EZH2 PTMs in particular involved in phosphorylation [31], acetylation [32], ubiquitylation [33], sumoylation [34] and O-GlcNAcylation [35]. However, the amount of relevant reports is largely limited and it remains obscure that how different kinds of PTMs orchestrate in EZH2 and whether other unknown types of EZH2 PTMs are also implicated in, indicating that research in this aspect is still in its early inception. In this review, we provide a current overview of the molecular mechanisms and biological functions of EZH2 PTMs in cancer development.
Overview of polycomb group (PcG) proteins and EZH2 in cancer
Initially verified in Drosophila as crucial epigenetic regulators, PcG and Trithorax Group (TrxG) proteins act antagonistically to control body segmentation by silencing or activating the expression patterns of homeotic genes (HOX genes) throughout development, respectively [36,37]. Later, both of these two types of proteins have been shown to be highly conserved from Drosophila to mammals and exert their efforts in many essential epigenetic regulatory processes implicated in embryogenesis, X chromosome inactivation, chromatin modification, stem cell development and tumor progression [38]. PcG proteins usually serve as a set of transcriptional repressors to regulate gene silencing related to cellular development, cell cycle and cell fate decision, whereas their counterparts TrxG proteins principally act in opposition to sustain a status of transcriptional activation [13]. Taken together, they corporately maintain a delicate balance and fine tuning of HOX gene activity and expression in the process of embryogenesis and development.
In mammals, PcG proteins are known to perform their repressive functions by forming two different protein multimeric complexes named PRC1 and PRC2 [13]. PRC1 composition is often variable and the mammalian core PRC1 consists of Bmi1, RING1 proteins (RING1A and RING1B), CBX, PH1, PH2, NSPC1 (Pcgf1) and MEL18 (Pcgf2) proteins. The RING1 proteins have the E3 ligase activity to specifically catalyze the monoubiquitylation of histone H2A at lysine119 (H2AK119ub), leading to a repressive chromatin structure with gene silencing [39]. Notably, RING1B-mediated chromatin compaction also functions to repress gene transcription and expression independent on its histone ubiquitination activity [40]. In addition, unlike previously published that PRC1-mediated blockage of chromatin remodeling is controlled by SWI/SNF complex [41], it has been reported that activation of SWI/SNF activity displaces PRC1-induced chromatin silencing in malignant rhabdoid tumor(MRT) cells [42]. Other relevant mechanism about dissociation of RNA polymerase II preinitiation complexes (PICs) by PRC1 also participates in PRC1-induced transcriptional silencing [43]. Altogether, these findings highlight that it remains poorly understood about PRC1-dependent transcriptional regulation.
The core subunits of mammalian PRC2 are generally composed of EZH2 or EZH1, EED, SUZ12 and RbAp46/48. Other components accessory to regulate PRC2 enzymatic activity and function include AEBP2, PCLs and JARID2 [14]. EZH2, as the human homolog 2 of Drosophila protein Enhancer of Zeste (E(z)), is a HMTase and the core catalytic subunit of PRC2 which catalyzes H3K27me3 via a conserved SET domain to initiate transcriptional repression of PcG target genes [44]. Currently, it has been well-identified that EZH2-mediated H3K27me3 might serve as a docking site for PRC1 chromo-domain containing protein CBX and facilitate the initial recruitment of PRC1 that catalyzes H2AK119ub to keep a repressed state of target genes [39,45-47], indicating a common and classic regulatory model that PRC1 functions downstream of PRC2. However, it was challenged by the findings that some genes are targeted by PRC2 without PRC1-mediated H2AK119ub [48] and others are triggered by PRC1 in a PRC2-independent manner [49,50]. Additionally, histone deacetylation is also beneficial to reinforce PcG-directed gene repression [51-53]. Thus, further explorations will be necessary to shed light on the relationship between PRC1, PRC2 and other factors in the maintenance of transcriptional regulation in different context-specific situations.
Epigenetic alternation leads to abnormal gene expressions that may result in dysregulated physiological functions in human diseases such as cancers. As a master epigenetic player, accumulated evidence confirm that overexpression of EZH2 is widely implicated in a broad array of aggressive and metastatic malignancies especially first recognized prostate and breast cancer [17,18]. Later, a large amount of human cancers such as hepatocellular carcinoma, gastric cancer, non-small-cell lung cancer and hematopoietic malignancies are detected with EZH2 alteration [19-22]. Furthermore, high EZH2 expression is frequently associated with tumor progression, metastasis and poor prognosis [54-57]. In addition, EZH2 overexpression is also involved in malfunction of some key signaling pathways in cancers, including Wnt/β-catenin signaling [22,58], Ras and NF-kB signaling pathways [59], PI3K/AKT pathway [60], β-adrenergic receptor signaling [61], bone morphogenetic protein [62] and Notch signaling pathways [63]. Given these findings above, EZH2 was supposed to act as an oncogene.
It is widely acknowledged that EZH2 can be regulated in a broad spectrum of tumors at both transcriptional and post-transcriptional levels. For example, transcriptional factors E2Fs can bind to the promoter of EZH2 and transactivate its expression, essential for EZH2-mediated cell proliferation in cancers [24]. Remarkably, ANCCA, as AAA+ ATPase-containing nuclear coactivator for the estrogen and androgen receptors, is critical for E2F-induced EZH2 transcription in both triple negative/basal-like cancer and prostate cancer cells [64,65]. Moreover, Myc oncoprotein either directly interacts with EZH2 promoter and activates its transcription, or increases EZH2 expression by inhibition of miR-26a and miR-26b in prostate cancer cells [24]. Additionally, EWS-FLI1 fusion oncoprotein and several well-known transcription factors including SOX4, NF-Y, STAT3 and ETS are also demonstrated to directly regulate EZH2 transcription and expression in Ewing tumors, epithelial breast, ovarian, colorectal, and prostate cancer cells, respectively [66-70].
In addition to transcriptional regulation, a large amount of miRNAs such as miR-26a, miR-32, miR-101, miR-137, miR-138 and miR-506 [25-30] have recently been reported to be implicated in the regulation of EZH2 expression directly in cancers at post-transcriptional level. What’s more, tumor microenvironment, hypoxia for instance, can effectively enhance HIF-α-mediated transactivation of EZH2 in the expansion of breast tumor initiating cell (BTIC) and breast cancer progression [71]. However, by comparison, research on PTMs associated with modulation of EZH2 itself is still less widely concerned.
PTMs of EZH2
Although an increasingly number of studies have been focused on the role of EZH2 in cancer pathogenesis, few have been devoted to investigation of its regulation at post-translational level. Recently it has been reported that EZH2 is modulated by multiple PTMs including phosphorylation, acetylation, ubiquitination, sumoylation and O-GlcNAcylation. Herein, we reviewed these dominant EZH2 PTMs and their relevant biological consequences in carcinogenesis. The effects of PTMs on EZH2 functions are summarized in Table 1.
Table 1.
Regulation of EZH2 through PTMs and their functions
| Type of PTMs | Condition | Modifying enzyme | Site | Biological Functions | Ref. |
|---|---|---|---|---|---|
| Phosphorylation | IGF-induced | AKT | S21 | Decreases H3K27me3 level, releases EZH2 silenced target genes and promotes tumor development. | [31] |
| Non-genomic ER signaling | AKT | S21 | Reduces the activity of H3K27me3 and HMTase. | [77] | |
| Tat | AKT | S21 | Decreases H3K27me3 level and increases EZH2 cytoplasmic translocation, activating HIV-1 transactivation. | [80,81] | |
| As3+ | AKT | S21 | Enhances Akt activation by upregulation of negative Akt regulator miR-21 via STAT3 S27 phosphorylation, contributing to pS21 EZH2 and oncogenesis. | [82,83] | |
| GSCs | AKT | S21 | Promotes EZH2-mediated STAT3 methylation, enhances EZH2-STAT3 interaction and STAT3 activity | [84] | |
| - | CDK1 and CDK2 | T350 in human | Enhances the silencing of target genes without affecting intrinsic HMTase activity or core PRC2 complex formation, promotes EZH2-mediated cell proliferation and migration. | [86] | |
| - | CDK1 | T345 in Mouse | Increases Ezh2 interaction with HOTAIR and the 5’end of Xist, further mediating PRC2 recruitment to its target loci and tumor progression. | [87] | |
| - | CDK1 | T492 in human (T487 in Mouse) | 1. Inhibit HMTase activity, disrupt binding to other PRC2 components | [87,91,92] | |
| 2. Promote ubiquitination and degradation by proteasome pathway | |||||
| - | CDK1 | T487 in Human | Suppresses EZH2 HMTase activity and disassociates EZH2 from other PRC2 complex components, finally inhibiting cancer cell migration and invasion and promoting human mesenchymal stem cells differentiation into osteoblasts. | [91] | |
| MKK6 or TNFα | p38α Kinase | T372 in Mouse | Enhances interaction of YY1 and PRC2, represses Pax7 expression and impair satellite cell proliferation | [96] | |
| Acetylation | - | PCAF | K348 | Enhances EZH2 stability, promotes lung cancer cell migration and invasion | [32] |
| Ubiquitination | - | Smurf2 | L421 | Upregulates target gene PPARγ, essential for neuron differentiation of hMSCs and functional regeneration of CNS repair after ischaemic stroke | [33] |
| Jak2-induced pY461 EZH2 | β-TrCP | - | Reduces EZH2 protein stability and H3K27me3 activity | [110] | |
| YC-1, PKA and Src-Raf-1-MEK-ERK pathways | c-Cbl | T731 and T774 | Leads to Src and ERK activation, resulting in formation of c-Cbl-ERK-EZH2 complex and enhancement of EZH2 uniquitination and degradation. | [111] | |
| DZNep treatment | PRAJA1 | - | Ub-mediated proteasomal degradation of individual PRC2 subunits including EZH2, SUZ12 and EED | [112] | |
| FOXP3 | PRAJA1 | - | Facilitates EZH2 protein degradation through K48-linkage polyubiquitination and decreases cell proliferation, migration and formation in breast cancer cells | [113] | |
| - | ω-3 PUFAs | - | Decreases EZH2 expression and H3K27me3 level, upregulates EZH2 downstream target genes E-cadherin and IGFBP3, leading to suppression of tumor invasion and metastasis. | [114] | |
| Sumoylation | - | - | - | EZH2 are sumoylated in vitro and vivo, but the biological functions remain unknown. | [34] |
| O-GlcNAcylation | OGT-dependent | - | S75 | Maintains EZH2 protein stability H3K27me3 activity, eventually contributing to tumorigenesis | [35] |
Phosphorylation
As one of the best studied PTMs, phosphorylation of proteins is an essential regulatory mechanism that occurs in both prokaryotic and eukaryotic organisms. It is a reversible regulatory network which can potentially be regulated by more than 500 human protein kinases and at least 150 phosphatases [72,73]. Phosphorylation usually occurs when protein kinases add phosphate groups in an ATP-dependent manner to serine (Ser), threonine (Thr), tyrosine (Tyr) and histidine (His) residues of substrates [74] (Figure 1), which finally results in a conformational change in the structure of many proteins, causing them to be activated or deactivated and consequently creating differences in the biological properties of their targets and binding affinities [75]. Of note, there are multiple distinct phosphorylation sites on a given protein and phosphorylation on any site can probably change the function or localization of that protein. The total known phosphorylation sites of EZH2 at present are illustrated in Figure 2. Here we summarize several main phosphorylation sites at EZH2 according to available data and relevant influence to their biological functions.
Figure 1.
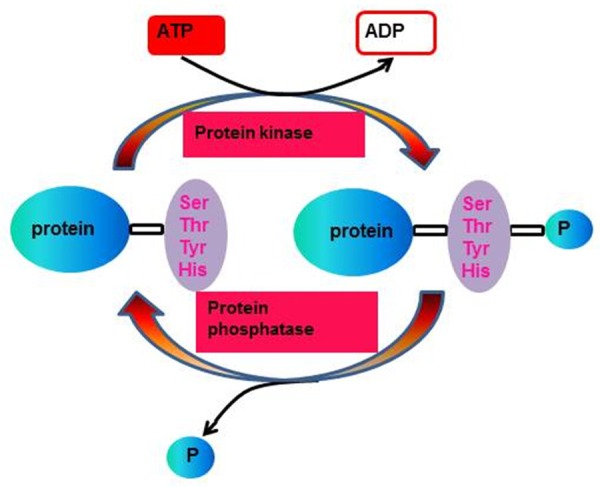
The general process of protein phosphorylation and dephosphorylation.
Figure 2.
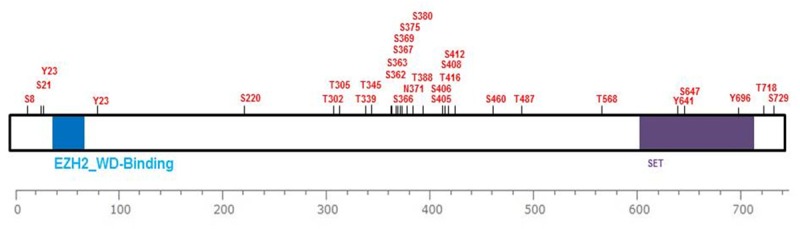
Schematic representation of the currently total known phosphorylation sites at human EZH2 (from www.phosphosite.org).
Phosphorylation of EZH2 at Ser21 (pS21 EZH2)
Several recent studies have identified that EZH2 Ser21 is known to be phosphorylated under different conditions. Cha et al. (2005) [31] firstly reported phosphorylation of EZH2 at Ser21 (pS21 EZH2) by insulin-like growth factor (IGF)-induced activation of Akt in breast cancer cells. Akt suppresses EZH2 HMTase activity by disrupting the affinity for histone H3 rather than changing its subcellular localization or its interaction with other PcG proteins Suz12 and Eed, eventually leading to a decreased H3K27me3 level, release of EZH2 silenced target genes and promote tumor development. Notably, Akt-mediated pS21 EZH2 did not compromise PRC complex composition, suggesting that phosphorylated-EZH2 complex may contribute to tumorigenicity by targeting other crucial non-histone substrates. Instead of transcriptional repressive role of EZH2, pS21 EZH2 by PI3K/AKT signaling has recently been confirmed to perform as a transcriptional coactivator in castration-resistant prostate cancer (CRPC) [76], Intriguingly, either silencing of PRC2 subunits SUZ12 or EED or lack of H3K27me3 has no effects on EZH2-activated genes, implying a positive role of EZH2 for both gene activation and androgen-independent tumor growth without relying on PRC2-mediated methyltransferase activity. Moreover, Akt-dependent pS21 EZH2 can also be observed in MCF-7 breast cancer cells by non-genomic estrogen receptor (ER)-mediated signaling in response to both 17β-estradiol (E2) and the xenoestrogen diethylstilbestrol (DES), resulting in the reduction of H3K27me3 and HMTase activity [77]. Activation of non-genomic ER signaling gave rise to reprogramming of the expression profile of estrogen-responsive genes in uterine myometrial cells, indicating a novel regulatory mechanism that nuclear hormone receptor signaling-related epigenetically modulation of chromatin structure during tissue development. Remarkably, previous reports have revealed that the association of DES exposure with poor reproductive outcome and neoplasia, indicating the increased risk of tumorigenesis when exposure with estrogen [78,79]. However, it remains to be further elucidated the progressive or suppressive role of ER-mediated pS21 EZH2 in cancer development. Additionally, the HIV-1 transactivator of transcription (Tat) protein [80]-mediated pS21 EZH2 by ROS/Akt signaling pathway may lead to decreased levels of H3K27me3 and increased EZH2 cytoplasmic translocation, acting as a potential activator for HIV-1 transactivation [81].
Besides IGF or estrogen-induced Akt activation, another recent report [82,83] has demonstrated that arsenic (As3+) is responsible for triggering Akt-dependent pS21 EZH2 through activation of JNK-STAT3-Akt signaling axis in human bronchial epithelial cells, revealing a novel mechanism underlying arsenic and other related metal carcinogens-induced carcinogenesis. Mechanistically, As3+-mediated growth-promoting role of JNK pathways can be achieved by STAT3 S27 phosphorylation, which in turns enhanced Akt activation by upregulation of negative Akt regulator miR-21, eventually contributing to pS21 EZH2 and oncogenesis. Interestingly, As3+-induced S21-phosphorylated EZH2 is largely localized in the cytoplasm, which is opposite from our traditional notion that EZH2 is predominantly a nuclear protein. This discrepancy possibly indicates that pS21 EZH2 by As3+ induction may have the ability to dissociation of the PRC2 complex from chromatin and translocation of EZH2 protein from the nucleus to the cytoplasm, where they can exert novel functions by interacting with other cytoplasmic proteins.
Furthermore, Akt-induced pS21 EZH2 can facilitate EZH2-STAT3 interaction, promote EZH2-mediated STAT3 methylation and enhance STAT3 activity in Gliblastoma multiforme stem-like cells (GSCs), suggesting that Akt-pS21 EZH2-STAT3 signaling axis is a potential regulator for GSCs tumor malignancy and a promising cancer therapeutic target for Gliblastoma multiforme [84].
Phosphorylation of EZH2 by CDKs
Emerging evidence suggests that the activities of specific cyclin-dependent kinases (CDKs) are required for proliferation of tumor cells [85], while elevated EZH2 expression is highly associated with tumor initiation and progression, implying a closed relationship between EZH2 functions and CDKs.
Phosphorylation of EZH2 at Thr350 (Thr345 in mouse)
Consistent with the hypothesis above, Chen et al. (2010) reported that EZH2 indeed harbored one perfectly matched (Thr350) and two imperfectly matched (Thr421 and Thr492) CDKs phosphorylation motifs (K(R)S(T)PXK(R) which are highly evolutionally conserved from fruit flies to humans [86]. By in vitro protein-kinase assays, they confirmed that mutation of Thr350 to alanine (T350A) resulted in approximately 60% reduction in CDK1-mediated EZH2 phosphorylation, whereas only about 30% or no reduction in phosphorylation was observed in T421A and T492A mutants, highlighting that Thr350 is a major CDKs-mediated phosphorylation site. Furthermore, they further illustrated that phosphorylation of Thr350 in human EZH2 (pT350 EZH2) can be mediated by CDK1 and CDK2 under physiological conditions, which is essential for recruitment of EZH2 and maintenance of H3K27me3 level at EZH2 target loci in cells. pT350 EZH2 significantly enhances its strength for silencing of target genes without affecting intrinsic HMTase activity or core PRC2 complex formation, but probably affects effectively recruitment of other PRC2 components to its target loci. Moreover, pT350 EZH2 is also responsible for promoting EZH2-mediated cell proliferation and migration. Blockage of its phosphorylation attenuates EZH2 oncogenic activity and its genome-wide repression of gene transcription [86]. Taken together, these data imply that CDK-induced phosphorylation plays a key role in governing EZH2 function and it links cell-cycle regulatory mechanism to epigenetic gene silencing.
Indeed, Thr345 residue in mouse Ezh2, a homologue site of Thr350 in human EZH2, can also be phosphorylated by CDK1 in a cell-cycle dependent manner in vitro and in vivo [87]. The amount of phosphorylation of Thr345 residue in mouse Ezh2 (pT345 Ezh2) was limited in only 1%. Its level was increased in G2/M phase, then was declined to a basal G1 phase level, consistent with the characteristics of CDK1 activity which is a G2/M specific kinase [85,88]. Similar to the role of pT350 EZH2, pT345 Ezh2 carries out its program neither affects HMTase activity nor disrupts PRC2 complex formation, nevertheless it increases Ezh2 interaction with noncoding RNAs (ncRNAs) HOTAIR and the 5’end of Xist (X-inactive specific transcript) which further mediate PRC2 recruitment to its target loci [87]. It is suggested that there is a closed correlation between ncRNA and EZH2 phosphorylation, opening a brand new avenue for exploring the role of ncRNA in PRC2-mediated regulation. In conclusion, both pT350 EZH2 and its counterpart pT345 Ezh2 function by affecting PRC2 components to its target loci, further demonstrating that this phosphorylation site is critical and highly conserved.
Phosphorylation of EZH2 at Thr492 (Thr487 in mouse) and Thr487
In addition to pT350 EZH2, phosphorylation of EZH2 Thr492 (pT492 EZH2) can also be mediated by CDK1 in a cell-cycle regulatory manner which was identified by stable isotope labeling along with phosphorpeptide enrichment and high mass accuracy mass spectrometry [89]. Importantly, the same phosphorpeptide by CDK1-cyclinB1 complex is also demonstrated in vitro by GST-fusion assay [86], implying that Thr492 in human EZH2 is indeed phosphorylated by CDK1. In contrast, pT492 EZH2 disassociates EZH2 from other PRC2 subunits SUZ12 and EED and consequently reduces the activity of EZH2 HMTase [90]. Thr487 in mouse Ezh2, the counterpart of pT492 EZH2, can also be phosphorylated in CDK1-involved cell-cycle regulation [87,91]. Given that Thr487 and Thr345 have common phosphorylation kinase and function in the same manner, we hypothesized that there are some similarities between phosphorylation of these two sites. However, different from pT345 Ezh2, phosphorylation of Thr487 in mouse Ezh2 (pT487 Ezh2) suppresses trimethylation of H3K27 through inhibiting EZH2 HMTase activity as well as disrupting the interaction of EZH2 to other PRC2 complex components SUZ12 and EED, which finally reduces the ability of EZH2 to silence target genes and inhibits cancer cell migration and invasion. Hence, pT487 Ezh2 prefers to perform tumor suppressive function.
Nevertheless, a similar discovery about CDK1-dependent phosphorylation of EZH2 but at a different residue Thr487 (pT487 EZH2) [91] arrives at a completely distinct conclusion that pT487 EZH2 by CDK1 has a negative impact on its HMTase activity-suppresses trimethylation of H3K27 through inhibiting EZH2 HMTase activity as well as disrupting the interaction of EZH2 to other PRC2 complex components, SUZ12 and EED, which in turn reduces the ability of EZH2 to silence target genes and finally inhibits cancer cell migration and invasion and promote human mesenchymal stem cells differentiation into osteoblasts.
In addition, another investigation [92] which also focuses on CDK1-mediated phosphorylation of EZH2 reveals that pT345 Ezh2 in mouse (pT350 EZH2 in human) and pT487 Ezh2 in mouse (pT492 EZH2 in human) are not very indispensable for global levels of H3K27me3, because they neither affect intrinsic HMTase activity nor disrupt interaction of EZH2 with other PRC2 complex subunits but promote EZH2 ubiquitination and subsequent degradation by proteasome pathway.
Several recent studies regarding closed correlation between ncRNAs and EZH2 has opened a brand new avenue for modulation of EZH2 with ncRNAs [93-95]. Remarkably, Kaneko et al. stated that phosphorylation of Ezh2 facilitates its binding with ncRNA in a cell-cycle dependent manner, of which the amount is limited even in G2/M phase [87]. Mechanistically, pT345 Ezh2 (pT350 EZH2 in human) enhances the recruitment of PRC2 other components to Polycomb target genes mainly through reinforcing EZH2 interaction with ncRNAs including HOTAIR and the 5’end of Xist, and ncRNA-binding domain(ncRBD1) is considered as critical motif for EZH2-ncRNAs interaction. Whereas pT487 Ezh2 was ineffectual for the association with EZH2 and ncRNAs, probably owing to the reason that Thr487 residue is located inside the SUZ12 recognition motif of EZH2 [88]. pT487 Ezh2 in mouse (pT490 EZH2 in human) blocks Ezh2 activity and deposes H3K27 trimethylation by potentially interfering with PRC2 complex formation, which might suppresses carcinogenesis. Taken together, these seemingly controversial but plausible explanations about the consequences of phosphorylation of EZH2 at different residues might indicate that further studies should be undertaken to understand subtle differences of their functions between phosphorylation of EZH2 different residues in the exquisite and specific regulation of their downstream target genes and their completely relevant biological mechanisms.
Phosphorylation of Ezh2 at T372 in mouse (pT372 Ezh2)
Using mass spectrometry (MS) for biological analysis, T372 in a peptide (363-LPN NSS RPS TPT INV LES K-381) of human EZH2 is detected to be phosphorylated in cells synchronized at the mitotic phase of the cell cycle [89,90]. More recently, a novel finding regarding the epigenetic control of muscle regeneration uncovers that activation of mitogen-activated protein kinase p38α by its upstream activator MKK6 or the inflammation cytokine tumor necrosis factorα (TNFα) can induce T372 phosphorylation of Ezh2 (pT372 Ezh2) in mouse C2C12 myoblasts [96]. Mechanistically, TNF-activated p38α kinase promotes the interaction between EHZ2 and Yin Yang 1 (YY1) through pT372 Ezh2, leading to the formation of repressive chromatin on Pax7 promoter and consequently repressed expression of Pax7, a gene essential for muscle stem (satellite) cell proliferation and impaired satellite cell proliferation. Therefore, this report provides a mechanism which links inflammation to the epigenetic control of muscle regeneration and establishes the biological relationship between p38/PRC2 signaling to Pax7 and muscle satellite cell decision to proliferate or differentiate [90,96].
Phosphorylation of EZH2 at other sites
Besides functional phosphorylation sites described above, phospho-proteomic analyses [97] (The PhosphoSitePlus database (www.phosphosite.org) is a curated collection of phosphorylation sites with 13,000 human sites from the literature) in both human EZH2 and mouse Ezh2 have identified many other phosphorylated residues. However, most of those functions have not been fully elucidated, suggesting that further studies are extremely urgent to explore other biological functions of EZH2 phosphorylation.
Phosphorylation of Akt-1 at Ser473 and BRCA1 at Ser1423 by EZH2
In addition to be phosphorylated substrate by different kinases, EZH2 can also function as a kinase to enhance phosphorylation level of its substrates. It has been presently confirmed that overexpression of EZH2 protein is highly associated with increased Akt-isoform 1 phosphorylation at Ser473 and decreased nuclear localization of phospho-BRCA1 tumor suppressor protein at Ser1423 in 39% human invasive breast carcinomas [98]. Collectively, these findings add a novel dimension to understand the functions of EZH2 in breast tumorigenesis: EZH2 control BRCA1 intracellular localization and genomic stability mediated by PI3K/Akt-1 pathway.
Acetylation
Acetylation is a reversible and important form of PTMs, playing a key role in regulating gene expression mainly through the modulation of core histone tails by histone acetyltransferases (HATs) or histone deacetylase s(HDACs) [99,100] and controlling a series of cellular processes including proliferation, apoptosis, differentiation, metabolism and transcriptional regulation [101-103]. It has been well-recognized that besides histones, other non-histones such as nuclear, cytoplasmic, mitochondrial proteins and a large number of transcription factors have also been reported to be targeted and modulated by acetylation [104-106]. However, whether and how EZH2 can be regulated by acetylation remains largely unclear. It was not until recently that Wan and colleagues [32] firstly provided convincing evidence that EZH2 is acetylated by acetyltransferase P300/CBP-associated factor (PCAF) and is deacetylated by deacetylase SIRT1. Mechanistically, PCAF can interact with EZH2 and lead to EZH2 acetylation mainly at lysine348 (K348), which decreases EZH2 phosphorylation at T345 and T487 and enhances EZH2 stability without either changing its interaction with other PRC2 complex members SUZ12 and EED or affecting its location and HMTase activity. Functionally, increased EZH2-K48 acetylation enhances its suppressive effects on the target genes by reinforcing H3K27me3 binding capacity to the target gene promoters, which promotes lung cancer cell migration and invasion and eventually results in a poor prognosis in lung adenocarcinoma patients. Strikingly, EZH2-K48 acetylation gives rise to lower phosphorylation level of T345 and/or T487 at EZH2 may be attributed to the restricted access of CDK1 by a local conformational change, but not vice versa. Moreover, it seems to be difficulty to understand how EZH2-K48 acetylation enhances its stability without influencing its ubiquitination. Therefore, further investigations will be urgent to shed light upon the roles of EZH2 acetylation at other sites and how they orchestrate their crosstalk relationship with other PTMs.
Ubiquitination
Ubiquitin (Ub), consisting of 76 amino acid residues, is an evolutionarily conserved protein dedicated to tagging target proteins for degradation post-translationally [107]. Ubiquitination is a well-recognized PTM process, which covalently attaches Ub to the modified proteins and regulates their stability, functions and localizations involved in multiple cell functions and diseases, especially in cancer development [108,109]. It occurs by activating a cascade of enzymatic reactions dependent on three indispensable enzymes: ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme (E2) and ubiquitin ligase (E3). Recently several studies have identified that Smad ubiquitination regulatory factor-2 (Smurf2) [33], β-TrCP(FBXW1) [110], Casitas B-lineage lymphoma (c-Cbl) protein [111] and PRAJA1 [112,113] serve as dynamic EZH2 ubiquitin E3 ligases. Smurf2 interacts with EZH2 and contributes to the ubiquitination and proteasome-mediated degradation of EZH2 at lysine 421 and consequently upregulates its target gene PPARγ, which are essential for neuron differentiation of human mesenchymal stem cells (hMSCs) and functional regeneration of central nervous system (CNS) repair after ischaemic stroke [33]. It indicates that EZH2 ubiquitination is important for its protein stability and subsequent functions and is probably an attractive approach for clinical use in treatment of neurodegenerative diseases. Moreover, EZH2 has recently been demonstrated as a novel substrate of Skp/cullin/F-box protein (SCF) E3 ubiquitin ligase β-TrCP (FBXW1). EZH2 specially interacts with β-TrCP and undergoes β-TrCP-mediated EZH2 ubiquitination [110]. Remarkably, Jak2-induced phosphorylation at EZH2 Y641 (pY461 EZH2) promotes EZH2-β-TrCP interaction and subsequent β-TrCP-mediated EZH2 ubiquitination and proteasomal degradation and leads to the reduced EZH2 protein stability and H3K27me3 hypoactivity, suggesting a phosphorylation-dependent EZH2 ubiquitination. Considered the findings that EZH2 mutation at Y641 abrogates β-TrCP-mediated degradation and phosphorylation of EZH2 at Y641 promotes β-TrCP-mediated EZH2 ubiquitination, we raise a question that whether EZH2 Y641 is a regulatory site with both phosphorylation and ubiquitination, and if that is the case, what is the sequence. Other possibility include that Jak2-mediated phosphorylation of EZH2 Y641 may cause conformational changes in EZH2, which results in either disruption of PRC2 complex and/or exposure of recognizable site for β-TrCP-mediated degradation. Furthermore, YC-1 promotes EZH2 uniquitination and proteasomal degradation through the PKA and Src-Raf-1-MEK-ERK pathways in triple-negative breast cancer cells without changing H3K27me3 level, eventually inhibiting cell proliferation and inducing cell apoptosis [111]. Of note, YC-1 induces the activation of c-Cbl, a known EGFR E3 Ub ligase that may function as a novel EZH2 ubiquitin E3 ligase. Rapid phosphorylation of c-Cbl at T731 and T774 leads to Src and ERK activation, resulting in formation of c-Cbl-ERK-EZH2 complex and consequently enhancement of EZH2 uniquitination and degradation. These findings indicate that YC-1 is a potential and promising drug candidate for in triple-negative breast cancer therapy.
In addition, two recent studies reported that Ub E3 ligase PRAJA1 plays key roles in ubiquitination-proteasome pathway-mediated EZH2 protein degradation. PRAJA1, induced by the protein-methylation inhibitor 3-deazaneplanocin A (DZNep) treatment, has been reported for Ub-mediated proteasomal degradation of individual PRC2 subunits including EZH2, SUZ12 and EED in a RING finger-dependent manner [112]. It has been shown a negative feedback loop that PRC2 inhibition by DZNep induces the expression of Ub ligase, which in turn leads to the degradation of PRC2 proteins. Strikingly, PRAJA1 targets individual PRC2 subunits instead of PRC2 complex, underlying that it is mainly required for degradation of excess but not endogenous PRC2 components as a quality control. Another report has shown that nuclear localization of transcription factor FOXP3 directly interacted with the promoter of PRAJA1 and promoted its mRNA transcription, which facilitated EZH2 protein degradation through K48-linkage polyubiquitination and decreased cell proliferation, migration and formation in breast cancer cells [113]. This finding provides new evidence for FOXP3 as a tumor suppressor gene in breast cancer. Intriguingly, knockdown of PRAJA1 by shRNA cannot reverse EZH2 protein level and attenuate its ubiquitination, highlighting that other unknown E3 ligases and other PTMs are involved in EZH2 ubiquitination.
Besides definite Ub E3 ligase implicated in EZH2 ubiquitination, chemopreventive agents such as omega-3 (ω-3) polyunsaturated fatty acids (PUFAs) has been discovered to execute its anti-cancer functions by decreasing EZH2 expression through induction of EZH2 ubiquitination via proteasome-mediated degradation in breast cancer cells [114]. It also results in reduced H3K27me3 level and upregulation of EZH2 downstream target genes E-cadherin and IGFBP3, leading to suppression of tumor invasion and metastasis.
Taken together, an increasing number of regulators have been recognized as Ub E3 ligases to play pivotal roles in EZH2 ubiquitination and subsequently proteasome-mediated degradation, raising some pending problems that whether different Ub E3 ligases are fine tuned to carry out their functions dependent on cancer cell-specific microenvironment and diverse initiating signals, or various kinds of PTMs with mutual competitions or a series of cascade reactions are implicated in EZH2 regulation and at last it displays a final pattern of PTM that we can detect. Thus, future investigations will be urgent to explore other ubiquitination sites and their crosstalk and mutual regulation with other EZH2 PTMs.
Sumoylation
Similar to ubiquitylation, sumoylation is a highly conserved enzymatic cascade in which a small ubiquitin-like modifier (SUMO) protein is enzymatically conjugated to the ε-amino group of certain lysine residues [115]. Different from ubiquitin modification, SUMO modification is primarily responsible for modifying their substrates instead of directly targeting them for degradation [116]. As another essential type of PTMs, sumoylation also plays indispensable roles in the regulation of various biological processes and functions including in gene regulation, cell differentiation, apoptosis, protein stability, tissue development and disease progression [117-120]. Recently, sumoylation was firstly validated to be associated with the regulation of EZH2 activity [34]. Being two major subunits of PRC2 complex, EZH2 and SUZ12 can be sumoylated both in vitro and in vivo. However, other than SUZ12 containing a single known sumoylation site at lysine 75 with definite E2-conjugating enzyme UBC9 and E3-ligase PIASXβ, EZH2 performs multiple bands of modifications both in western blot analysis and in vitro sumoylation assay, indicating that EZH2 might have various SUMO-modified sites or different patterns of sumoylations on the same site. Unfortunately, there is little research on the field of EZH2 sumoylation in the last several years, thus it remains to be further explored the precise sumoylation site on EZH2 and the biological mechanism about how EZH2 is implicated in sumoylation and its functional significance. Taken together, understanding the underlying mechanisms of sumoylation at exact sites and other novel PTMs manipulated by EZH2 will be necessary for further clarification of EZH2 functions in the repression of downstream target genes, which will break a new path for exploring the mechanisms involved in PcG protein activity and the role of EZH2 in PcG protein-involved epigenetic regulation.
O-GlcNAcylation
Protein glycosylation with β-N-acetyl-D-glucosamine, also referred as to O-GlcNAcylation, is a reversible and dynamic PTM process ubiquitously in both cytosol and nucleus which was originally discovered in 1984 [121,122]. O-GlcNAcylation of proteins is catalyzed by the only known enzyme O-linked N-acetylglucosamine (GlcNAc) transferase(OGT) at side chain hydroxyl group of serine or threonine residue, and de-O-GlcNAcylation is achieved by the glycosidase O-GlcNAcase(OGA) [123-125]. To date, accumulating studies have illuminated that O-GlcNAcylation exerts its functions in a wide variety of fields including protein reorganization [126], competition with phosphorylation [127], modulation of protein-protein interaction [128], protein recruitment [129] and regulation of protein stability [130]. Dysregulation of O-GlcNAcylation has been reported to be widely associated with carcinogenesis such as cervical cancer [131], hepatocellular carcinoma [132], breast cancer [133] and so on. More recently, it has been firstly elucidated that EZH2 can be regulated by O-GlcNAcylation in breast cancer cells. EZH2 was identified to be physically interact with OGT and OGT-dependent O-GlcNAcylation of EZH2 at serine 75 (S75) is essential for the maintenance of EZH2 protein stability and subsequently the formation of H3K27me3, eventually contributing to tumorigenesis [35]. Intriguingly, it has been observed that OGT depletion decreased not only EZH2 protein expression but also the expression of all other PRC2 complex components such as EED, SUZ12 and RbAp46/48, consistent with previous studies that disruption of a crucial subunit may result in destabilization of the whole complex [24,134,135]. However, it remains poorly understood how S75 O-GlcNAcylation regulates EZH2 protein stability. In consideration of the ability of O-GlcNAcylation to compete with phosphorylation mentioned above, it is hypothesized that O-GlcNAcylation of EZH2 at serine75 prevents phosphorylation at the same site required for EZH2 degradation or protects EZH2 from other modifications at other sites that are beneficial for EZH2 degradation. Furthermore, it had been previously reported the CDK1-triggered EZH2 phosphorylation at T345 and T487 is targeted for ubiquitin-mediated degradation due to reduced protein stability [92] and OCT expression can decrease CDK1 activity [136], we speculate that EZH2 phosphorylation at T345 is inhibited by OCT-induced reduced CDK1 activity, which is conducive to its O-GlcNAcylation at S75. Collectively, better understanding of the crosstalk regulatory network implicated in OCT, CDK1 and EZH2 will be feasible for exploration of other known and unknown EZH2 PTMs and their complicated relationship and interaction.
Conclusions and perspectives
As the methyltransferase core subunit of PRC2, EZH2 is one of the central players in the epigenetic regulation of gene expression during embryogenesis, tissue regeneration and carcinogenesis. EZH2 activity was often upregulated in human cancers, thus targeting EZH2 was proposed as one novel and feasible approach of cancer treatment and prevention. The effects of EZH2 on the expression of target genes are influenced by its transcriptional and post-transcriptional level as well as the PTMs that affect the activity, stability, localization and protein-protein interactions of EZH2. Our understanding of the regulation of EZH2 PTMs in the development of cancer has made significant progress in the past several years. So far, it has been reported several PTMs of EZH2 mainly including phosphorylation, acetylation, ubiquitination, sumoylation and O-GlcNAcylation, which have been demonstrated in detail. However, there are still a lot of puzzles need to be figured out. Firstly, many modifying enzymes and precise sites of PTMs at EZH2 remain largely unknown. Moreover, whether other rare types of PTMs such as succinylation, malonylation, crotonylation, propionylation and butyrylation also exist in EZH2 remain mysterious and require further investigations. More importantly, until now there are not any clinical trials targeting EZH2 PTMs for cancer therapeutics to be developed. Thus, there is a long way to go before clinical application of anti-EZH2 PTMs relevant strategies for cancer therapy.
Collectively, better understanding of the regulation of different types of PTMs on EZH2 and their crosstalk modifications in carcinogenesis and clarifying their intrinsic molecular mechanisms will open a potential and promising avenue for the development of novel cancer therapeutic intervention.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81502386), Zhejiang Provincial Natural Science Foundation of China (No. LQ15H160005, LQ16H160015) and Zhejiang Provincial Medicine & Health Science and Technology General Project (No. 2016KYA109, No. 2016KYA045).
Disclosure of conflict of interest
None.
Abbreviations
- PTMs
post-translational modifications
- PRC2
Polycomb repressive complex 2
- HMTase
histone methyltransferase
- H3K27me3
histone H3 lysine 27 trimethylation
- PRC1
Polycomb repressive complex 1
- PcG
Polycomb Group
- TrxG
Trithorax Group
- HOX genes
homeotic genes
- H2AK119ub
monoubiquitylation of histone H2A at lysine 119
- MRT
malignant rhabdoid tumor
- PICs
preinitiation complexes
- E(z)
Enhancer of Zeste
- BTIC
breast tumor initiating cell
- Ser
serine
- Thr
threonine
- Tyr
tyrosine
- His
histidine
- pS21 EZH2
phosphorylation of EZH2 at Ser21
- IGF
insulin-like growth factor
- CRPC
castration-resistant prostate cancer
- ER
estrogen receptor
- E2
17β-estradiol
- DES
diethylstilbestrol
- Tat
transactivator of transcription
- As3+
arsenic
- GSCs
Gliblastoma multiforme stem-like cells
- CDKs
cyclin-dependent kinases
- pT350 EZH2
phosphorylation of Thr350 in human EZH2
- pT345 Ezh2
phosphorylation of Thr345 residue in mouse Ezh2
- ncRNAs
noncoding RNAs
- Xist
X-inactive specific transcript
- pT492 EZH2
phosphorylation of EZH2 Thr492
- pT487 Ezh2
phosphorylation of Thr487 in mouse Ezh2
- pT487 EZH2
phosphorylation of EZH2 at Thr487
- ncRBD1
ncRNA-binding domain
- pT372 Ezh2
phosphorylation of Ezh2 at T372 in Mouse
- MS
mass spectrometry
- TNFα
tumor necrosis factorα
- YY1
Yin Yang 1
- HATs
histone acetyltransferases
- HDACs
histone deacetylases
- PCAF
P300/CBP-associated factor
- K348
lysine348
- Ub
Ubiquitin
- Smurf2
Smad ubiquitination regulatory factor-2
- FBXW1
β-TrCP
- c-Cbl
Casitas B-lineage lymphoma
- hMSCs
human mesenchymal stem cells
- CNS
central nervous system
- SCF
Skp/cullin/F-box protein
- pY461 EZH2
phosphorylation at EZH2 Y641
- DZNep
3-deazaneplanocin A
- ω-3
omega-3
- PUFAs
polyunsaturated fatty acids
- SUMO
small ubiquitin-like modifier
- OGT
O-linked N-acetylglucosamine (GlcNAc) transferase
- OGA
O-GlcNAcase
References
- 1.Walsh CT, Garneau-Tsodikova S, Gatto GJ Jr. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl. 2005;44:7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 2.Mateo Sanchez S, Freeman SD, Delacroix L, Malgrange B. The role of post-translational modifications in hearing and deafness. Cell Mol Life Sci. 2016;73:3521–33. doi: 10.1007/s00018-016-2257-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He M, Zhou Z, Shah AA, Hong Y, Chen Q, Wan Y. New insights into posttranslational modifications of Hippo pathway in carcinogenesis and therapeutics. Cell Div. 2016;11:4. doi: 10.1186/s13008-016-0013-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liddy KA, White MY, Cordwell SJ. Functional decorations: post-translational modifications and heart disease delineated by targeted proteomics. Genome Med. 2013;5:20. doi: 10.1186/gm424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Witze ES, Old WM, Resing KA, Ahn NG. Mapping protein post-translational modifications with mass spectrometry. Nat Methods. 2007;4:798–806. doi: 10.1038/nmeth1100. [DOI] [PubMed] [Google Scholar]
- 6.Chandrasekaran AP, Suresh B, Kim HH, Kim KS, Ramakrishna S. Concise Review: Fate Determination of Stem Cells by Deubiquitinating Enzymes. Stem Cells. 2016 doi: 10.1002/stem.2446. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 7.Okamoto S, Lipton SA. S-Nitrosylation in neurogenesis and neuronal development. Biochim Biophys Acta. 2015;1850:1588–1593. doi: 10.1016/j.bbagen.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xuan Q, Zhang YX, Liu DG, Chan P, Xu SL, Cui YQ. Post-translational modifications of alpha-synuclein contribute to neurodegeneration in the colon of elderly individuals. Mol Med Rep. 2016;13:5077–5083. doi: 10.3892/mmr.2016.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang E, Abe J. Kinase-SUMO networks in diabetes-mediated cardiovascular disease. Metabolism. 2016;65:623–633. doi: 10.1016/j.metabol.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisenberg-Lerner A, Ciechanover A, Merbl Y. Post-translational modification profiling-A novel tool for mapping the protein modification landscape in cancer. Exp Biol Med (Maywood) 2016;241:1475–82. doi: 10.1177/1535370216651732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther. 2009;8:1409–1420. doi: 10.1158/1535-7163.MCT-08-0860. [DOI] [PubMed] [Google Scholar]
- 12.Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7:299–313. doi: 10.1016/j.stem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 14.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitcomb SJ, Basu A, Allis CD, Bernstein E. Polycomb Group proteins: an evolutionary perspective. Trends Genet. 2007;23:494–502. doi: 10.1016/j.tig.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 16.Joshi P, Carrington EA, Wang L, Ketel CS, Miller EL, Jones RS, Simon JA. Dominant alleles identify SET domain residues required for histone methyltransferase of Polycomb repressive complex 2. J Biol Chem. 2008;283:27757–27766. doi: 10.1074/jbc.M804442200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Li Y, Lin C, Ding J, Liao G, Tang B. Aberrant upregulation of 14-3-3sigma and EZH2 expression serves as an inferior prognostic biomarker for hepatocellular carcinoma. PLoS One. 2014;9:e107251. doi: 10.1371/journal.pone.0107251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huqun , Ishikawa R, Zhang J, Miyazawa H, Goto Y, Shimizu Y, Hagiwara K, Koyama N. Enhancer of zeste homolog 2 is a novel prognostic biomarker in nonsmall cell lung cancer. Cancer. 2012;118:1599–1606. doi: 10.1002/cncr.26441. [DOI] [PubMed] [Google Scholar]
- 21.Lund K, Adams PD, Copland M. EZH2 in normal and malignant hematopoiesis. Leukemia. 2014;28:44–49. doi: 10.1038/leu.2013.288. [DOI] [PubMed] [Google Scholar]
- 22.Lu H, Sun J, Wang F, Feng L, Ma Y, Shen Q, Jiang Z, Sun X, Wang X, Jin H. Enhancer of zeste homolog 2 activates wnt signaling through downregulating CXXC finger protein 4. Cell Death Dis. 2013;4:e776. doi: 10.1038/cddis.2013.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer. 2012;106:243–247. doi: 10.1038/bjc.2011.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, Moller P, Stilgenbauer S, Pollack JR, Wirth T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112:4202–4212. doi: 10.1182/blood-2008-03-147645. [DOI] [PubMed] [Google Scholar]
- 26.Ma YB, Song DW, Nie RH, Mu GY. MicroRNA-32 functions as a tumor suppressor and directly targets EZH2 in uveal melanoma. Genet Mol Res. 2016;15 doi: 10.4238/gmr.15027935. [DOI] [PubMed] [Google Scholar]
- 27.Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, Brenner JC, Yu J, Kim JH, Han B, Tan P, Kumar-Sinha C, Lonigro RJ, Palanisamy N, Maher CA, Chinnaiyan AM. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322:1695–1699. doi: 10.1126/science.1165395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Z, Tang J, Wang J, Duan G, Zhou L, Zhou X. MiR-138 Acts as a Tumor Suppressor by Targeting EZH2 and Enhances Cisplatin-Induced Apoptosis in Osteosarcoma Cells. PLoS One. 2016;11:e0150026. doi: 10.1371/journal.pone.0150026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Lin C, Liao G, Liu S, Ding J, Tang F, Wang Z, Liang X, Li B, Wei Y, Huang Q, Li X, Tang B. MicroRNA-506 suppresses tumor proliferation and metastasis in colon cancer by directly targeting the oncogene EZH2. Oncotarget. 2015;6:32586–32601. doi: 10.18632/oncotarget.5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun J, Zheng G, Gu Z, Guo Z. MiR-137 inhibits proliferation and angiogenesis of human glioblastoma cells by targeting EZH2. J Neurooncol. 2015;122:481–489. doi: 10.1007/s11060-015-1753-x. [DOI] [PubMed] [Google Scholar]
- 31.Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 32.Wan J, Zhan J, Li S, Ma J, Xu W, Liu C, Xue X, Xie Y, Fang W, Chin YE, Zhang H. PCAF-primed EZH2 acetylation regulates its stability and promotes lung adenocarcinoma progression. Nucleic Acids Res. 2015;43:3591–3604. doi: 10.1093/nar/gkv238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu YL, Chou RH, Shyu WC, Hsieh SC, Wu CS, Chiang SY, Chang WJ, Chen JN, Tseng YJ, Lin YH, Lee W, Yeh SP, Hsu JL, Yang CC, Hung SC, Hung MC. Smurf2-mediated degradation of EZH2 enhances neuron differentiation and improves functional recovery after ischaemic stroke. EMBO Mol Med. 2013;5:531–547. doi: 10.1002/emmm.201201783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riising EM, Boggio R, Chiocca S, Helin K, Pasini D. The polycomb repressive complex 2 is a potential target of SUMO modifications. PLoS One. 2008;3:e2704. doi: 10.1371/journal.pone.0002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chu CS, Lo PW, Yeh YH, Hsu PH, Peng SH, Teng YC, Kang ML, Wong CH, Juan LJ. O-GlcNAcylation regulates EZH2 protein stability and function. Proc Natl Acad Sci U S A. 2014;111:1355–1360. doi: 10.1073/pnas.1323226111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grossniklaus U, Paro R. Transcriptional silencing by polycomb-group proteins. Cold Spring Harb Perspect Biol. 2014;6:a019331. doi: 10.1101/cshperspect.a019331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steffen PA, Ringrose L. What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat Rev Mol Cell Biol. 2014;15:340–356. doi: 10.1038/nrm3789. [DOI] [PubMed] [Google Scholar]
- 38.Schuettengruber B, Cavalli G. Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development. 2009;136:3531–3542. doi: 10.1242/dev.033902. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Brown JL, Cao R, Zhang Y, Kassis JA, Jones RS. Hierarchical recruitment of polycomb group silencing complexes. Mol Cell. 2004;14:637–646. doi: 10.1016/j.molcel.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 40.Eskeland R, Leeb M, Grimes GR, Kress C, Boyle S, Sproul D, Gilbert N, Fan Y, Skoultchi AI, Wutz A, Bickmore WA. Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol Cell. 2010;38:452–464. doi: 10.1016/j.molcel.2010.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shao Z, Raible F, Mollaaghababa R, Guyon JR, Wu CT, Bender W, Kingston RE. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell. 1999;98:37–46. doi: 10.1016/S0092-8674(00)80604-2. [DOI] [PubMed] [Google Scholar]
- 42.Kia SK, Gorski MM, Giannakopoulos S, Verrijzer CP. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol Cell Biol. 2008;28:3457–3464. doi: 10.1128/MCB.02019-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lehmann L, Ferrari R, Vashisht AA, Wohlschlegel JA, Kurdistani SK, Carey M. Polycomb repressive complex 1 (PRC1) disassembles RNA polymerase II preinitiation complexes. J Biol Chem. 2012;287:35784–35794. doi: 10.1074/jbc.M112.397430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deb G, Singh AK, Gupta S. EZH2: not EZHY (easy) to deal. Mol Cancer Res. 2014;12:639–653. doi: 10.1158/1541-7786.MCR-13-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003;17:1823–1828. doi: 10.1101/gad.269603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- 47.Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, Adli M, Kasif S, Ptaszek LM, Cowan CA, Lander ES, Koseki H, Bernstein BE. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4:e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sing A, Pannell D, Karaiskakis A, Sturgeon K, Djabali M, Ellis J, Lipshitz HD, Cordes SP. A vertebrate Polycomb response element governs segmentation of the posterior hindbrain. Cell. 2009;138:885–897. doi: 10.1016/j.cell.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 50.Schoeftner S, Sengupta AK, Kubicek S, Mechtler K, Spahn L, Koseki H, Jenuwein T, Wutz A. Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J. 2006;25:3110–3122. doi: 10.1038/sj.emboj.7601187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Vlag J, Otte AP. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat Genet. 1999;23:474–478. doi: 10.1038/70602. [DOI] [PubMed] [Google Scholar]
- 52.Reynolds N, Salmon-Divon M, Dvinge H, Hynes-Allen A, Balasooriya G, Leaford D, Behrens A, Bertone P, Hendrich B. NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb Repressive Complex 2 to direct gene repression. EMBO J. 2012;31:593–605. doi: 10.1038/emboj.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A, Diaz MO, Scacheri PC, Harte PJ. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 2009;136:3131–3141. doi: 10.1242/dev.037127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drelon C, Berthon A, Mathieu M, Ragazzon B, Kuick R, Tabbal H, Septier A, Rodriguez S, Batisse-Lignier M, Sahut-Barnola I, Dumontet T, Pointud JC, Lefrancois-Martinez AM, Baron S, Giordano TJ, Bertherat J, Martinez A, Val P. EZH2 is overexpressed in adrenocortical carcinoma and is associated with disease progression. Hum Mol Genet. 2016;25:2789–2800. doi: 10.1093/hmg/ddw136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun R, Shen J, Gao Y, Zhou Y, Yu Z, Hornicek F, Kan Q, Duan Z. Overexpression of EZH2 is associated with the poor prognosis in osteosarcoma and function analysis indicates a therapeutic potential. Oncotarget. 2016;7:38333–38346. doi: 10.18632/oncotarget.9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu F, Gu L, Cao Y, Fan X, Zhang F, Sang M. Aberrant overexpression of EZH2 and H3K27me3 serves as poor prognostic biomarker for esophageal squamous cell carcinoma patients. Biomarkers. 2016;21:80–90. doi: 10.3109/1354750X.2015.1118537. [DOI] [PubMed] [Google Scholar]
- 57.Lv YF, Yan GN, Meng G, Zhang X, Guo QN. Enhancer of zeste homolog 2 silencing inhibits tumor growth and lung metastasis in osteosarcoma. Sci Rep. 2015;5:12999. doi: 10.1038/srep12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen JF, Luo X, Xiang LS, Li HT, Zha L, Li N, He JM, Xie GF, Xie X, Liang HJ. EZH2 promotes colorectal cancer stem-like cell expansion by activating p21cip1-Wnt/beta-catenin signaling. Oncotarget. 2016;7:41540–41558. doi: 10.18632/oncotarget.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T, Guney I, Strochlic DE, Macconaill LE, Beroukhim R, Bronson RT, Ryeom S, Hahn WC, Loda M, Cichowski K. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med. 2010;16:286–294. doi: 10.1038/nm.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riquelme E, Behrens C, Lin HY, Simon G, Papadimitrakopoulou V, Izzo J, Moran C, Kalhor N, Lee JJ, Minna JD, Wistuba II. Modulation of EZH2 Expression by MEK-ERK or PI3K-AKT Signaling in Lung Cancer Is Dictated by Different KRAS Oncogene Mutations. Cancer Res. 2016;76:675–685. doi: 10.1158/0008-5472.CAN-15-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu J, Cao Q, Mehra R, Laxman B, Yu J, Tomlins SA, Creighton CJ, Dhanasekaran SM, Shen R, Chen G, Morris DS, Marquez VE, Shah RB, Ghosh D, Varambally S, Chinnaiyan AM. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell. 2007;12:419–431. doi: 10.1016/j.ccr.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 62.Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, Kim M, Totonchy M, Cusack T, Ene C, Ma H, Su Q, Zenklusen JC, Zhang W, Maric D, Fine HA. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell. 2008;13:69–80. doi: 10.1016/j.ccr.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manning CS, Hooper S, Sahai EA. Intravital imaging of SRF and Notch signalling identifies a key role for EZH2 in invasive melanoma cells. Oncogene. 2015;34:4320–4332. doi: 10.1038/onc.2014.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalashnikova EV, Revenko AS, Gemo AT, Andrews NP, Tepper CG, Zou JX, Cardiff RD, Borowsky AD, Chen HW. ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2. Cancer Res. 2010;70:9402–9412. doi: 10.1158/0008-5472.CAN-10-1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duan Z, Zou JX, Yang P, Wang Y, Borowsky AD, Gao AC, Chen HW. Developmental and androgenic regulation of chromatin regulators EZH2 and ANCCA/ATAD2 in the prostate Via MLL histone methylase complex. Prostate. 2013;73:455–466. doi: 10.1002/pros.22587. [DOI] [PubMed] [Google Scholar]
- 66.Richter GH, Plehm S, Fasan A, Rossler S, Unland R, Bennani-Baiti IM, Hotfilder M, Lowel D, von Luettichau I, Mossbrugger I, Quintanilla-Martinez L, Kovar H, Staege MS, Muller-Tidow C, Burdach S. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci U S A. 2009;106:5324–5329. doi: 10.1073/pnas.0810759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, Meyer-Schaller N, Schubeler D, van Nimwegen E, Christofori G. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell. 2013;23:768–783. doi: 10.1016/j.ccr.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 68.Garipov A, Li H, Bitler BG, Thapa RJ, Balachandran S, Zhang R. NF-YA underlies EZH2 upregulation and is essential for proliferation of human epithelial ovarian cancer cells. Mol Cancer Res. 2013;11:360–369. doi: 10.1158/1541-7786.MCR-12-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin YW, Ren LL, Xiong H, Du W, Yu YN, Sun TT, Weng YR, Wang ZH, Wang JL, Wang YC, Cui Y, Sun DF, Han ZG, Shen N, Zou W, Xu J, Chen HY, Cao W, Hong J, Fang JY. Role of STAT3 and vitamin D receptor in EZH2-mediated invasion of human colorectal cancer. J Pathol. 2013;230:277–290. doi: 10.1002/path.4179. [DOI] [PubMed] [Google Scholar]
- 70.Kunderfranco P, Mello-Grand M, Cangemi R, Pellini S, Mensah A, Albertini V, Malek A, Chiorino G, Catapano CV, Carbone GM. ETS transcription factors control transcription of EZH2 and epigenetic silencing of the tumor suppressor gene Nkx3.1 in prostate cancer. PLoS One. 2010;5:e10547. doi: 10.1371/journal.pone.0010547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, Woodward WA, Hsu JM, Hortobagyi GN, Hung MC. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell. 2011;19:86–100. doi: 10.1016/j.ccr.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson SA, Hunter T. Kinomics: methods for deciphering the kinome. Nat Methods. 2005;2:17–25. doi: 10.1038/nmeth731. [DOI] [PubMed] [Google Scholar]
- 73.Denu JM, Dixon JE. Protein tyrosine phosphatases: mechanisms of catalysis and regulation. Curr Opin Chem Biol. 1998;2:633–641. doi: 10.1016/s1367-5931(98)80095-1. [DOI] [PubMed] [Google Scholar]
- 74.Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci U S A. 2004;101:12130–12135. doi: 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 76.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, Xu H, Cato L, Thornton JE, Gregory RI, Morrissey C, Vessella RL, Montironi R, Magi-Galluzzi C, Kantoff PW, Balk SP, Liu XS, Brown M. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bredfeldt TG, Greathouse KL, Safe SH, Hung MC, Bedford MT, Walker CL. Xenoestrogen-induced regulation of EZH2 and histone methylation via estrogen receptor signaling to PI3K/AKT. Mol Endocrinol. 2010;24:993–1006. doi: 10.1210/me.2009-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Newbold RR, Moore AB, Dixon D. Characterization of uterine leiomyomas in CD-1 mice following developmental exposure to diethylstilbestrol (DES) Toxicol Pathol. 2002;30:611–616. doi: 10.1080/01926230290105839. [DOI] [PubMed] [Google Scholar]
- 79.Newbold RR, Jefferson WN, Grissom SF, Padilla-Banks E, Snyder RJ, Lobenhofer EK. Developmental exposure to diethylstilbestrol alters uterine gene expression that may be associated with uterine neoplasia later in life. Mol Carcinog. 2007;46:783–796. doi: 10.1002/mc.20308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Victoriano AF, Okamoto T. Transcriptional control of HIV replication by multiple modulators and their implication for a novel antiviral therapy. AIDS Res Hum Retroviruses. 2012;28:125–138. doi: 10.1089/AID.2011.0263. [DOI] [PubMed] [Google Scholar]
- 81.Zhang HS, Liu Y, Wu TC, Du GY, Zhang FJ. EZH2 phosphorylation regulates Tat-induced HIV-1 transactivation via ROS/Akt signaling pathway. FEBS Lett. 2015;589:4106–4111. doi: 10.1016/j.febslet.2015.11.033. [DOI] [PubMed] [Google Scholar]
- 82.Chen B, Liu J, Chang Q, Beezhold K, Lu Y, Chen F. JNK and STAT3 signaling pathways converge on Akt-mediated phosphorylation of EZH2 in bronchial epithelial cells induced by arsenic. Cell Cycle. 2013;12:112–121. doi: 10.4161/cc.23030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rojanasakul Y. Linking JNK-STAT3-Akt signaling axis to EZH2 phosphorylation: a novel pathway of carcinogenesis. Cell Cycle. 2013;12:202–203. doi: 10.4161/cc.23419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, Lee C, Joo KM, Rich JN, Nam DH, Lee J. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23:839–852. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 86.Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat Cell Biol. 2010;12:1108–1114. doi: 10.1038/ncb2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010;24:2615–2620. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sharif J, Endoh M, Koseki H. Epigenetic memory meets G2/M: to remember or to forget? Dev Cell. 2011;20:5–6. doi: 10.1016/j.devcel.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 89.Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zeng X, Chen S, Huang H. Phosphorylation of EZH2 by CDK1 and CDK2: a possible regulatory mechanism of transmission of the H3K27me3 epigenetic mark through cell divisions. Cell Cycle. 2011;10:579–583. doi: 10.4161/cc.10.4.14722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, Yang CC, Yang JY, Lin CY, Lai CC, Hung MC. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol. 2011;13:87–94. doi: 10.1038/ncb2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J Biol Chem. 2011;286:28511–28519. doi: 10.1074/jbc.M111.240515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hirata H, Hinoda Y, Shahryari V, Deng G, Nakajima K, Tabatabai ZL, Ishii N, Dahiya R. Long Noncoding RNA MALAT1 Promotes Aggressive Renal Cell Carcinoma through Ezh2 and Interacts with miR-205. Cancer Res. 2015;75:1322–1331. doi: 10.1158/0008-5472.CAN-14-2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu Y, Zhang L, Zhang L, Wang Y, Li H, Ren X, Wei F, Yu W, Liu T, Wang X, Zhou X, Yu J, Hao X. Long non-coding RNA HOTAIR promotes tumor cell invasion and metastasis by recruiting EZH2 and repressing E-cadherin in oral squamous cell carcinoma. Int J Oncol. 2015;46:2586–2594. doi: 10.3892/ijo.2015.2976. [DOI] [PubMed] [Google Scholar]
- 95.Kong R, Zhang EB, Yin DD, You LH, Xu TP, Chen WM, Xia R, Wan L, Sun M, Wang ZX, De W, Zhang ZH. Long noncoding RNA PVT1 indicates a poor prognosis of gastric cancer and promotes cell proliferation through epigenetically regulating p15 and p16. Mol Cancer. 2015;14:82. doi: 10.1186/s12943-015-0355-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, Marquez VE, Valente S, Mai A, Forcales SV, Sartorelli V, Puri PL. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell. 2010;7:455–469. doi: 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gonzalez ME, DuPrie ML, Krueger H, Merajver SD, Ventura AC, Toy KA, Kleer CG. Histone methyltransferase EZH2 induces Akt-dependent genomic instability and BRCA1 inhibition in breast cancer. Cancer Res. 2011;71:2360–2370. doi: 10.1158/0008-5472.CAN-10-1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 100.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 101.Mawatari T, Ninomiya I, Inokuchi M, Harada S, Hayashi H, Oyama K, Makino I, Nakagawara H, Miyashita T, Tajima H, Takamura H, Fushida S, Ohta T. Valproic acid inhibits proliferation of HER2-expressing breast cancer cells by inducing cell cycle arrest and apoptosis through Hsp70 acetylation. Int J Oncol. 2015;47:2073–2081. doi: 10.3892/ijo.2015.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arboleda VA, Lee H, Dorrani N, Zadeh N, Willis M, Macmurdo CF, Manning MA, Kwan A, Hudgins L, Barthelemy F, Miceli MC, Quintero-Rivera F, Kantarci S, Strom SP, Deignan JL UCLA Clinical Genomics Center. Grody WW, Vilain E, Nelson SF. De novo nonsense mutations in KAT6A, a lysine acetyl-transferase gene, cause a syndrome including microcephaly and global developmental delay. Am J Hum Genet. 2015;96:498–506. doi: 10.1016/j.ajhg.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen S, Yao X, Li Y, Saifudeen Z, Bachvarov D, El-Dahr SS. Histone deacetylase 1 and 2 regulate Wnt and p53 pathways in the ureteric bud epithelium. Development. 2015;142:1180–1192. doi: 10.1242/dev.113506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 105.Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mortenson JB, Heppler LN, Banks CJ, Weerasekara VK, Whited MD, Piccolo SR, Johnson WE, Thompson JW, Andersen JL. Histone deacetylase 6 (HDAC6) promotes the pro-survival activity of 14-3-3zeta via deacetylation of lysines within the 14-3-3zeta binding pocket. J Biol Chem. 2015;290:12487–12496. doi: 10.1074/jbc.M114.607580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. 2014;20:1242–1253. doi: 10.1038/nm.3739. [DOI] [PubMed] [Google Scholar]
- 108.Voutsadakis IA. Ubiquitin- and ubiquitin-like proteins-conjugating enzymes (E2s) in breast cancer. Mol Biol Rep. 2013;40:2019–2034. doi: 10.1007/s11033-012-2261-0. [DOI] [PubMed] [Google Scholar]
- 109.Zhou MJ, Chen FZ, Chen HC. Ubiquitination involved enzymes and cancer. Med Oncol. 2014;31:93. doi: 10.1007/s12032-014-0093-6. [DOI] [PubMed] [Google Scholar]
- 110.Sahasrabuddhe AA, Chen X, Chung F, Velusamy T, Lim MS, Elenitoba-Johnson KS. Oncogenic Y641 mutations in EZH2 prevent Jak2/beta-TrCP-mediated degradation. Oncogene. 2015;34:445–454. doi: 10.1038/onc.2013.571. [DOI] [PubMed] [Google Scholar]
- 111.Chang LC, Lin HY, Tsai MT, Chou RH, Lee FY, Teng CM, Hsieh MT, Hung HY, Huang LJ, Yu YL, Kuo SC. YC-1 inhibits proliferation of breast cancer cells by down-regulating EZH2 expression via activation of c-Cbl and ERK. Br J Pharmacol. 2014;171:4010–4025. doi: 10.1111/bph.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zoabi M, Sadeh R, de Bie P, Marquez VE, Ciechanover A. PRAJA1 is a ubiquitin ligase for the polycomb repressive complex 2 proteins. Biochem Biophys Res Commun. 2011;408:393–398. doi: 10.1016/j.bbrc.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 113.Shen Z, Chen L, Yang X, Zhao Y, Pier E, Zhang X, Yang X, Xiong Y. Downregulation of Ezh2 methyltransferase by FOXP3: new insight of FOXP3 into chromatin remodeling? Biochim Biophys Acta. 2013;1833:2190–2200. doi: 10.1016/j.bbamcr.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 114.Dimri M, Bommi PV, Sahasrabuddhe AA, Khandekar JD, Dimri GP. Dietary omega-3 polyunsaturated fatty acids suppress expression of EZH2 in breast cancer cells. Carcinogenesis. 2010;31:489–495. doi: 10.1093/carcin/bgp305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang XJ, Chiang CM. Sumoylation in gene regulation, human disease, and therapeutic action. F1000Prime Rep. 2013;5:45. doi: 10.12703/P5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhao J. Sumoylation regulates diverse biological processes. Cell Mol Life Sci. 2007;64:3017–3033. doi: 10.1007/s00018-007-7137-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hannoun Z, Greenhough S, Jaffray E, Hay RT, Hay DC. Post-translational modification by SUMO. Toxicology. 2010;278:288–293. doi: 10.1016/j.tox.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 118.Chen Z, Lu W. Roles of ubiquitination and SUMOylation on prostate cancer: mechanisms and clinical implications. Int J Mol Sci. 2015;16:4560–4580. doi: 10.3390/ijms16034560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Enserink JM. Sumo and the cellular stress response. Cell Div. 2015;10:4. doi: 10.1186/s13008-015-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sarangi P, Zhao X. SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem Sci. 2015;40:233–242. doi: 10.1016/j.tibs.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem. 1984;259:3308–3317. [PubMed] [Google Scholar]
- 122.Holt GD, Hart GW. The subcellular distribution of terminal N-acetylglucosamine moieties. Localization of a novel protein-saccharide linkage, O-linked GlcNAc. J Biol Chem. 1986;261:8049–8057. [PubMed] [Google Scholar]
- 123.Vocadlo DJ. O-GlcNAc processing enzymes: catalytic mechanisms, substrate specificity, and enzyme regulation. Curr Opin Chem Biol. 2012;16:488–497. doi: 10.1016/j.cbpa.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 124.Nagel AK, Ball LE. O-GlcNAc transferase and O-GlcNAcase: achieving target substrate specificity. Amino Acids. 2014;46:2305–2316. doi: 10.1007/s00726-014-1827-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cieniewski-Bernard C, Lambert M, Dupont E, Montel V, Stevens L, Bastide B. O-GlcNAcylation, contractile protein modifications and calcium affinity in skeletal muscle. Front Physiol. 2014;5:421. doi: 10.3389/fphys.2014.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S, Imai Y, Kim J, He HH, Igarashi K, Kanno J, Ohtake F, Kitagawa H, Roeder RG, Brown M, Kato S. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature. 2011;480:557–560. doi: 10.1038/nature10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yao H, Li A, Wang M. Systematic Analysis and Prediction of In Situ Cross Talk of O-GlcNAcylation and Phosphorylation. Biomed Res Int. 2015;2015:279823. doi: 10.1155/2015/279823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lambert M, Richard E, Duban-Deweer S, Krzewinski F, Deracinois B, Dupont E, Bastide B, Cieniewski-Bernard C. O-GlcNAcylation is a key modulator of skeletal muscle sarcomeric morphometry associated to modulation of protein-protein interactions. Biochim Biophys Acta. 2016;1860:2017–30. doi: 10.1016/j.bbagen.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 129.Koo J, Bahk YY. In vivo putative O-GlcNAcylation of human SCP1 and evidence for possible role of its N-terminal disordered structure. BMB Rep. 2014;47:593–598. doi: 10.5483/BMBRep.2014.47.10.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jiang K, Bai B, Ta Y, Zhang T, Xiao Z, Wang PG, Zhang L. O-GlcNAc regulates NEDD4-1 stability via caspase-mediated pathway. Biochem Biophys Res Commun. 2016;471:539–544. doi: 10.1016/j.bbrc.2016.02.037. [DOI] [PubMed] [Google Scholar]
- 131.Kim M, Kim YS, Kim H, Kang MY, Park J, Lee DH, Roh GS, Kim HJ, Kang SS, Cho GJ, Park JK, Cho JW, Shin JK, Choi WS. O-linked N-acetylglucosamine transferase promotes cervical cancer tumorigenesis through human papillomaviruses E6 and E7 oncogenes. Oncotarget. 2016;7:44596–44607. doi: 10.18632/oncotarget.10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhu G, Tao T, Zhang D, Liu X, Qiu H, Han L, Xu Z, Xiao Y, Cheng C, Shen A. O-GlcNAcylation of histone deacetylases 1 in hepatocellular carcinoma promotes cancer progression. Glycobiology. 2016;26:820–33. doi: 10.1093/glycob/cww025. [DOI] [PubMed] [Google Scholar]
- 133.Tao T, He Z, Shao Z, Lu H. TAB3 O-GlcNAcylation promotes metastasis of triple negative breast cancer. Oncotarget. 2016;7:22807–18. doi: 10.18632/oncotarget.8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Montgomery ND, Yee D, Chen A, Kalantry S, Chamberlain SJ, Otte AP, Magnuson T. The murine polycomb group protein Eed is required for global histone H3 lysine-27 methylation. Curr Biol. 2005;15:942–947. doi: 10.1016/j.cub.2005.04.051. [DOI] [PubMed] [Google Scholar]
- 135.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang Z, Udeshi ND, Slawson C, Compton PD, Sakabe K, Cheung WD, Shabanowitz J, Hunt DF, Hart GW. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci Signal. 2010;3:ra2. doi: 10.1126/scisignal.2000526. [DOI] [PMC free article] [PubMed] [Google Scholar]