

Abstract
Uveal melanoma (UM) is the most common primary ocular malignancy in adults. Currently, no beneficial systemic therapy is available; therefore, there is an urgent need for effective targeted therapeutic drugs. As verteporfin has shown anti-neoplastic activity in several types of cancers, here we hypothesized and investigated the efficacy of verteporfin against UM cells without light activation. MTS assay, flow cytometry analysis of apoptosis, Western blotting of relevant proteins, transwell migration and invasion assay, melanosphere culture, and measurement of ALDH+ populations, were used to evaluate the effects of verteporfin on UM cells. We found that verteporfin disrupted the interaction between YAP and TEAD4 in UM cells and decreased the expression of YAP targeted downstream genes. Verteporfin treatment decreased the cytoplasmic and nuclear levels of YAP and induced lysosome-dependent degradation of YAP protein. Verteporfin exhibited distinct inhibitory effect on the proliferation of four lines of UM cells (e.g., 92.1, Mel 270, Omm 1 and Omm 2.3), and induced apoptosis through the intrinsic pathway. Additionally, verteporfin suppressed migration and invasion of UM cells, impaired the traits of cancer stem-like cells (e.g., melanosphere formation capacity, and ALDH+ cell population). This study demonstrated the anti-neoplastic activity of verteporfin against UM cells in vitro, providing a rationale for evaluating this agent in clinical investigation.
Keywords: Apoptosis, cancer stem-like cells, uveal melanoma, verteporfin, YAP
Introduction
Melanomas derived from the choroid, ciliary body and iris of the eye are defined as uveal melanomas (UM) [1]. UM is the most common primary ocular malignancy in adult with an incidence of 5.1 per million per year [2]. Although several relative risk factors have been identified, the etiology of this neoplasm remains largely unclear [3,4]. Despite the relatively low incidence rate, UM results in high mortality, mostly due to the frequent metastasis to the liver by means of blood circulation [5]. Nearly half of patients develop metastatic diseases over a 15 year period [6]. Although there are effective therapies to eradicate primary UM and prevent local recurrence, including radio-plaque, proton beam and enucleation, the median survival of patients with advanced UM is only 4-6 months after diagnosis [7], largely due to lack of beneficial systemic therapy.
The genetic studies of UM have helped understand such aggressive cancer. For instance, activating mutations in BRAF and NRAS are common in cutaneous melanoma, but are rare in UM [8]. The reported mutations of UM include Gα subunits GNAQ and GNA11, BAP1 (BRCA 1-associated protein-1), and splicing factor 3B subunit 1 (SF3B1). While SF3B1 and BAP1 mutations occur later in tumor progression, mutations in GNAQ and GNA11 are early and initiating events [9]. These mutations decrease the guanosine triphosphatase activity of the G proteins, leading to constitutive downstream signaling, with PLCβ as one of the best-known downstream molecules [10].
The Hippo signaling pathway controls organ size by regulating cell proliferation and apoptosis [11]. The deregulation of the Hippo pathway has been reported in various types of cancer, including breast, lung and colorectal cancers [12-14]. Being a core component of the Hippo pathway, YAP translocates to the nucleus when it is not phosphorylated by LATS1/2 and binds with corresponding transcriptional factors TEAD1-4, promoting the expression of target genes such as connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61) [15]. YAP has been considered as an important oncoprotein in UM [16-18]. It has been reported recently that YAP can be activated by actin polymerization and partial LATS1/2 inhibition in UM harboring mutations in GNAQ or GNA11 [19].
The benzoporphyrine derivative, verteporfin, is a photosensitizer used in photodynamic therapy for the treatment of age-related macular degeneration and neovascularization when this agent is stimulated by irradiation at a wavelength of 693 nm [20]. Photodynamic therapy with verteporfin has been tested for treatment of several human cancers including pancreatic cancer, metastatic breast cancer, and posterior uveal melanoma [21-23]. Notably, verteporfin has been used to inhibit YAP-TEAD association and YAP-induced liver overgrowth alone without irradiation [24]. Moreover, verteporfin showed anti-tumor effects in certain types of cancers in the absence of light activation [25-27]. Considering the photodynamic-independent inhibition of YAP by verteporfin, here we sought to determine whether verteporfin possesses cytotoxicity against UM cells. Reports have shown that verteporfin reduced UM cells tumorigenesis and proliferation in mouse model [17,19]; here we found that verteporfin can effectively suppress the malignant phenotypes such as migration, invasion and cancer stem-like cells (CSCs) of UM cells in the absence of light activation. Our study suggests that verteporfin holds promise to be a therapeutic agent for UM.
Materials and methods
Chemicals and antibodies
Verteporfin was purchased from Selleck (Shanghai, China) and prepared as a 20 mmol/L stock solution in DMSO. The stock solution was stored in aliquots at -20°C. Annexin-V was from Sigma-Aldrich (Shanghai, China). Antibody against cytochrome c oxidase subunit II (COX II) was from Invitrogen (Shanghai, China). Antibodies against BAX, survivin, proliferating cell nuclear antigen (PCNA), CYR61, CTGF were purchased from Santa Cruz Biotech (Santa Cruz, CA). Antibodies against PARP (clone 4C10-5), caspase-3, cytochrome c (clone 6H2.B4), XIAP, Bcl-2 were from BD Biosciences (San Jose, CA). Antibodies against MMP-2, YAP, phospho-YAP (S127) were from Cell Signaling Tech. (Beverly, MA). MG 132 was from EMD Biosciences. Cycloheximide and chloroquine were from Sigma-Aldrich. Anti-mouse immunoglobulin G and anti-rabbit immunoglobulin G horseradish peroxidase-conjugated antibodies were from LI-COR Biotechnology (Nebraska, USA).
Cell culture
The UM cell lines, Mel 270, 92.1, Omm 1 and Omm 2.3, were generous gifts from Dr. MJ Jager of Leiden University Medical Center, Leiden, The Netherlands [28-30]. The cells were cultured in RPMI 1640 supplemented with 10% FBS in a 37°C humidified incubator containing 5% CO2.
Cell viability assay
The MTS assay (CellTiter 96 Aqueous One Solution reagent; Promega) was used to assess cell viability [31]. Briefly, UM cells seeded in 96-well plates at a density of 5,000 cells per well were exposed to increasing concentrations of verteporfin for 72 hours, MTS was added to each well and incubated for 4 hours. The absorbance density was measured at a wavelength of 490 nm. The drug concentration resulting in 50% inhibition of cell growth (IC50) was calculated. The combinations were done in serial fixed-ratio dilutions of the two-drug mixtures. The effects of combinations were estimated using the CalcuSyn software [32]. The combination index (CI) was the ratio of the combination dose to the sum of the single-agent doses at an isoeffective level. CI<1 indicates synergy; CI>1, antagonism; and CI=1, additive.
Colony-formation assay
UM cells pretreated with increasing concentration of verteporfin (0-1 μM) or vehicle (DMSO, control) for 24 hours were harvested and washed. The cells (5,000/sample) were then incubated for 10-14 days in a modified double layer soft agar system in the absence of verteporfin. Colony composed with more than 50 cells was counted under an inverted phase-contrast microscope [33].
Dual luciferase reporter assay
The Gal4-TEAD4 reporter plasmid was from Addgene (Cat# 24640). pG5luc plasmid and the Renilla luciferase reporter construct, pRLTK were purchased from Promega. UM 92.1 cells were co-transfected with plasmids Gal4-TEAD4 (0.5 μg), pG5luc (0.5 μg), Renilla (1 ng) using polyethyleneimine (Polysciences, Inc., Warrington, PA), and then treated with indicated concentrations of verteporfin for 18 hours. The dual luciferase reporter assay was performed according to the manufacturer’s instructions [34].
Western blotting analysis
Total cell lysates were prepared in RIPA buffer (1 × PBS, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with 1 × protease inhibitor cocktail (Roche, Nutley, NJ), 10 mM β-glycerophosphate, 1 mM sodium orthovanadate, 10 mM sodium fluoride, and 1 mM phenylmethylsulfonyl fluoride unless otherwise stated. As for cytochrome c detection, cytosolic fraction was prepared with digitonin extraction buffer (10 mM PIPES, 0.015% digitonin, 300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, 5 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride). For extractions of sub-cellular cytoplasmic and nuclear fractions, cells were pelleted, washed by PBS, and then re-suspended in 200 μL of ice-cold lysis buffer (10 mM 4-[2-hydroxyethyl]-1-piperazine ethane-sulfonic acid (HEPES) PH 7.9, 10 mM KCl, 0.1 mM sodium orthovanadate and Complete Protease Inhibitor cocktail. The lysates were incubated on ice for 10 minutes followed by centrifugation at 10,000 g. The supernatants were transferred to fresh tubes and these were cytoplasmic extracts. Remaining pellets were washed with the lysis buffer, vigorously re-suspended in nuclear protein extraction buffer with inhibitors (20 mM HEPES PH 7.9, 0.4 M NaCl, 1 mM EDTA with 1 mM DTT, 0.5 mM PMSF, 0.2 mM sodium orthovanadate and Complete Protease Inhibitor Cocktail) and centrifuged at 14,000 g for 10 minutes at 4°C. The supernatants were collected as nuclear fractions [35]. Protein samples were subjected to SDS-PAGE and then transferred to nitrocellulose membranes. Membranes were subsequently incubated with the primary antibodies overnight before incubation with appropriate second antibodies. Actin was used as a loading control. The immunoblots were recorded with the Odyssey infrared imaging system (LI-COR).
Apoptosis assay
Apoptosis was measured by Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide apoptosis detection kit (Sigma-Aldrich, Shanghai) according to the instructions of the manufacturer. Briefly, the cells treated with different concentrations of verteporfin (0 μM, 1.5 μM, 2.5 μM or 5 μM) or 5 μM verteporfin for various periods of time were pelleted, washed in PBS, and re-suspended in 100 μL of annexin V binding buffer (10 mM HEPES/NaOH, [pH 7.4], 4 M NaCl, 1 M CaCl2). After addition of 0.3 μL annexin V-FITC (Sigma), the mixtures were incubated for 30 minutes at room temperature in dark. At the end of incubation, the cells were washed and re-suspended in 0.5 mL of binding buffer. Immediately after staining with propidium iodide, the samples were run and analyzed on BD C6 flow cytometer.
Melanosphere formation assay
92.1 and Omm 2.3 cells were harvested and washed after treatment with 0 μM or 1 μM verteporfin for 24 hours. Primary tumor spheres were derived by plating 5,000 cells in the DMEM/F-12 medium (HyClone, containing B27 1 ml, basic Fibroblast growth factor 10 ng/ml, Epidermal growth factor 20 ng/ml) in each well of 24-well CorningTM Ultra-Low Attachment Plates (Thermo Fisher Scientific Inc., Waltham, MA) and incubating for 10~14 days. At the end of incubation, melanospheres were counted under an inverted phase-contrast microscope. The cells were then collected, and 5,000 cells were replated for the secondary and tertiary rounds of melanosphere culture; colonies were counted on day 14 after each round of culture.
Aldehyde dehydrogenase (ALDH) assay
Mel 270 and Omm 2.3 were treated with 0 μM or 1 μM verteporfin for 24 hours, and then analyzed for ALDH activity following the manufacturer’s instructions [36]. In brief, 1 × 105 UM cells were incubated with 5 μL ALDH reagent in the absence of 5 μL DEAB or not for 1 hour in 37°C. Then cells were washed with ALDH assay buffer, and ALDH activity was measured using flow cytometry.
In vitro transwell migration and invasion assays
After pretreated with 0 μM or 1 μM verteporfin for 24 hours, 92.1 and Omm 2.3 cells were collected and washed with PBS. For transwell migration assays, 1 × 104 cells in FBS-free medium were plated in the upper chamber with the non-coated membrane (24-well insert; pore size, 8 µm; Corning, NY). For invasion assays, 1 × 105 cells in FBS-free medium were plated in the upper chamber with a Matrigel-coated membrane. In both assays, medium supplemented with 10% FBS in the lower chambers served as chemo-attractant. After 24 or 48 hours of incubation at 37°C, the cells that had migrated or invaded to the lower surface of membrane were fixed with 4% methanol, stained with crystal violet, and counted in 3 random microscopic fields (200 ×).
Cell transfection
pBABE YAP1 (Cat# 15682) was obtained from Addgene (Cambridge, MA). Omm 2.3 cells were transfected with HA-YAP plasmid or empty vector using polyethyleneimine (Polysciences, Inc., Warrington, PA). For establishment of stable expression of HA-YAP, the transfected cells were selected with puromycin (0.5 μg/mL) for 4 days.
Statistical analysis
All experimental results were presented as mean ± standard error (SEM). For statistical analysis, GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA) was used. Differences between two groups were analyzed by 2-tailed Student’s t test while differences among multiple groups were analyzed by one-way analysis of variance (ANOVA) with post-hoc intergroup comparisons with Turkey test. P<0.05 was considered as statistically significant.
Results
Verteporfin inhibits YAP-TEAD signaling pathway through accelerating YAP turnover in uveal melanoma cells
We first determined the effect of verteporfin on YAP-TEAD4 complex in UM cells. Twenty-four hours after co-transfection with plasmids of Gal4-TEAD4, pG5luc and Renilla luciferase, 92.1 cells were exposed to verteporfin treatment for 18 hours, followed by dual luciferase activity assay. The results showed that verteporfin treatment significantly reduced the TEAD-dependent reporter luciferase activity (Figure 1B). We next assessed the expression of YAP and its downstream targets, and found that the levels of YAP, phospho-YAP (S127), CYR61 and CTGF were appreciably decreased in UM cells exposed to verteporfin (Figure 1C). To explore the underlying regulation of YAP by verteporfin, we performed pulse-chase experiments using 92.1 cells [37]. Figure 1D shows that verteporfin accelerated the turnover rate of YAP. Further, immunoblotting analysis showed that YAP protein in both of the cytoplasmic and nuclear fractionations was remarkably reduced (Figure 1E). Because YAP phosphorylation by the Lats1/2 kinases was reported to induce its cytosolic retention and subsequent protein degradation by the ubiquitin-proteasome pathway, we examined the cellular YAP levels in UM cells in the presence of MG132, a proteasome inhibitor. The results showed that the verteporfin-mediated decrease of YAP protein was not rescued by MG132 but by chloroquine, an inhibitor of lysosomal degradation (Figure 1F). These data hint that verteporfin may trigger lysosome-dependent degradation of YAP protein in UM cells.
Figure 1.
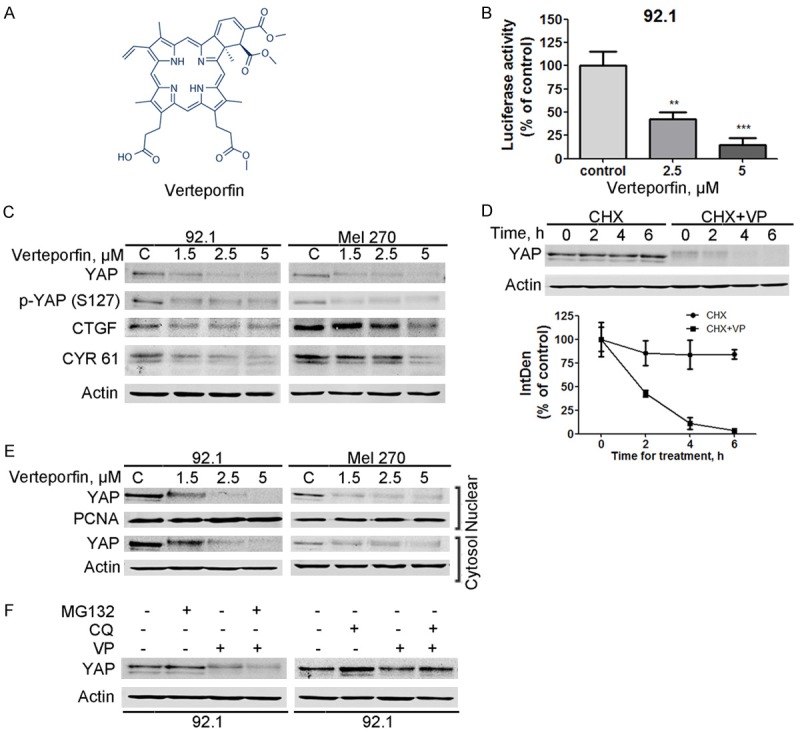
Verteporfin disrupts YAP-TEAD4 interaction through promoting YAP turnover in uveal melanoma cells. A: The chemical structure of verteporfin. B: Verteporfin treatment inhibited the TEAD4-dependent luciferase activity in uveal melanoma (UM) cells. Twenty-four hours after 92.1 cells were transfected with Luc-TEAD4-reporter, the cells were then exposed to verteporfin for another 18 hours. The luciferase activity was then detected. C: The levels of YAP, phospho-YAP (S127), and its downstream targeted molecules CTGF and CYR61 were analyzed by Western blotting after UM cells were exposed to verteporfin for 24 hours. D: The turnover rate of YAP protein was accelerated by verteporfin treatment. 92.1 cells were exposed to 1.5 μM verteporfin for 2 hours, and then cycloheximide was added (50 μg/ml). Cells were harvested at the indicated time points and YAP expression was analyzed by Western blotting. Data shown are one representative Western blotting and the curves of integrated density 100% normalized to the control band obtained by Image J from three independent experiments. E: Verteporfin decreased the levels of YAP in both cytoplasm and nucleus fractionations. F: Chloroquine, not MG132, blocked YAP degradation in the verteporfin-treated cells. After a 2-hour pretreatment with 1.5 μM MG132 or a 6-hour pretreatment with 15 μM chloroquine, 92.1 cells were treated with verteporfin for 12 or 2 hours, respectively. Whole cell lysates were then collected for Western blotting.
Verteporfin suppresses growth of uveal melanoma cells
To assess the inhibitory effect of verteporfin on cellar proliferation, four lines of UM cells were treated with various concentrations of verteporfin for 72 hours, followed by MTS assay. Figure 2A shows that verteporfin significantly decreased the cell viability of UM cells in a dose-dependent manner. The IC50 values for 92.1, Mel 270, Omm 1, Omm 2.3 cells were 4.67 μM, 6.43 μM, 5.89 μM, and 7.27 μM, respectively (Figure 2A). We also carried out colony formation assay to investigate the effect of verteporfin on the anchorage-independent growth of UM cells. Figure 2B reveals that verteporfin pretreatment led to a concentration-dependent decrease of colony formation ability. These experiments demonstrated that verteporfin can effectively inhibit the growth of UM cells.
Figure 2.
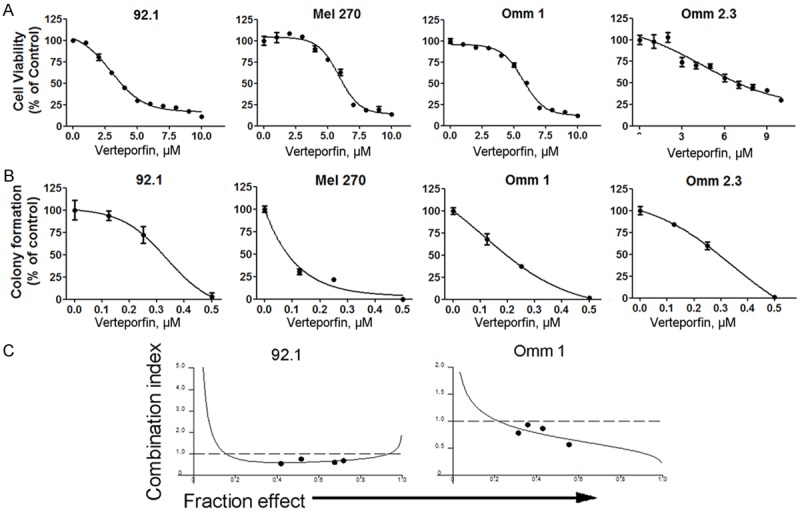
Verteporfin suppresses the growth of uveal melanoma cells. A: Cell viability was determined by MTS assay after the UM cells were exposed to verteporfin for 72 hours. B: After treated with various concentrations of verteporfin for 24 hours, the UM cells (e.g., 92.1, Mel 270, Omm 1, Omm 2.3) were seeded in drug-free soft agar culture for 14 days. Colonies were counted. C: The UM cells were exposed to escalating concentrations (serial fixed-ratio dilutions) of the mixture of vinblastine and verteporfin, synergistic effect of the combination was evaluated by CalcuSyn software. Combination Index (CI) <1 indicates synergy.
Synergistic cytotoxic effect between verteporfin and vinblastine
Vinblastine, an anti-microtubule chemotherapeutic agent, is used clinically for metastatic UM patients [38]. We found that there was a synergistic effect between vinblastine and verteporfin in killing the tumor cells, based on the combination index (CI), which was less than 1 (Figure 2C) [32].
Verteporfin induces apoptosis in UM cells
We next determined whether verteporfin induced apoptosis in UM cells. The UM cells were stained with Annexin V/PI after treatment with various concentrations of verteporfin or 5 μM verteporfin for different durations, and then were analyzed by flow cytometry. The results showed that verteporfin caused a drastic apoptic cell death in UM cells in a concentration- or time-dependent fashion (Figure 3A-C). Treatment with verteporfin also resulted in a dose- or time-dependent cleavage of poly ADP-ribose polymerase (PARP), decline of caspase-9, and activation of caspase-3 (Figure 3D). In the UM cells treated with verteporfin, the level of the cytoplasmic cytochrome c was increased (Figure 3E), suggesting that this agent triggers mitochondrial release of cytochrome c. In addition, the levels of anti-apoptotic proteins Bcl-2, Bcl-XL, XIAP and Survivin were down-regulated, whereas the pro-apoptotic Bax was up-regulated in the tumor cells subjected to verteporfin treatment (Figure 4A and 4B). These results indicate that verteporfin can activate apoptosis in UM cells without light activation.
Figure 3.
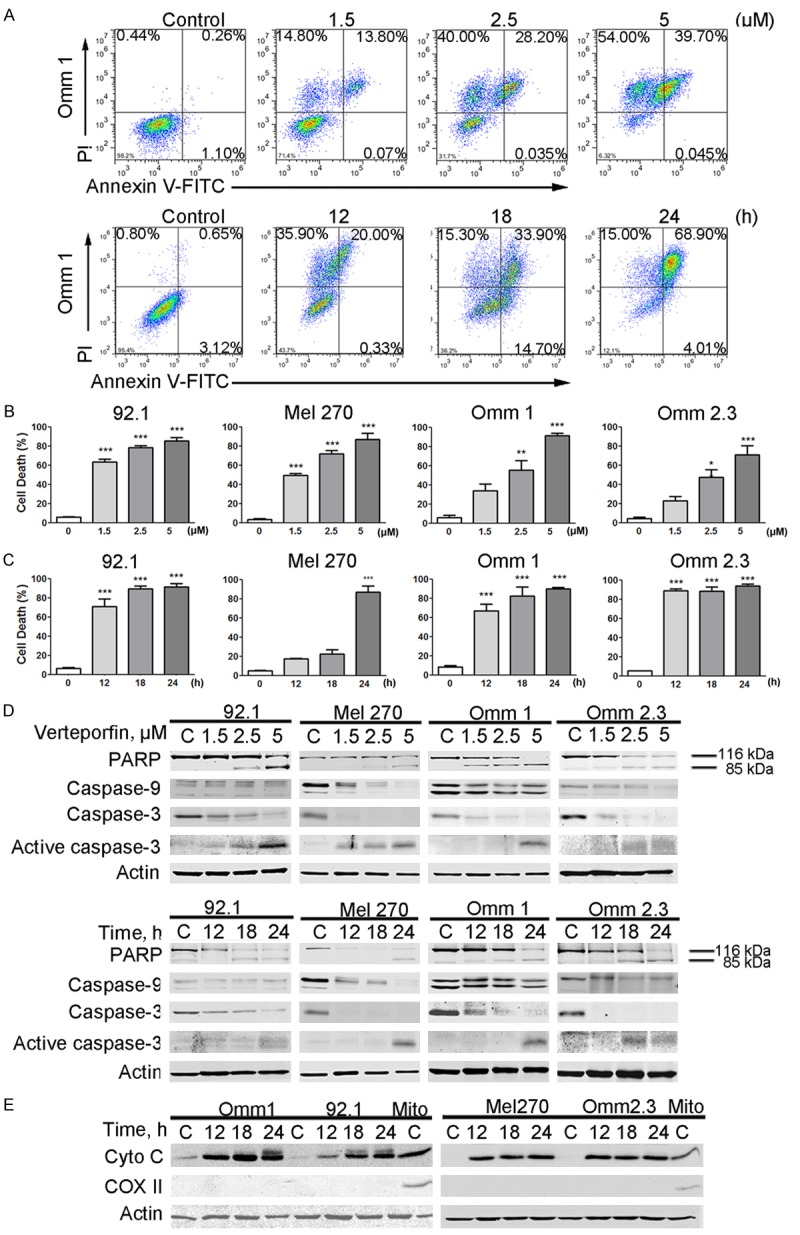
Verteporfin induces apoptosis in uveal melanoma cells. (A-D) UM cells were treated with various concentrations of verteporfin for 24 hours or with 5 μM verteporfin for different periods of time. Apoptosis was measured by flow cytometry following Annexin V-FITC/PI dual staining (A-C) or analyzed by Western blotting (D). (A) Representative histograms are shown. (B and C) Results from 3 independent experiments are shown. The Y-axis presents the sum of the top left, top right, and bottom right quadrants. Columns, mean; bars, SEM. *, P<0.05; **, P<0.01; ***, P<0.001. One-way ANOVA with post hoc intergroup comparison by the Tukey test. (D) Data shows immunoblotting of PARP, caspase-3 and caspase-9. (E) After treatment with verteporfin for 24 hours, cytochrome c in the cytosolic fractionations was detected with Western blotting. COX II served as a mitochondria marker to exclude mitochondrial contamination in the cytosolic fractionations.
Figure 4.
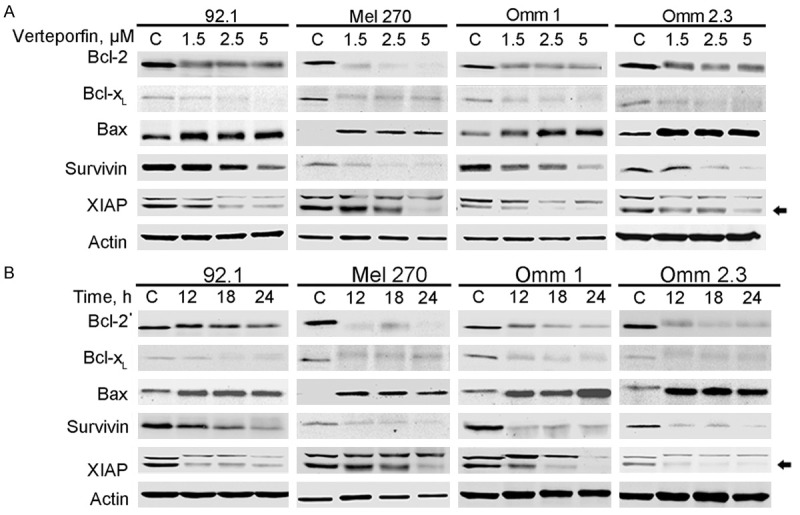
Verteporfin affects expression of the apoptosis-related proteins. A: Western blotting analysis of Bcl-2, Bax, Bcl-XL, Survivin and XIAP in the whole cell lysates after the UM cells were exposed to increasing concentrations of verteporfin. B: UM cells were treated with 5 μM verteporfin for various time periods. Bcl-2, BAX, Bcl-XL, Survivin and XIAP expression were detected by immunoblotting.
Verteporfin inhibits migration and invasion of UM cells
Although approximately half of UM patients develop metastatic disease [7], so far no systemic chemotherapy has been proved to be clinically beneficial for patients with the advanced disease. We tested whether or not verteporfin had inhibitory effect on migration and invasion of UM cells. 92.1 and Omm 2.3 cells were exposed to DMSO-containing medium or 1 μM verteporfin for 24 hours, then equal numbers of cells were seeded in Boyden chambers. Migrated cells were quantified after 24 hours’ culture. We observed that the numbers of migrated cells were significantly reduced in the presence of verteporfin (Figure 5A). Verteporfin treatment also decreased the invaded cells in the matrigel invasion assay (Figure 5B). Matrix metalloproteinase-2 (MMP-2), a critical molecule in metastasis and invasion [39], was also down-regulated in the tumor cells treated with verteporfin (Figure 5C). These results indicate that verteporfin can effectively hinder migration and invasion of UM cells.
Figure 5.
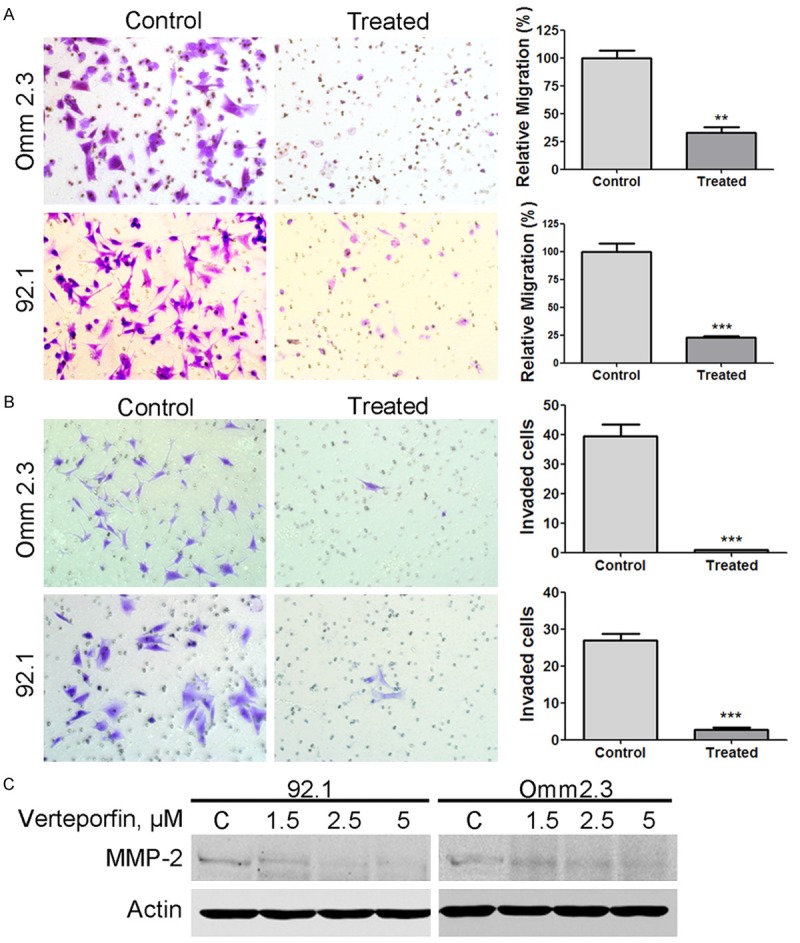
Verteporfin inhibits the migration and invasion of uveal melanoma cells. A: After pretreated with 1 μM verteporfin or control medium for 24 hours, the Omm 2.3 and 92.1 cells (1 × 104 cells) were inoculated in the transwell inserts, and then the migrated cells were counted in 3 random microscopic fields. Statistical graphs were shown. B: The cells invaded through matrigel-embedded upper chamber were quantified and statistical graphs were shown. Columns, mean; bars, SEM. *, P<0.05; **, P<0.01; ***, P<0.001. 2 tailed-student’s t-test. C: MMP-2 expression was analyzed after 92.1 and Omm 2.3 cells were exposed to verteporfin for 24 hours.
Verteporfin reduces cancer stem-like cells in UM
CSCs are a subset of cancerous cells with abilities of self-renewal and multi-lineage differentiation. CSCs have been identified and isolated in various types of cancers [40,41]. UM also contains stem-like cells that survive chemotherapy [42]. To determine the effect of verteporfin on CSCs, we cultured UM cell line, 92.1 and Omm 2.3, with 0 μM or 1 μM verteporfin for 24 hours. Cells were then collected and seeded in 24-well ultra-low attachment plates in the absence of verteporfin. The number of melanosphere consisting of more than 50 cells was counted 10-14 days later. The results of three rounds of serially replating experiments demonstrated that the self-renewal ability of UM CSCs was reduced by pre-treatment with verteporfin (Figure 6A and 6B). ALDH+ cells in UM have enhanced tumorigenicity over ALDH- cells and superior self-renewal capacity [43]. We observed that following treatment of Mel 270 and Omm 2.3 cells with verteporfin, the percentage of ALDH+ cells was markedly decreased (Figure 6C and 6D). These data imply that verteporfin is able to eliminate CSCs in UM; however, the underlying mechanisms remain to be elucidated.
Figure 6.
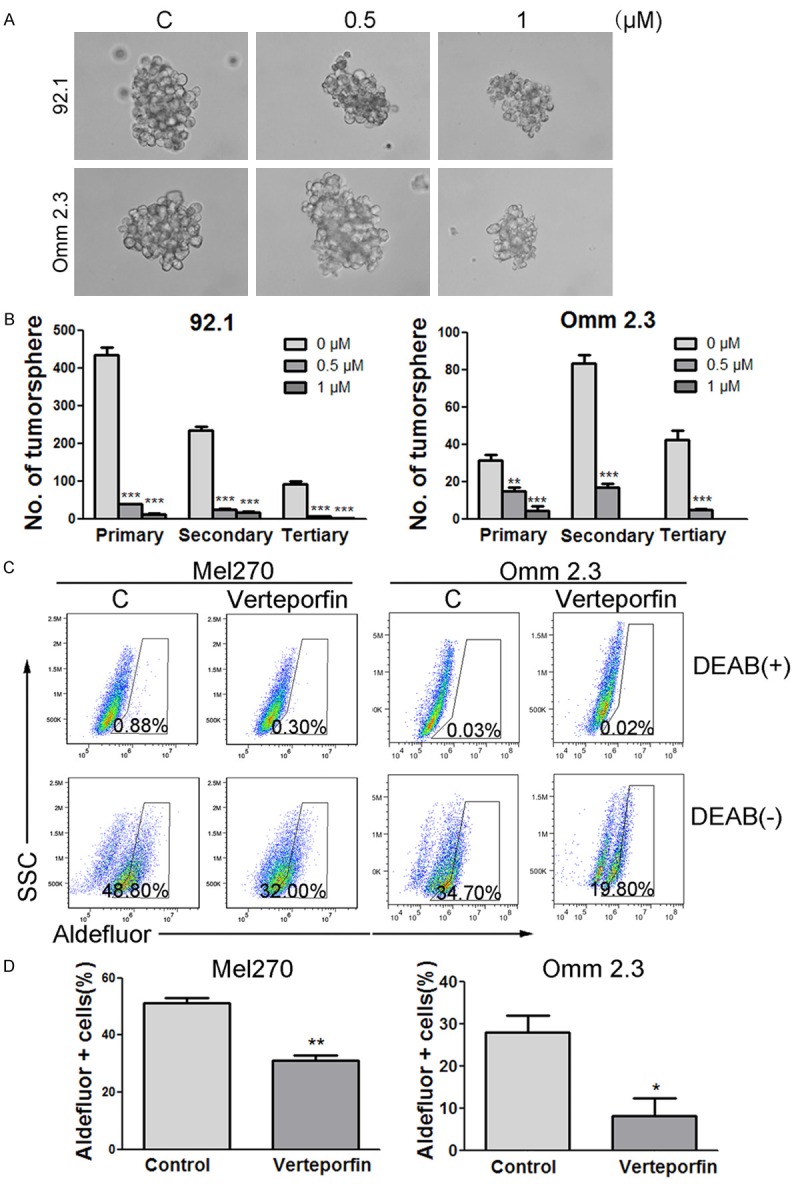
Verteporfin impairs self-renewal and eliminates uveal melanoma stem-like cells. A: The pictures of typical melanospheres. B: 92.1 and Omm 2.3 cells were exposed to 0 μM, 0.5 μM, or 1 μM verteporfin for 24 hours, and then cultured in DMEM/F-12 medium for 14 days. Melanospheres containing more than 50 cells were counted. The cells were then harvested for another two rounds of culture for melanospheres. C: 92.1 and Omm 2.3 cells were treated with control or verteporfin (1 μM) for 24 hours, ALDH activity was analyzed with flow cytometry. D: Bar charts summarizing the statistics were shown. Columns, mean; bars, SEM. *, P<0.05; **, P<0.01; ***, P<0.001. One-way ANOVA with post hoc intergroup comparison by the Tukey test.
Ectopic overexpression of YAP partially decreases verteporfin’s efficacy against UM cells
Omm 2.3 cells stably expressing HA-YAP showed less apoptosis compared with vector control in response to verteporfin treatment as indicated by activation of caspase-3 and trypan blue exclusion assay (Figure 7A). Moreover, stable expression of YAP partially restored the migration and invasion of Omm 2.3 cells (Figure 7B and 7C), as well as the capacity of melanosphere formation (Figure 7D). We found that cells expressing HA-YAP showed increased number of migrated cells, invaded cells and melanospheres in comparison with empty vector control after verteporfin exposure. These data indicate that verteporfin exhibits anti-tumor activity partially through targeting YAP.
Figure 7.
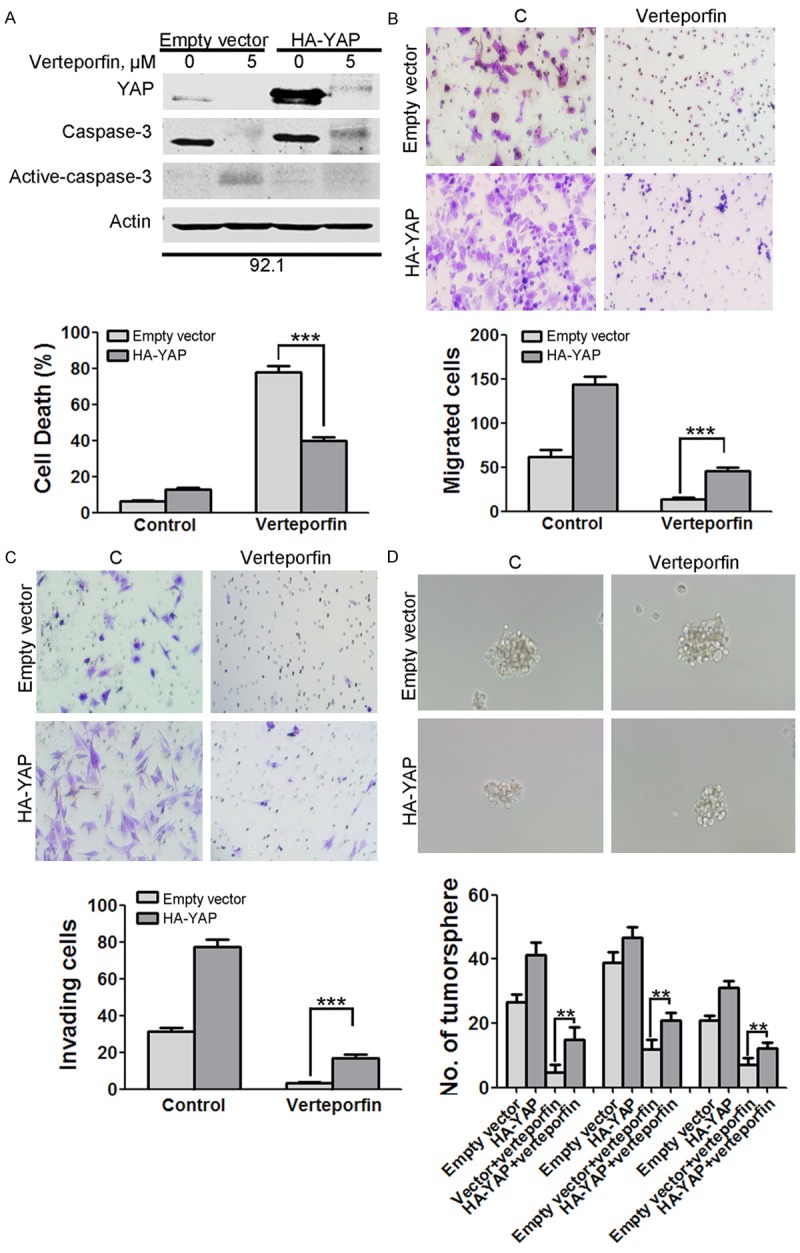
Over-expression of YAP attenuates the antitumor activity of verteporfin. (A) YAP was critical in verteporfin-induced apoptosis in UM cells. After treated with control or 5 μM verteporfin for 24 hours, Omm 2.3 cells stably expressing HA-YAP or empty vector underwent trypan blue exclusion assay and Western blotting analysis. (B, C) YAP promoted migration of UM cells. After treated with or without 0.5 μM verteporfin, HA-YAP-overexpressing Omm 2.3 cells underwent migration (B) and invasion (C) assay. (D) Overexpression of YAP rescued the verteporfin-induced decrease in capacity of melanosphere formation. Omm 2.3 cells stably expressing HA-YAP or empty vector were exposed to 0.25 μM verteporfin, and inoculated for melanosphere assay in drug-free DMEM/F-12 medium for 14 days. Columns, mean; bars, SEM. *, P<0.05; **, P<0.01; ***, P<0.001. One-way ANOVA with post hoc intergroup comparison by the Tukey test.
Discussion
In the present study, we demonstrate that verteporfin, a photosensitizer in common clinical use, has anti-neoplastic activity against UM cells, likely through inhibiting the YAP-TEAD4 interaction and triggering intrinsic apoptosis pathway.
Different from that in photodynamic therapy, without light activation verteporfin alone can significantly suppress the proliferation of UM cells and induce apoptosis. Our findings are in accordance with previous reports [17,19]. In addition, verteporfin inhibits migration and invasion of UM cells. Also, verteporfin exhibited synergistic effect with vinblastine, a conventional chemotherapeutic agent. The anti-tumor capacity of verteporfin might be associated with its role in blocking of canonical Hippo-YAP pathway, as previously demonstrated in other types of cancers [25-27,44].
Verteporfin was first identified as a YAP-inhibitory compound by Liu-Chittenden et al in HEK293 cells [24]. Indeed, we showed that verteporfin disrupted the interaction between YAP and TEAD4 in UM cells. CTGF and CYR61 are two downstream targets of YAP. Verteporfin reduced their expression in a dose-dependent manner. Moreover, verteporfin reduced the total expression of YAP including phospho-YAP (S127), suggesting that LATS1/2 is not involved in this process. We further demonstrated that this reduction was a consequence of enhanced turnover of YAP. Cytoplasmic and nuclear YAP distributions were also altered, and this prompted us to explore the underlying mechanism of verteporfin-promoted turnover of YAP. Interestingly, we found that in UM cells, inhibition of autophagy by chloroquine led to accumulation of YAP; in contrast, inhibition of the proteasome did not affect the amount of YAP. It was reported that verteporfin can inhibit autophagosome by promoting oligomerization of p62 in pancreatic ductal adenocarcinoma [45]. We found that verteporfin decreased YAP expression in UM. In addition, YAP expression was restored when cells were subjected to combined treatment with chloroquine and verteporfin. Based on our data, we propose that verteporfin promotes YAP degradation through the lysosomes in UM cells.
Existence of CSCs is believed to be a major contributor to tumorigenesis, drug resistance and local or distant recurrence. High YAP activity has been observed in the cancer stem and progenitor cells of multiple tissues, suggesting a role for YAP in CSC maintenance [46]. CSCs also exist in UM, but no validated biomarkers have been available so far [42]. The capacity of melanosphere formation and activity of ALDH are the two universal features of CSCs. Our data showed that verteporfin treatment can impair the melanosphere formation ability and reduce the population of ALDH+ cells in UM, suggesting an inhibitory effect of this drug on CSCs in uveal melanoma.
Furthermore, ectopic expression of YAP antagonized verteporfin in inducing apoptosis and inhibiting migration, invasion as well as tumor sphere formation. It is reasonable to conclude that verteporfin achieves its anti-neoplastic effect at least in part by targeting YAP.
In summary, we demonstrate that verteporfin can potently inhibit UM without light activation and we report for the first time that verteporfin impairs CSCs in UM. These observations warrant further studies of the anti-uveal melanoma activity of verteporfin in patients with this malignancy.
Acknowledgements
This study was supported by grants from National Natural Science Funds (no. U1301226, no. 81373434, no. 81025021, and no. 91213304 to J. Pan); the National Basic Research Program of China (973 Program grant no. 2009CB825506 to J. Pan), the Research Foundation of Education Bureau of Guangdong Province, China (Grant cxzd1103 to J. Pan), the Research Foundation of Guangzhou Bureau of Science and Technology, and the Fundamental Research Funds for the Central Universities (to J. Pan). The authors thank Dr. Sai-Ching J. Yeung (The University of Texas MD Anderson Cancer Center, Houston, TX, USA) for a critical reading of the manuscript.
Disclosure of conflict of interest
None.
Authors’ contribution
Y.M. designed and performed the experiments and wrote the manuscript; Y.L. reviewed the data; J.P. designed, performed research, analyzed data, and wrote the manuscript.
References
- 1.Chattopadhyay C, Kim DW, Gombos DS, Oba J, Qin Y, Williams MD, Esmaeli B, Grimm EA, Wargo JA, Woodman SE, Patel SP. Uveal melanoma: From diagnosis to treatment and the science in between. Cancer. 2016;122:2299–2312. doi: 10.1002/cncr.29727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh AD, Topham A. Incidence of uveal melanoma in the United States: 1973-1997. Ophthalmology. 2003;110:956–961. doi: 10.1016/S0161-6420(03)00078-2. [DOI] [PubMed] [Google Scholar]
- 3.Saornil MA. Iris colour and uveal melanoma. Can J Ophthalmol. 2004;39:448–452. doi: 10.1016/s0008-4182(04)80018-8. [DOI] [PubMed] [Google Scholar]
- 4.Logan P, Bernabeu M, Ferreira A, Burnier MN Jr. Evidence for the Role of Blue Light in the Development of Uveal Melanoma. J Ophthalmol. 2015;2015:386986. doi: 10.1155/2015/386986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kujala E, Makitie T, Kivela T. Very long-term prognosis of patients with malignant uveal melanoma. Invest Ophthalmol Vis Sci. 2003;44:4651–4659. doi: 10.1167/iovs.03-0538. [DOI] [PubMed] [Google Scholar]
- 6.Luke JJ, Triozzi PL, McKenna KC, Van Meir EG, Gershenwald JE, Bastian BC, Gutkind JS, Bowcock AM, Streicher HZ, Patel PM, Sato T, Sossman JA, Sznol M, Welch J, Thurin M, Selig S, Flaherty KT, Carvajal RD. Biology of advanced uveal melanoma and next steps for clinical therapeutics. Pigment Cell Melanoma Res. 2015;28:135–147. doi: 10.1111/pcmr.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woodman SE. Metastatic uveal melanoma: biology and emerging treatments. Cancer J. 2012;18:148–152. doi: 10.1097/PPO.0b013e31824bd256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van den Bosch T, Kilic E, Paridaens D, de Klein A. Genetics of uveal melanoma and cutaneous melanoma: two of a kind? Dermatol Res Pract. 2010;2010:360136. doi: 10.1155/2010/360136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harbour JW. Genomic, prognostic, and cell-signaling advances in uveal melanoma. Am Soc Clin Oncol Educ Book. 2013:388–391. doi: 10.1200/EdBook_AM.2013.33.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shoushtari AN, Carvajal RD. GNAQ and GNA11 mutations in uveal melanoma. Melanoma Res. 2014;24:525–534. doi: 10.1097/CMR.0000000000000121. [DOI] [PubMed] [Google Scholar]
- 11.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 12.Maugeri-Sacca M, De Maria R. Hippo pathway and breast cancer stem cells. Crit Rev Oncol Hematol. 2016;99:115–122. doi: 10.1016/j.critrevonc.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Z, Hao Y, Liu N, Raptis L, Tsao MS, Yang X. TAZ is a novel oncogene in non-small cell lung cancer. Oncogene. 2011;30:2181–2186. doi: 10.1038/onc.2010.606. [DOI] [PubMed] [Google Scholar]
- 14.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13:246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- 15.Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30:1–17. doi: 10.1101/gad.274027.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu FX, Zhang K, Guan KL. YAP as oncotarget in uveal melanoma. Oncoscience. 2014;1:480–481. doi: 10.18632/oncoscience.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z, Zhao L, Peyman G, Ouyang H, Jiang W, Zhao J, Chen X, Zhang L, Wang CY, Bastian BC, Zhang K, Guan KL. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell. 2014;25:822–830. doi: 10.1016/j.ccr.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Field MG, Harbour JW. GNAQ/11 mutations in uveal melanoma: is YAP the key to targeted therapy? Cancer Cell. 2014;25:714–715. doi: 10.1016/j.ccr.2014.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, Zaidi MR, Ksander BR, Merlino G, Sodhi A, Chen Q, Gutkind JS. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell. 2014;25:831–845. doi: 10.1016/j.ccr.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Battaglia Parodi M, La Spina C, Berchicci L, Petruzzi G, Bandello F. Photosensitizers and Photodynamic Therapy: Verteporfin. Dev Ophthalmol. 2016;55:330–336. doi: 10.1159/000434704. [DOI] [PubMed] [Google Scholar]
- 21.Huggett MT, Jermyn M, Gillams A, Illing R, Mosse S, Novelli M, Kent E, Bown SG, Hasan T, Pogue BW, Pereira SP. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br J Cancer. 2014;110:1698–1704. doi: 10.1038/bjc.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akens MK, Wise-Milestone L, Won E, Schwock J, Yee AJ, Wilson BC, Whyne CM. In vitro and in vivo effects of photodynamic therapy on metastatic breast cancer cells pre-treated with zoledronic acid. Photodiagnosis Photodyn Ther. 2014;11:426–433. doi: 10.1016/j.pdpdt.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Rundle P. Treatment of posterior uveal melanoma with multi-dose photodynamic therapy. Br J Ophthalmol. 2014;98:494–497. doi: 10.1136/bjophthalmol-2013-304432. [DOI] [PubMed] [Google Scholar]
- 24.Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brodowska K, Al-Moujahed A, Marmalidou A, Meyer Zu Horste M, Cichy J, Miller JW, Gragoudas E, Vavvas DG. The clinically used photosensitizer Verteporfin (VP) inhibits YAP-TEAD and human retinoblastoma cell growth in vitro without light activation. Exp Eye Res. 2014;124:67–73. doi: 10.1016/j.exer.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng H, Zhang Z, Rodriguez-Barrueco R, Borczuk A, Liu H, Yu J, Silva JM, Cheng SK, Perez-Soler R, Halmos B. Functional genomics screen identifies YAP1 as a key determinant to enhance treatment sensitivity in lung cancer cells. Oncotarget. 2016;7:28976–28988. doi: 10.18632/oncotarget.6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slemmons KK, Crose LE, Rudzinski E, Bentley RC, Linardic CM. Role of the YAP Oncoprotein in Priming Ras-Driven Rhabdomyosarcoma. PLoS One. 2015;10:e0140781. doi: 10.1371/journal.pone.0140781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Waard-Siebinga I, Blom DJ, Griffioen M, Schrier PI, Hoogendoorn E, Beverstock G, Danen EH, Jager MJ. Establishment and characterization of an uveal-melanoma cell line. Int J Cancer. 1995;62:155–161. doi: 10.1002/ijc.2910620208. [DOI] [PubMed] [Google Scholar]
- 29.Chen PW, Murray TG, Uno T, Salgaller ML, Reddy R, Ksander BR. Expression of MAGE genes in ocular melanoma during progression from primary to metastatic disease. Clin Exp Metastasis. 1997;15:509–518. doi: 10.1023/a:1018479011340. [DOI] [PubMed] [Google Scholar]
- 30.Luyten GP, Naus NC, Mooy CM, Hagemeijer A, Kan-Mitchell J, Van Drunen E, Vuzevski V, De Jong PT, Luider TM. Establishment and characterization of primary and metastatic uveal melanoma cell lines. Int J Cancer. 1996;66:380–387. doi: 10.1002/(SICI)1097-0215(19960503)66:3<380::AID-IJC19>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 31.Jin B, Wang C, Li J, Du X, Ding K, Pan J. Anthelmintic niclosamide disrupts the interplay of p65 and FOXM1/beta-catenin and eradicates leukemia stem cells in chronic myelogenous leukemia. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-0226. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 32.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 33.Lu Z, Jin Y, Qiu L, Lai Y, Pan J. Celastrol, a novel HSP90 inhibitor, depletes Bcr-Abl and induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation. Cancer Lett. 2010;290:182–191. doi: 10.1016/j.canlet.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 34.Jin Y, Zhou J, Xu F, Jin B, Cui L, Wang Y, Du X, Li J, Li P, Ren R, Pan J. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J Clin Invest. 2016;126:3961–3980. doi: 10.1172/JCI85239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu Z, Jin Y, Chen C, Li J, Cao Q, Pan J. Pristimerin induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation by blocking NF-kappaB signaling and depleting Bcr-Abl. Mol Cancer. 2010;9:112. doi: 10.1186/1476-4598-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen Y, Shi X, Pan J. The conformational control inhibitor of tyrosine kinases DCC-2036 is effective for imatinib-resistant cells expressing T674I FIP1L1-PDGFRalpha. PLoS One. 2013;8:e73059. doi: 10.1371/journal.pone.0073059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melichar B, Voboril Z, Lojik M, Krajina A. Liver metastases from uveal melanoma: clinical experience of hepatic arterial infusion of cisplatin, vinblastine and dacarbazine. Hepatogastroenterology. 2009;56:1157–1162. [PubMed] [Google Scholar]
- 39.Che YL, Luo SJ, Li G, Cheng M, Gao YM, Li XM, Dai JM, He H, Wang J, Peng HJ, Zhang Y, Li WY, Wang H, Liu B, Linghu H. The C3G/Rap1 pathway promotes secretion of MMP-2 and MMP-9 and is involved in serous ovarian cancer metastasis. Cancer Lett. 2015;359:241–249. doi: 10.1016/j.canlet.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 40.Geng SQ, Alexandrou AT, Li JJ. Breast cancer stem cells: Multiple capacities in tumor metastasis. Cancer Lett. 2014;349:1–7. doi: 10.1016/j.canlet.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lundin A, Driscoll B. Lung cancer stem cells: progress and prospects. Cancer Lett. 2013;338:89–93. doi: 10.1016/j.canlet.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalirai H, Damato BE, Coupland SE. Uveal melanoma cell lines contain stem-like cells that self-renew, produce differentiated progeny, and survive chemotherapy. Invest Ophthalmol Vis Sci. 2011;52:8458–8466. doi: 10.1167/iovs.11-7379. [DOI] [PubMed] [Google Scholar]
- 43.Boonyaratanakornkit JB, Yue L, Strachan LR, Scalapino KJ, LeBoit PE, Lu Y, Leong SP, Smith JE, Ghadially R. Selection of tumorigenic melanoma cells using ALDH. J Invest Dermatol. 2010;130:2799–2808. doi: 10.1038/jid.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perra A, Kowalik MA, Ghiso E, Ledda-Columbano GM, Di Tommaso L, Angioni MM, Raschioni C, Testore E, Roncalli M, Giordano S, Columbano A. YAP activation is an early event and a potential therapeutic target in liver cancer development. J Hepatol. 2014;61:1088–1096. doi: 10.1016/j.jhep.2014.06.033. [DOI] [PubMed] [Google Scholar]
- 45.Donohue E, Thomas A, Maurer N, Manisali I, Zeisser-Labouebe M, Zisman N, Anderson HJ, Ng SS, Webb M, Bally M, Roberge M. The autophagy inhibitor verteporfin moderately enhances the antitumor activity of gemcitabine in a pancreatic ductal adenocarcinoma model. J Cancer. 2013;4:585–596. doi: 10.7150/jca.7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]