

Abstract
Paediatric glioneuronal tumour with neuropil-like islands (GTNI) is a rare neoplasm of neuronal differentiation and diffusely infiltrating astroglial and oligodendrocyte-like components. The 2007 World Health Organization classification of central nervous system tumours considered it as a pattern variation of anaplastic astrocytoma. There are few data on paediatric GTNI probably both for their rarity and variable clinical aggressiveness. We studied by SNP/CGH array four tumour samples of GTNI from two males and two females (one new-born and three children aged from 4 to 8 years), in order to identify any possible common genomic alteration. All patients received chemo- and radiotherapy after their surgical treatment. No genomic instability nor recurrent alterations have been demonstrated in two of our GTNI cases. In the remaining two, we detected a mosaic trisomy 8 (15-20%) in one case, and an amplification at 5q14.1 involving DMGDH (partially), BHMT2 and BHMT genes, with the distal breakpoint falling at 23 Kbp from the 5’UTR of JMY, a p53 cofactor. Although the smallness of the sample impairs any clinical-histological correlation, GTNI appear different at the molecular level, with genomic imbalances playing a possible role in at least part of them. Our work gives an important contribution in knowledge and classification of this family of tumours.
Keywords: Glioneuronal tumour with neuropil-like islands (GTNI), paediatric brain tumours, central nervous tumours (CNS), copy number variations (CNVs), SNP/CGH array, Database of Genomic Variants (DGV), mosaicism, amplification, common genomic alteration, variation of anaplastic astrocytoma
Introduction
The neuronal and mixed glioneuronal tumours are a group of central nervous system (CNS) neoplasms with a spectrum of clinical aggressiveness that spans from indolent to highly aggressive tumours. Glioneuronal tumour with neuropil-like islands (GTNI), also known as ‘rosetted glioneuronal tumour’, is a novel representative of this type of neoplasms that was described for the first time about 16 years ago [1,2]. GTNI currently is considered a variant of astrocytoma, with a WHO-grade II or III [2]. It is characterized by infiltrating growth of astrocytic cells punctuated by foci of neuronal differentiation consisting of neuropil-like islands rimmed by neuronal cells. They are typically tumours of adult age and only very rare paediatric cases have been documented, mostly involving the spinal cord [3]. More recently, it has been reported one case in which a GTNI was identified at autopsy of an in-utero demise of a 38-week-gestation female foetus [4]. Any specific signature has been so far highlighted either in adults or paediatric GTNI [5-7].
In recent years, cytogenetic and molecular investigations have dramatically improved our understanding of the biology of CNS tumours, identifying relevant molecular features.
Although point mutations, loss of heterozigosity (LOH), gene amplifications are most commonly described as one of the key factors in the cancer pathogenesis, recently it is known that common genomic copy number variations (CNVs) and CNVs with low frequencies in the population (rare CNVs) may contain cancer related genes contributing to carcinogenesis [8-10].
Since in our previous studies we identified a strong genomic instability with recurrent CNVs in paediatric Glioblastoma Multiforme (pGBMs) [8], we decided to use the same approach (array platforms) to investigate 4 GTNI, first treated with surgery, then followed by high doses chemotherapy and radiotherapy, in order to identify the presence of numerical and structural rearrangements. In all cases, we compared the tumour biopsy with blood sample of the same patient. We could not find any recurrent CNVs although in two of the cases we detected a mosaic trisomy 8 (15-20%) in one case, and an amplification, inherited from the mother, at 5q14.1 involving DMGDH (partially), BHMT2 and BHMT genes, with the distal breakpoint falling at 23 Kbp from the 5’UTR of JMY, a p53 cofactor, in the last case.
Materials and methods
Patients
Four paediatric patients with GTNI were enrolled at our institution (Meyer Children’s University Hospital, Florence). Histological assessments were done by two independent pathologists, according to the WHO criteria. The study was approved from the Institutional Ethics Committee, and in all cases informed consent was obtained from parents.
Their main clinical characteristics are summarized in Table 1 and the MR scans showing GTNI lesions in the Figure 1. Median age at the time of diagnosis was 60 months (range, 0-96 months). All subjects underwent surgery for resection of CNS main lesion, which turned to be complete in 1 of 4 cases. All cases had been treated with high doses of chemotherapy with autologous stem cells transplant (HDCT/ASCT). Three patients underwent radiotherapy before HDCT/ASCT. Only case 4 underwent to HDCT/ASCT without radiotherapy, according to infant CNS tumours protocol of the Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP) [11].
Table 1.
Clinical characteristics of GTNI patients
| ID | Sex (M/F) | Age | Site | Disseminated | Surgery | Therapy | Response | Follow-up (Months) |
|---|---|---|---|---|---|---|---|---|
| Case 1 | M | 4 y | Fronto-parietal | NO | GTR | HDCT-ACST/focal RT | CR | 73 |
| Case 2 | F | 6 y | Spinal | YES | PTR | HDCT-ACST/cranio-spinal RT | PR | 76 |
| Case 3 | F | 8 y | Spinal | NO | PTR | HDCT-ACST/focal RT | PR | 70 |
| Case 4 | M | At birth | Intra-ventricular | NO | PTR | HDCT-ACST | PR | 23 |
GTR: gross total removal; PTR: partial total removal; HDCT: high dose chemotherapy; ACST: autologous stem cell transplantation; RT: radiotherapy; CR: Complete response; PR: Partial response.
Figure 1.
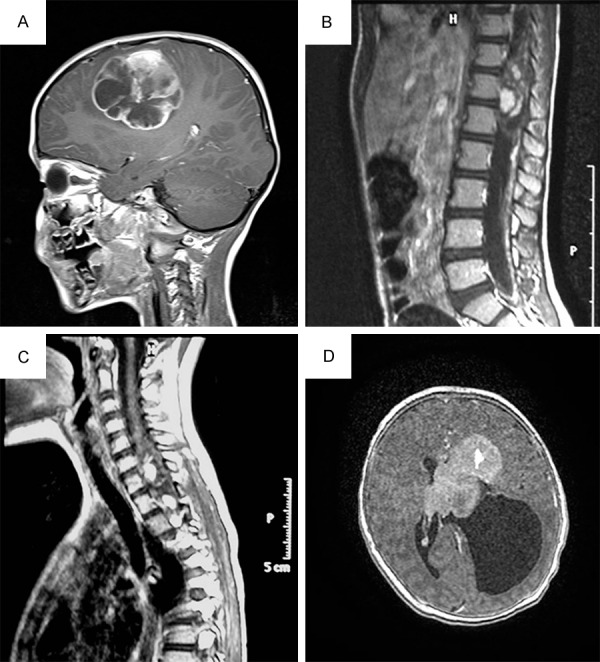
Preoperative Gd-enhanced T1-weighted MR scans showing GTNI lesions. A: Sagittal emispheric scan of case 1; B: Sagittal cervical spinal scan of patient 2; C: Sagittal dorso-lumbar scan of patient 3; D: Axial emispheric scan of patient 4.
All patients tolerated well the chemotherapy regimen, 3 of them with partial response, and 1 with a complete one. After a follow up time ranging from 23 to 76 months (median 71, 5 months), all are alive.
Histology
Surgical samples were routinely fixed in neutral buffered formalin and embedded in paraffin. Five μm sections were stained with hematoxylin-eosin (H&E) for the morphological evaluation, and further 5 μm sections of the most representative specimens were mounted on electrostatic slides and used for the immunohistochemical analysis. Immunohistochemical studies were performed using the standard streptavidin-biotin technique and commercially available antibodies (glial fibrillary acidic protein [GFAP], clone ZCG29, Zymed Lab., San Francisco, California, USA; S-100 protein, policlonal, DAKO CYTOMATION, Glostrup, Denmark; synaptophysin [SP] polyclonal, Cell Marque, Rocklin, California; neurofilaments [NF], clone 2F11, Cell Marque; EMA, clone E29, DAKO CYTOMATION; and Ki-67, clone Mib-1, DAKO CYTOMATION).
DNA extraction
Tumour, peripheral blood and buccal swab DNAs were extracted using QIAamp Mini Kit (QIAGEN®, Hilden, Germany) according to manufacturers’ instructions and quantified by NanoDROP 2000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA).
SNP/CGH array
SNP/CGH array was performed using the Agilent Human Genome CGH Microarray Kit 180 K (Agilent Technologies, Santa Clara, California, USA). This platform is an oligonucleotide-based microarray with an average resolution of about 100 kb to detect CNVs and 4 Mb to detect LOH. Labelling and hybridization were performed following the protocols provided by Agilent. 500 ng of purified DNA of the patient and of a control of the same sex (Coriell) were double digested with RsaI and AluI enzymes (Promega) for 2 h at 37°C, obtaining products between 200 bp and 500 bp in length. Each digested sample was labelled for 2 h, minimizing light exposure, using the Agilent Genomic DNA Labelling Kit, using Cy5-dUTP for the patient DNA and Cy3-dUTP for the reference DNA. Labelled products were column purified (Amicon Ultra, Millipore) and prepared combining test and control sample according to the Agilent protocol. After probe denaturation and pre-annealing with 50 μg of Human Cot-1 DNA (Invitrogen), hybridization was performed at 65°C for 24 h in a rotating oven at 20 rpm. Images of the arrays were acquired with the Agilent C Scanner (Agilent Technologies, Santa Clara, CA, USA). Each hybridization produced a pair of 16-bit images, which were processed using the Agilent Feature Extraction 10.5 software. Row data were analysed using the Genomic Workbench Standard Edition 5.0 software by the ADM-2 algorithm (breakpoint positions were reported according to Hg19, build 37). In order to take into account sample heterogeneity, for each experiment, we set the cellularity parameter c equal to 0.7, assuming 70% tumour purity.
Validation of CNVs by qPCR
Validation of CNVs by qPCR was performed using the Roche LightCycler® 480 Detection System with DNA-binding dye SYBR Green I (Roche) according to the manufacturers’ instructions. The primers were designed using Primer 3 software (http://biotools.umassmed.edu/bioapps/primer3_www.cgi).
Statistical analysis
Statistical significance of events of deletion and duplication in GTNI was evaluated using the t-test [12].
Cytogenetic analysis
Chromosomal analysis was performed on phytohemagglutinin-stimulated peripheral lymphocyte cultures using standard cytogenetic methods (Chromosome Kit P EuroClone), incubated 72-hours at 37°C and investigated by QFQ -banding analysis.
Results
Histology
Case 1
Microscopic examination revealed a biphasic cyto-architectural pattern of the lesion. There were some areas composed by neurocytic-like cells delimiting islands of fine eosinophilic neurofibrillary matrix (true rosettes) and areas consisted of elongated cells forming prominent ependymal perivascular rosettes (Figure 2A). The tumour showed large areas of necrosis, haemorrhage, vascular proliferation and rare micro-calcifications with mitotic counts of 5 × 10 high-power fields (HPF). The neoplastic cells diffusely stained for GFAP and S-100. The neurofibrillary islands were SP positive punctate dot-like intracytoplasmic staining for EMA in the elongated cells was observed. NF were negative. Ki-67, in the most positive areas, was 12%. Morphological features and immunohistochemical results were consistent with the diagnosis of WHO grade III glioneuronal tumor with neuropil-like islands and prominent ependymal component.
Figure 2.
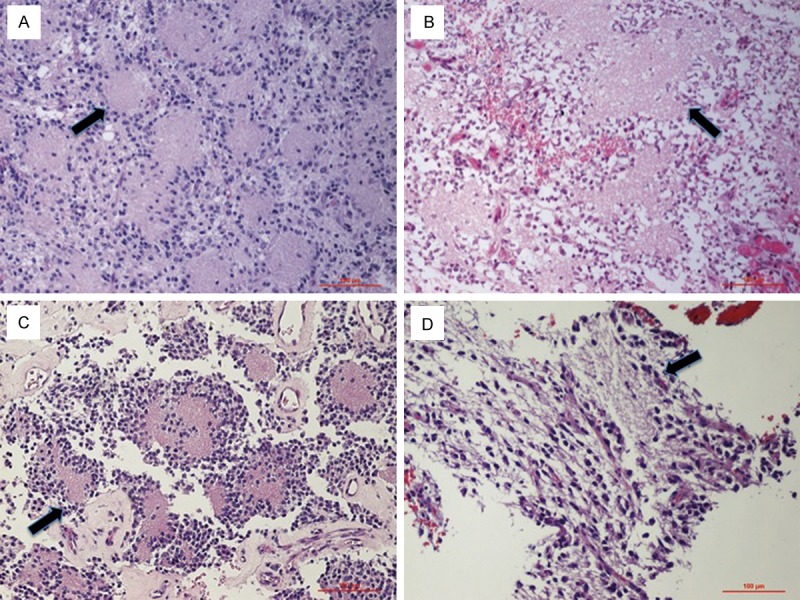
Histopathological features of our GTNI tumors: neurocytic-like cells delimiting islands of fine eosinophilic neurofibrillary matrix (A-D). The arrows indicate the rosette glioneuronal islands.
Case 2
Histological examination showed a neoplasm with some areas composed by elongated pleomorphic cells and other ones composed by neurocytic-like cells disposed in “rosettes” delimiting a core of fine neurofibrillary matrix (Figure 2B). Neither vascular proliferation nor necrosis were present. Several focal haemorrhagic areas were observed. Mitotic activity was 6 mitoses × 10 HPF. The tumour diffusely stained for S-100 protein. Elongated cells were GFAP positive. The neuropil islands and adjacent neurocytic-like cells were SP positive. No EMA and NF immunoreactivity were detected. Up to 18% of tumour cells were Ki-67 positive. Morphological features along with immunoistochemical results were consistent with the diagnosis of WHO grade III glioneuronal tumour with neuropil-like islands.
Case 3
The specimen obtained from the operation showed a densely cellular tumour composed of roundish neurcytic-like cells delimiting islands of neurofibrillary matrix (Figure 2C). Notably, the blood vessels showed hyaline thickened walls. No necrosis, haemorrhage or vascular proliferation were observed. There was no significant mitotic activity. The tumour diffusely stained for SP and S-100 protein. GFAP was focally positive. Few NF positive cells were appreciable. No EMA immunoreactivity was detected. The proliferation index, evaluated at the immunoistochemistry (Ki-67), was 16%. Morphological features and immunoistochemical results were consistent with the diagnosis of WHO grade III glioneuronal tumour with neuropil-like islands.
Case 4
Histological examination showed a neoplasm composed of pleomorphic cells delimiting cores of fine neurofibrillary matrix (very small bioptic specimens) (Figure 2D). Neither vascular proliferation nor necrosis were present. There was no significant mitotic activity. The tumour diffusely stained for S-100 protein and GFAP. The neuropil islands were SP positive. No EMA and NF immunoreactivity was detected. About 8% of tumor cells were Ki-67 positive. Morphological features and immunoistochemical results were suggestive of WHO grade III glioneuronal tumour with neuropil-like islands.
SNP/CGH array
We performed array analysis in the peripheral blood and tumour samples from all cases. The CNVs present in the Database of Genomic Variants (DGV: http://projects.tcag.ca/variation/) were taken into consideration only if with a frequency < to 5%.
In all tumor’s samples we did not find any specific and recurrent CNV. Cases 2 and 3 did not show any difference in the CNV pattern between blood and cancer. Case 4 has a duplication of the entire chromosome 8 with a dosage suggestive of mosaicism of 15-20% (log2 ratio of +0.3), and we also have shown that the supernumerary chromosome 8 was of maternal origin (Figure 3B). No mosaicism of supernumerary chromosome 8 was detected in the buccal swab analysis (data available on request).
Figure 3.

Human Genome SNP/CGH array hybridization profile of chromosome 5 and 8 for the case 1 (A) and case 4 (B). The size of the amplification is indicated by the blue bars and the red dots represent probes with positive Log2 fluorescence ratios. Chromosome 5 (A) and 8 (B) view are exhibited on the left-side panels and gene views of the proximal and distal amplification breakpoint regions are shown on the right-side panels (A). In the bottom (B) is showed, in the right-side panel, the mosaic amplification of chromosome 8. The order of sample from left to right is: tumour and blood of patients, father and mother blood. (A) Cases 1 with amplification at 5q14.1 including partially DMGDH, BHMT2 and BHMT genes. This amplification is present in the tumour and blood of proband and in the mother blood. (B) In case 4 the level of mosaicism of the entire chromosome 8 duplication is reflected in reduced log2 values, along the x-axis compared with normal samples and is present only in the tumour sample. The maternal origin of duplication is indicated from blue line that in the table reveals one of the significant SNPs in which it is confirmed the presence of a supernumerary maternal allele.
Case 1 presented an about 291 kb amplification in 5q14.1 (chr5: 78,316,935-78,508,370) containing the DMGDH (partially), BHMT2 and BHMT genes (Figure 3A).
qPCR
All CNVs were validated with Real Time-PCR method (data available on request), that confirmed array data.
Cytogenetic analysis
Chromosome examination on 50 metaphases in cases 1-3 and 100 metaphases in case 4 provided normal results. This result is generally sufficient to exclude a constitutional chromosome 8 mosaicism, demonstrating a plausible somatic trisomy 8 in GTNI.
Discussion
GTNI is a recently characterized type of primary glioneuronal tumour described mainly in adults [1,2] with very few cases so far studied from the molecular point of view. Differential diagnoses include ependymomas, other astrocytoma variants, and oligodendromas. Preferential localization in adults is the cerebrum, while brain and the spinal cord GTNI are almost equally represented in paediatric cases [3,13]. Although most tumours are histologically low grade, there is an appreciable risk of progression with time, with a poor prognosis despite their low-grade morphology [5,14]. No treatment strategy may currently be defined as a “gold standard” for patient with GTNI. Radiotherapy and chemotherapy as adjuvant cures after resection are a cornerstone of the treatment.
Numerous studies have shown that GTNI differ from gliomas on the morphological and immunohistochemical characteristics. Histologically, they are characterized by biphasic neurocytic and glial population. The neuronal component demonstrates immunoreactivity with neurocytic markers, such as synaptophysin, whereas the glial component displays strong immunoreactivity for GFAP and S-100 protein [13,15-18].
We report 4 cases of GTNI studied by SNP/CGH array approach in order to identify possible recurrent CNVs, as demonstrated for our previous findings on pGBMs [8]. We did not identify any recurrent CNV, whereas in cases 1 and 4 a 5q14.1 amplification and a mosaic trisomy 8 without any specific clinical signature were respectively detected.
Patient 4 showed a 15-20% of mosaic duplication of the entire chromosome 8 (Figure 3B). Mosaic trisomy 8 (MT8) is a rare condition with prevalence estimated in the range of 1:25,000-1:50,000 births and a preponderance in males. It was found both in syndromic (Warkany syndrome) and healthy people, and it has been proposed that could predispose to hematologic neoplastic disorders [19] and childhood cancer, such as Wilms tumour [20]. In case 4 we could not get any evidence in favour or against of a meiotic origin of the trisomy, but we are able to establish that in tumour tissue supernumerary chromosome 8 is of maternal origin. The patient, a new-born male, looks as a bright child without any signs of the Warkany syndrome including deep plantar furrows [21].
Case 1 was a supratentorial GNTI who underwent to gross total removal and a complete response to an intensive adjuvant chemo/radiotherapy program. The patient is alive with a follow-up of 73 months. SNP/CGH array displayed an amplification at 5q14.1 (Figure 3A), containing the partially DMGDH, BHMT2 and BHMT genes. It has to be emphasized that the distal breakpoint falls at 23 Kbp from the 5’UTR of JMY gene, encoding for a p53 cofactor.
Regarding DMGDH gene, it encodes a mitochondrial dimethylglycine dehydrogenase related with oxidative demethylation of dimethylglycine in vitro and formation of sarcosine, hydrogen peroxide and formaldehyde [22]. Recently this gene was associated by genome wide studies (WAS) with the juvenile papillary thyroid carcinoma (PTC), a rare tumour of the thyroid gland. It seems that DMGDH gene interacting with a non-coding RNA, promotes carcinoma development and progression [22].
BHMT gene encodes a cytosolic enzyme that catalyses the conversion of betaine and homocysteine to dimethylglycine and methionine, respectively. Defects in this gene might lead to hyperhomocyst(e)inemia, but such a defect has not yet been observed. Pellanda et al described that a transcription variant of exon 4 of this gene produces a loss of function of BHMT in human hepatocarcinoma, suggesting that this abnormal transcription of BHMT could be part or consequence of liver carcinogenesis [23]. No duplication or amplification of this gene have so far been reported in cancer. At the moment, we have no information about a possible involvement of the BHMT2 gene in the tumorigenesis.
Concerning the 5q rearrangement inheritance, literature data report different inherited genomic regions that influence susceptibility to cancer [24]. Understanding the mechanisms by which inherited genetic variants predispose to cancer only in some family members is partially understood for deletions and duplications. For example, recurrent inherited rearrangements in 9p21, including the CDKN2A/CDKN2B genes, with expression variability/incomplete penetrance are reported in multiple types of cancer (including breast cancer, melanoma, glioma, and leukemia), as well as non cancer-related diseases, such as type 2 diabetes and myocardial infarction [25-28].
In contrast, no clear data have been so far reported for the role of inherited amplification in cancer whereas incomplete penetrance/expression variability is well documented for some constitutional disorders [29-31].
It must be said that in the case 1, the association between tumour and 5q14.1 amplification may not reside in the amplification per se, but rather in the destruction of the JMY gene regulation. This gene, that is located about 23 Kbp from the distal breakpoints of the amplification, acts as a nuclear p53/TP53-cofactor increasing p53/TP53-dependent transcription and apoptosis [32-35]. The distal breakpoint at 5q14.1 might interferes with the normal expression and regulation of JMY, determining the loss of tumour suppressive functions of p53. However, in that case it is not clear why any possible misexpression of this gene is associated with cancer in the child and not the mother.
Conclusions
Clinical understanding of the GTNI is currently in evolution. Although our study deals with is a very limited sample of cases, given the rarity of the disorder, it may provide a preliminary understanding of the molecular basis of this family of tumours, showing that no specific imbalances are involved in paediatric GTNI. MT8, to our knowledge, was never described in brain tumours. Since in this patient the tumour was diagnosed at birth, the supernumerary 8 could really have had a role in the onset of cancer. Similarly, the 5q14.1 amplification points on specific cancer-associated pathways, whose involvement remains entirely to be proven.
Certainly, from our preliminary data, we can argue that GTNI is not a genetically homogeneous entity, with chromosome imbalances present only in 2 of the 4 cases. The two cases are distinguished by a brain localization, at variance of the remaining two with a spinal localization. Further studies are required to show if this is a coincidence or not.
Acknowledgements
This work was supported by grants from AMICO DI VALERIO ASSOCIAZIONE ONLUS and CON LORENZO PER MANO ONLUS.
Disclosure of conflict of interest
None.
References
- 1.Teo JG, Gultekin SH, Bilsky M, Gutin P, Rosenblum M. A distinctive glioneuronal tumor of the adult cerebrum with neuropil-like (including “rosetted”) islands: report of 4 cases. Am J Surg Pathol. 1999;23:502–10. doi: 10.1097/00000478-199905000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Comunoglu N, Kilickesmez O, Oz B. Spinal cord glioneuronal tumor with rosetted neuropil-like islands in pediatric age group. Case Rep Pathol. 2014;2014:471645. doi: 10.1155/2014/471645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chesser JD, Friedman NR, Prayson RA. Congenital glioneuronal tumor with neuropil-like islands. J Clin NeuroscI. 2016;24:156–7. doi: 10.1016/j.jocn.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 5.Allende DS, Prayson RA. The expanding family of glioneuronal tumors. Adv Anat Pathol. 2009;16:33–9. doi: 10.1097/PAP.0b013e3181915e3b. [DOI] [PubMed] [Google Scholar]
- 6.Barbashina V, Salazar P, Ladanyi M, Rosenblum MK, Edgar MA. Glioneuronal tumor with neuropil-like islands (GNTI): a report of 8 cases with chromosome 1p/19q deletion analysis. Am J Surg Pathol. 2007;31:1196–202. doi: 10.1097/PAS.0b013e3180335f65. [DOI] [PubMed] [Google Scholar]
- 7.Keyvani K, Rickert CH, von Wild K, Pailus W. Rosetted glioneuronal tumor: a case with proliferating neuronal nodules. Acta Neuropathol. 2001;101:525–8. doi: 10.1007/s004010000303. [DOI] [PubMed] [Google Scholar]
- 8.Giunti L, Pantaleo M, Sardi I, Provenzano A, Magi A, Cardellicchio S, Castiglione F, Tattini L, Novara F, Buccoliero AM, de Martino M, Genitori L, Zuffardi O, Giglio S. Genome-wide copy number analysis in pediatric glioblastoma multiforme. Am J Cancer Res. 2014;4:293–303. [PMC free article] [PubMed] [Google Scholar]
- 9.Carvalho CM, Lupski JR. Copy number variation at the breakpoint region of isochromosome 17q. Genome Res. 2008;18:1724–32. doi: 10.1101/gr.080697.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan X, Yu G, Hou X, Shih IeM, Clarke R, Zhang J, Hoffman EP, Wang RR, Zhang Z, Wang Y. Genome-wide identification of significant aberrations in cancer genome. BMC Genomics. 2012;13:342. doi: 10.1186/1471-2164-13-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Massimino M, Gandola L, Luksch R, Spreafico F, Riva D, Solero C, Giangaspero F, Locatelli F, Podda M, Bozzi F, Pignoli E, Collini P, Cefalo G, Zecca M, Casanova M, Ferrari A, Terenziani M, Meazza C, Polastri D, Scaramuzza D, Ravagnani F, Fossati-Bellani F. Sequential chemotherapy, high-dose thiotepa, circulating progenitor cell rescue, and radiotherapy for childhood high-grade glioma. Neuro Oncol. 2005;7:41–8. doi: 10.1215/S1152851704000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Box JF. Guinness, Gosset, Fisher, and Small Samples. Statist Sci. 1987;2:1–104. [Google Scholar]
- 13.Poliani PL, Sperli D, Valentini S, Armentano A, Bercich L, Bonetti MF, Corriero G, Brisigotti M, Quattrone A, Lanza PL. Spinal glioneuronal tumor with neuropil-like islands and meningeal dissemination: histopathological and radiological study of a pediatric case. Neuropathology. 2009;29:574–8. doi: 10.1111/j.1440-1789.2008.00988.x. [DOI] [PubMed] [Google Scholar]
- 14.Schlamann A, von Bueren AO, Hagel C, Zwiener I, Seidel C, Kortmann RD, Müller K. An individual patient data meta-analysis on characteristics and outcome of patients with papillary glioneuronal tumor, rosette glioneuronal tumor with neuropil-like islands and rosette forming glioneuronal tumor of the fourth ventricle. PLoS One. 2014;9:e101211. doi: 10.1371/journal.pone.0101211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agarwal S, Suri V, Rishi A, Shukla B, Garg A, Sharma MC, Sinha S, Sarkar C. Glioneuronal tumor with neuropil like islands: a new entity. Neuropathology. 2009;29:96–100. doi: 10.1111/j.1440-1789.2008.00933.x. [DOI] [PubMed] [Google Scholar]
- 16.Ruppert B, Welsh CT, Hannah J, Giglio P, Rumboldt Z, Johnson I, Fortney J, Jenrette JM, Patel S, Scheithauer BW. Glioneuronal tumor with neuropil-like islands of the spinal cord with diffuse leptomeningeal neuraxis dissemination. J Neurooncol. 2011;104:529–33. doi: 10.1007/s11060-010-0505-1. [DOI] [PubMed] [Google Scholar]
- 17.Buccoliero AM, Castiglione F, Degl’innocenti DR, Moncini D, Paglierani M, Sardi I, Giunti L, Giordano F, Sanzo M, Mussa F, Aricò M, Genitori L, Taddei GL. Glioneuronal tumor with neuropil-like islands: clinical, morphologic, immunohistochemical, and molecular features of three pediatric cases. Pediatr Dev Pathol. 2012;15:352–60. doi: 10.2350/12-01-1147-OA.1. [DOI] [PubMed] [Google Scholar]
- 18.Serra SM, Dabdoub CB, da Cunha AH, Salazar B, Lima TP, Azevedo-Filho HC. Disseminated glioneuronal tumor with neuropil-like islands of the spinal cord: a distinctive entity. World Neurosurg. 2013;80:655, e1–5. doi: 10.1016/j.wneu.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 19.Maserati E, Aprili F, Vinante F, Locatelli F, Amendola G, Zatterale A, Milone G, Minelli A, Bernardi F, Lo Curto F, Pasquali F. Trisomy 8 in myelodysplasia and acute leukemia is constitutional in 15-20% of cases. Genes Chromosomes Cancer. 2002;33:93–7. doi: 10.1002/gcc.1214. [DOI] [PubMed] [Google Scholar]
- 20.Valind A, Pal N, Asmundsson J, Gisselsson D, Holmquist Mengelbier L. Confined trisomy 8 mosaicism of meiotic origin: a rare cause of aneuploidy in childhood cancer. Genes Chromosomes Cancer. 2014;53:634–8. doi: 10.1002/gcc.22173. [DOI] [PubMed] [Google Scholar]
- 21.Secker-Walker LM, Fitchett M. Constitutional and acquired trisomy 8. Leuk Res. 1995;19:737–40. doi: 10.1016/0145-2126(95)00051-o. [DOI] [PubMed] [Google Scholar]
- 22.Binzak BA, Wevers RA, Moolenaar SH, Lee YM, Hwu WL, Poggi-Bach J, Engelke UF, Hoard HM, Vockley JG, Vockley J. Cloning of dimethylglycine dehydrogenase and a new human inborn error of metabolism, dimethylglycine dehydrogenase deficiency. Am J Hum Genet. 2001;68:839–47. doi: 10.1086/319520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pellanda H, Namour F, Fofou-Caillierez M, Bressenot A, Alberto JM, Chéry C, Ayav A, Bronowicki JP, Guéant JL, Forges T. A splicing variant leads to complete loss of function of betaine-homocysteine methyltransferase (BHMT) gene in hepatocellular carcinoma. Int J Biochem Cell Biol. 2012;44:385–92. doi: 10.1016/j.biocel.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 24.Pomerantz MM, Freedman ML. The genetics of cancer risk. Cancer J. 2011;17:416–22. doi: 10.1097/PPO.0b013e31823e5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng J, Kim ST, Liu W, Kim JW, Zhang Z, Zhu Y, Berens M, Sun J, Xu J. An integrated analysis of germline and somatic, genetic and epigenetic alterations at 9p21.3 in glioblastoma. Cancer. 2012;118:232–40. doi: 10.1002/cncr.26250. [DOI] [PubMed] [Google Scholar]
- 26.Iolascon A, Faienza MF, Coppola B, Rosolen A, Basso G, Della Ragione F, Schettini F. Analysis of cyclin-dependent kinase inhibitor genes (CDKN2A, CDKN2B, and CDKN2C) in childhood rhabdomyosarcoma. Genes Chromosomes Cancer. 1996;15:217–22. doi: 10.1002/(SICI)1098-2264(199604)15:4<217::AID-GCC3>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 27.Landman GW, van Vliet-Ostaptchouk JV, Kleefstra N, van Hateren KJ, Drion I, Groenier KH, Gans RO, Snieder H, Hofker MH, Bilo HJ. Association between 9p21 genetic variants and mortality risk in a prospective cohort of patients with type 2 diabetes (ZODIAC-15) Cardiovasc Diabetol. 2012;11:138. doi: 10.1186/1475-2840-11-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wahlstrand B, Orho-Melander M, Delling L, Kjeldsen S, Narkiewicz K, Almgren P, Hedner T, Melander O. The myocardial infarction associated CDKN2A/CDKN2B locus on chromosome 9p21 is associated with stroke independently of coronary events in patients with hypertension. J Hypertens. 2009;27:769–73. doi: 10.1097/HJH.0b013e328326f7eb. [DOI] [PubMed] [Google Scholar]
- 29.Patil SJ, Salian S, Bhat V, Girisha KM, Shrivastava Y, Vs K, Sapare A. Familial 7q11.23 duplication with variable phenotype. Am J Med Genet A. 2015;167A:2727–30. doi: 10.1002/ajmg.a.37226. [DOI] [PubMed] [Google Scholar]
- 30.Ou Z, Berg JS, Yonath H, Enciso VB, Miller DT, Picker J, Lenzi T, Keegan CE, Sutton VR, Belmont J, Chinault AC, Lupski JR, Cheung SW, Roeder E, Patel A. Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet Med. 2008;10:267–77. doi: 10.1097/GIM.0b013e31816b64c2. [DOI] [PubMed] [Google Scholar]
- 31.Fernandez BA, Roberts W, Chung B, Weksberg R, Meyn S, Szatmari P, Joseph-George AM, Mackay S, Whitten K, Noble B, Vardy C, Crosbie V, Luscombe S, Tucker E, Turner L, Marshall CR, Scherer SW. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J Med Genet. 2010;47:195–203. doi: 10.1136/jmg.2009.069369. [DOI] [PubMed] [Google Scholar]
- 32.Shikama N, Lee CW, France S, Delavaine L, Lyon J, Krstic-Demonacos M, La Thangue NB. A novel cofactor for p300 that regulates the p53 response. Mol Cell. 1999;4:365–76. doi: 10.1016/s1097-2765(00)80338-x. [DOI] [PubMed] [Google Scholar]
- 33.Coutts AS, Boulahbel H, Graham A, La Thangue NB. Mdm2 targets the p53 transcription cofactor JMY for degradation. EMBO Rep. 2007;8:84–90. doi: 10.1038/sj.embor.7400855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326:1208–12. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuchero JB, Coutts AS, Quinlan ME, Thangue NB, Mullins RD. p53-cofactor JMY is a multifunctional actin nucleation factor. Nat Cell Biol. 2009;11:451–9. doi: 10.1038/ncb1852. [DOI] [PMC free article] [PubMed] [Google Scholar]