

Abstract
The vast majority of multicellular organisms coexist with bacterial symbionts that may play various roles during their life cycle. Parasitoid wasp Megaphragma amalphitanum (Hymenoptera: Trichogrammatidae) belongs to the smallest known insects whose size is comparable with some bacteria. Using 16S rRNA gene sequencing and Whole Genome Sequencing (WGS), we described microbiota diversity for this arthropod and its potential impact on their lifecycle. Metagenomic sequences were deposited to SRA database which is available at NCBI with accession number SRX2363723 and SRX2363724. We found that small body size and limited lifespan do not lead to a significant reduction of bacterial symbionts diversity. At the same time, we show here a specific feature of microbiota composition in M. amalphitanum – the absence of the Rickettsiaceae family representatives that are known to cause sex-ratio distortion in arthropods and well represented in other populations of parasitoid wasps.
| Specifications | |
|---|---|
| Organism/cell line/tissue | Metagenome of parasitoid wasp Megaphragma amalphitanum |
| Sex | Not applicable |
| Sequencer or array type | Roche GS FLX instrument |
| Data format | Raw data: FASTAQ file |
| Experimental factors | Insect sample |
| Experimental features | 16S rRNA genes amplified from the metagenome using Illumina platform followed by bacterial community analysis using QIIME |
| Consent | Not applicable |
| Sample source location | Santa Margherita, Northern Italy |
1. Direct link to deposited data
M. amalphitanum: M1 bulk: https://www.ncbi.nlm.nih.gov/sra/SRX2363724
M. amalphitanum: M2 bulk: https://www.ncbi.nlm.nih.gov/sra/SRX2363723
2. Experimental design, materials and methods
Megaphragma amalphitanum specimens were reared from thrips eggs collected in Santa Margherita, Northern Italy. DNA was extracted using NucleoSpin Tissue XS kit (Macherey-Nagel, Germany). DNA quantity was measured with a Qubit® double-stranded DNA (dsDNA) High Sensitivity (HS) Assay Kit (Life Technologies, Eugene, OR, USA) and read a by Qubit® 2.0 Fluorometer (Life Technologies, Eugene, OR, USA). The amounts of extracted DNA were very low: M1 bulk (10 individuals M. amalphitanum) – 1.98 ng and M2 bulk (10 individuals M. amalphitanum) – 1.24 ng. Reagent and laboratory contamination can critically impact on the metagenome analyze. All works were conducted in clean conditions of the laminar airflow bench to avoid any contamination.
To analyze the composition of the microbial communities, we used the method based on pyrosequencing of the fragments of 16S ribosomal RNA genes [1]. PCR fragments of 16S rRNA genes were obtained using the “universal” primers 11F (5′ GTTTGATC MTGGCTCAG 3′) and 519R (5′ GWATTAC CGCGGCKGCTG 3′) [2]. The PCR fragments were purified using the Agencourt AMPure beads (Beckman Coulter Inc., USA) according to the manufacturer's instructions and sequenced in the GS FLX instrument (Roche, Switzerland) using the Titanium protocol.
The 16S rRNA sequencing data was filtered by quality and analyzed using QIIME software [3]. The sequences were clustered into Operational Taxonomic Units (OTUs) using uclust with 97% threshold based on their similarity [4]. Before that we assigned the appropriate taxonomy for our data using uclust and rdp methods with Naive Bayes classification [5]. The final tables and histograms with basic OTUs were generated using biom-format package [6].
Previously generated whole-genomic sequencing data (35,043,964 Illumina paired-end reads) were used for searching Wolbachia strains in Megaphragma [7]. At first step these reads were analyzed using Metaphlan2 [8].
Microbiota of M. amalphitanum presents a diverse variety of bacteria strains. In fact, the decrease of body size and reduction of structures do not affect microbiome diversity (Fig. 1). Nevertheless, its composition differs from much bigger parasitoid wasps - Nasonia, Asobara, Megastigmus [9], [10], [11]. Moreover, microbiota samples of M. amalphitanum from Northern Italy do not have representatives of Rickettsia and Wolbachia genus. Interestingly, we did not find a presence of sex ratio distortion and the numbers of reared Wolbachia-free males and Wolbachia-free females of this parasitoid wasp were similar to those specimens shown infected by Wolbachia, which were collected and analyzed previously [12]. Our results are consistent with the data published by Nguyen with colleagues [13] who showed that global population of greenhouse thrips (hosts of Megaphragma species) was not infected with Wolbachia. This also suggests that Wolbachia specimen presence that was found by Pintureau et al. [12] in Megaphragma species from Portugal and France was most likely due to unspecific primer binding or contamination artefact formation as we have not identified Wolbachia-strains in M. amalphitanum using 16S rRNA sequencing and by searching in WGS data of M. amalphitanum that were generated previously [7].
Fig. 1.
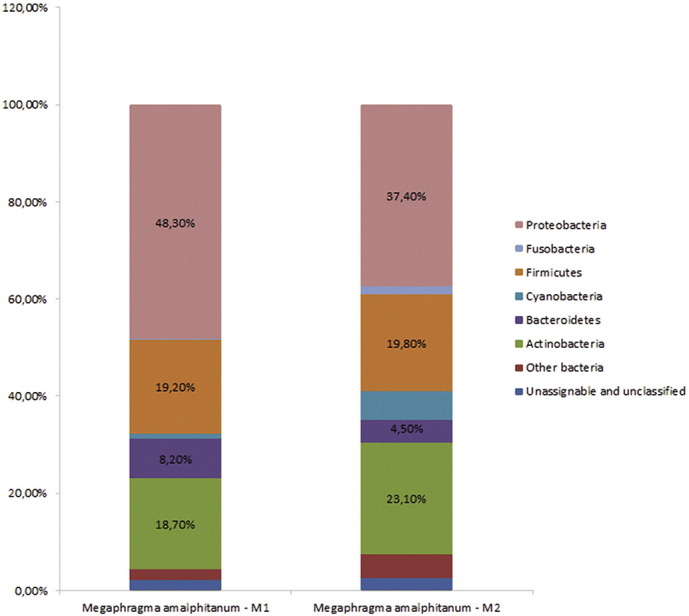
Relative abundance of major bacterial OTUs associated with M. amalphitanum based on 16 rRNA fragment from pyrosequencing.
In this work, we have clearly shown that despite the fact of the miniaturization, parasitoid wasp Megaphragma amalphitanum has a rich microbiota composition. However, although having the phylogenetic proximity, the diversity of microbiota representatives in these two arthropods has distinctive features related to specifics of their lifecycle.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by Russian Scientific Foundation (RSF) grant #14-24-00175.
References
- 1.Sogin M.L., Morrison H.G., Huber J.A., Welch M.D., Huse S.M., Neal P.R., Arrieta J.M., Herndl G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. U. S. A. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lane D.J. Wiley; Chichester: 1991. 16S/23S rRNA Sequencing, nucleic acid techniques in bacterial systematics; pp. 115–175. [Google Scholar]
- 3.Caporaso J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K., Fierer N., Peña A.G., Goodrich J.K., Gordon J.I., Huttley G.A., Kelley S.T., Knights D., Koenig J.E., Ley R.E., Lozupone C.A., McDonald D., Muegge B.D., Pirrung M., Reeder J., Sevinsky J.R., Turnbaugh P.J., Walters W.A., Widmann J., Yatsunenko T., Zaneveld J., Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edgar R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 5.Wang Q., Garrity G.M., Tiedje J.M., Cole J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDonald D., Clemente J.C., Kuczynski J., Rideout J.R., Stombaugh J., Wendel D., Wilke A., Huse S., Hufnagle J., Meyer F., Knight R., Caporaso J.G. The biological observation matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome. Gigascience. 2012;1:7. doi: 10.1186/2047-217X-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nedoluzhko A.V., Sharko F.S., Boulygina E.S., Tsygankova S.V., Sokolov A.S., Mazur A.M., Polilov A.A., Prokhortchouk E.B., Skryabin K.G. Mitochondrial genome of Megaphragma amalphitanum (Hymenoptera: Trichogrammatidae) Mitochondrial DNA. 2015:1–2. doi: 10.3109/19401736.2015.1101546. [DOI] [PubMed] [Google Scholar]
- 8.Segata N., Waldron L., Ballarini A., Narasimhan V., Jousson O., Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods. 2012;9:811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brucker R.M., Bordenstein S.R. The roles of host evolutionary relationships (genus: Nasonia) and development in structuring microbial communities. Evolution. 2012;66:349–362. doi: 10.1111/j.1558-5646.2011.01454.x. [DOI] [PubMed] [Google Scholar]
- 10.Paulson A.R., von Aderkas P., Perlman S.J. Bacterial associates of seed-parasitic wasps (Hymenoptera: Megastigmus) BMC Microbiol. 2014;14:224. doi: 10.1186/s12866-014-0224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zouache K., Voronin D., Tran-Van V., Mavingui P. Composition of bacterial communities associated with natural and laboratory populations of Asobara tabida infected with Wolbachia. Appl. Environ. Microbiol. 2009;75:3755–3764. doi: 10.1128/AEM.02964-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pintureau B., Lassabliere F., Khatchadourian C., Daumal J. Parasitoides oophages et symbiotes de deux thrips Europeens. Ann. Soc. Entomol. Fr. 1999;35:416–420. [Google Scholar]
- 13.Nguyen D.T., Spooner-Hart R.N., Riegler M. Polyploidy versus endosymbionts in obligately thelytokous thrips. BMC Evol. Biol. 2015;15:23. doi: 10.1186/s12862-015-0304-6. [DOI] [PMC free article] [PubMed] [Google Scholar]