

Abstract
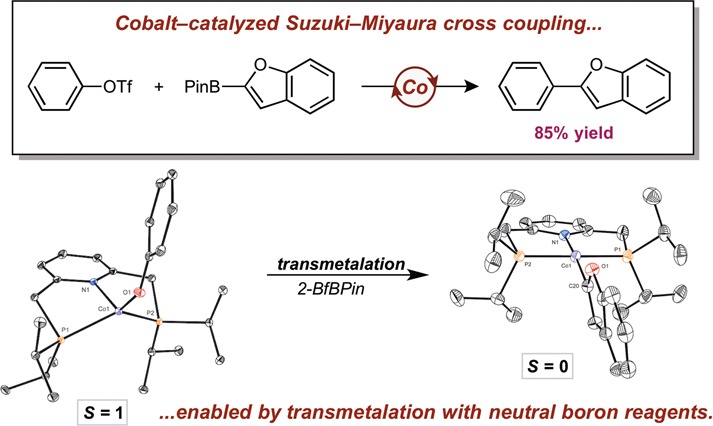
Among the fundamental transformations that comprise a catalytic cycle for cross coupling, transmetalation from the nucleophile to the metal catalyst is perhaps the least understood. Optimizing this elementary step has enabled the first example of a cobalt-catalyzed Suzuki–Miyaura cross coupling between aryl triflate electrophiles and heteroaryl boron nucleophiles. Key to this discovery was the preparation and characterization of a new class of tetrahedral, high-spin bis(phosphino)pyridine cobalt(I) alkoxide and aryloxide complexes, (iPrPNP)CoOR, and optimizing their reactivity with 2-benzofuranylBPin (Pin = pinacolate). Cobalt compounds with small alkoxide substituents such as R = methyl and ethyl underwent swift transmetalation at 23 °C but also proved kinetically unstable toward β–H elimination. Secondary alkoxides such as R = iPr or CH(Ph)Me balanced stability and reactivity. Isolation and structural characterization of the product following transmetalation, (iPrPNP)Co(2-benzofuranyl), established a planar, diamagnetic cobalt(I) complex, demonstrating the high- and low-spin states of cobalt(I) rapidly interconvert during this reaction. The insights from the studies in this elementary step guided selection of appropriate reaction conditions to enable the first examples of cobalt-catalyzed C–C bond formation between neutral boron nucleophiles and aryl triflate electrophiles, and a model for the successful transmetalation reactivity is proposed.
Short abstract
Transmetalation from neutral boron reagents to high spin cobalt(I) alkoxides was examined. Understanding this unprecendented reactivity enabled the development of the first example of cobalt-catalyzed Suzuki−Miyaura cross coupling.
Transition metal-catalyzed cross coupling has revolutionized carbon–carbon formation by enabling selective and efficient reactions of organic electrophiles and various nucleophiles.1 Among these methods, the palladium-catalyzed Suzuki–Miyaura reaction of organic halides electrophiles and organoboron nucleophiles is one of the most commonly applied strategies for C–C bond formation in chemical synthesis.2,3 The safe handling, relative stability, and broad availability of boron-based nucleophiles distinguishes the Suzuki–Miyaura reaction from other cross coupling methods that rely on reactive organolithium, Grignard, or zinc nucleophiles that can be incompatible with functional groups found in late stage intermediates and are dangerous to handle on a large scale.4 As a result, Suzuki–Miyaura cross coupling has become one of the preferred methods for making C–C bonds in the pharmaceutical, agricultural, and fine chemicals industries.5 The distinction between boron and other nucleophiles is particularly apparent in a recent analysis of reactions employed by the pharmaceutical industry that found Suzuki–Miyaura coupling accounted for approximately 40% of C–C bond-forming reactions, while coupling with other nucleophiles represented less than 5%.6
Palladium compounds are state-of-the-art catalysts for the Suzuki–Miyaura cross coupling. Their widespread use raises concerns about the sustainability7 and economics of these processes, the latter being influenced not only by cost of goods but by the purification steps required to meet strict regulations (typically <5 ppm) for palladium content in active pharmaceutical ingredients.8 These practical motivations, coupled with opportunities for new modes of reactivity and potential expansion of reaction scope, have prompted exploration of Earth abundant, first row transition metals as potential catalysts for the Suzuki–Miyaura reaction. Considerable progress has been made with nickel-9−18 and copper-19−25 catalyzed variants, but reactivity with earlier first row transition metals such as iron and cobalt has remained elusive. State-of-the-art iron-26−38 and cobalt-39−48 catalyzed C–C cross coupling methods rely on more reactive nucleophiles rather than the preferable neutral boron reagents associated with Suzuki–Miyaura chemistry (Scheme 1). Specifically, several recent examples of C(sp2)–C(sp3) and C(sp3)–C(sp3) iron-catalyzed cross coupling rely on stoichiometric addition of organolithium reagents to form activated borinate nucleophiles because the corresponding neutral boron partners do not participate.31−34 As such, the advantages of neutral boron nucleophiles are lost, and the reliance on radical capture for C–C bond formation has limited the scope to principally pure hydrocarbyl products such as cycloheptylbenzene. Understanding transmetalation reactivity is therefore key for developing improved iron- and cobalt-catalysts for Suzuki–Miyaura cross coupling. Here we describe a cobalt pincer complex that promotes Suzuki–Miyaura cross coupling of aryl triflate electrophiles and organoboron nucleophiles. Stoichiometric reactions designed to mimic elementary steps of the catalytic reaction provided crucial insight about the optimal nucleophile, electrophile, base, and conditions for the desired reactivity. A combination of these observations ultimately enabled successful catalytic turnover.
Scheme 1. C–C Bond Formation by Fe- and Co-Catalyzed Cross Coupling.
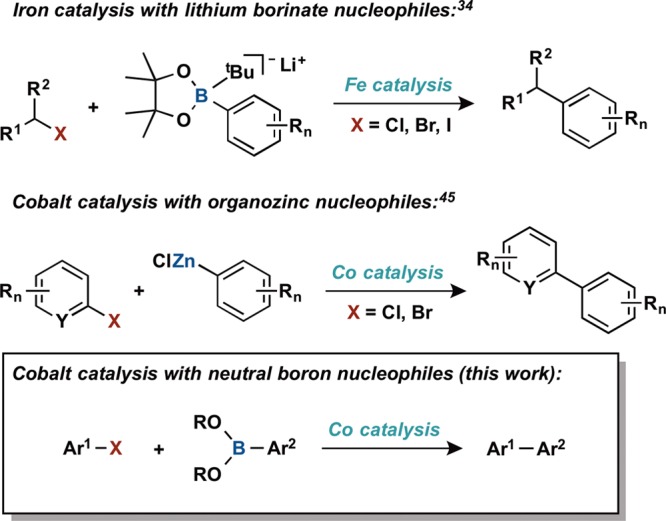
Scheme 2 presents a plausible cycle for cobalt-catalyzed C(sp2)−C(sp2) Suzuki–Miyaura coupling. Guiding principals from this pathway are (i) transmetalation of an aryl group from a neutral boron reagent to a CoOR complex to generate a cobalt(I) aryl, (ii) interaction with the aryl electrophile to promote carbon–carbon bond formation and release of product, generating a cobalt(I) halide, and (iii) exchange of the halide ligand with exogenous base to regenerate the CoOR species. One notable difference with first row transition metals is the likelihood of variable spin states and coordination geometries and kinetically accessible one-electron chemistry.49 Depending on the field strength of the supporting ligands, cobalt(I) complexes like those shown in Scheme 2 may be high (S = 1) or low (S = 0) spin with tetrahedral or planar geometries, respectively. With these considerations in mind, the flexible diisopropyl-substituted bis(phosphino)pyridine pincer ligand (iPrPNP) was selected for this study due to its known ability to support both tetrahedral (X = Cl) and planar (X = alkyl, aryl) cobalt(I) complexes.50−54 Furthermore, the (iPrPNP)cobalt platform promotes two-electron oxidative addition53 and has been applied to catalytic C–H borylation.50,52
Scheme 2. Plausible Catalytic Cycle for Suzuki–Miyaura Coupling with Cobalt.
The requirement for highly reactive nucleophiles in current iron and cobalt-catalyzed C–C coupling methods suggests sluggish transmetalation prevents catalytic turnover with neutral boron reagents. While transmetalation from boron is well established55 and mechanistically understood56,57 for palladium, analogous reactivity with iron58,59 or cobalt60,61 has not been demonstrated.62 Precedent with palladium63−65 suggested a transmetalation study would be best approached with the corresponding (iPrPNP)CoOR derivatives, a new class of bis(phosphine)pyridine cobalt complexes with unknown spin states, geometries and substitution chemistry.
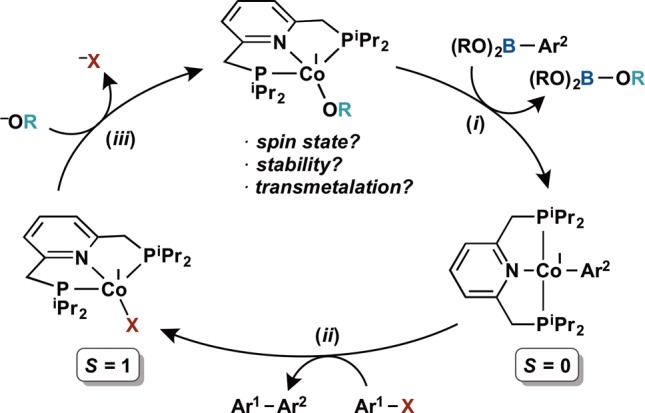
Synthesis of the target (iPrPNP)CoOR complexes was accomplished by protonolysis of the cobalt(I) alkyl complex (iPrPNP)CoCH2SiMe352 with a stoichiometric quantity of the appropriate alcohol (ROH, Scheme 3). In this manner, a series of paramagnetic cobalt(I) aryloxides (iPrPNP)CoOR and alkoxides were prepared. The cobalt aryloxide complexes (R = Ph, C6H4(4-OMe) and C6H4(3-F)) were isolated as purple powders, and solid state magnetic measurements established S = 1 ground states for each, consistent with high spin Co(I) derivatives. Despite their paramagnetism, these compounds were reliably identified by 1H NMR spectroscopy, exhibiting the number of peaks expected for compounds with C2v molecular symmetries. The cobalt phenoxide, (iPrPNP)CoOPh, was also characterized by X-ray diffraction (Figure 1). The pseudotetrahedral geometry is consistent with the high spin cobalt(I) ground state. To accommodate this geometry, the pincer ligand is significantly distorted from planarity, as evidenced by a P–Co–P bond angle of 123.751(13)° and the N(1)–Co(1)–O(1) bond angle of 128.18(4)°. Accordingly, the cobalt is deviated by 1.032 Å from the idealized plane of the PNP pincer. Other metrical parameters are as expected for a tetrahedral cobalt(I) center (see Supporting Information for complete details).
Scheme 3. Preparation and Decomposition of (iPrPNP)CoOR Complexes.
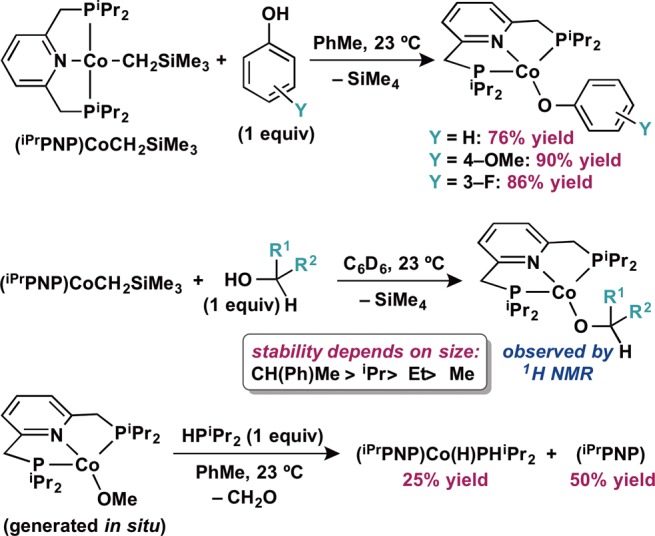
Figure 1.
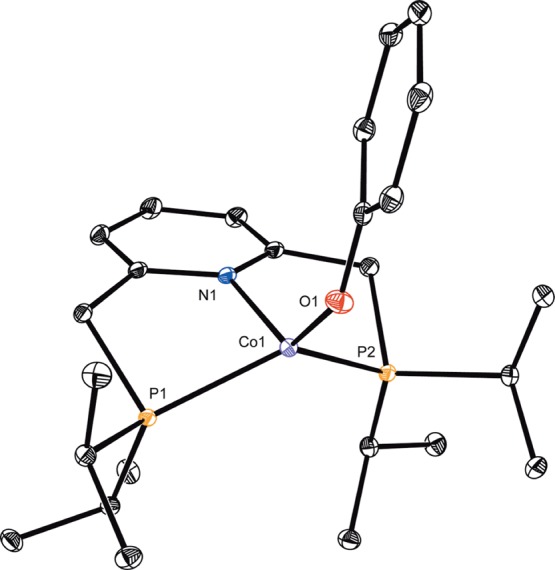
Solid state molecular structure of (iPrPNP)CoOPh at 30% probability ellipsoids. Hydrogen atoms omitted for clarity.
Several cobalt(I) alkoxides (iPrPNP)CoOR (R = CH(Ph)Me, iPr, Et and Me) were also prepared by protonolysis (Scheme 3). Attempts to prepare the tert-butoxide derivative (iPrPNP)CoOtBu have been unsuccessful, resulting in recovery of the starting cobalt alkyl. Like the aryloxides, the cobalt(I) alkoxides were paramagnetic but observable by 1H NMR spectroscopy. These complexes proved unstable in benzene-d6 solution at 23 °C with relative rates of decomposition decreasing from R = Me > Et > iPr > CH(Me)Ph. Approximate times for decomposition at 23 °C vary from over the course of 2 h for R = Me to 4 h for Et, 24 h for iPr, and 72 h for CH(Me)Ph. These times are approximate because the decomposition reactions do not yield a single cobalt product, and the complexity of the reaction mixture precluded detection of the expected aldehydes or ketones. In general, the relative stability is consistent with decomposition of the alkoxide complex by β-hydride elimination, where small alkyl substituents with more β-hydrogens decompose more rapidly. The putative cobalt hydride product, (iPrPNP)CoH, is known to decompose by P–C bond cleavage and is likely the source of the product mixtures.53 The formation of (iPrPNP)Co(PHiPr2)H53 in 25% yield by 1H NMR, accompanied by 50% free (iPrPNP) ligand, by methanolysis of (iPrPNP)CoCH2SiMe3 in the presence of HPiPr2 provides support for this hypothesis (Scheme 3).
Carboxylates were also explored as possible oxygen-based ligands for cobalt to promote transmetalation with boron reagents. These ligands were of interest due to precedent from our laboratory66 and Nagashima’s67 that cobalt carboxylate derivatives are bench-stable catalyst precursors. Addition of 1 equiv of benzoic acid (BzOH, Bz = C(O)Ph) to (iPrPNP)CoCH2SiMe3 at room temperature produced a complex mixture of cobalt products from which the cobalt(II) bis(carboxylate) (iPrPNP)Co(OBz)2 was identified as the major component. Addition of 2 equiv of BzOH to (iPrPNP)CoCH2SiMe3 resulted in clean formation of (iPrPNP)Co(OBz)2 isolated as a purple solid in 87% yield and was characterized by X-ray diffraction (see Figure S6). The target cobalt(I) benzoate (iPrPNP)CoOBz was prepared by slow addition of BzOH to a thawing toluene solution of (iPrPNP)CoCH2SiMe3 (see Supporting Information). The solid-state structure of this S = 1 complex (μeff = 2.8 μB, 23 °C) established a κ1-benzoate ligand. The P–Co–P bond angle of 126.081(14)° and the N(1)–Co(1)–O(1) bond angle of 119.65(4)° as well as the deviation of the cobalt by 1.000 Å from the metal chelate plane establish an idealized tetrahedral geometry, similar to (iPrPNP)CoOPh (Figure 2).68
Figure 2.
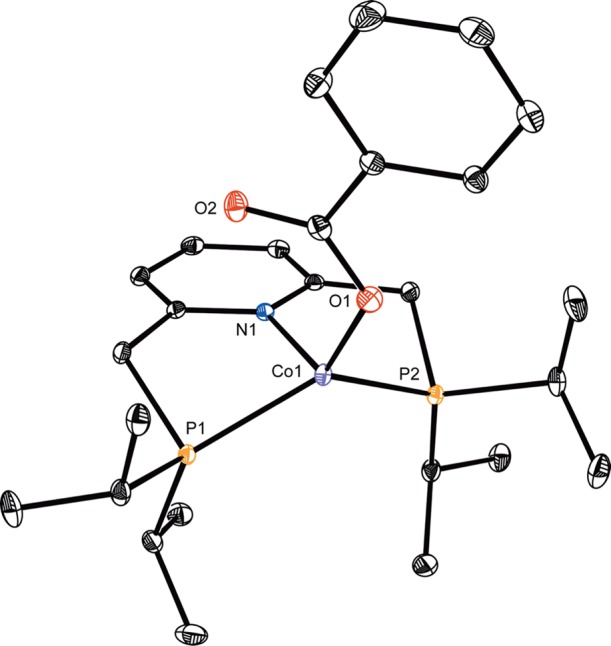
Solid state molecular structure of (iPrPNP)CoOBz at 30% probability ellipsoids. Hydrogen atoms omitted for clarity.
With a series of cobalt aryloxide, alkoxide, and carboxylate compounds in hand, transmetalation with neutral boron reagents was explored (Scheme 4). Addition of 1 equiv of phenylboronic acid pinacol ester (PhBPin) to a benzene-d6 solution of (iPrPNP)CoOPh at room temperature resulted in no reaction as judged by 1H NMR spectroscopy. By contrast, the corresponding reaction with 2-benzofuranylBPin resulted in gradual appearance of a new diamagnetic cobalt compound over the course of 24 h at 23 °C. Characterization by 1H, 13C, and 31P NMR spectroscopies as well as X-ray diffraction identified the product as (iPrPNP)Co(2-benzofuranyl), arising from transmetalation of the heteroaryl group from boron to cobalt (Figure 3). The solid state structure of (iPrPNP)Co(2-benzofuranyl) was determined by X-ray diffraction and confirmed the idealized planar geometry about the metal center, consistent with the observed diamagnetic ground state. While the analogous reaction with (iPrPNP)CoOBz generated an intractable mixture, clean formation of the desired organometallic complex was observed upon addition of 2-benzofuranylBPin to the cobalt alkoxides (iPrPNP)CoOR (R = CH(Me)Ph, iPr, Et, and Me) generated in situ. In these cases, transmetalation occurred immediately, signaled by a color change to a red brown solution, and 1H NMR spectroscopy confirmed clean formation of (iPrPNP)Co(2-benzofuranyl) within the time required for 1H NMR analysis. Notably, transmetalation occurred rapidly from S = 1 cobalt alkoxides to form diamagnetic cobalt heteroarene products, a difference from palladium chemistry where catalysis occurs predominantly on the S = 0 spin surface. Use of 2-benzofuranyl boronic acid as the boron reagent in the reaction of (iPrPNP)CoOCH(Me)Ph led to partial formation of the desired product within a complex mixture, indicating boronic acids are not optimal organoboron nucleophiles for transmetalation in this system.
Scheme 4. Transmetalation to Cobalt from Neutral Boron Reagents.
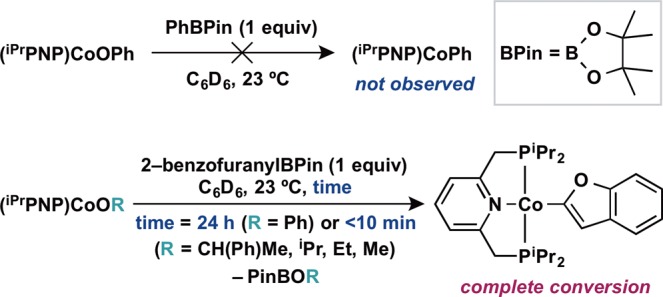
Figure 3.
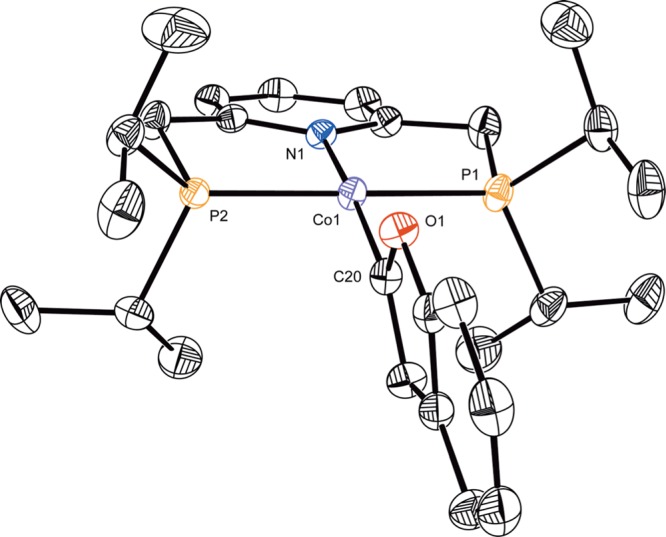
Solid state molecular structure of (iPrPNP)Co(2-benzofuranyl) at 30% probability ellipsoids. Hydrogen atoms omitted for clarity.
To gain further insight about this unprecendented reactivity, transmetalation of various heteroaryl boron reagents was explored (Scheme 5). Addition of a solution of 2-methylfuranylBPin or benzothiophenylBPin to (iPrPNP)CoOR (R = CH(Ph)Me, generated in situ) resulted in rapid formation of the corresponding organometallic product within the time required for analysis by 1H NMR spectroscopy, determined by analogy to the benzofuranyl complex (vide supra). When N-methylindolylBPin was used as the boron reagent, transmetalation did not proceed. These observations suggest the lone pair of the heteroaryl group is essential for transmetalation reactivity. This influence may arise from initial coordination of the lone pair to cobalt, bringing the boron atom in proximity of the alkoxide ligand and initiating the transmetalation process (Scheme 5). This coordination event may also induce a change in the spin state from S = 1 to S = 0, opening up a coordination site and hence a vacant cobalt orbital for enabling heteroaryl group transfer.
Scheme 5. Influence of Heteroaryl and Aryloxide Substituent on Reactivity.
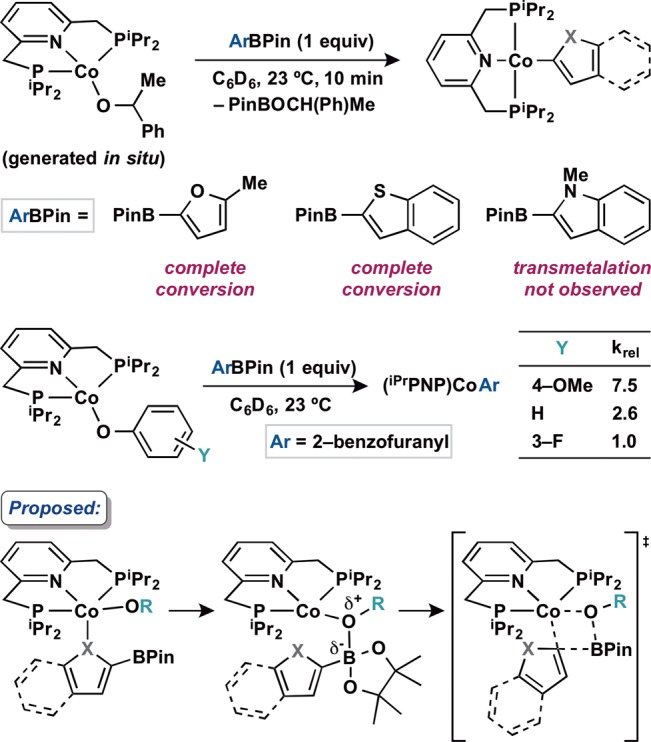
Given the observed difference in the reactivity of cobalt phenoxide and alkoxide complexes toward 2-benzofuranylBPin, experiments were conducted to establish the relative rates of transmetalation as a function of the oxygen substituent in (iPrPNP)CoOR (Scheme 5). Cobalt aryloxides with varied electronic properties (R = C6H4(4-OMe) and C6H4(3-F)) were synthesized and subjected to transmetalation with 2-benzofuranylBPin. Relative rate constants (krel) were measured by comparing initial rates of these and the parent cobalt phenoxide complex (see Figure S1). This kinetic analysis established that the relative rate constant for transmetalation increases with increasing electron donation from the aryl substituent (C6H4(4-OMe) > Ph > C6H4(3-F)). This behavior is consistent with accumulation of positive charge within the aryloxide ligand during the transmetalation process. Additional kinetic experiments established an overall second-order reaction indicated by a linear slope of inverse product concentration versus time (see Figure S2). Measurement of the initial rates of transmetalation at varying concentrations of (iPrPNP)CoOPh or 2-benzofuranylBPin established the reaction to be first order in both cobalt and boron reagents (see Figures S3 and S4). These results support an association between boron and the cobalt complex en route to heteroaryl group transfer (Scheme 5).69
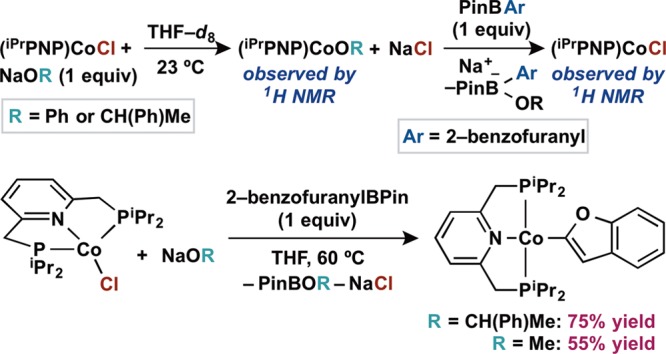
In a typical cross coupling reaction, for example, as illustrated in Scheme 2, the aryl or alkoxide ligand (OR) enters the coordination sphere of the cobalt catalyst by salt metathesis rather than by protonation. Thus, the catalytically relevant route to the (iPrPNP)CoOR species was explored using the known cobalt(I) chloride (iPrPNP)CoCl.53 Addition of 1 equiv of NaOPh in THF-d8 solution resulted in formation of (iPrPNP)CoOPh as the major product observed by 1H NMR spectroscopy (Scheme 6). Addition of 2-benzofuranylBPin to the resulting THF-d8 solution produced a mixture containing (iPrPNP)CoCl as the only cobalt species observable by 1H NMR spectroscopy. These results suggest a preference for the cobalt chloride and boronate species over the cobalt alkoxide and neutral boronic ester at room temperature, potentially complicating the combination of salt metathesis and transmetalation steps that is necessary for catalytic turnover. Indeed, addition of 1 equiv each of NaOCH(Ph)Me and 2-benzofuranylBPin in THF to (iPrPNP)CoCl resulted in slow formation of (iPrPNP)Co(2-benzofuranyl) at room temperature (incomplete conversion after 3 h). However, heating the same reaction mixture to 60 °C resulted in complete conversion to the desired organometallic complex after 90 min (Scheme 6). Replacing the bulky alkoxide base with NaOMe resulted in a decreased yield of (iPrPNP)Co(2-benzofuranyl) (55% versus 75%), highlighting the importance of the steric profile of the alkoxide ligand substituent and providing key insight for base selection in the catalytic cross coupling reaction.
Scheme 6. Salt Metathesis/Transmetalation Studies.
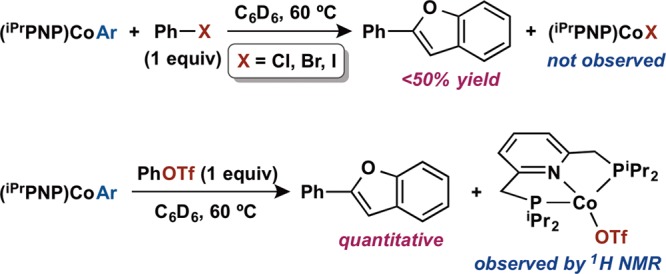
These observations provided an understanding of salt metathesis and transmetalation steps of the cobalt-catalyzed Suzuki–Miyaura cross coupling reaction proposed in Scheme 2. Aiming to complete the catalytic cycle, interaction of the organometallic complex (iPrPNP)Co(2-benzofuranyl) and potential electrophilic coupling partners was investigated (Scheme 7). Heating a mixture of 1 equiv of phenyl halide (PhX, X = Cl, Br, I) and (iPrPNP)Co(2-benzofuranyl) in benzene-d6 resulted in partial conversion (<50%) to 2-phenylbenzofuran after 90 min at 60 °C. Formation of the anticipated cobalt(I) halide species (iPrPNP)CoX was not observed in any case. Performing the same reaction at 60 °C using phenyl triflate (X = OTf) as the electrophile generated a quantitative amount of 2-phenylbenzofuran and a new paramagnetic species (Scheme 7). Though difficulties in isolation prevented additional characterization, this compound was identified as (iPrPNP)CoOTf by 1H NMR spectroscopy by analogy to the related cobalt(I) chloride complex.53 Oxidation with Ph3CCl yielded the stable cobalt(II) species (iPrPNP)CoOTf(Cl) (see Supporting Information) that was characterized by X-ray diffraction (see Figure S9).
Scheme 7. Electrophile-Dependent Reactivity of (iPrPNP)Co(2-benzofuranyl).
Results from these stoichiometric studies offered guidelines for realization of a successful Co-catalyzed Suzuki–Miyaura coupling method. The stability and transmetalation reactivity of (iPrPNP)CoOR complexes pointed to NaOCH(Ph)Me as the optimal base. Reactions of the organometallic complex and different electrophiles indicated that the use of aryl triflates, rather than aryl halides, would allow for catalytic turnover. Combining salt metathesis and transmetalation processes suggested the reaction might require elevated temperature, and entry to the catalytic cycle could be accomplished from the isolable complex (iPrPNP)CoCl. Thus, heating a 1:1:1 mixture of PhOTf, 2-benzofuranylBPin, and NaOCH(Ph)Me in the presence of 5 mol % (iPrPNP)CoCl in THF at 60 °C for 24 h resulted in formation of 2-phenylbenzofuran in 85% yield (Table 1). Analysis by GC and 1H NMR spectroscopy indicated a trace amount (<4%) of bis(benzofuran) and no observable amount of biphenyl in the reaction mixture, amounting to >20:1 selectivity for the cross coupled product. Preliminary evaluation of the reaction scope established tolerance for CF3, OMe, C(O)Me, and F functional groups on the aryl electrophile (Table 1). Heteroaryl cross coupling with 3-pyridinyl triflate was also successful, albeit in low yield.
Table 1. Preliminary Aryl Triflate Scope.

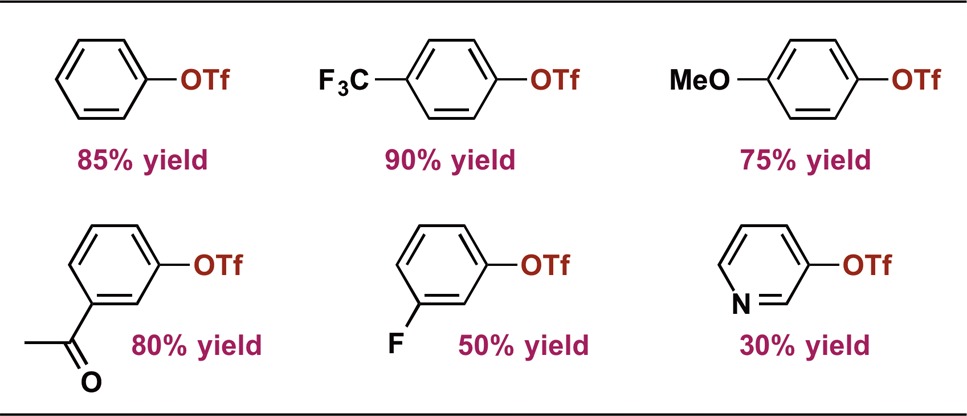
Aryl triflate (0.05 mmol), boron reagent (1 equiv), NaOCH(Ph)Me (1 equiv), and (iPrPNP)CoCl (5 mol %) in THF (1 mL) heated at 60 °C for 24 h. > 20:1 cross selectivity in all cases. Yields determined by 1H NMR.
The scope of nucleophilic partner was also examined (Table 2). Furanyl derivatives containing CHO and BPin groups in the 2-position underwent selective cross coupling with PhOTf. Only a trace amount of product was observed in the reaction of 2-methylfuranylBPin but use of the more electron-rich 4-pyrrolidinyl-(iPrPNP)CoCl complex50 allowed for catalytic turnover, suggesting that ligand modification may overcome current limitations with substrate scope. While benzothiophenylBPin participated in stoichiometric transmetalation with (iPrPNP)CoOR, catalytic reactivity was very low, suggesting that interaction with the aryl triflate electrophile is also substrate dependent. Cross coupling of PhOTf and N-methylindolylBPin did not proceed, as expected due to the lack of transmetalation reactivity observed in the stoichiometric reaction with the nucleophilic partner.
Table 2. Preliminary Boron Reagent Scope.
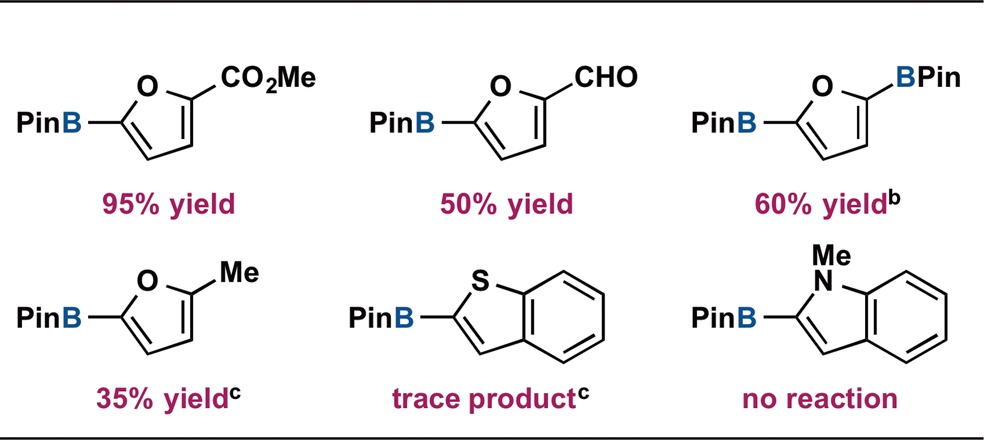
Fundamental insights gained by stoichiometric reactions of proposed transition metal intermediates have enabled the first example of a cobalt-catalyzed C(sp2)–C(sp2) Suzuki–Miyaura cross coupling. Specifically addressing the transmetalation step in the catalytic cycle provided access to crucial information regarding turnover and base selection, ultimately enabling catalytic reactivity. The flexibility of the supporting PNP pincer allows rapid interconversion between high-spin tetrahedral and low-spin planar catalytic intermediates, a distinguishing feature for the first row transition metal over state-of-the-art palladium catalysts. Importantly, these findings provide insight into the interaction of Earth abundant first row transition metals and neutral boron reagents, and this mechanistic foundation may enable new catalytic reactions involving this important class of nucleophiles.
Experimental Section
Preparation of (iPrPNP)CoOPh
A scintillation vial was charged with (iPrPNP)CoCH2SiMe3 (82 mg, 0.17 mmol), 1.7 mL of toluene, and a magnetic stirbar. A solution of phenol (16 mg, 0.17 mmol) in 1.7 mL of toluene was added, and the mixture was stirred at room temperature for 15 min. Removal of the volatiles in vacuo and recrystallization from Et2O afforded pure (iPrPNP)CoOPh as purple crystals (63 mg, 76% yield). Crystals suitable for X-ray diffraction were obtained by vapor diffusion of pentane into a saturated Et2O solution of (iPrPNP)CoOPh. Anal. Calcd for C25H40CoNOP2: C, 61.10; H, 8.20; N, 2.85. Found: C, 60.82; H, 8.31; N, 2.79. 1H NMR (benzene-d6, 23 °C): δ 51.4 (bs, Δv1/2 = 254 Hz, 1H, 4-pyridinyl CH), 41.3 (bs, Δv1/2 = 628 Hz, 4H, PCH(CH3)2), 32.4 (bs, Δv1/2 = 1209 Hz, 4H, CH2PiPr2), 20.5 (s, 2H, 3-pyridinyl CH), 17.1 (s, 2H, 2-aryl CH), 3.90 (bs, Δv1/2 = 160 Hz, 12H, PCH(CH3)2), 1.69 (bs, Δv1/2 = 148 Hz, 12H, PCH(CH3)2), −8.28 (s, 1H, 4-aryl CH), −10.3 (bs, Δv1/2 = 512 Hz, 2H, 3-aryl CH). Magnetic moment (magnetic susceptibility balance, 23 °C): μeff = 2.8 μB.
Transmetalation with 2-BenzofuranylBPin
A solution of 2-benzofuranylBPin (7.8 mg, 0.032 mmol) in 0.22 mL of benzene-d6 was added to a solution containing (iPrPNP)CoOPh (16 mg, 0.032 mmol) in 0.28 mL of benzene-d6 in a scintillation vial. The mixture was transferred to a J Young tube, and the reaction mixture was analyzed by 1H NMR spectroscopy. Isolation of (iPrPNP)Co(2-benzofuranyl) was accomplished using the following procedure. A solution of 2-benzofuranylBPin (59 mg, 0.240 mmol) in 4.8 mL of PhMe was added to (iPrPNP)CoCH2SiMe3 (117 mg, 0.240 mmol) in a scintillation vial. MeOH (9.7 μL, 0.240 mmol) was added, and the mixture was stirred at room temperature for 15 min. Removal of the volatiles in vacuo and recrystallization from PhMe/pentane afforded pure (iPrPNP)Co(2-benzofuranyl) as dark brown block crystals (100 mg, 81% yield). Crystals suitable for X-ray diffraction were obtained by slow diffusion of pentane into a saturated PhMe solution of (iPrPNP)Co(2-benzofuranyl). Anal. Calcd for C27H40CoNOP2: C, 62.91; H, 7.82; N, 2.72. Found: C, 62.61; H, 7.53; N, 2.63. 1H NMR (benzene-d6, 23 °C): δ 7.51 (d, 3JHH = 7.88 Hz, 1H, 8-benzofuranyl CH), 7.45 (d, 3JHH = 7.56 Hz, 1H, 5-benzofuranyl CH), 7.42 (t, 3JHH = 7.39 Hz, 1H, 4-pyridinyl CH), 7.13 (t, 3JHH = 7.14 Hz, 1H, 6-benzofuranyl CH), 6.92 (t, 3JHH = 7.55 Hz, 1H, 7-benzofuranyl CH), 6.40 (s, 1H, 3-benzofuranyl CH), 6.01 (d, 3JHH = 7.48 Hz, 2H, 3-pyridinyl CH), 2.15–2.11 (m, 8H, CH2PiPr2 and PCH(CH3)2), 1.22 (dd, 3JPH = 14.96 Hz, 3JHH = 7.23 Hz, 12H, PCH(CH3)2), 1.11 (dd, 3JPH = 13.04 Hz, 3JHH = 6.60 Hz, 12H, PCH(CH3)2). {1H} 13C NMR (benzene-d6, 23 °C): δ 193.4 (bs, 2-benzofuranyl C), 160.7 (s, 9-benzofuranyl C), 160.1 (apparent t, 2JPC = 8.5 Hz, 2-pyridinyl C), 132.6 (s, 4-benzofuranyl C), 121.9 (s, 4-pyridinyl C), 120.6 (s, 6-benzofuranyl C), 120.6 (apparent t, 3JPC = 6.2 Hz, 3-pyridinyl C), 117.5 (s, 7-benzofuranyl C), 115.1 (s, 5-benzofuranyl C), 115.0 (s, 3-benzofuranyl C), 108.1 (s, 8-benzofuranyl C), 35.2 (apparent t, 1JPC = 5.8 Hz, CH2PiPr2), 23.8 (apparent t, 1JPC = 9.8 Hz, PCH(CH3)2), 19.3 (apparent t, 2JPC = 2.4 Hz, PCH(CH3)2), 18.2 (s, PCH(CH3)2). {1H} 31P NMR (benzene-d6, 23 °C): δ 54.7 (bs, PCH(CH3)2).
Cobalt-Catalyzed Suzuki–Miyuara Cross Coupling
A solution of 2-benzofuranylBPin (12 mg, 0.05 mmol) and NaOCH(Ph)Me (7.4 mg, 0.05 mmol) in 0.5 mL of THF was added to a scintillation vial containing (iPrPNP)CoCl (1.0 mg, 0.0025 mmol) and a magnetic stirbar. A solution of phenyl triflate (11 mg, 0.05 mmol) in 0.5 mL of THF was added to this mixture. The reaction was sealed and heated in a heating block at 60 °C for 24 h. Trimethoxybenzene was added as a standard, and the volatiles were removed in vacuo. The residue was dissolved in CDCl3, filtered through a glass frit, and analyzed by 1H NMR spectroscopy. This data matched that previously reported for 2-phenylbenzofuran.70
Acknowledgments
We acknowledge the American Chemical Society Green Chemistry Institute Pharmaceutical Roundtable for funding. J.M.N. acknowledges the National Institute of General Medical Sciences of the National Institutes of Health for funding under Award Number F32GM115219. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. M.J.B. thanks the Natural Sciences and Engineering Research Council of Canada for a predoctoral fellowship (PGS-D). We would also like to thank Drs. David Leahy, Yi Xiao, Kenneth Fraunhoffer, and Eric Simmons (Bristol-Myers Squibb) for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00283.
The authors declare no competing financial interest.
Supplementary Material
References
- Shen H. C.Selected Application of Transition Metal-Catalyzed Carbon–Carbon Cross-Coupling Reactions in the Pharmaceutical Industry. In Applications of Transition Metal Catalysis in Drug Discovery and Development; Crawley M. L., Trost B. M., Eds; Wiley: Hoboken, NJ, 2012; pp 25–43. [Google Scholar]
- Valente C.; Organ M. G.. The Contemporary Suzuki–Miyaura Reaction. In Boronic Acids; Hall D. G., Ed; Wiley: Weinheim, Germany, 2011; pp 213–262. [Google Scholar]
- Miyaura N.; Suzuki A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. 10.1021/cr00039a007. [DOI] [Google Scholar]
- Lennox A. J. J.; Lloyd-Jones G. C. Selection of boron reagents for Suzuki–Miyaura coupling. Chem. Soc. Rev. 2014, 43, 412–443. 10.1039/C3CS60197H. [DOI] [PubMed] [Google Scholar]
- Torborg C.; Beller M. Recent Applications of Palladium-Catalyzed Coupling Reactions in the Pharmaceutical, Agrochemical, and Fine Chemical Industries. Adv. Synth. Catal. 2009, 351, 3027–3043. 10.1002/adsc.200900587. [DOI] [Google Scholar]
- Roughley S. D.; Jordan A. M. The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451–3479. 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- Holzwarth M. S.; Plietker B. Biorelevant Metals in Sustainable Metal Catalysis–A Survey. ChemCatChem 2013, 5, 1650–1679. 10.1002/cctc.201200592. [DOI] [Google Scholar]
- Garrett C. E.; Prasad K. The Art of Meeting Palladium Specifications in Active Pharmaceutical Ingredients Produced by Pd-Catalyzed Reactions. Adv. Synth. Catal. 2004, 346, 889–900. 10.1002/adsc.200404071. [DOI] [Google Scholar]
- For a review of nickel-catalyzed Suzuki–Miyaura cross coupling, see:Han F.-S. Transition-metal-catalyzed Suzuki–Miyaura cross-coupling reactions: a remarkable advance from palladium to nickel catalysts. Chem. Soc. Rev. 2013, 42, 5270–5298. 10.1039/c3cs35521g. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Cobb K. M.; Tan T.; Watson M. P. Stereospecific Cross Couplings To Set Benzylic, All-Carbon Quaternary Stereocenters in High Enantiopurity. J. Am. Chem. Soc. 2016, 138, 12057–12060. 10.1021/jacs.6b08075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastalir M.; Stöger B.; Pittenauer E.; Allmaier G.; Kirchner K. Air-Stable Triazine-Based Ni(II) PNP Pincer Complexes As Catalysts for the Suzuki–Miyaura Cross-Coupling. Org. Lett. 2016, 18, 3186–3189. 10.1021/acs.orglett.6b01398. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Berthel J. H. J.; Kuntze-Fechner M. W.; Friedrich A.; Marder T. B.; Radius U. NHC Nickel-Catalyzed Suzuki–Miyaura Cross-Coupling Reactions of Aryl Boronate Esters with Perfluorobenzenes. J. Org. Chem. 2016, 81, 5789–5794. 10.1021/acs.joc.6b01041. [DOI] [PubMed] [Google Scholar]
- Shi S.; Meng G.; Szostak M. Synthesis of Biaryls through Nickel-Catalyzed Suzuki–Miyaura Coupling of Amides by Carbon–Nitrogen Bond Cleavage. Angew. Chem., Int. Ed. 2016, 55, 6959–6963. 10.1002/anie.201601914. [DOI] [PubMed] [Google Scholar]
- Malan F. P.; Singleton E.; van Rooyen P. H.; Landman M. Facile Suzuki–Miyaura coupling of activated aryl halides using new CpNiBr(NHC) complexes. J. Organomet. Chem. 2016, 813, 7–14. 10.1016/j.jorganchem.2016.03.017. [DOI] [Google Scholar]
- Jiang X.; Kulbitski K.; Nisnevich G.; Gandelman M. Enantioselective assembly of tertiary stereocenters via multicomponent chemoselective cross-coupling of geminal chloro(iodo)alkanes. Chem. Sci. 2016, 7, 2762–2767. 10.1039/C5SC04378F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Garcia P. M.; Di Franco T.; Epenoy A.; Scopelliti R.; Hu X. From Dimethylamine to Pyrrolidine: The Development of an Improved Nickel Pincer Complex for Cross-Coupling of Nonactivated Secondary Alkyl Halides. ACS Catal. 2016, 6, 258–261. 10.1021/acscatal.5b02324. [DOI] [Google Scholar]
- Weires N. A.; Baker E. L.; Garg N. K. Nickel-catalysed Suzuki–Miyaura coupling of amides. Nat. Chem. 2015, 8, 75–79. 10.1038/nchem.2388. [DOI] [PubMed] [Google Scholar]
- Shields J. D.; Gray E. E.; Doyle A. G. A Modular, Air-Stable Nickel Precatalyst. Org. Lett. 2015, 17, 2166–2169. 10.1021/acs.orglett.5b00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review of copper-catalyzed Suzuki–Miyaura cross coupling, see:Thapa S.; Shrestha B.; Gurung S. K.; Giri R. Copper-catalysed cross-coupling: an untapped potential. Org. Biomol. Chem. 2015, 13, 4816–4827. 10.1039/C5OB00200A. [DOI] [PubMed] [Google Scholar]
- Basnet P.; Thapa S.; Dickie D. A.; Giri R. The copper-catalysed Suzuki–Miyaura coupling of alkylboron reagents: disproportionation of anionic (alkyl) (alkoxy)borates to anionic dialkylborates prior to transmetalation. Chem. Commun. 2016, 52, 11072–11075. 10.1039/C6CC05114F. [DOI] [PubMed] [Google Scholar]
- Gurung S. K.; Thapa S.; Shrestha B.; Giri R. Copper-catalysed cross-couplings of arylboronate esters with aryl and heteroaryl iodides and bromides. Org. Chem. Front. 2015, 2, 649–653. 10.1039/C4QO00331D. [DOI] [Google Scholar]
- Zhang Z.-Q.; Yang C.-T.; Liang L.-J.; Xiao B.; Lu X.; Liu J.-H.; Sun Y.-Y.; Marder T. B.; Fu Y. Copper-Catalyzed/Promoted Cross-coupling of gem-Diborylalkanes with Nonactivated Primary Alkyl Halides: An Alternative Route to Alkylboronic Esters. Org. Lett. 2014, 16, 6342–6345. 10.1021/ol503111h. [DOI] [PubMed] [Google Scholar]
- Sun Y.-Y.; Yi J.; Lu X.; Zhang Z.-Q.; Xiao B.; Fu Y. Cu-Catalyzed Suzuki–Miyaura reactions of primary and secondary benzyl halides with arylboronates. Chem. Commun. 2014, 50, 11060–11062. 10.1039/C4CC05376A. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; You W.; Smith K. B.; Brown M. K. Copper-Catalyzed Cross-Coupling of Boronic Esters with Aryl Iodides and Application to the Carboboration of Alkynes and Allenes. Angew. Chem., Int. Ed. 2014, 53, 3475–3479. 10.1002/anie.201310275. [DOI] [PubMed] [Google Scholar]
- Gurung S. K.; Thapa S.; Kafle A.; Dickie D. A.; Giri R. Copper-Catalyzed Suzuki–Miyaura Coupling of Arylboronate Esters: Transmetalation with (PN)CuF and Identification of Intermediates. Org. Lett. 2014, 16, 1264–1267. 10.1021/ol500310u. [DOI] [PubMed] [Google Scholar]
- Toriyama F.; Cornella J.; Wimmer L.; Chen T.-G.; Dixon D. D.; Creech G.; Baran P. S. Redox-Active Esters in Fe-Catalyzed C–C Coupling. J. Am. Chem. Soc. 2016, 138, 11132–11135. 10.1021/jacs.6b07172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tindall D. J.; Krause H.; Fürstner A. Iron-Catalyzed Cross-Coupling of 1-Alkynylcyclopropyl Tosylates and Related Substrates. Adv. Synth. Catal. 2016, 358, 2398–2403. 10.1002/adsc.201600357. [DOI] [Google Scholar]
- Rivera A. C. P.; Still R.; Frantz D. E. Iron-Catalyzed Stereoselective Cross-Coupling Reactions of Stereodefined Enol Carbamates with Grignard Reagents. Angew. Chem., Int. Ed. 2016, 55, 6689–6693. 10.1002/anie.201601899. [DOI] [PubMed] [Google Scholar]
- Jia Z.; Liu Q.; Peng X.-S.; Wong H. N. C. Iron-catalysed cross-coupling of organolithium compounds with organic halides. Nat. Commun. 2016, 7, 10614. 10.1038/ncomms10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua Y.-Y.; Duong H. A. Iron(II) triflate/N-heterocyclic carbene-catalysed cross-coupling of arylmagnesiums with aryl chlorides and tosylates. Chem. Commun. 2016, 52, 1466–1469. 10.1039/C5CC08302H. [DOI] [PubMed] [Google Scholar]
- Bedford R. B.; Brenner P. B.; Carter E.; Carvell T. W.; Cogswell P. M.; Gallagher T.; Harvey J. N.; Murphy D. M.; Neeve E. C.; Nunn J.; Pye D. R. Expedient Iron-Catalyzed Coupling of Alkyl, Benzyl and Allyl Halides with Arylboronic Esters. Chem. - Eur. J. 2014, 20, 7935–7938. 10.1002/chem.201402174. [DOI] [PubMed] [Google Scholar]
- Hatakeyama T.; Hashimoto T.; Kathriarachchi K. K. A. D. S.; Zenmyo T.; Seike H.; Nakamura M. Iron-Catalyzed Alkyl-Alkyl Suzuki–Miyaura Coupling. Angew. Chem., Int. Ed. 2012, 51, 8834–8837. 10.1002/anie.201202797. [DOI] [PubMed] [Google Scholar]
- Hashimoto T.; Hatakeyama T.; Nakamura M. Stereospecific Cross-Coupling between Alkenylboronates and Alkyl Halides Catalyzed by Iron-Bisphosphine Complexes. J. Org. Chem. 2012, 77, 1168–1173. 10.1021/jo202151f. [DOI] [PubMed] [Google Scholar]
- Hatakeyama T.; Hashimoto T.; Kondo Y.; Fujiwara Y.; Seike H.; Takaya H.; Tamada Y.; Ono T.; Nakamura M. Iron-Catalyzed Suzuki–Miyaura Coupling of Alkyl Halides. J. Am. Chem. Soc. 2010, 132, 10674–10676. 10.1021/ja103973a. [DOI] [PubMed] [Google Scholar]
- Daifuku S. L.; Kneebone J. L.; Snyder B. E. R.; Neidig M. L. Iron(II) Active Species in Iron-Bisphosphine Catalyzed Kumada and Suzuki–Miyaura Cross-Couplings of Phenyl Nucleophiles and Secondary Alkyl Halides. J. Am. Chem. Soc. 2015, 137, 11432–11444. 10.1021/jacs.5b06648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daifuku S. L.; Al-Afyouni M. H.; Snyder B. E. R.; Kneebone J. L.; Neidig M. L. A Combined Mössbauer, Magnetic Circular Dichroism, and Density Functional Theory Approach for Iron Cross-Coupling Catalysis: Electronic Structure, In Situ Formation, and Reactivity of Iron-Mesityl-Bisphosphines. J. Am. Chem. Soc. 2014, 136, 9132–9143. 10.1021/ja503596m. [DOI] [PubMed] [Google Scholar]
- Bedford R. B.; Brenner P. B.; Carter E.; Clifton J.; Cogswell P. M.; Gower N. J.; Haddow M. F.; Harvey J. N.; Kehl J. A.; Murphy D. M.; Neeve E. C.; Neidig M. L.; Nunn J.; Snyder B. E. R.; Taylor J. Iron Phosphine Catalyzed Cross-Coupling of Tetraorganoborates and Related Group 13 Nucleophiles with Alkyl Halides. Organometallics 2014, 33, 5767–5780. 10.1021/om500518r. [DOI] [Google Scholar]
- Bedford R. B.; Hall M. A.; Hodges G. R.; Huwe M.; Wilkinson M. C. Simple mixed Fe-Zn catalysts for the Suzuki couplings of tetraarylborates with benzyl halides and 2-halopyridines. Chem. Commun. 2009, 6430–6432. 10.1039/b915945b. [DOI] [PubMed] [Google Scholar]
- Wu L.; Zhong J.-C.; Liu S.-K.; Liu F.-P.; Gao Z.-D.; Wang M.; Bian Q.-H. Asymmetric synthesis of (R)-ar-curcumene, (R)-4,7-dimethyl-l- tetralone, and their enantiomers via cobalt-catalyzed asymmetric Kumada cross-coupling. Tetrahedron: Asymmetry 2016, 27, 78–83. 10.1016/j.tetasy.2015.11.009. [DOI] [Google Scholar]
- Gonnard L.; Guérinot A.; Cossy J. Cobalt-Catalyzed Cross-Coupling of 3- and 4-Iodopiperidines with Grignard Reagents. Chem. - Eur. J. 2015, 21, 12797–12803. 10.1002/chem.201501543. [DOI] [PubMed] [Google Scholar]
- Frlan R.; Sova M.; Gobec S.; Stavber G.; Časar Z. Cobalt-Catalyzed Cross-Coupling of Grignards with Allylic and Vinylic Bromides: Use of Sarcosine as a Natural Ligand. J. Org. Chem. 2015, 80, 7803–7809. 10.1021/acs.joc.5b01156. [DOI] [PubMed] [Google Scholar]
- Kuzmina O. M.; Steib A. K.; Fernandez S.; Boudot W.; Markiewicz J. T.; Knochel P. Practical Iron- and Cobalt-Catalyzed Cross-Coupling Reactions between N-Heterocyclic Halides and Aryl or Heteroaryl Magnesium Reagents. Chem. - Eur. J. 2015, 21, 8242–8249. 10.1002/chem.201500747. [DOI] [PubMed] [Google Scholar]
- Hammann J.; Haas D.; Steib A.; Knochel P. Highly Diastereoselective Cobalt-Mediated C(sp3)–C(sp2) Cross-Coupling Reactions of Cyclic Halohydrins with (Hetero)Aryl Grignard Reagents. Synthesis 2015, 47, 1461–1468. 10.1055/s-0034-1380407. [DOI] [Google Scholar]
- Mao J.; Liu F.; Wang M.; Wu L.; Zheng B.; Liu S.; Zhong J.; Bian Q.; Walsh P. J. Cobalt-Bisoxazoline-Catalyzed Asymmetric Kumada Cross-Coupling of Racemic α-Bromo Esters with Aryl Grignard Reagents. J. Am. Chem. Soc. 2014, 136, 17662–17668. 10.1021/ja5109084. [DOI] [PubMed] [Google Scholar]
- Haas D.; Hammann J. M.; Lutter F. H.; Knochel P. Mild Cobalt-Catalyzed Negishi Cross-Couplings of (Hetero)arylzinc Reagents with (Hetero)aryl Halides. Angew. Chem., Int. Ed. 2016, 55, 3809–3812. 10.1002/anie.201510665. [DOI] [PubMed] [Google Scholar]
- Benischke A. D.; Knoll I.; Rérat A.; Gosmini C.; Knochel P. A practical cobalt-catalyzed cross-coupling of benzylic zinc reagents with aryl and heteroaryl bromides or chlorides. Chem. Commun. 2016, 52, 3171–3174. 10.1039/C5CC10272C. [DOI] [PubMed] [Google Scholar]
- Hammann J. M.; Haas D.; Knochel P. Cobalt-catalyzed negishi cross-coupling reactions of (hetero)arylzinc reagents with primary and secondary alkyl bromides and iodides. Angew. Chem., Int. Ed. 2015, 54, 4478–4481. 10.1002/anie.201411960. [DOI] [PubMed] [Google Scholar]
- Corpet M.; Bai X.-Z.; Gosmini C. Cobalt-Catalyzed Cross-Coupling of Organozinc Halides with Bromoalkynes. Adv. Synth. Catal. 2014, 356, 2937–2942. 10.1002/adsc.201400369. [DOI] [Google Scholar]
- Hartwig J. F.Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, 2010. [Google Scholar]
- Obligacion J. V.; Semproni S. P.; Pappas I.; Chirik P. J. Cobalt-Catalyzed C(sp2)-H Borylation: Mechanistic Insights Inspire Catalyst Design. J. Am. Chem. Soc. 2016, 138, 10645–10653. 10.1021/jacs.6b06144. [DOI] [PubMed] [Google Scholar]
- Semproni S. P.; Milsmann C.; Chirik P. J. Four-Coordinate Cobalt Pincer Complexes: Electronic Structure Studies and Ligand Modification by Homolytic and Heterolytic Pathways. J. Am. Chem. Soc. 2014, 136, 9211–9224. 10.1021/ja504334a. [DOI] [PubMed] [Google Scholar]
- Obligacion J. V.; Semproni S. P.; Chirik P. J. Cobalt-Catalyzed C-H Borylation. J. Am. Chem. Soc. 2014, 136, 4133–4136. 10.1021/ja500712z. [DOI] [PubMed] [Google Scholar]
- Semproni S. P.; Atienza C. C. H.; Chirik P. J. Oxidative addition and C–H activation chemistry with a PNP pincer-ligated cobalt complex. Chem. Sci. 2014, 5, 1956–1960. 10.1039/c4sc00255e. [DOI] [Google Scholar]
- Khaskin E.; Diskin-Posner Y.; Weiner L.; Leitus G.; Milstein D. Formal loss of an H radical by a cobalt complex via metal-ligand. Chem. Commun. 2013, 49, 2771–2773. 10.1039/c3cc39049g. [DOI] [PubMed] [Google Scholar]
- Partyka D. V. Transmetalation of Unsaturated Carbon Nucleophiles from Boron-Containing Species to the Mid to Late d-Block Metals of Relevance to Catalytic C–X Coupling Reactions (X = C, F, N, O, Pb, S, Se, Te). Chem. Rev. 2011, 111, 1529–1595. 10.1021/cr1002276. [DOI] [PubMed] [Google Scholar]
- Ridgway B. H.; Woerpel K. A. Transmetalation of Alkylboranes to Palladium in the Suzuki Coupling Reaction Proceeds with Retention of Stereochemistry. J. Org. Chem. 1998, 63, 458–460. 10.1021/jo970803d. [DOI] [PubMed] [Google Scholar]
- Matos K.; Soderquist J. A. Alkylboranes in the Suzuki–Miyaura coupling: Stereochemical and mechanistic studies. J. Org. Chem. 1998, 63, 461–470. 10.1021/jo971681s. [DOI] [PubMed] [Google Scholar]
- Ingleson and coworkers reported the formation of an iron(I) aryl species from the corresponding iron(II) dibromide and a potassium tolyl amido(dialkoxy)boronate reagent. See:Dunsford J. J.; Clark E. R.; Ingleson M. J. Highly nucleophilic dipropanolamine chelated boron reagents for aryl-transmetallation to iron complexes. Dalton Trans. 2015, 44, 20577–20583. 10.1039/C5DT03835A. [DOI] [PubMed] [Google Scholar]
- Transmetalation from BEt3 to an iron hydride has been demonstrated by Holland and coworkers. See:Yu Y.; Brennessel W. W.; Holland P. L. Borane B–C Bond Cleavage by a Low-Coordinate Iron Hydride Complex and N–N Bond Cleavage by the Hydridoborate Product. Organometallics 2007, 26, 3217–3226. 10.1021/om7003805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Only two examples of cobalt catalysis with neutral boron reagents exist to our knowledge and each propose a transmetalation step. See:Huang Y.; Huang R.-Z.; Zhao Y. Cobalt-Catalyzed Enantioselective Vinylation of Activated Ketones and Imines. J. Am. Chem. Soc. 2016, 138, 6571–6576. 10.1021/jacs.6b02372. [DOI] [PubMed] [Google Scholar]
- See also:Kobayashi T.; Yorimitsu H.; Oshima K. Cobalt-Catalyzed Addition of Styrylboronic Acids to 2-Vinylpyridine Derivatives. Chem. - Asian J. 2011, 6, 669–673. 10.1002/asia.201000275. [DOI] [PubMed] [Google Scholar]
- The reaction of an iron(II) halide and an aryl tris(alkoxy)boronate results in alkoxide transfer to iron over aryl group transfer:Dunsford J. J.; Cade I. A.; Fillman K. L.; Neidig M. L.; Ingleson M. J. Reactivity of (NHC)2FeX2 Complexes toward Arylborane Lewis Acids and Arylboronates. Organometallics 2014, 33, 370–377. 10.1021/om401105k. [DOI] [Google Scholar]
- Lennox A. J. J.; Lloyd-Jones G. C. Transmetalation in the Suzuki–Miyaura Coupling: The Fork in the Trail. Angew. Chem., Int. Ed. 2013, 52, 7362–7370. 10.1002/anie.201301737. [DOI] [PubMed] [Google Scholar]
- Amatore C.; Le Duc G.; Jutand A. Mechanism of Palladium-Catalyzed Suzuki–Miyaura Reactions: Multiple and Antagonistic Roles of Anionic “Bases” and Their Countercations. Chem. - Eur. J. 2013, 19, 10082–10093. 10.1002/chem.201300177. [DOI] [PubMed] [Google Scholar]
- Carrow B. P.; Hartwig J. F. Distinguishing Between Pathways for Transmetalation in Suzuki–Miyaura Reactions. J. Am. Chem. Soc. 2011, 133, 2116–2119. 10.1021/ja1108326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster C. H.; Diao T.; Pappas I.; Chirik P. J. Bench-Stable, Substrate-Activated Cobalt Carboxylate Pre-Catalysts for Alkene Hydrosilylation with Tertiary Silanes. ACS Catal. 2016, 6, 2632–2636. 10.1021/acscatal.6b00304. [DOI] [Google Scholar]
- Noda D.; Tahara A.; Sunada Y.; Nagashima H. Non-Precious-Metal Catalytic Systems Involving Iron or Cobalt Carboxylates and Alkyl Isocyanides for Hydrosilylation of Alkenes with Hydrosiloxanes. J. Am. Chem. Soc. 2016, 138, 2480–2483. 10.1021/jacs.5b11311. [DOI] [PubMed] [Google Scholar]
- For the related cobalt(I) formate complex, see:Scheuermann M. L.; Semproni S. P.; Pappas I.; Chirik P. J. Carbon Dioxide Hydrosilylation Promoted by Cobalt Pincer. Inorg. Chem. 2014, 53, 9463–9465. 10.1021/ic501901n. [DOI] [PubMed] [Google Scholar]
- Thomas A. A.; Denmark S. E. Pre-transmetalation intermediates in the Suzuki–Miyaura reaction revealed: The missing link. Science 2016, 352, 329–332. 10.1126/science.aad6981. [DOI] [PubMed] [Google Scholar]
- Geary L. M.; Hultin P. G. 2-Substituted Benzo[b]furans from (E)-1,2-Dichlorovinyl Ethers and Organoboron Reagents: Scope and Mechanistic Investigations into the One-Pot Suzuki Coupling/Direct Arylation. Eur. J. Org. Chem. 2010, 2010, 5563–5573. 10.1002/ejoc.201000787. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.