

Abstract
Selective degradation of proteins by proteolysis targeting chimeras (PROTACs) offers a promising potential alternative to protein inhibition for therapeutic intervention. Current PROTAC molecules incorporate a ligand for the target protein, a linker, and an E3 ubiquitin ligase recruiting group, which bring together target protein and ubiquitinating machinery. Such hetero-bifunctional molecules require significant linker optimization and possess high molecular weight, which can limit cellular permeation, solubility, and other drug-like properties. We show here that the hetero-bifunctional molecule can be formed intracellularly by bio-orthogonal click combination of two smaller precursors. We designed a tetrazine tagged thalidomide derivative which reacts rapidly with a trans-cyclo-octene tagged ligand of the target protein in cells to form a cereblon E3 ligase recruiting PROTAC molecule. The in-cell click-formed proteolysis targeting chimeras (CLIPTACs) were successfully used to degrade two key oncology targets, BRD4 and ERK1/2. ERK1/2 degradation was achieved using a CLIPTAC based on a covalent inhibitor. We expect this approach to be readily extendable to other inhibitor-protein systems because the tagged E3 ligase recruiter is capable of undergoing the click reaction with a suitably tagged ligand of any protein of interest to elicit its degradation.
Short abstract
Tetrazine-tagged thalidomide derivatives undergo click reaction with trans-cyclo-octene-tagged protein ligands in cells: the resulting click-formed proteolysis-targeting chimeric molecule elicits ubiquitination and degradation of the target protein.
Introduction
Proteolysis targeting chimeras (PROTACs) are hetero-bifunctional molecules which incorporate a ligand for an intracellular target protein and an E3 ubiquitin ligase recruiting group, joined by a linker of a length appropriate to bring together target protein and ubiquitinating machinery and thereby elicit the ubiquitination of the protein of interest and its subsequent degradation in the proteasome. The methodology may provide a powerful alternative approach to classical protein inhibition for therapeutic intervention for several reasons. Small molecule inhibitors need to achieve sustained target occupancy, typically requiring high systemic concentrations, while PROTACs offer a long lasting effect by suppressing the target until resynthesis,1 which can take hours or days. While a classical inhibitor typically only blocks one function of a protein, the degradation of the protein perturbs all functions including allosteric regulatory sites and scaffolding/protein–protein interaction sites, which may lead to a more pronounced phenotype. In addition, since the PROTAC should be able to function when binding to any part of the target protein, the approach may provide additional opportunities to address less druggable proteins by allowing allosteric or even nonfunctional binding sites to be targeted.2,3
The first PROTACs used natural peptide substrate sequences as ligands to recruit the Skp1-Cullin-F box complex or the von-Hippel-Lindau (VHL) E3 ubiquitin ligases,4−6 with obvious limitations in the cell permeability of the resulting bifunctional molecules. Nonpeptidic VHL ligands were subsequently identified with improved physicochemical properties,7 offering the possibility to design more drug-like PROTACs. In addition, the phthalimide immunomodulatory drug (IMiD) thalidomide was recently characterized as a ligand of the E3 ubiquitin ligase cereblon (CRBN).8 These discoveries have enabled several groups to develop PROTACs targeting the efficient degradation of several biologically important proteins including BRD4,9−11 BCR-ABL,12 ERRα, and RIPK2.13 Of the reported PROTACs eliciting the degradation of BRD4, two (dBET19 and ARV-82510) incorporate the BRD4 ligand JQ1 and the ligase recruiter thalidomide. These PROTACs differ only in the nature and length of their linker, which affects the efficiency of BRD4 degradation. The third, MZ1,11 also incorporates JQ1 as the BRD4 ligand, but linked to VHL-1 as the ligase recruiter.
Although protein degradation is an attractive concept in drug discovery, the properties of current PROTACs are likely to limit their potential as therapeutics. The need to incorporate both a target protein ligand and an E3 ligase recruiting element leads to hetero-bifunctional molecules possessing high molecular weight and polar surface area, typically in the range 800–1000 Da and ∼200 Å2, respectively (see Table 1).1 This combination of properties can limit cellular permeation and solubility, and compromise bioavailability and pharmacokinetics, especially distribution to the CNS.14 In addition, bespoke fine-tuning of the linker is required for each target protein–E3 ligase pairing: an overly short linker may sterically prevent the target protein and E3 ligase from simultaneously binding to the PROTAC, while an overly long linker may fail to bring ligase and target protein into sufficient proximity to elicit ubiquitination.2,15
Table 1. Properties of PROTACs Targeting BRD4 for Degradation Compared with CLIPTAC Components.
| dBET1 | ARV-825 | MZ1 | Tz-thalidomide | JQ1-TCO | Probe 1 | ||
|---|---|---|---|---|---|---|---|
| MW | 785 | 924 | 1003 | 572 | 609 | 586 | |
| ClogP | 2.5 | 4.8 | 4.9 | 1.2 | 5.9 | 6.5 | |
| PSA (Å2) | 194 | 205 | 211 | 173 | 111 | 130 | |
| IC50 (μM) | BRD4-1 | 0.020 | 0.090 | 0.38 | 46.3 | 0.016 | |
| BRD4-2 | 0.028 | 0.12 | 62.3 | 0.063 | |||
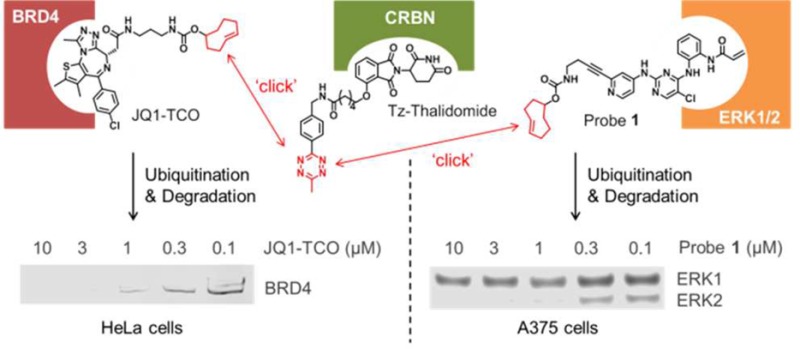
As an alternative to treating cells with a high molecular weight PROTAC, we have evaluated the use of click chemistry to generate the hetero-bifunctional PROTAC intracellularly from two smaller precursors that are expected to be more permeable (Figure 1 and Table 1). Among the reported bio-orthogonal reactions, we elected to evaluate the inverse electron demand Diels–Alder (IEDDA) cycloaddition between tetrazine and trans-cyclo-octene (TCO).16 This reaction has been shown to be fast and high yielding,17 does not require the presence of a catalyst,18 and has found numerous biological applications, especially in the optical imaging field.19−22 We designed and synthesized a tetrazine tagged thalidomide derivative (Tz-thalidomide 1, Figure 1) that can self-assemble with a TCO-tagged inhibitor of the protein of interest: the resulting click-formed proteolysis targeting chimera (CLIPTAC) recruits the E3 ligase CRBN to the protein of interest resulting in its ubiquitination and then degradation. This approach was used for the degradation of BRD4 using JQ1-TCO (2), allowing comparison of the methodology with previously described JQ1-PROTACs.9−11 A series of control experiments were performed to validate the CLIPTAC approach including disruption of binding to CRBN using methylated Tz-thalidomide (4, Supplementary Figure 1); disruption of the self-assembly of the CLIPTAC components by using untagged equivalents; and the use of the inactive enantiomer of JQ1-TCO as a negative control. The method was further extended to elicit degradation of ERK1/2 by treating cells with the TCO-tagged covalent ERK1/2 inhibitor Probe 1 (5, Figure 3a)23 and Tz-thalidomide to form the corresponding ERK-CLIPTAC. Together these results validate the CLIPTAC approach and suggest that a Tz-tagged E3 ligase recruiting ligand such as Tz-thalidomide could provide a convenient way to exploit a wide range of TCO-tagged ligands to elicit the degradation of their target proteins.
Figure 1.

Representative scheme explaining the mode of action of click-formed PROTACs (CLIPTACs). Cells are treated sequentially with the two CLIPTAC precursors: the TCO-tagged ligand of the protein of interest (in this example, BRD4), followed by a tetrazine-tagged E3 ligase recruiting ligand, in this case thalidomide (which recruits CRBN). Click reaction inside cells forms the heterobifunctional CLIPTAC molecule which brings the E3 ligase in close proximity to the protein of interest for ubiquitination and subsequent proteasomal degradation.
Figure 3.

(a) Chemical structures of Probe 1 (5), ERK-CLIPTAC (6), and a covalent ERK1/2 inhibitor (7). (b) Immunoblot for ERK1/2 and actin showing Probe 1 concentration-dependent downregulation of ERK1/2 protein levels. A375 cells were treated with Probe 1 for 4 h followed by Tz-thalidomide (1 μM) for 18 h. (c) Immunoblot for ERK1/2 and actin showing time dependent downregulation of ERK1/2 protein levels. A375 cells were treated with Probe 1 (10 μM) for 18 h followed by Tz-thalidomide (10 μM) for the indicated time. (d) Immunoblot for ERK1/2 and actin showing reduced ERK1/2 degradation when the interaction between thalidomide and CRBN is perturbed. A375 cells were treated with Probe 1 for 4 h followed by methyl-Tz-thalidomide (10 μM) for 18 h. (e) Immunoblot for ERK1/2 andactin showing the effects of Probe 1 and Tz-thalidomide alone, the effects of preventing the click reaction using a covalent ERK1/2 inhibtor, and the effects of a 4-h pretreatment with carfilzomib (1 μM) on ERK1/2 protein levels. Experiments performed on A375 cells. (f) Immunoblot for phospho-ERK1/2, phospho-RSK, and actin showing Probe 1 concentration-dependent downregulation of phospho-ERK1/2 and phospho-RSK protein levels. A375 cells were treated with Probe 1 for 8 h followed by Tz-thalidomide (10 μM) for 18 h.
Results
Design and Synthesis of the CLIPTAC Precursors
Design of the two click partners suitable for formation of CLIPTAC hetero-bifunctional molecules required careful consideration of the positions and lengths of the linking and tagging groups. Comparison of the published data on the BRD4 degrading PROTACs dBET19 and ARV-82510 suggests that the linker length between the target protein ligand and the E3 ligase recruiting thalidomide plays a major role in determining the efficiency of protein degradation: dBET1, which shows a 50% degradation concentration (DC50) of ∼430 nM, contains eight bonds separating the linking atoms from JQ1 and thalidomide, while ARV-825 contains 17 bonds in its linker and shows exquisite potency with a DC50 of ∼1 nM. Analysis of the X-ray crystal structure of thalidomide in complex with CRBN24 suggested that a 5-methylene chain linker appended to the aromatic ring of thalidomide would be long enough to position the clickable tetrazine moiety into the solvent without perturbing binding to CRBN (Supplementary Figure 1), so we designed and synthesized the tetrazine tagged Tz-thalidomide 1 (Figure 1). The design of the corresponding TCO-tagged JQ1 2 used the JQ1 carboxy group as a growth point, as this is well precedented to be tolerated without perturbing BRD4 binding, with a short 3-methylene and carbamate linker to position the TCO group in solvent (Supplementary Figure 2). Click combination of the TCO and tetrazine tagged ligands generates a JQ1-thalidomide CLIPTAC (3) in which the linking atoms are separated by 25 bonds. Although somewhat longer than the linker in the highly potent ARV-825, computational modeling of the CLIPTAC in the absence of protein indicated that the nonlinear shape of the clicked linker would allow the two protein-interacting groups to occupy a similar range of distances as the fully flexible linker in ARV-825 (Supplementary Figure 3). As negative control CLIPTAC precursors, we also synthesized the inactive enantiomer of JQ1-TCO (−)2 and the N-methylated Tz-thalidomide 4 (Supplementary Figure 1) which should perturb binding to CRBN by disrupting the key hydrogen bond with His380 and causing a steric clash with the protein. The syntheses are outlined in Supplementary Scheme 1 and the accompanying description.
JQ1-TCO and JQ1-CLIPTAC Interaction with BRD4
To confirm successful click formation of the JQ1-thalidomide CLIPTAC (JQ1-CLIPTAC 3), we investigated the bio-orthogonal reaction between Tz-thalidomide 1 and JQ1-TCO 2 in solution. By mixing the two reagents in a 1:1 ratio, the reaction was shown by liquid chromatography–mass spectrometry (LC-MS) to be complete after 15 min in DMSO (Supplementary Figure 4). Next, using AlphaScreen histone peptide displacement assays,25 we evaluated the ability of JQ1-TCO 2 and JQ1-CLIPTAC 3 to bind to BRD4-1 and BRD4-2 bromodomains (Figure 2a and Supplementary Figure 5). These experiments showed low nanomolar IC50’s vs both bromodomains, confirming that addition of either the TCO tag alone, or the fully clicked linker and thalidomide, did not significantly affect the binding affinity to BRD4, thereby supporting the use of JQ1-TCO as a CLIPTAC precursor. In the same assay, (−)JQ1-TCO showed ∼1000 fold weaker binding, validating its use as a negative control (Figure 2a). To exclude any possibility of interference when interpreting the results of our experiments, the binding affinity of Tz-thalidomide to BRD4 was also assessed and shown to be negligible (IC50 (BRD4-1) = 46.25 ± 1.91 μM and IC50 (BRD4-2) = 62.55 ± 5.73 μM, Supplementary Figure 6). Finally, the selectivity profile of JQ1-TCO against the BET family was determined by thermal shift assay. As expected, JQ1-TCO was found to bind selectively to subfamily II demonstrating that the addition of the TCO tag did not compromise the selectivity of the compound (Figure 2b and Supplementary Figure 7).26
Figure 2.

(a) Affinity of JQ1 (n = 1), JQ1-TCO (n = 2), JQ1-CLIPTAC (n = 2), and (−)JQ1-TCO (n = 2) for BRD4-1 bromdomain as determined by AlphaScreen histone peptide displacement assays. (b) Selectivity of JQ1-TCO (10 μM, duplicate, thermal shift) across the bromodomain family. (c) Immunoblot for BRD4 and actin showing JQ1-TCO concentration-dependent downregulation of BRD4 protein levels. HeLa cells were treated with JQ1-TCO for 18 h followed by treatment with Tz-thalidomide (10 μM) for 18 h. (d) Immunoblot for BRD4 and actin showing Tz-thalidomide concentration-dependent downregulation of BRD4 protein levels. HeLa cells were treated with JQ1-TCO (10 μM) for 18 h followed by treatment with Tz-thalidomide for 18 h. (e) Immunoblot for BRD4 and actin showing time-dependent downregulation of BRD4 protein levels. HeLa cells were treated with JQ1-TCO (10 μM) for 18 h followed by treatment with Tz-thalidomide (10 μM) for the indicated time. (f) Immunoblot for BRD4 and actin showing no BRD4 degradation when the interaction between JQ1 and BRD4 is perturbed. HeLa cells were treated with (−)JQ1-TCO for 18 h followed by treatment with Tz-thalidomide for 18 h. (g) Immunoblot for BRD4 and actin showing no BRD4 degradation when the interaction between thalidomide and CRBN is perturbed. HeLa cells were treated with JQ1-TCO for 18 h followed by treatment with methyl-Tz-thalidomide for 18 h. (h) Immunoblot for BRD4 and actin showing the effects of JQ1-TCO and Tz-thalidomide alone, the effects of preventing the click reaction using JQ1, and the effects of a 4-h pretreatment with carfilzomib (1 μM) on BRD4 protein levels. Experiments performed on HeLa cells.
BRD4 Degradation by CLIPTAC
To evaluate the CLIPTAC approach for protein degradation, we first treated HeLa cells with JQ1-TCO for 18 h followed by Tz-thalidomide for a further 18 h. BRD4 protein levels were assessed by SDS-PAGE followed by Western Blot using a specific BRD4 antibody. At a fixed concentration of 10 μM Tz-thalidomide, JQ1-TCO elicited concentration-dependent degradation of BRD4, with complete degradation at 10 and 3 μM and partial degradation at 1 and 0.3 μM (Figure 2c). We then repeated the experiment varying the concentration of Tz-thalidomide, while the concentration of JQ1-TCO remained fixed (10 μM). Again, BRD4 was completely degraded at high concentrations of Tz-thalidomide (10 and 3 μM) and partially at lower concentrations (Figure 2d). These two experiments demonstrate that BRD4 degradation is dependent on the concentration of each CLIPTAC precursor, JQ1-TCO and Tz-thalidomide.
Next, we performed a time-course experiment in which HeLa cells were treated with JQ1-TCO (10 μM) for 18 h followed by Tz-thalidomide (10 μM) for a range of 1–24 h (Figure 2e). The immunodetection signal indicated no change to BRD4 levels up to 8 h following the addition of Tz-thalidomide. After 16 h, BRD4 was still detected, but the abundance of protein had clearly dropped compared to untreated cells. After 24 h, BRD4 levels were undetectable consistent with 100% degradation. This time course experiment shows that the effect of CLIPTACs on BRD4 levels is visible after 16 h.
To confirm that degradation of BRD4 occurs according to the proposed mechanism, we tested whether perturbing the interaction with either BRD4 or CRBN would ablate protein degradation. HeLa cells were treated with the inactive enantiomer (−)JQ1-TCO followed by Tz-thalidomide (10 μM). No BRD4 degradation was observed at any of the concentrations of (−)JQ1-TCO tested (Figure 2f), confirming that binding of the CLIPTAC to BRD4 is required for protein degradation.
We then treated HeLa cells with JQ1-TCO (10 μM) followed by Tz-thalidomide-Me 4 (10 μM). The level of BRD4 remained unchanged across the experiment (Figure 2g), indicating that interfering with the binding to CRBN blocks BRD4 degradation. With these two experiments, we showed that degradation of BRD4 is dependent on the CLIPTAC binding to both BRD4 and CRBN in order to promote spatial proximity between the two proteins.
In additional control experiments, we showed that the level of BRD4 was unaffected by treatment with either JQ1-TCO or Tz-thalidomide alone (Figure 2h). We also observed no BRD4 degradation when HeLa cells were treated with untagged JQ1 (10 μM) followed by Tz-thalidomide (10 μM) (Figure 2h). In this situation, JQ1 binds to BRD4 but is unable to “click” with Tz-thalidomide, preventing spatial proximity between BRD4 and CRBN.
Finally, we tested the effect of the proteasome inhibitor carfilzomib on the ability of JQ1-TCO and Tz-thalidomide treatment to elicit BRD4 degradation. In this experiment BRD4 levels were rescued, confirming that the CLIPTAC effects on BRD4 levels proceed via proteasomal degradation (Figure 2h).
ERK1/2 Degradation by CLIPTAC
Having validated the CLIPTAC approach for BRD4 degradation, we explored the broader application of this new methodology by studying a TCO-tagged inhibitor of a target from different protein family. ERK1/2 are closely related serine/threonine kinases and are part of the RAS/RAF/MEK/ERK signaling cascade which is implicated in numerous cancers.27,28 Although this signaling pathway is currently the subject of intense efforts in cancer research, exemplified by the number of ERK1/2 inhibitors under development,29−32 to the best of our knowledge ERK1/2 degradation has not been explored as a potential therapeutic strategy. Modulation of ERK1/2 levels could be a particularly advantageous approach in comparison with pharmacological inhibition, since it has been demonstrated that a significant proportion of signaling by ERK1 and 2 arises from protein–protein interactions of their phosphorylated forms, in addition to their catalytic activity.33
We recently published the design and synthesis of a covalent TCO-tagged inhibitor of ERK1/2 kinases23 (Probe 1, Figure 3a), which we used in click experiments for in-gel fluorescence affinity-based protein profiling experiments. Since Probe 1 shows sub-micromolar activity in cells (A375 GI50 = 0.47 ± 0.16 μM), we decided to use this compound to study the CLIPTAC-mediated degradation of ERK1/2. We first evaluated the bio-orthogonal click reaction between Probe 1 and Tz-thalidomide in buffer by LC-MS, which indicated complete reaction after 15 min to form the CLIPTAC 6 (Figure 3a and Supplementary Figure 8). Next, we evaluated the inhibition of ERK2 by Tz-thalidomide in a bioassay, in order to rule out potentially confounding effects in the cellular studies. As expected, the inhibition of ERK2 by Tz-thalidomide was found to be negligible (46 ± 4% inhibition @ 30 μM, Supplementary Figure 9).
Having established suitable pharmacology for Probe 1 and Tz-thalidomide, we explored their potential to form a CLIPTAC and degrade ERK1/2 in two cell lines in which RAS/RAF/MEK/ERK cascade activation leads to upregulation of ERK1/2 signaling. We first treated BRAFV600E mutant melanoma A375 cells with Probe 1 for 24 h followed by Tz-thalidomide (10 μM) for a further 18 h. ERK1 and 2 levels were assessed by SDS-PAGE followed by Western Blot using a total ERK1/2 antibody. The immunodetection signal revealed that ERK1/2 abundance was dependent on Probe 1 concentration (Supplementary Figure 10). This experiment was also performed in KRAS mutant colorectal HCT116 cells, where complete degradation of ERK1/2 was also observed at higher concentrations of Probe 1 (Supplementary Figure 11). We then studied ERK1/2 degradation in time-course experiments. A375 cells were treated with Probe 1 (10 μM) for 18 h followed by Tz-thalidomide (10 μM) for a range of 1–24 h. From the immunodetection signal, ERK1/2 degradation is observed partially after 4 h and is complete after 16 h (Figure 3b). A similar degradation profile was obtained when the cells were pretreated with Probe 1 for a shorter period of 8 or 4 h followed by treatment with Tz-thalidomide (Supplementary Figure 10). We then investigated the impact of Tz-thalidomide concentration on ERK1/2 degradation (Supplementary Figure 12). Degradation was retained with lower concentrations of Tz-thalidomide, and treating A375 cells with 1 μM Probe 1 for 4 h followed by Tz-thalidomide (1 μM) for 18 h led to the best degradation profile (Figure 3c). At lower concentrations of Tz-thalidomide (0.1 and 0.3 μM), ERK1/2 degradation was not observed. We also studied the influence of a washout step on ERK1/2 degradation. Before the addition of Tz-thalidomide, A375 cells were washed with media to remove excess of unreacted Probe 1 (Supplementary Figure 13). Surprisingly, washing the cells partially ablated the ERK1/2 degradation, suggesting that at least part of the degradation observed with Probe 1 and Tz-thalidomide arises from noncovalent binding of Probe 1 to ERK1/2.
To ensure that the ERK1/2 degradation observed in these experiments is due to the action of CRBN, we undertook comparative experiments using the inactive CLIPTAC precursor Tz-thalidomide-Me 4. A375 cells were treated with Probe 1 followed by Tz-thalidomide-Me (10 μM). Under these conditions, insignificant degradation of ERK1/2 was observed (Figure 3d). In addition, we conducted a competition experiment in which A375 cells were first treated with Probe 1 followed by untagged thalidomide (10 μM) and Tz-thalidomide (1 μM). As expected, degradation of ERK1/2 was suppressed (Supplementary Figure 14), confirming that binding to CRBN is key to induce spatial proximity and therefore degradation.
Next, we performed a similar set of control experiments as for the studies on BRD4 degradation. We showed that the level of ERK1/2 was unchanged when A375 cells were treated with Probe 1 or Tz-thalidomide alone (Figure 3e). Treatment with Tz-thalidomide and the untagged covalent ERK1/2 inhibitor 7 did not elicit protein degradation (Figure 3e), confirming the requirement for the two binding groups to be covalently attached in the same molecule for degradation efficacy. We also observed that ERK1/2 degradation was prevented when carfilzomib was added in the experiment, confirming that the degradation is proteasome dependent (Figure 3e).
Finally, we evaluated cellular levels of phosphorylated ERK1/2 and phosphorylated RSK (ribosomal s6 kinase) after sequential treatment with Probe 1 and Tz-thalidomide. Downregulation of phospho-ERK1/2 was observed (Figure 3f) demonstrating target engagement of Probe 1 with ERK1/2 and the ability of the ERK-CLIPTAC 6 to degrade ERK1/2 and elicit downregulation of phospho-ERK1/2 signal. RSK is a direct substrate of ERK1/2: consistent with the degradation of ERK1/2 by ERK-CLIPTAC 6, phospho-RSK downregulation was observed to be dependent on Probe 1 concentration, with no phospho-RSK detected at high concentrations (Figure 3f).
Formation of CLIPTACs in Cells Drives Targeted Protein Degradation
To confirm that the observed protein degradation arises from in-cell click formation of the heterobifunctional molecule, we performed analogous experiments in which each CLIPTAC was preformed by reaction of the two click precursors prior to addition to cells. We combined Tz-thalidomide with JQ1-TCO or Probe 1 in a 1:1 ratio to form the clicked molecules JQ1-CLIPTAC 3 or ERK-CLIPTAC 6, respectively. HeLa or A375 cells were then treated with JQ1-CLIPTAC 3 or ERK-CLIPTAC 6, respectively, for 18 h (Figure 4). The immunodetection signals were identical across the experiments, indicating no degradation of BRD4 or ERK1/2, respectively. These results are consistent with a lack of cell permeability of the preclicked heterobifunctional CLIPTAC and confirm that the observed protein degradation results from click formation of the CLIPTAC from its two precursor molecules subsequent to their entry into cells.
Figure 4.

(a) Immunoblot for BRD4 and actin showing no BRD4 degradation when preclicked JQ1-CLIPTAC 3 was used. HeLa cells were treated with JQ1-CLIPTAC for 18 h. (b) Immunoblot for ERK1/2 and actin showing no ERK1/2 degradation when preclicked ERK-CLIPTAC 6 was used. A375 cells were treated with ERK-CLIPTAC for 18 h.
Discussion
Recent successes in the design of PROTAC molecules have raised the possibility that, with further refinement, the methodology could be used to downregulate protein function in a therapeutic setting. The need for PROTACs to enter cells in order to elicit protein degradation means that their physicochemical properties are of paramount importance. Current PROTACs show molecular weights in a significantly higher range than Lipinski guidelines—for example, the three recently published BRD4-targeting PROTACs range from 785 to 1002 (Table 1). In addition, these molecules possess a high polar surface area (PSA range 194–211 Å2) that is normally associated with poor cellular penetration. With these shortcomings in mind, we conceived the idea of CLIPTACs in which two small precursor molecules with the ability to click in cells will pass through cellular membranes more easily than one large compound. A comparison of physicochemical properties of the published BRD4 PROTACs with the CLIPTAC precursors in this paper indicates that MW and PSA have indeed been significantly reduced (Table 1). The successful degradation of BRD4 and ERK1/2 in three different cell lines and the accompanying control experiments confirm the hypothesis that CLIPTACs can be formed in cells from the click reaction between Tz-thalidomide and TCO-ligand, and that the observed degradation occurs through proteasomal action following the binding of the CLIPTAC to CRBN and the protein of interest.
A potentially significant pitfall for the CLIPTAC approach is that the two click partners may undergo their bio-orthogonal combination reaction outside cells, leading to a hetero-bifunctional with high molecular weight and PSA that is unable to enter cells. Indeed, we showed that prior combination reaction of the precursors to form CLIPTACs outside cells did not result in protein degradation in cells. The observation that sequential treatment of cells with one partner followed by the other is effective suggests that a proportion of the second partner is able to enter cells before the click reaction takes place. This raises the possibility that tuning the click reaction to proceed at a slower rate, or indeed use of an alternative bio-orthogonal chemistry with slower kinetics, might result in a greater proportion of the two partners reacting inside cells, with a concomitant improvement in the DC50.
In the time course experiments, we observed that BRD4 levels were completely downregulated after 24 h, whereas ERK1/2 abundance was shown to be suppressed after 16 h. We speculate that this difference in the kinetics of protein degradation might result from the more facile access of the CLIPTAC ligand to ERK1/2 compared with BRD4 due to the latter protein’s complexation to chromatin. By contrast, BRD4 degradation by PROTACs has been described to be complete within a few hours.9,10
The observation that the CLIPTAC 6 derived from a covalent ERK1/2 inhibitor is capable of eliciting ERK degradation is interesting because until now the vast majority of PROTACs have been based on noncovalent reversible ligands for their target proteins. Indeed, this is likely to have been motivated by the proposed potential for catalytic degradation of proteins by reversible PROTACs.13 Clearly, with a PROTAC based on a covalent warhead, one would expect a stoichiometric amount of PROTAC to be required to elicit protein degradation, which might link the achievable potency to the cellular concentration of the protein in question.
An important feature in the design of PROTACs, in addition to selection of appropriate target protein and E3 ligase recruiting ligands, is the length and chemical nature of the linker.9,10 A comprehensive exploration of linkers is beyond the scope of this work; however, we believe it is reasonable to expect that such a study might uncover linkers eliciting more efficient degradation, leading to improved DC50 values and, potentially, to catalytic degradation as reported recently.10 However, the successful use of Tz-thalidomide for the degradation of both BRD4 and ERK1/2 highlights its potential as a general tool compound to study protein degradation using the CLIPTAC methodology. Clearly, Tz-thalidomide is capable of undergoing the click reaction with any TCO-tagged ligand and could be used to induce degradation of that ligand’s target protein. Mindful that control experiments are strongly recommended in this field, we also designed an inactive CLIPTAC precursor, Tz-thalidomide-Me, whose binding affinity to CRBN was considerably reduced.
Finally, a not insignificant additional benefit of these results is that having developed a target protein ligand tagged with a clickable group such as TCO, the same chemical probe may be exploited for a suite of chemical biology experiments including cell imaging, ABPP, pulldown and protein degradation, simply by in-cell click reaction with an appropriate partner, containing a fluorescent, affinity or E3 ligase recruiting group, respectively. It is worth mentioning that a ligand with moderate affinity for its target is sufficient enough to induce degradation as illustrated with Probe 1 which showed low micromolar affinity for ERK2. We hope that these findings will enhance chemical biologists’ ability to extract maximum understanding from their hard-won chemical probes.
Methods
LC-MS Method
The compounds were solubilized in DMSO to generate a 10 mM solution. Pure and mixed samples (TCO ligand/Tz thalidomide 1:1) were then analyzed by LC-MS on a Shimadzu Nexera UPLC coupled with a Shimadzu LCMS-2020 single-quadrupole MS using a YMC-Triart C18 column (50 × 2.0 mm, 1.9 μm) at 45 °C. Gradient elution was performed from 3% acetonitrile to 99% acetonitrile in 10 mM ammonium bicarbonate pH 9.4 over 0.7 min.
ERK2 Bioassay
Activity of ERK2 enzyme (Life Technologies) was determined using a time-resolved fluorescence format measuring the phosphorylation of a truncated version of Activating transcription factor 2 labeled with green fluorescent protein (ATF2-GFP) (Life Technologies). Assay reactions containing 50 mM Tris pH 7.5, 10 mM MgCl2, 1 mM EGTA, 0.01% Triton X-100, 1 mM DTT, 2.5% DMSO, 0.4 μM ATF2-GFP, 20 μM ATP, and 0.25 nM ERK2 were set up in the presence of Tz-thalidomide and allowed to proceed for 30 min at room temperature. Reactions were then stopped using TR-FRET dilution buffer (Life Technologies), 25 mM EDTA, and 2 nM Tb-Anti-pATF2 (Thr71) (Life Technologies). After a further incubation period of at least 30 min, fluorescence was read on a Pherastar reader (Lanthascreen optic module; excitation 340 nm, emission 520 nm (channel A), 495 nm (channel B)). The ratio between A and B counts was used to calculate signal. IC50 values were calculated using a sigmoidal dose response equation (Prism GraphPad software, La Jolla, CA, USA).
Bromodomain Bioassays
The AlphaScreen binding assays (Ex/Em = 680/520–620 nm, in Envision) were performed at Reaction Biology Corp. The compounds were tested in 10-dose IC50 mode with 3-fold serial dilution starting at 10, 30, or 100 μM in duplicate against BRD4-1 and BRD4-2, using a histone H4 peptide (1–21) K5/8/12/16Ac-Biotin.
Cell Culture
HeLa cells (purchased from ATCC) were cultured in Eagle’s minimum essential medium (EMEM) supplemented with 10% FBS (Gibco, Life Technologies), 0.1% NEAA (non-essential amino acid, Gibco, Life Technologies), and 2 mM glutamine and were grown at 37 °C with 5% CO2. A375 and HCT116 cells (purchased from ATCC) were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% FBS (Gibco, Life Technologies) and were grown at 37 °C with 5% CO2.
Immunoblotting
The medium was removed and the cells were washed with PBS (2×). A375 and HCT116 cells were lysed with TG Lysis buffer (150 μL per well) and kept on ice for 20 min. The cell lysates were centrifuged at 14 000 rpm for 10 min at 4 °C, and the protein concentration was determined by a Pierce BCA Protein Assay kit. Samples were normalized, separated on 4–12% NuPAGE gels (Life Technologies), and transferred onto a nitrocellulose membrane (Novex). The membrane was blocked in blocking buffer (Odyssey) at r.t. for 1 h and subjected to immunodetection using a total ERK1/2 primary antibody (p44/42 MAPK ERK1/2, Cell Signaling Technologies, 1:1000) and antiactin antibody (Abcam ab6276, 1:10000) in blocking buffer, at r.t. for 1 h. After being washed 3× with a Tris-buffered saline (TBS) with 0.1% Tween-20 solution (TBST), the membrane was incubated with fluorescently labeled secondary antibody (IRDye800CW Donkey Anti-Rabbit, 1:10000 and IRDye680RD Donkey Anti-Mouse, 1:10 000) for 1 h at r.t. in the dark. After being washed 2× with a TBS solution, the membrane was imaged on an Li-Cor Biosciences Odyssey system in the 800 and 700 nm channels.
The same procedure was followed with HeLa cells using RIPA Lysis buffer (150 μL per well) and 5% nonfat milk in TBST as blocking buffer. Immunodetection was performed using an anti-BRD4 antibody (Bethyl Laboratories, A301/985A100, 1:1000) and antiactin antibody (Abcam ab6276, 1:10000) in 5% nonfat milk in TBST, at 4 °C for 48 h.
Cell Treatment with CLIPTACs
HeLa, A375, and HCT116 cells were seeded in six-well plates at 1.5 × 105 cells/mL with 2 mL/well and allowed to attach overnight before being incubated with the appropriate compounds. TCO-ligand was added from a 1000× stock in DMSO-d6 (2 μL) to the plates. The cells were incubated at 37 °C in an atmosphere of 5% CO2 and air for the indicated time. Tz-Thalidomide was added from a 1000× stock in DMSO-d6 (2 μL) to the plates. The cells were incubated at 37 °C in an atmosphere of 5% CO2 and air for 18 h. When carfilzomib was used, the compound was added from a 1000× stock in DMSO-d6 (2 μL) 4 h before the addition of Tz-thalidomide. For control experiments, untagged ligands were added instead of the TCO-compound, and methyl-Tz-thalidomide was used instead of Tz-thalidomide. For the time course experiments, the cells were incubated for the indicated time after the addition of Tz-thalidomide and consequently lysed.
Acknowledgments
We thank Drs. Aurélie Courtin, Julia Kristensson, Brent Graham, Keisha Hearn, George Ward, and David C. Rees for helpful discussions. We thank Charlotte E. East for the bioassay evaluation of Tz-thalidomide against ERK2. The authors also thank Dr. Torren M. Peakman for the NMR characterizations of JQ1-TCO, Stuart Whibley for his assistance with LC-MS studies, and Dr. Paul Mortenson for the calculations using MOE.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00280.
Full experimental procedures for the synthesis of CLIPTAC precursors and additional figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Deshaies R. J. Protein degradation: Prime time for PROTACs. Nat. Chem. Biol. 2015, 11, 634–635. 10.1038/nchembio.1887. [DOI] [PubMed] [Google Scholar]
- Corson T. W.; Aberle N.; Crews C. M. Design and Applications of Bifunctional Small Molecules: Why Two Heads Are Better Than One. ACS Chem. Biol. 2008, 3, 677–692. 10.1021/cb8001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toure M.; Crews C. M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem., Int. Ed. 2016, 55, 1966–1973. 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- Sakamoto K. M.; Kim K. B.; Kumagai A.; Mercurio F.; Crews C. M.; Deshaies R. J. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 8554–8559. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneekloth J. S.; Fonseca F. N.; Koldobskiy M.; Mandal A.; Deshaies R.; Sakamoto K.; Crews C. M. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- Bargagna-Mohan P.; Baek S.-H.; Lee H.; Kim K.; Mohan R. Use of PROTACS as molecular probes of angiogenesis. Bioorg. Med. Chem. Lett. 2005, 15, 2724–2727. 10.1016/j.bmcl.2005.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C.; Gadd M. S.; Soares P.; Scaffidi S.; Van Molle I.; Birced I.; Hewitt S.; Dias D. M.; Ciulli A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 2014, 57, 8657–8663. 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T.; Ando H.; Suzuki T.; Ogura T.; Hotta K.; Imamura Y.; Yamaguchi Y.; Handa H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K.-H.; Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. C.; Toure M.; Hellerschmied D.; Salami J.; Jaime-Figueroa S.; Ko E.; Hines J.; Crews C. M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem., Int. Ed. 2016, 55, 807–810. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajouhesh H.; Lenz G. R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyrus K.; Wehenkel M.; Choi E.-Y.; Han H.-J.; Lee H.; Swanson H.; Kim K.-B. Impact of linker length on the activity of PROTACs. Mol. BioSyst. 2011, 7, 359–364. 10.1039/C0MB00074D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels–Alder Reactivity. J. Am. Chem. Soc. 2008, 130, 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj R.; Fox J. M. trans-Cyclooctene - a stable, voracious dienophile for bioorthogonal labeling. Curr. Opin. Chem. Biol. 2013, 17, 753–760. 10.1016/j.cbpa.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson D. M.; Nazarova L. A.; Prescher J. A. Finding the Right (Bioorthogonal) Chemistry. ACS Chem. Biol. 2014, 9, 592–605. 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- Tsai Y.-H.; Essig S.; James J. R.; Lang K.; Chin J. W. Selective, rapid and optically switchable regulation of protein function in live mammalian cells. Nat. Chem. 2015, 7, 554–561. 10.1038/nchem.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. S.; Budin G.; Reiner T.; Vinegoni C.; Weissleder R. Bioorthogonal Imaging of Aurora Kinase A in Live Cells. Angew. Chem., Int. Ed. 2012, 51, 6598–6603. 10.1002/anie.201200994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner T.; Earley S.; Turetsky A.; Weissleder R. Bioorthogonal Small-Molecule Ligands for PARP1 Imaging in Living Cells. ChemBioChem 2010, 11, 2374–2377. 10.1002/cbic.201000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keinänen O.; Li X.-G.; Chenna N. K.; Lumen D.; Ott J.; Molthoff C. F. M.; Sarparanta M.; Helariutta K.; Vuorinen T.; Windhorst A. D.; Airaksinen A. J. A New Highly Reactive and Low Lipophilicity Fluorine-18 Labeled Tetrazine Derivative for Pretargeted PET Imaging. ACS Med. Chem. Lett. 2016, 7, 62–66. 10.1021/acsmedchemlett.5b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebraud H.; Wright D. J.; East C. E.; Holding F. P.; O’Reilly M.; Heightman T. D. In-gel activity-based protein profiling of a clickable covalent ERK1/2 inhibitor. Mol. BioSyst. 2016, 12, 2867–2874. 10.1039/C6MB00367B. [DOI] [PubMed] [Google Scholar]
- Fischer E. S.; Bohm K.; Lydeard J. R.; Yang H.; Stadler M. B.; Cavadini S.; Nagel J.; Serluca F.; Acker V.; Lingaraju G. M.; Tichkule R. B.; Schebesta M.; Forrester W. C.; Schirle M.; Hassiepen U.; Ottl J.; Hild M.; Beckwith R. E. J.; Harper J. W.; Jenkins J. L.; Thoma N. H. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaction Biology Corp. website, http://www.reactionbiology.com/webapps/site/Bromodomain-Assay-AlphaScreen.aspx, accessed November 2016.
- Liu L.; Zhen X. T.; Denton E.; Marsden B. D.; Schapira M. ChromoHub: a data hub for navigators of chromatin-mediated signalling. Bioinformatics 2012, 28, 2205–2206. 10.1093/bioinformatics/bts340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R. Jr ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Samatar A. A.; Poulikakos P. I. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discovery 2014, 13, 928–942. 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Shipps G. W.; Cooper A.; English J. M.; Annis D. A.; Carr D.; Nan Y.; Wang T.; Zhu H. Y.; Chuang C.-C.; Dayananth P.; Hruza A. W.; Xiao L.; Jin W.; Kirschmeier P.; Windsor W. T.; Samatar A. A. Discovery of Novel, Dual Mechanism ERK Inhibitors by Affinity Selection Screening of an Inactive Kinase. J. Med. Chem. 2014, 57, 8817–8826. 10.1021/jm500847m. [DOI] [PubMed] [Google Scholar]
- Bagdanoff J. T.; Jain R.; Han W.; Zhu S.; Madiera A.-M.; Lee P. S.; Ma X.; Poon D. Tetrahydropyrrolo-diazepenones as inhibitors of ERK2 kinase. Bioorg. Med. Chem. Lett. 2015, 25, 3788–3792. 10.1016/j.bmcl.2015.07.091. [DOI] [PubMed] [Google Scholar]
- Ren L.; Grina J.; Moreno D.; Blake J. F.; Gaudino J. J.; Garrey R.; Metcalf A. T.; Burkard M.; Martinson M.; Rasor K.; Chen H.; Dean B.; Gould S. E.; Pacheco P.; Shahidi-Latham S.; Yin J.; West K.; Wang W.; Moffat J. G.; Schwarz J. B. Discovery of Highly Potent, Selective, and Efficacious Small Molecule Inhibitors of ERK1/2. J. Med. Chem. 2015, 58, 1976–1991. 10.1021/jm501921k. [DOI] [PubMed] [Google Scholar]
- Ward R. A.; Colclough N.; Challinor M.; Debreczeni J. E.; Eckersley K.; Fairley G.; Feron L.; Flemington V.; Graham M. A.; Greenwood R.; Hopcroft P.; Howard T. D.; James M.; Jones C. D.; Jones C. R.; Renshaw J.; Roberts K.; Snow L.; Tonge M.; Yeung K. Structure-Guided Design of Highly Selective and Potent Covalent Inhibitors of ERK1/2. J. Med. Chem. 2015, 58, 4790–4801. 10.1021/acs.jmedchem.5b00466. [DOI] [PubMed] [Google Scholar]
- Rodríguez J.; Crespo P. Working Without Kinase Activity: Phosphotransfer-Independent Functions of Extracellular Signal–Regulated Kinases. Sci. Signaling 2011, 4, 1–6. 10.1126/scisignal.2002324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.