

Abstract
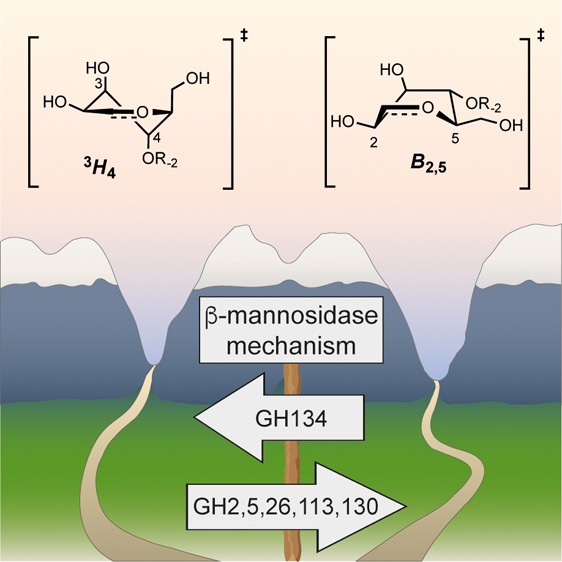
The enzymatic cleavage of β-1,4-mannans is achieved by endo-β-1,4-mannanases, enzymes involved in germination of seeds and microbial hemicellulose degradation, and which have increasing industrial and consumer product applications. β-Mannanases occur in a range of families of the CAZy sequence-based glycoside hydrolase (GH) classification scheme including families 5, 26, and 113. In this work we reveal that β-mannanases of the newly described GH family 134 differ from other mannanase families in both their mechanism and tertiary structure. A representative GH family 134 endo-β-1,4-mannanase from a Streptomyces sp. displays a fold closely related to that of hen egg white lysozyme but acts with inversion of stereochemistry. A Michaelis complex with mannopentaose, and a product complex with mannotriose, reveal ligands with pyranose rings distorted in an unusual inverted chair conformation. Ab initio quantum mechanics/molecular mechanics metadynamics quantified the energetically accessible ring conformations and provided evidence in support of a 1C4 → 3H4‡ → 3S1 conformational itinerary along the reaction coordinate. This work, in concert with that on GH family 124 cellulases, reveals how the lysozyme fold can be co-opted to catalyze the hydrolysis of different polysaccharides in a mechanistically distinct manner.
Short abstract
Mannosidases of the new glycosidase hydrolase (GH) family 134 are lysozyme-like enzymes that hydrolase β-1,4-mannans, industrially important plant polymers, and use a mechanism that is distinct to all β-mannosidases hailing from other GH families.
Introduction
β-1,4-Mannans are a diverse group of polysaccharides present within the plant cell wall and endosperm.1 Their degradation is catalyzed by β-1,4-mannanases and β-mannosidases, and assorted auxiliary enzymes, which act in concert to deconstruct the β-1,4-mannan backbone to liberate, ultimately, d-mannose.2 β-Mannanases are of increasing industrial significance in the food, detergent, and biofuels industries,3 which process β-mannan-rich plant material, and, in the oil and gas industries, which deploy β-1,4-mannan materials to modulate fluid rheology in fracking processes.4,5 Nature has evolved a range of elegant strategies to overcome the stereochemical challenges associated with catalyzing mannosyl transfer,6,7 and which continue to inspire methods in organic synthesis for nucleophilic substitution at the anomeric center of mannose.8−10
endo-β-1,4-Mannanase activity was originally reported for three sequence-based glycoside hydrolase (GH) families of the CAZy classification scheme; GH families 5, 26, and 113 (see www.cazy.org; www.cazypedia.org).11 These families belong to clan GH-A and all share a (β/α)8-barrel protein fold, similar catalytic machinery, and a stereochemically retaining double displacement mechanism involving a covalent glycosyl-enzyme intermediate.12−14 GH family 5, 26, and 113 β-mannanases,14 along with GH family 2 and 130 β-mannosidases, operate through conserved 1S5 → B2,5‡ → OS2 conformational itineraries for the glycosylation half-reaction;15,16 distortion of the pyranose ring to a 1S5 conformation in the Michaelis complex provides an unfettered trajectory for approach of the catalytic nucleophile (Figure S1). Recently, a new family of fungal and bacterial endo-β-1,4-mannanases was reported, GH family 134.17 Here, we delineate the mechanism, substrate preference, and tertiary structure of a ligand-free and complexed GH134 enzyme. Utilizing these atomic resolution data we perform quantum mechanical calculations for the free energy landscape of a β-mannopyranose molecule within the confines of the active site, which enable assignment of a 1C4 → 3H4‡ → 3S1 “Southern hemisphere” conformational itinerary (see Figure S1 for details); the first observed for a β-mannanase.6,7
Results and Discussion
Streptomyces sp. NRRL B-24484 possesses an operon encoding a series of proteins predicted to facilitate the deconstruction and metabolism of β-mannan, including a GH2 β-mannosidase, two GH5 β-mannanases, one GH134 β-mannanase, sugar ABC transporters, and a mannose-6-phosphate isomerase (Figure 1a). Hypothetical protein WP_030268297.1, the putative GH134 β-mannanase, possesses a signal peptide and CBM10 domain (carbohydrate binding modules, CBMs, reviewed in ref (18)) attached through a short GlySer linker to the GH134 catalytic domain (Figure S2a). A gene encoding the catalytic domain, hereafter termed SsGH134, was synthesized and expressed in Escherichia coli with an N-terminal hexahistidine-tagged maltose binding protein (MBP) fusion partner (Figure S2b). This fusion protein was assayed for activity on β-1,4-oligomannosides, revealing an ability to cleave mannopentaose (M5), to yield mannotriose (M3) and mannobiose (M2), and to cleave mannohexaose (M6), to give predominantly M3, with smaller amounts of mannotetraose (M4) and M2, with optimal activity (kcat/KM 2.7 × 105 M–1 min–1) toward M6 at pH 5 and a strong preference for unsubstituted linear β-mannans over gluco- and galactomannans (Figure S3); a specificity rarely seen for β-mannanases. To define the cleavage site within these repeating oligosaccharides, enzymatic hydrolysis was performed in 18O-labeled water, which resulted in incorporation of 18O into the nonreducing end fragment.19−21 Mass spectrometric analysis of the products formed revealed that M5 is cleaved to provide M2 and M3 + 2 quasi-molecular ions, demonstrating 18O incorporation into the latter fragment, and consistent with hydrolysis of M5 across the −3 → +2 subsites (Figure S4).22 Similar analysis of the products formed from cleavage of M6 revealed a preferred cleavage of the hexasaccharide across the −3 → + 3 subsites, as well as some cleavage across the −4 → +2 subsites (Figure S5).
Figure 1.

Family GH134 β-mannanases act with inversion of anomeric stereochemistry. (a) Genomic context of SsGH134 within the genome of Streptomyces sp. NRRL B-24484. (b) Summary of derivatization and stereochemistry determination: β-1,4-mannohexaose (M6) was converted to the glycosyl hydrazide and then purified. The mannohexaosyl benzoylhydrazide was digested with SsGH134 and the stereochemical outcome was determined. (c) M6-benzoyl hydrazide was incubated with SsGH134 and the stereochemistry of the reaction was monitored by 1H NMR spectroscopy. At t = 0 there are no anomeric protons corresponding to the hemiacetal. At t = 2 min, two new peaks, assigned as H1α and H1β of M3 at δ 5.2 and 4.92 ppm, respectively. Mutarotation is at equilibrium by t = 40 min.
We assessed the stereochemistry of substrate cleavage using 1H NMR spectroscopy. Owing to spectral overlap of the substrate reducing end anomeric protons with those of the newly formed reducing end upon enzyme cleavage, we converted M6 into the glycosyl benzoylhydrazide derivative (Figure 1b) to provide a clear spectral region between δ 4.85–5.25 ppm to facilitate the detection of the enzymatic hydrolysis product. SsGH134-catalyzed cleavage of mannohexaosyl benzoylhydrazide resulted in formation of the α-anomer of M3, which over time underwent mutarotation, demonstrating that SsGH134 is an inverting glycoside hydrolase (Figures 1c and S6).
Family GH134 comprises proteins of approximately 200 amino acids in length, which are among the shortest of any glycoside hydrolase family. Motivated by the desire to see how such a small protein acts as a glycoside hydrolase, possibly reflecting unusual or unique strategies, we sought to determine the three-dimensional structure of SsGH134. After removal of the MBP fusion partner, SsGH134 crystals were obtained that diffracted to atomic resolution (approximately 1 Å), allowing the native structure to be solved by a novel “no-prior knowledge” molecular replacement method23 using only an isolated standard helix as the search model (Tables 1 and S1). The SsGH134 structure revealed a mixed α-helix/β-sheet fold with a strong superficial resemblance to family GH19 chitinases, GH22 C-type lysozymes, GH23 G-type lysozymes, and GH124 cellulases (Figures 2a, 3a–d, and S8).
Table 1. 3-D Structure Data for SsGH134.
| native | SsGH134-mannotriose | SsGH134 E45Q-mannopentaose | |
|---|---|---|---|
| PDB Code | 5JTS | 5JU9 | 5JUG |
| space group | P212121 | P212121 | P212121 |
| resolution (Å) | 34 (1.09) | 45 (1.18) | 45 (0.96) |
| Rmerge | 0.066 (0.86) | 0.056 (0.56) | 0.054 (0.52) |
| I/σI | 12.2 (1.8) | 16.6 (2.4) | 12.4 (1.1) |
| completeness (%) | 99.9 (99.9) | 97.5 (80.2) | 84.1 (29.2)a |
| rmsd bond lengths (Å) | 0.014 | 0.012 | 0.012 |
| rmsd bond angles (deg) | 1.56 | 1.57 | 1.67 |
The low completeness for the outer shell is due to the integration of data into the corners of the square detector. Data in the penultimate shell are 99% complete.
Figure 2.

Family GH134 β-mannanases adopt a lysozyme-like fold. SsGH134 E45Q variant in complex with β-1,4-mannopentaose. (a) Overview of complex rainbow-colored from the N- to C-terminus. (b) Surface representation of complex colored by sequence conservation according to the sequence alignment shown in Figure S9 (cyan = variable, burgundy = conserved). The figure was prepared using the CONSURF server.24
Figure 3.

Pairwise comparison of the fold, active site clefts, and key catalytic residues of SsGH134 and representative glycoside hydrolases with lysozyme folds. SsGH134 E45Q (PDB 5JUG, light green) with M5 bound (blue) overlaid with (a) retaining GH22 family HEWL (PDB 2WAR, yellow) with chitopentaose bound (plum), (b) inverting GH19 family chitinase A (PDB 3WH1, pink) with chitobiose and chitotriose bound (magenta), (c) inverting GH124 family cellulose CtCel124 (PDB 2XQO, lavender) with two molecules of cellotriose bound (chocolate), (d) inverting GH23 G-type lysozyme (PDB 3GXR, dark green) with chitobiose and chitotriose bound (brown). (e) Close up overlay of the catalytic residues of SsGH134 (light green), HEWL (yellow), chitinase A (pink), CtCel124 (lavender), and G-type lysozyme (dark green).
Structural similarity searches using PDBeFold25 revealed that SsGH134 is most similar to the GH22 C-type lysozyme family, albeit with a relatively low Z score of 3.3 reflecting 88 Cα carbons overlapping with an rmsd of approximately 2.6 Å (Figure 3a). The GH22 C-type lysozyme family is exemplified by hen egg white lysozyme (HEWL),26 the archetypal glycoside hydrolase that performs catalysis with net retention of anomeric configuration, via the formation and subsequent hydrolysis of a covalent intermediate bonded to D52 and with E35 acting as a general acid/base.27 In contrast, SsGH134 performs catalysis with inversion of anomeric configuration, presumably through the direct attack of water in a single displacement mechanism, assisted by residues acting as a general acid and general base. Comparison of the location of the catalytic residues of the SsGH134 E45Q structure in complex with M5 (vide infra) with that of a chitohexaose complex of HEWL28 reveals that the general acid/base of HEWL, E35, overlays with E45 (Q in the mutated variant) of SsGH134, whereas the nucleophile D52 of HEWL overlays with N65 of SsGH134. As Asn is a poor nucleophile, this alternative residue in SsGH134 is consistent with its opposite stereochemical outcome versus HEWL (Figure 3e). In the case of the inverting chitinase A of family GH19,29 the general acid E61 of chitinase A overlays with E45 in SsGH134, and the putative nucleophilic water coordinated by N65 and D57 in SsGH134 is located 4.3 Å away from an equivalent water in chitinase A held by E70 and S102. For the inverting G-type lysozyme,30 the essential catalytic residue E73 overlays with E45 of SsGH134. Finally, the catalytic general acid in Clostridium thermocellum CtCel124A overlays with E45 of SsGH134.31
In order to probe the molecular basis of catalysis, and confirm catalytic acid and base assignments inferred from the overlaps described above, the structure of SsGH134 in complex with M3 was solved at a resolution of 1.2 Å (Figure 4a, Table 1). M3 binds in the −3 → −1 subsites32 with the −1 subsite occupied by an α-mannose group. This represents a product complex, with the stereochemistry of the α-mannose group matching that formed through catalysis with inversion of anomeric configuration. Remarkably, the −1 subsite mannose in the product complex was in a ring-flipped 1C4 conformation, hinting at a novel conformational itinerary for GH family 134. The location of E45 is consistent with its role as a general acid catalyst (as expected from the overlaps above), and the spatial location of D57 implicates it as a general base, positioned to deprotonate a water nucleophile in a single displacement inverting mechanism. These structure-based assignments were supported by mutagenesis: the D57N and E45Q variants were inactive against M5 and M6.
Figure 4.

Complexes of (a) wild-type SsGH134 with M3 and (b) E45Q variant with M5 showing the proposed general acid (E45/Q45) suitably positioned near the fissile glycosidic bond, and on the opposing side the proposed general base D57, coordinating the nucleophilic water molecule. Electron density meshes are σA-weighted 2F0 – Fc contoured at 1σ (0.58 and 0.47 electrons per Å3, respectively), assembled using CCP4mg.33 (c) Cartoon showing major protein substrate interactions for the SsGH134–mannopentaose complex. Interactions are shown for the model of the SsGH134 bound to the α-anomer of β-1,4-mannopentaose. The red spheres denote water molecules.
A “Michaelis” complex was obtained by soaking M5 into crystals of the catalytically inactive mutant SsGH134-E45Q. Clear electron density at 0.96 Å resolution revealed M5 bound from the −3 → +2 subsites (Figure 4b), consistent with the 18O-water incorporation experiments. With the exception of the −1 subsite, discussed below, all the remaining sugar rings are in ground state 4C1 conformations. Residues lying within a major loop region (44–72) form contacts with the substrate down the length of the active site, and a contiguous stretch of residues within the C-terminal segment (168–173) seals the active center from −2 to +2. Notably, the C-terminal carboxylate of I173 is hydrogen bonded to O6 of the −2 mannosyl unit and forms a salt-bridge with the highly conserved K59 (Figure 4c, residues highlighted in green). This interaction presumably locks the carboxylate in place and completes an active center tunnel now reminiscent of processive glycoside hydrolases (Figure 2b). Completion of the tunnel occurs only in the Michaelis complex (when the positive subsites are occupied) as the C-terminal segment is disordered in both “apo” and M3 product-bound forms. Alignment of SsGH134 with other family member sequences in the NCBI nonredundant database reveals a highly conserved C-terminus ending with an isoleucine (Figure S9). Two exceptions are the putative proteins KFZ17922.1 and KFZ19809.1 from Pseudogymnoascus sp. (VKM F-4519), which are fused to a predicted GH27 α-galactosidase linked through the GH134 C-terminus and which we postulate may act on galactomannan, a β-1,4-mannan polysaccharide modified with α-1,6-galactosyl residues. This suggests a role for the C-terminal Ile in distinguishing β-1,4-mannans lacking a substituent at the −2 mannose from more complex mannans (Figure 4c). In support of this hypothesis, it is notable that SsGH134 is only weakly active on carob galactomannan and konjac glucomannan, but is unusual in its ability to process highly purified β-1,4-mannan and ivory nut mannan extensively (Figure S3). The structure of the Michaelis complex contains a well-ordered water molecule positioned to act as a nucleophile and coordinated to the general base D57, as well as K59 and N65 on the α-face of the mannoside in the −1 subsite. The side chain carbonyl oxygen of Q45 is 3.4 Å away from the leaving-group oxygen, consistent with the assignment of E45 as the general acid. Notably, the −1 mannoside resides in a ring flipped 1C4 conformation, similar to that seen in the SsGH134-M3 product complex, with the leaving group C1–O oriented axially, and the nucleophilic water positioned for in-line nucleophilic attack with a water–C1–O1 angle of 160°. The structures of the Michaelis and product complexes are suggestive of a Southern hemisphere 1C4 → 3H4‡ → 3S1 conformational itinerary along the reaction coordinate, with the product relaxing to a Michaelis-mimicking 1C4 conformation (possibly via a 3H4 conformation) after the reaction is complete. Such a conformational itinerary has never been reported for a β-mannosidase or β-mannanase; however, a reversed Southern hemisphere 3S1 → 3H4‡ → 1C4 conformational itinerary has been assigned to inverting α-mannosidases from family GH47.34−36
The unusual conformations observed in the Michaelis and product complexes prompted study of the conformational coordinate by classical molecular dynamics (MD) and QM/MM metadynamics.37 Three collective variables (CV), including all bonds to be broken/cleaved by the enzyme, were considered. CV1 accounts for the nucleophilic attack of the catalytic water molecule; CV2 accounts for proton transfer between D57 and the water molecule; and CV3 accounts for the transfer of the E45 proton to the glycosidic oxygen atom (Figure S11). The shape of the reconstructed free energy surface, projected onto CV1/CV2 (Figure 5a), is indicative of a concerted one-step reaction. The reaction free energy barrier (17 kcal mol–1) is commensurate with the measured reaction rate.17
Figure 5.
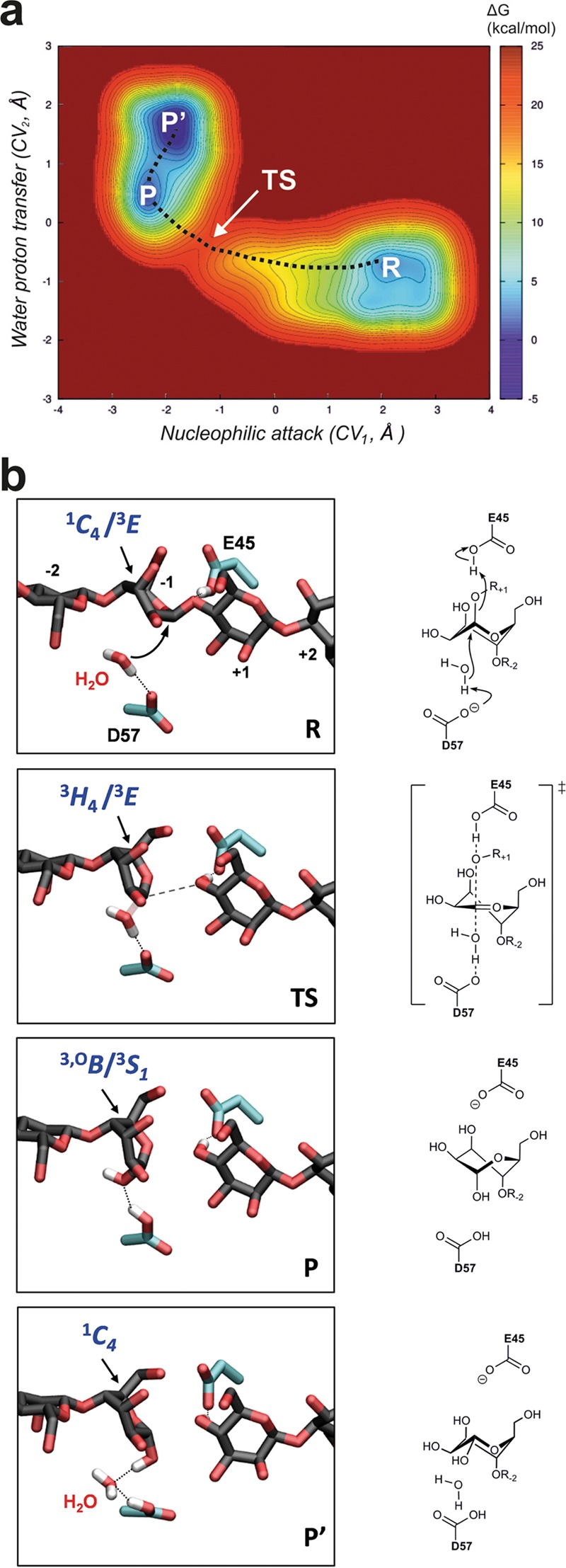
QM/MM metadynamics simulations of the mechanism of SsGH134. (a) Free energy landscape of the SsGH134-catalyzed reaction obtained by QM/MM metadynamics. (b) Reaction coordinate for SsGH134 inverting endo-β-1,4-mannanase. R denotes reactants (Michaelis complex), TS the transition state, and P the products. From P to P′, a solvent water molecule enters the active site and fills the space previously occupied by the anomeric hydroxyl group of the mannose in the −1 subsite. Hydrogen atoms have been omitted for clarity, except those of the catalytic water and the carboxylate group of E45.
Representative states along the lowest free energy pathway are shown in Figure 5b. The −1 sugar at the reactants state (R) adopts a conformation intermediate between 1C4 and 3E. The reaction commences with elongation of the glycosidic bond simultaneous with the transfer of the carboxylic hydrogen atom of the acid residue to the glycosidic oxygen. At the transition state (TS), the −1 mannopyranose ring distorts from 1C4/3E to 3H4/3E (Figures 5 and S12), a conformation compatible with the requirement of an oxocarbenium ion like TS.6,7 At this stage, the E45 proton is already transferred, the glycosidic bond is completely broken (3.3 Å; Table S2), and the bond between the nucleophilic water and the anomeric carbon is partially formed (2.0 Å; Table S2). Proton transfer from the water to D57 then takes place, while the −1 sugar changes to a 3,OB/3S1 conformation (P) (Figures 5 and S12). Thereafter, the anomeric OH loses its interaction with D57 (transition from P to P′, ΔG‡ = 2 kcal mol–1), and the −1 mannopyranose spontaneously undergoes relaxation to a 1C4 conformation (see also Figure S13), matching the conformation observed in the product complex of SsGH134 with M3 (Figure 4a). It is expected that once the product exits the active site it relaxes to its most stable 4C1 conformation in solution.36 The computed mechanism can be considered an electrophilic migration of the anomeric carbon from the departing sugar residue to the nucleophilic water, assisted by E45 as general acid and D57 as general base (Figure 5).
Concluding Remarks
The present work reveals the emergence of a new enzyme activity from the lysozyme fold, a fold known since the first-determined X-ray structure of an enzyme, that of HEWL.26 This work provides an elegant example of how tertiary structures can be co-opted during evolution to bind different substrates and process them using entirely different mechanisms. While the majority of mannoside hydrolases appear to act through mechanisms involving a transition state in a B2,5 conformation (β-mannosidases/mannanases: GH2, 5, 26, 113, 130; α-mannosidases/mannanases: GH38, 76, 92)7 the β-mannanases of family GH134 stand with the α-mannosidases of family GH47 as GHs operating through a 3H4 transition state. Collectively these groups illustrate two distinct solutions to the problem of nucleophilic attack on a β-mannopyranoside residue.
Acknowledgments
Australian Research Council (FT130100103), the European Research Council (ERC-2012-AdG-322942), the Ramaciotti Foundation and VESKI with additional support from the Australian Cancer Research Foundation and Victorian State Government Operational Infrastructure Support, NHMRC IRIISS Grant 9000220, the Spanish Ministry of Economy and Competitiveness (MINECO, CTQ2014-55174-P) and the Generalitat de Catalunya (2014SGR-987). G.J.D. is supported by the Royal Society. We acknowledge the staff of the Diamond Light Source (UK) for provision of I02 beamline facilities (Proposal Number mx-9948), and the support, technical expertise, and assistance provided by the Barcelona Supercomputing Center: Centro Nacional de Supercomputación (BSC–CNS). L.R. acknowledges a Ph.D. fellowship from the University of Barcelona (APIF-UB).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00232.
Supplementary figures (Figures S1–S13), Supplementary Tables (Tables S1 and S2), all experimental details, X-ray data collection, processing and refinement statistics (PDF)
Accession Codes
The coordinates and structure factors have been deposited in the Protein Data Bank (accession codes 5JTS, 5JU9, 5JUG).
The authors declare no competing financial interest.
Supplementary Material
References
- Rodríguez-Gacio M. d. C.; Iglesias-Fernández R.; Carbonero P.; Matilla Á. J. Softening-up mannan-rich cell walls. J. Exp. Bot. 2012, 63, 3976–3988. 10.1093/jxb/ers096. [DOI] [PubMed] [Google Scholar]
- Malgas S.; van Dyk J. S.; Pletschke B. I. A review of the enzymatic hydrolysis of mannans and synergistic interactions between β-mannanase, β-mannosidase and α-galactosidase. World J. Microbiol. Biotechnol. 2015, 31, 1167–1175. 10.1007/s11274-015-1878-2. [DOI] [PubMed] [Google Scholar]
- Dhawan S.; Kaur J. Microbial mannanases: an overview of production and applications. Crit. Rev. Biotechnol. 2007, 27, 197–216. 10.1080/07388550701775919. [DOI] [PubMed] [Google Scholar]
- Moreira L. R.; Filho E. X. An overview of mannan structure and mannan-degrading enzyme systems. Appl. Microbiol. Biotechnol. 2008, 79, 165–178. 10.1007/s00253-008-1423-4. [DOI] [PubMed] [Google Scholar]
- van Zyl W. H.; Rose S. H.; Trollope K.; Görgens J. F. Fungal β-mannanases: Mannan hydrolysis, heterologous production and biotechnological applications. Process Biochem. 2010, 45, 1203–1213. 10.1016/j.procbio.2010.05.011. [DOI] [Google Scholar]
- Davies G. J.; Planas A.; Rovira C. Conformational analyses of the reaction coordinate of glycosidases. Acc. Chem. Res. 2012, 45, 308–316. 10.1021/ar2001765. [DOI] [PubMed] [Google Scholar]
- Speciale G.; Thompson A. J.; Davies G. J.; Williams S. J. Dissecting conformational contributions to glycosidase catalysis and inhibition. Curr. Opin. Struct. Biol. 2014, 28, 1–13. 10.1016/j.sbi.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crich D. Mechanism of a chemical glycosylation reaction. Acc. Chem. Res. 2010, 43, 1144–1153. 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- Crich D.; Chandrasekera N. S. Mechanism of 4,6-O-benzylidene-directed β-mannosylation as determined by α-deuterium kinetic isotope effects. Angew. Chem., Int. Ed. 2004, 43, 5386–5389. 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- Huang M.; Garrett G. E.; Birlirakis N.; Bohe L.; Pratt D. A.; Crich D. Dissecting the mechanisms of a class of chemical glycosylation using primary 13C kinetic isotope effects. Nat. Chem. 2012, 4, 663–667. 10.1038/nchem.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V.; Golaconda Ramulu H.; Drula E.; Coutinho P. M.; Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–495. 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G.; Henrissat B. Structures and mechanisms of glycosyl hydrolases. Structure 1995, 3, 853–859. 10.1016/S0969-2126(01)00220-9. [DOI] [PubMed] [Google Scholar]
- Vocadlo D. J.; Davies G. J. Mechanistic insights into glycosidase chemistry. Curr. Opin. Chem. Biol. 2008, 12, 539–555. 10.1016/j.cbpa.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Williams R. J.; Iglesias-Fernandez J.; Stepper J.; Jackson A.; Thompson A. J.; Lowe E. C.; White J. M.; Gilbert H. J.; Rovira C.; Davies G. J.; Williams S. J. Combined inhibitor free-energy landscape and structural analysis reports on the mannosidase conformational coordinate. Angew. Chem., Int. Ed. 2014, 53, 1087–1091. 10.1002/anie.201308334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailford L. E.; Offen W. A.; Smith N. L.; Dumon C.; Morland C.; Gratien J.; Heck M. P.; Stick R. V.; Bleriot Y.; Vasella A.; Gilbert H. J.; Davies G. J. Structural and biochemical evidence for a boat-like transition state in β-mannosidases. Nat. Chem. Biol. 2008, 4, 306–312. 10.1038/nchembio.81. [DOI] [PubMed] [Google Scholar]
- Tankrathok A.; Iglesias-Fernández J.; Williams R. J.; Pengthaisong S.; Baiya S.; Hakki Z.; Robinson R. C.; Hrmova M.; Rovira C.; Williams S. J.; Ketudat Cairns J. R. A single glycosidase harnesses different pyranoside ring transition state conformations for hydrolysis of mannosides and glucosides. ACS Catal. 2015, 5, 6041–6051. 10.1021/acscatal.5b01547. [DOI] [Google Scholar]
- Shimizu M.; Kaneko Y.; Ishihara S.; Mochizuki M.; Sakai K.; Yamada M.; Murata S.; Itoh E.; Yamamoto T.; Sugimura Y.; Hirano T.; Takaya N.; Kobayashi T.; Kato M. Novel β-1,4-mannanase belonging to a new glycoside hydrolase family in Aspergillus nidulans. J. Biol. Chem. 2015, 290, 27914–27927. 10.1074/jbc.M115.661645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boraston A. B.; Bolam D. N.; Gilbert H. J.; Davies G. J. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 2004, 382, 769–781. 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier M.; Roussel A.; Rosengren A.; Leone P.; Stålbrand H.; Berrin J.-G. Structural and biochemical analyses of glycoside hydrolase families 5 and 26 β-1,4-mannanases from Podospora anserina reveal differences upon manno-oligosaccharide catalysis. J. Biol. Chem. 2013, 288, 14624–14635. 10.1074/jbc.M113.459438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor N.; Morar M.; Fenger T. H.; Stogios P.; Lenfant N.; Yin V.; Xu X.; Evdokimova E.; Cui H.; Henrissat B.; Savchenko A.; Brumer H. Structure-function analysis of a mixed-linkage β-glucanase/xyloglucanase from the key ruminal bacteroidetes Prevotella bryantii B14. J. Biol. Chem. 2016, 291, 1175–1197. 10.1074/jbc.M115.691659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagerlof H.; Nilsson C.; Gorton L.; Tjerneld F.; Stalbrand H.; Cohen A. Use of 18O water and ESI-MS detection in subsite characterisation and investigation of the hydrolytic action of an endoglucanase. Anal. Bioanal. Chem. 2009, 394, 1977–1984. 10.1007/s00216-009-2891-9. [DOI] [PubMed] [Google Scholar]
- Davies G. J.; Wilson K. S.; Henrissat B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 1997, 321, 557–559. 10.1042/bj3210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greive S. J.; Fung H. K.; Chechik M.; Jenkins H. T.; Weitzel S. E.; Aguiar P. M.; Brentnall A. S.; Glousieau M.; Gladyshev G. V.; Potts J. R.; Antson A. A. DNA recognition for virus assembly through multiple sequence-independent interactions with a helix-turn-helix motif. Nucleic Acids Res. 2016, 44, 776–789. 10.1093/nar/gkv1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celniker G.; Nimrod G.; Ashkenazy H.; Glaser F.; Martz E.; Mayrose I.; Pupko T.; Ben-Tal N. ConSurf: Using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 2013, 53, 199–206. 10.1002/ijch.201200096. [DOI] [Google Scholar]
- Krissinel E.; Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2256–2268. 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Blake C. C. F.; Koenig D. F.; Mair G. A.; North A. C. T.; Phillips D. C.; Sarma V. R. Structure of hen egg-white lysozyme. Nature 1965, 206, 757–761. 10.1038/206757a0. [DOI] [PubMed] [Google Scholar]
- Vocadlo D. J.; Davies G. J.; Laine R.; Withers S. G. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature 2001, 412, 835–838. 10.1038/35090602. [DOI] [PubMed] [Google Scholar]
- Davies G. J.; Withers S. G.; Vocadlo D. J. The chitopentaose complex of a mutant hen egg-white lysozyme displays no distortion of the – 1 sugar away from a 4C1 chair conformation. Aust. J. Chem. 2009, 62, 528–532. 10.1071/CH09038. [DOI] [Google Scholar]
- Ohnuma T.; Umemoto N.; Nagata T.; Shinya S.; Numata T.; Taira T.; Fukamizo T. Crystal structure of a ″loopless″ GH19 Chitinase in complex with chitin tetrasaccharide spanning the catalytic center. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844, 793–802. 10.1016/j.bbapap.2014.02.013. [DOI] [PubMed] [Google Scholar]
- Helland R.; Larsen R. L.; Finstad S.; Kyomuhendo P.; Larsen A. N. Crystal structures of g-type lysozyme from Atlantic cod shed new light on substrate binding and the catalytic mechanism. Cell. Mol. Life Sci. 2009, 66, 2585–2598. 10.1007/s00018-009-0063-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bras J. L.; Cartmell A.; Carvalho A. L.; Verze G.; Bayer E. A.; Vazana Y.; Correia M. A.; Prates J. A.; Ratnaparkhe S.; Boraston A. B.; Romao M. J.; Fontes C. M.; Gilbert H. J. Structural insights into a unique cellulase fold and mechanism of cellulose hydrolysis. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 5237–5242. 10.1073/pnas.1015006108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G. J.; Wilson K. S.; Henrissat B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 1997, 321, 557–559. 10.1042/bj3210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaveg K.; Siriwardena A.; Tempel W.; Liu Z. J.; Glushka J.; Wang B. C.; Moremen K. W. Mechanism of class 1 (glycosylhydrolase family 47) α-mannosidases involved in N-glycan processing and endoplasmic reticulum quality control. J. Biol. Chem. 2005, 280, 16197–16207. 10.1074/jbc.M500119200. [DOI] [PubMed] [Google Scholar]
- Vallée F.; Karaveg K.; Herscovics A.; Moremen K. W.; Howell P. L. Structural basis for catalysis and inhibition of N-glycan processing class I α-1,2-mannosidases. J. Biol. Chem. 2000, 275, 41287–41298. 10.1074/jbc.M006927200. [DOI] [PubMed] [Google Scholar]
- Thompson A. J.; Dabin J.; Iglesias-Fernandez J.; Ardevol A.; Dinev Z.; Williams S. J.; Bande O.; Siriwardena A.; Moreland C.; Hu T. C.; Smith D. K.; Gilbert H. J.; Rovira C.; Davies G. J. The reaction coordinate of a bacterial GH47 α-mannosidase: A combined quantum mechanical and structural approach. Angew. Chem., Int. Ed. 2012, 51, 10997–11001. 10.1002/anie.201205338. [DOI] [PubMed] [Google Scholar]
- Iannuzzi M.; Laio A.; Parrinello M. Efficient exploration of reactive potential energy surfaces using Car-Parrinello molecular dynamics. Phys. Rev. Lett. 2003, 90, 238302. 10.1103/PhysRevLett.90.238302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.