

Abstract
Background:
Congenital nystagmus (CN) is characterized by conjugated, spontaneous, and involuntary ocular oscillations. It is an inherited disease and the most common inheritance pattern is X-linked CN. In this study, our aim is to identify the disease-causing mutation in a large sixth-generation Chinese family with X-linked CN.
Methods:
It has been reported that mutations in four-point-one, ezrin, radixin, moesin domain-containing 7 gene (FRMD7) and G protein-coupled receptor 143 gene (GPR143) account for the majority patients of X-linked nystagmus. We collected 8 ml blood samples from members of a large sixth-generation pedigree with X-linked CN and 100 normal controls. FRMD7 and GPR143 were scanned by polymerase chain reaction (PCR)-based DNA sequencing assays, and multiplex PCR assays were applied to detect deletions.
Results:
We identified a previously unreported deletion covering 7 exons in GPR143 in a Chinese family. The heterozygous deletion from exon 3 to exon 9 of GPR143 was detected in all affected males in the family, while it was not detected in other unaffected relatives or 100 normal controls.
Conclusions:
This is the first report of molecular characterization in GPR143 gene in the CN family. Our results expand the spectrum of GPR143 mutations causing CN and further confirm the role of GPR143 in the pathogenesis of CN.
Keywords: Four-point-one, ezrin, radixin, moesin domain-containing 7 gene, G protein-coupled receptor 143 gene, X-linked congenital nystagmus
Congenital nystagmus (CN) is characterized by conjugated, spontaneous, and involuntary ocular oscillations. It is an inherited disease and the most common inheritance pattern is X-linked CN. Four-point-one, ezrin, radixin, moesin domain-containing 7 (FRMD7) and G protein-coupled receptor 143 (GPR143) have been identified as the disease-causing genes for X-linked CN.[1,2] The FRMD7 gene comprises 12 exons and encodes a protein with 714 amino acids. The GPR143 gene consists of 9 exons and encodes a protein with 404 amino acids.
Numerous mutations in FRMD7 gene have been reported since it was first identified in 2006.[3,4,5,6,7,8] Mutations in FRMD7 gene are the major causes of Chinese familial X-linked CN and account for approximately 47% of Chinese patients with the disorder.[9]
GPR143 gene is also known as the ocular albinism type 1 (OA1) gene, as mutations in GPR143 also cause OA1.[10] Most patients with OA1 show nystagmus and poor visual acuity. Several reports confirmed the pathogenicity of GPR143 gene in CN family.[2,11,12,13,14]
In this study, we analyzed the variants in FRMD7 and GPR143 genes in a sixth-generation, nonconsanguineous CN Chinese family. We identified a deletion covering 3–9 exons of GPR143 gene resulting in a truncated protein of 120 residues. Our results expand the spectrum of GPR143 mutations causing CN. This is the first report of molecular characterization in the GPR143 gene in the CN family.
Methods
A sixth-generation Han family from Hebei Province in China including six male patients participated in this study [Fig. 1]. The patients underwent complete physical and ophthalmic examinations. The Institutional Review Board approved the project, and investigators followed the principles of the Declaration of Helsinki. Informed consent was obtained from each person. One hundred normal male controls mainly from the north of China were also analyzed.
Figure 1.

The pedigree of family. Squares and circles indicate males and females, the darkened symbols represent affected members, dot-marked symbols represent females who carried the mutation, and the patient above the arrow is the proband
Blood specimens (8 ml) of each family member available were collected in ethylenediaminetetraacetic acid, and genomic DNA was extracted from peripheral blood cells according to a standard protocol (Roche Diagnostics Corporation, Indianapolis, IN, USA). In brief, all the exons and exon-intron boundaries of FRMD7 and GPR143 genes were amplified using the standard polymerase chain reaction (PCR) buffer system with primers listed in Tables 1 and 2. PCR reactions were performed in a 10 μL volume, containing 1.5 mM MgCl2, 0.4 mM of each primer, 200 μM dNTPs, 1 U Taq DNA polymerase, and 10–20 ng template DNA. Amplification was performed with an initial denaturation for 3 min at 95°C, followed by 30 cycles of denaturation at 95°C for 1 min, annealing at 55°C for 1 min, extension at 72°C for 1 min, and a final extension at 72°C for 3 min. PCR products were purified using a PCR product purification kit (QIAquick; Qiagen, Valencia, CA, USA). The purified PCR products were sequenced using the BigDye Terminator Cycle Sequencing v3.1 kit (Applied Biosystems, Foster City, CA, USA). Briefly, about 10 ng of template DNA was added in each reaction using a temperature program which included 25 cycles of denaturation at 95°C for 30 s, annealing at 50°C for 15 s, and extension at 60°C for 4 min. All samples were analyzed in an ABI Prism 310 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The FRMD7 and GPR143 cDNA reference sequence with GenBank accession was used (National Center for Biotechnical Information, Bethesda, Md; available at: http://www.ncbi.nlm.nih.gov).
Table 1.
Primers used to amplify the exons of four-point-one, ezrin, radixin, moesin domain-containing 7

Table 2.
Primers used to amplify the exons of G protein-coupled receptor 143

Results
This sixth-generation pedigree showed a high possibility of an X-linked recessive inheritance pattern [Fig. 1]. All patients in this family had various reduced visual acuity with a similar pattern of nystagmus. The type of nystagmus in the family appeared to be jerk nystagmus that manifests as early as at birth. They all had reduced vision, amblyopia, and astigmatism at a different degree. There were normal pigmentation of skin and hair in all patients. Moreover, no retinal pathological changes, optic nerve lesions, any typical sign of OA1, and hypopigmentation of the iris and fundus were detected [Figs. 2–4 and Table 3].
Figure 2.
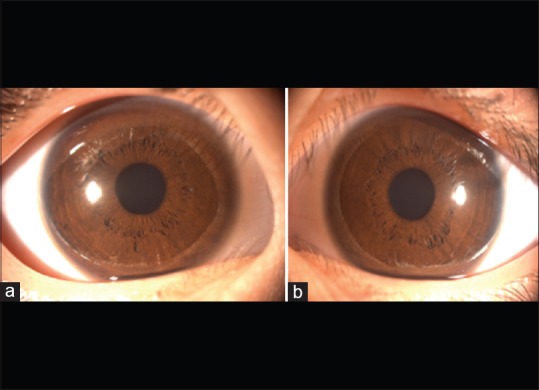
The iris of proband (VI1) in family, (a) right eye, (b) left eye
Figure 4.

Photographs of OCT from proband (VI1) in family, (a) right eye, (b) left eye
Table 3.
Summary of clinical features of some affected males and carriers

Figure 3.

Photographs of fundus from proband (VI1) in family, (a) right eye, (b) left eye
We sequenced 12 coding exons of FRMD7, 9 coding exons of GPR143, and the adjacent intronic regions of these two genes in the patients. After a complete analysis of the coding sequence of FRMD7 and GPR143, there was no mutation in FRMD7 and no PCR products for the DNA fragments spanning exon 3 to exon 9 of GPR143, suggesting a large deletion in this region [Fig. 5].
Figure 5.

The polymerase chain reaction results of GPR143 gene. M: marker; E1–E9: exon1–Exon9; C: Represents carrier member; P: Represents affected member
The mutation was detected in all patients but not found in other unaffected members or in the 100 normal controls. It was predicted to result in a premature stop codon emerged in exon 2, resulting in a truncated protein of only 120 amino acids.
Discussion
Bassi et al. cloned GPR143 gene for OA1 on chromosome Xp22.3.[15] Schiaffino et al. detected various mutations in GPR143 in one-third of X-linked ocular albinism.[10] Sometimes, OA1 can be ignored or misdiagnosed, the ocular disorders should be eliminated before the diagnosis of congenital motor nystagmus is made. X-linked OA1 is characterized by decreased ocular pigmentation, foveal hypoplasia, nystagmus, photodysphoria, and reduced visual acuity. All patients in this family had various reduced visual acuity with nystagmus but there were no retinal pathological changes, optic nerve lesions, or any typical sign of OA1.
We have characterized nystagmus in a sixth-generation Chinese family with a X-linked recessive inheritance pattern. No mutation was identified in the FRMD7 gene and a novel large deletion in exon 3 to exon 9 of the GPR143 gene. All the affected males were hemizygous for the mutation and female carriers were heterozygous for the mutation whereas other normal members of the family had no mutation. These results strongly suggested that the 3–9 exons deletion of GPR143 gene causes the disease in the family.
Until now, more than 100 mutations of GPR143 have been determined, including frameshift deletion and nonsense mutations. Most mutations were reported to cause ocular albinism, but some mutations were reported to cause CN without classical phenotype of ocular albinism.[12,13]
The human GPR143 gene consists of 9 exons which encode a 439-kDa protein of 404 amino acids with homology to seven transmembrane segments, a GPR. The GPR has been shown to participate in the most common signal transduction system at the plasma membrane. Thus, it suggests that GPR143 mediates signal transduction system and operates at the internal membranes in mammalian cells.[16] Giordano used siRNA inactivation of GPR143 and combined morphological and biochemical methods to investigate melanosomal ultrastructure, melanosomal protein localization, and expression in human pigmented melanocytic cells. The functional loss of GPR143 may lead to decreased pigmentation and causes formation of enlarged aberrant premelanosomes harboring disorganized fibrillar structures and displays proteins of mature melanosomes and lysosomes at their membrane.[17]
We revealed the 7-exons deletion of GPR143 in a Chinese pedigree. The presence of the deletion in all patients and its absence in unaffected members and other 100 unrelated controls indicate that the identified deletion causes CN. The deletion in GPR143 is predicted to result in a truncated protein of 120 amino acid residues, in which the C-terminus of GPR143 protein was deleted.
Conclusions
Our results expand the spectrum of GPR143 mutations causing CN and also confirm the role of GPR143 in the pathogenesis of CN.
Financial support and sponsorship
This study was supported by the National Natural Science Foundation (No. 81300789).
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
We are grateful to the patients and their family members for their cooperation in this study. This study was supported by the National Natural Science Foundation (No. 81250039, No. 81300789) and the Beijing Natural Science Foundation (No. 7102160) to L. J. W. The study was approved by the Ethics Committee of the Peking University Third Hospital and conformed to the Declaration of Helsinki.
References
- 1.Du W, Bu J, Dong J, Jia Y, Li J, Liang C, et al. A novel frame-shift mutation in FRMD7 causes X-linked idiopathic congenital nystagmus in a Chinese family. Mol Vis. 2011;17:2765–8. [PMC free article] [PubMed] [Google Scholar]
- 2.Hu J, Liang D, Xue J, Liu J, Wu L. A novel GPR143 splicing mutation in a Chinese family with X-linked congenital nystagmus. Mol Vis. 2011;17:715–22. [PMC free article] [PubMed] [Google Scholar]
- 3.Self JE, Ennis S, Collins A, Shawkat F, Harris CM, Mackey DA, et al. Fine mapping of the X-linked recessive congenital idiopathic nystagmus locus at Xq24-q26.3. Mol Vis. 2006;12:1211–6. [PubMed] [Google Scholar]
- 4.Zhang B, Liu Z, Zhao G, Xie X, Yin X, Hu Z, et al. Novel mutations of the FRMD7 gene in X-linked congenital motor nystagmus. Mol Vis. 2007;13:1674–9. [PubMed] [Google Scholar]
- 5.Li N, Wang L, Cui L, Zhang L, Dai S, Li H, et al. Five novel mutations of the FRMD7 gene in Chinese families with X-linked infantile nystagmus. Mol Vis. 2008;14:733–8. [PMC free article] [PubMed] [Google Scholar]
- 6.Fingert JH, Roos B, Eyestone ME, Pham JD, Mellot ML, Stone E. Novel intragenic FRMD7 deletion in a pedigree with congenital X-linked nystagmus. Ophthalmic Genet. 2010;31:77–80. doi: 10.3109/13816810903584989. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Pu J, Liu Z, Xu S, Jin F, Zhu L, et al. Identification of a novel FRMD7 splice variant and functional analysis of two FRMD7 transcripts during human NT2 cell differentiation. Mol Vis. 2011;17:2986–96. [PMC free article] [PubMed] [Google Scholar]
- 8.Radhakrishna U, Ratnamala U, Deutsch S, Bartoloni L, Kuracha MR, Singh R, et al. Novel homozygous, heterozygous and hemizygous FRMD7 gene mutations segregated in the same consanguineous family with congenital X-linked nystagmus. Eur J Hum Genet. 2012;20:1032–6. doi: 10.1038/ejhg.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He X, Gu F, Wang Z, Wang C, Tong Y, Wang Y, et al. A novel frameshift mutation in FRMD7 causing X-linked idiopathic congenital nystagmus. Genet Test. 2008;12:607–13. doi: 10.1089/gte.2008.0070. [DOI] [PubMed] [Google Scholar]
- 10.Schiaffino MV, Bassi MT, Galli L, Renieri A, Bruttini M, De Nigris F, et al. Analysis of the OA1 gene reveals mutations in only one-third of patients with X-linked ocular albinism. Hum Mol Genet. 1995;4:2319–25. doi: 10.1093/hmg/4.12.2319. [DOI] [PubMed] [Google Scholar]
- 11.Peng Y, Meng Y, Wang Z, Qin M, Li X, Dian Y, et al. A novel GPR143 duplication mutation in a Chinese family with X-linked congenital nystagmus. Mol Vis. 2009;15:810–4. [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou P, Wang Z, Zhang J, Hu L, Kong X. Identification of a novel GPR143 deletion in a Chinese family with X-linked congenital nystagmus. Mol Vis. 2008;14:1015–9. [PMC free article] [PubMed] [Google Scholar]
- 13.Liu JY, Ren X, Yang X, Guo T, Yao Q, Li L, et al. Identification of a novel GPR143 mutation in a large Chinese family with congenital nystagmus as the most prominent and consistent manifestation. J Hum Genet. 2007;52:565–70. doi: 10.1007/s10038-007-0152-3. [DOI] [PubMed] [Google Scholar]
- 14.Preising M, Op de Laak JP, Lorenz B. Deletion in the OA1 gene in a family with congenital X linked nystagmus. Br J Ophthalmol. 2001;85:1098–103. doi: 10.1136/bjo.85.9.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bassi MT, Schiaffino MV, Renieri A, De Nigris F, Galli L, Bruttini M, et al. Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat Genet. 1995;10:13–9. doi: 10.1038/ng0595-13. [DOI] [PubMed] [Google Scholar]
- 16.Schiaffino MV, d’Addio M, Alloni A, Baschirotto C, Valetti C, Cortese K, et al. Ocular albinism: Evidence for a defect in an intracellular signal transduction system. Nat Genet. 1999;23:108–12. doi: 10.1038/12715. [DOI] [PubMed] [Google Scholar]
- 17.Giordano F, Bonetti C, Surace EM, Marigo V, Raposo G. The ocular albinism type 1 (OA1) G-protein-coupled receptor functions with MART-1 at early stages of melanogenesis to control melanosome identity and composition. Hum Mol Genet. 2009;18:4530–45. doi: 10.1093/hmg/ddp415. [DOI] [PubMed] [Google Scholar]