

Abstract
Secreted Dickkopf (Dkk) proteins are major Wnt pathway modulators during organ development. Dkk1 has been widely studied and acts as a general Wnt inhibitor. However, the molecular function of other Dkks remains largely unknown. Here, we show that Dkk4 selectively inhibits a subset of Wnts, but is further inactivated by proteolytic cleavage. Meibomian gland (MG) formation is employed as a model where Dkk4 and its Wnt targets are expressed. Skin-specific expression of Dkk4 arrests MG growth at early germ phase, which is similar to that observed in Eda-ablated Tabby mice. Consistent with transient Dkk4 action, intact Dkk4 inhibits MG extension but the cleaved form progressively increases during MG development with a concomitant upswing in Wnt activity. Furthermore, both Dkk4 and its receptor (and Wnt co-receptor) Lrp6 are direct Eda targets during MG induction. In cell and organotypic cultures, Dkk4 inhibition is eliminated by elevation of Lrp6. Also, Lrp6 upregulation restores MG formation in Tabby mice. Thus, the dynamic state of Dkk4 itself and its interaction with Lrp6 modulates Wnt function during MG development, with a novel limitation of Dkk4 action by proteolytic cleavage.
KEY WORDS: Dkk4, Proteolytic cleavage, Meibomian gland, Wnt, Eda, Lrp6, Mouse
Summary: In the meibomian glands of the mouse eyelid, dynamic regulation of Dkk4 activity by proteolytic cleavage and interaction with Lrp6 modulates Wnt pathway activity and hence gland development.
INTRODUCTION
Wnt signaling is one of the fundamental mechanisms regulating embryonic development, adult homeostasis and disease progression (Clevers, 2006; Logan and Nusse, 2004). Canonical Wnt/β-catenin signaling is initiated when a Wnt ligand simultaneously complexes with the receptor Frizzled (Fzd) and co-receptor Lrp5/6, which regulates the amount and nuclear translocation of β-catenin and triggers gene expression (MacDonald et al., 2009). Wnt/β-catenin signaling is further regulated by a wide range of modulators. In contrast to agonistic Noggin, Norrin and R-spondin (Botchkarev et al., 1999; Kazanskaya et al., 2004; Xu et al., 2004), the Dickkopf proteins (Dkks) are considered to be antagonists. Thus far, studies have largely focused on Dkk1, which is regarded as a potent Wnt inhibitor in many cell types, blocking the Wnt/β-catenin signaling cascade (Bafico et al., 2001; Glinka et al., 1998; Mao et al., 2001; Semenov et al., 2001). Other Dkks are less extensively studied but have diverse molecular properties and functions (Cruciat and Niehrs, 2013). Among them, Dkk4 functionally acts like Dkk1 in ‘gain-of-function’ assays in Xenopus embryo development (Krupnik et al., 1999; Mao and Niehrs, 2003), but data from our group showed that, in mice, Dkk4 has much less potency than Dkk1 during hair development (Cui et al., 2010). Despite these findings, the basic molecular properties and detailed function of Dkk members remain largely unknown. Interestingly, Dkks are likely to be modified by post-translational modification including glycosylation and possibly by proteolytic processing (Niehrs, 2006). But whether the Dkk function is regulated by these modifications is unexplored.
The Wnt/β-catenin pathway has a central role in early skin development (Driskell and Watt, 2015; Lien and Fuchs, 2014) and skin appendage initiation (Fuchs, 2007; Lim and Nusse, 2013; Widelitz, 2008). Powerful Wnt-inhibitory effects of Dkk1 include blockage of feather bud formation in chicken (Chang et al., 2004) and of skin appendage germ induction in mouse (Andl et al., 2002). Therefore, initially, Dkk1 seemed to be the most likely candidate for involvement in any skin appendage formation. However, our previous findings also implicated Dkk4 in modulating hair follicle subtype formation in mice, and possibly in regulating the maturation of the eyelid skin appendage meibomian glands (MGs) (Cui et al., 2010), which produce oils to prevent excessively rapid evaporation of tears. Based on these findings, we hypothesized that (1) Dkk4 may have a ‘unique’ function, and limited activity of Dkk4 may result from some post-translational modification; and (2) MG formation may be a novel mouse model to study Dkk4 function and skin appendage development.
Here, we show that Dkk4 is expressed in nascent MGs, and specifically inhibits some Wnts when it binds to Lrp6. It thereby limits the extent of MG germ development, but is in turn inactivated by proteolytic cleavage during later development. The interaction and relative levels of Lrp6 and Dkk4 are thus crucial, and Lrp6 is a key mediator linking Dkk4 and Eda action to modulate the Wnt pathway during MG development.
RESULTS
Dkk4, unlike Dkk1, selectively inhibits a narrow group of Wnt ligands
To compare Dkk4 with Dkk1 for functional differences, super 8×Topflash assays (Veeman et al., 2003) were used to measure Wnt/β-catenin activity in mouse Kera308 cells. We used Wnt3a conditioned medium (CM) to check for inhibitory action by Dkk1 or Dkk4. Relative to controls, Wnt3a CM induced a 46.5-fold Topflash activity (Fig. 1A), an augmentation that was precluded by prior transfection of the cells with a vector construct expressing Dkk1. Next, we asked whether Dkk1 could inhibit β-catenin activity induced by other canonical Wnt. We added CM containing each of the other known canonical Wnts (Wnt1, Wnt2, Wnt3, Wnt8a, Wnt8b, Wnt10a and Wnt10b) in the same conditions for Topflash measurements. Dkk1 robustly inhibited β-catenin activity induced by each of these Wnts, although with differential efficacy (Fig. 1A). In sharp contrast to Dkk1, Dkk4 expression surprisingly showed no inhibition of β-catenin activity induced by the frequently used Wnt3a, either as Wnt3a CM (Fig. 1A) or recombinant Wnt3a (Fig. S1). However, it showed highly significant inhibition of Topflash activity specifically induced by Wnt1, Wnt10a or Wnt10b (Fig. 1A) – although its inhibitory efficacy was generally not more potent than Dkk1.
Fig. 1.

Dkk4 inhibits a subset of Wnt proteins and is sensitive to proteolytic cleavage. (A) Differential effect of Dkk1 and Dkk4 on canonical Wnt ligands. 8×Top/Fop flash assays measured Wnt/β-catenin activity. Kera308 cells were transfected with Topflash or Fopflash vector together with an empty vector, Dkk1-, or Dkk4-expressing vector. After stimulation with conditioned medium (CM) containing various Wnts for 24 h, luciferase activity of Topflash was normalized with Fopflash in all experiments. Transfection and luciferase assays were performed in triplicate. Error bars, mean±s.e.m. **P<0.01, Student's t-test. (B) Intact and cleaved forms of Dkk4 proteins. HEK293 or Kera308 cells were transiently transfected with expression vectors for Flag-tagged Dkk1 or Dkk4. The CM or cell lysate was used to detect Dkk1 or Dkk4 by western blotting with anti-Flag antibody. (C) Dkk4-Flag protein was purified from CM of HEK293 cells expressing Dkk4-Flag using anti-Flag affinity beads. Two protein bands ∼28 kDa and one lower band at 18 kDa are shown (left). Mass spectrometric analysis revealed 28 kDa bands as Dkk4 protein. Edman N-terminal sequencing of the smaller 18 kDa band identified the cleavage site at Asp88 (D88), indicated by red arrow (right). (D) Schematic structure of Dkk4 protein with signal peptide (SP), cysteine-rich domains (Cys-1 and Cys-2) and cleavage site of Dkk4 at D88 (red asterisk). (E) Plasmids expressing WT Dkk4, CL-Dkk4 and PM-Dkk4 (D88A) were transfected into Kera308 cells for 24 h culture. CM from each culture was collected for immunoblot analysis. Actin was used as a loading control.
Dkk4 is sensitive to proteolytic cleavage
The molecular state of Dkk1 and Dkk4 was examined to study their differential functions. Plasmids expressing Dkk1-Flag or Dkk4-Flag were transfected into HEK293 or Kera308 cells, respectively. After 48 h, culture medium and cell lysate from each sample were analyzed by western blotting with anti-Flag antibody. Both secreted Dkk1 in culture medium and the non-secreted form in cell lysates showed the expected molecular mass of ∼37 kDa in both cell types. In contrast, secreted Dkk4 protein showed two clear bands of 28 kDa and 18 kDa, suggesting that Dkk4 was subject to cleavage (Fig. 1B, top panel). Furthermore, non-secreted Dkk4 protein showed only the 28 kDa species, suggesting that cleavage occurred after protein secretion (Fig. 1B, middle panel).
To further detail the cleavage process, we purified Dkk4-Flag protein from Dkk4 CM using anti-Flag affinity beads. Silver-stained polyacrylamide gels showed three candidate Dkk4 protein species, a doublet band of ∼28 kDa and a third band of ∼18 kDa (Fig. 1C). A mass spectroscopic analysis of the upper two bands identified them as Dkk4, with or without its signal peptide (SP). Further analyses by Edman degradation of the smaller 18 kDa band showed that its N-terminal sequence was DVCTA, corresponding to the sequence at the C-terminal end of the Cys-1 domain of Dkk4. Fig. 1D illustrates the domains and inferred cleavage site of Dkk4 at Asp88.
We next cloned three Dkk4 isoforms and expressed them in Kera308 cells, including the N-terminal portion, the C-terminal portion of Dkk4 (CL-Dkk4) and an entire Dkk4 containing a D88A point mutation (PM-Dkk4, mutated to change the putative recognition sequence at the cleavage site). Protein bands of the expected size were present for CL-Dkk4 and PM-Dkk4 (Fig. 1E). However, we did not see a 10 kDa N-terminal fragment of Dkk4, possibly because it was degraded further (data not shown). Taken together, these results indicate that Dkk4, unlike Dkk1, has a selective function on a subset of Wnts and is sensitive to N-terminal proteolytic cleavage at residue 88.
Proteolytic cleavage of Dkk4 inactivates its Wnt-inhibitory capacity
To clarify whether the proteolytic cleavage of Dkk4 affected its capacity to inhibit Wnt activity, three Dkk4 isoforms were cloned into an alkaline phosphatase (AP)-tagged expression vector and transfected for CM collection. Results from AP cell surface binding assays showed that CM from both WT-Dkk4-AP and PM-Dkk4-AP had the same effect as CM from a positive control of Dkk1-AP and strongly attached to Lrp6-expressing cells, whereas CL-Dkk4-AP CM had no remaining capacity to bind (Fig. 2A). To confirm these results, we examined the receptor-binding capability of WT-Dkk4 CM containing both FL- and CL-Dkk4 proteins or CL-Dkk4 CM. Co-immunoprecipitation (co-IP) indicated that Flag antibody recognized both FL- and CL-Dkk4 and pulled down endogenous Lrp6; but anti-Lrp6 antibody only precipitated FL-Dkk4, not CL-Dkk4 (Fig. 2B, top panels). Consistent with these results, when we used CL-Dkk4 CM in Co-IP, neither CL-Dkk4 nor Lrp6 could co-precipitate the other (Fig. 2B, bottom panels). Thus, cleaved Dkk4 loses its capacity to interact with Lrp6 and putatively would no longer be able to inhibit Wnt action.
Fig. 2.

Cleaved Dkk4 does not bind to Lrp6 and loses Wnt-inhibitory capacity. (A) Cell surface binding assays for different Dkk4 isoforms. Dkk1-AP is a positive control. HEK293 cells were transfected with Lrp6-expressing vector and cultured for 24 h, incubated with CM from each Dkk isoform for another 2 h and fixed for AP staining. Scale bar: 50 µm. (B) Lrp6 binds full-length (FL) Dkk4 but not cleaved (CL) Dkk4. Kera308 cells incubated with WT-Dkk4 (top) or CL-Dkk4 CM (bottom) for 2 h were lysed for co-IP assays. Anti-Flag antibody and anti-Lrp6 antibody were used for co-IP experiments. (C) CL-Dkk4 loses Wnt-inhibitory function. HEK293 cells transfected with TopFlash or FopFlash vector were pre-incubated with WT-Dkk4, CL-Dkk4 or PM-Dkk4 CM for 4 h, followed by Wnt10b CM for a further 24 h. Luciferase activity was measured and Top/Fop ratio calculated. Error bars, mean±s.e.m. of triplicates. **P<0.01, ***P<0.001, Student's t-test. (D) CL-Dkk4 does not reduce the protein level of active β-catenin. Kera308 cells were transfected with empty vector, Dkk1, WT-Dkk4, CL-Dkk4 or PM-Dkk4 and cultured for 24 h, followed by application of CM containing each Wnt (as indicated) for a further 24 h. Cell lysates were collected and western blotting assays carried out. The histogram (below) shows calculated density of each protein band. Average protein level of active β-catenin in naïve cells is normalized to 1 and relative density of protein bands in other lanes is reported.
To compare the inhibitory potency of the Dkk4 species directly, we measured the activity of CL-Dkk4 and PM-Dkk4 against Wnt10b, the species most sensitive to Dkk4 (Fig. 1A). Dkk4 CM blocked 53.5% of the Topflash activity induced by Wnt10b (Fig. 2C). For comparison, the generally more potent Dkk1 CM blocked 93% of Topflash activity in the same assay. Consistent with the binding activities of Dkk4 isoforms to Lrp6, CL-Dkk4 failed to inhibit Wnt10b-induced Topflash activity, and PM-Dkk4 restored this capability, with a higher efficacy compared with WT-Dkk4 (Fig. 2C).
To confirm the effect of Dkk4 isoforms on β-catenin activity induced by Wnt ligands, we examined the protein level of active (non-phosphorylated) β-catenin using immunoblotting assays. We tested naïve Kera308 cells or cells transfected with either empty vector or vectors expressing Dkk1, Dkk4, CL-Dkk4 or PM-Dkk4. In keeping with the results of the Topflash assays, CM of Wnt3a, Wnt1, Wnt10a and Wnt10b all induced robust active β-catenin levels compared with naïve cells. Consistently, Dkk1 reduced active β-catenin levels stimulated by all four Wnts, whereas Dkk4 again effectively inhibited the other three Wnts but not Wnt3a. Importantly, CL-Dkk4 again had no capacity to inhibit Wnt1, Wnt10a or Wnt10b; and consistent with the other assays, PM-Dkk4 was able to rescue the inhibitory effect for the other three Wnts but not for Wnt3a (Fig. 2D).
In sum, the proteolytic cleavage rather functions to inactivate Dkk4 activity, while the selective effect of Dkk4 on Wnts precedes its cleavage. Thus, Dkk4, unlike Dkk1, transiently inhibits a restricted group of Wnts and is then disarmed by N-terminal protein cleavage.
Dkk4 and its Wnt targets are expressed at early germ phase of MG induction
To assess the relevance of Dkk4 metabolism to a developmental pathway, we turned to meibomian gland (MG) formation as a likely model. Our previous study showed that skin-specific expression of Dkk4 blocked mature MG formation in adult mice (Cui et al., 2010), suggesting that Dkk4 may regulate MG early development.
First, we analyzed the early morphological progression of MGs in WT mice at embryonic day (E)14.5, E15.5, E16.5 and E17.5. Hematoxylin and Eosin (H&E) staining indicated that no MG germs had appeared by E14.5. The first sign of germ formation was observed at E15.5, when a cluster of basal cells protruded toward the mesenchyme, at a time when eyelids were still open. Further growth of MG germs, with the clustering cells at the dermal condensation (DC), was detected at E16.5 and E17.5 (Fig. 3A, top row). To confirm the timing of germ formation, we examined the expression of Lef1 (a marker of skin appendage germ cells). Lef1-positive cells started to accumulate at E15.5 from basal cells that also stained for the keratin 14 (K14) marker. These cells then grew further into the mesenchyme, with multiple layers apparent at E17.5 (Fig. 3A, middle and bottom rows). Thus, germ induction of MGs occurred by E15.5, even before eyelid closure.
Fig. 3.

Endogenous Dkk4 localizes in MG germs and epidermal expression of Dkk4 arrests the growth of MG germs. (A) Time course of early progression of MG development in WT mice. H&E staining and K14 (green)/Lef1 (red) IHC staining of WT eyelids. Bottom panels indicate amplified images of MG germs (arrows) and dermal condensation (DC) (arrowheads). (B) Expression of Dkk4, Dkk1, Wnt1, Wnt10a and Wnt10b in WT MG pre-germs at E15.5. Sense probe of Dkk4 is used as negative control. (C) K14-driven Dkk4 expression inhibits MG growth at E18.5. H&E staining shows no MG germ formed in Dkk4Tg or Tabby mice. Lef1 staining indicates MG germ growth is impaired in Dkk4Tg or Tabby (Ta) compared with WT. In Dkk4Tg and Tabby, amplified images show MG germs arrested at pre-germ stage. Dotted line separates dorsal and ventral part of skin. Arrows in A-C, MG germs; arrowheads, DC; HF, hair follicle. Scale bars: 50 µm.
We next tested whether Dkk4 and its Wnt targets were normally expressed at the induction stage of MG development, by in situ hybridization. Indeed, Dkk4 mRNA was mainly localized in MG germs at E15.5, but Dkk1 expression was not detected in MGs. Of the three canonical Wnts specifically inhibited by Dkk4 (Wnt1, Wnt10a and Wnt10b; Fig. 1A), only Wnt10b, was found restricted to MG germs. By contrast, Wnt10a was widely expressed throughout the epidermis of eyelids, although its expression was slightly higher in MG germs; Wnt1 was not observed at all in eyelids at this stage (Fig. 3B).
Expression of Dkk4 transgene in skin arrests further growth of MG germs but does not affect initial MG induction
The defects in MG formation and resultant blindness in adult Dkk4Tg mice were similar to defects seen in Tabby (Eda-deficient) mice (Cui et al., 2010). Eda encodes the eponymous Ectodysplasin (Eda), which helps to initiate cascade induction of many skin appendages including MGs (Cui et al., 2005; Mikkola, 2008). To address whether Dkk4 affected MG formation at the induction stage, we first examined Dkk4 mRNA expression in Dkk4Tg and Tabby at E15.5 by in situ hybridization. When expressed from the skin-specific K14 promoter in transgenic mice, Dkk4 mRNA was observed, as expected, throughout the epidermal layer of eyelids. Notably, Dkk4 expression was remarkably reduced in MG germs in Tabby mice compared with WT (Fig. S2, left panel), which is reminiscent of findings in hair follicle development (Fliniaux et al., 2008). Interestingly, in hair follicle development, both Dkk4 itself and the Wnt pathway might be regulated somehow by Eda or Wnt signaling (Bazzi et al., 2007; Zhang et al., 2009). Regulation of crosstalk between the Wnt pathway and Eda signaling might thus underlie the similar arrest of MG development in Tabby and Dkk4Tg mice.
We next examined MG germ morphology in WT, Dkk4Tg and Tabby mice at E15.5. In both Dkk4Tg and Tabby mice, the initiation of MG germs remained normal compared with that in WT, indicated by Lef1-positive cell clusters (Fig. S2, right panel). To identify when MG development started to be affected, we observed MG germ morphology at E16.5 and E17.5 – subsequent to MG induction at E15.5 – in WT, Dkk4 and Tabby mice. In both Dkk4Tg and Tabby mice, the growth of MG germs was arrested at the stage of a single layer of Lef1-positive germ cells compared with WT (Fig. S3). Furthermore, by E18.5, when MG anlage in WT had invaded deep into the dermis, with a clear DC structure (Fig. 3C, left panel), MG germs in both Dkk4Tg and Tabby were still blocked with a single layer of Lef1-positive cells, without any appearance of DC formation (Fig. 3C, middle and right panels).
Thus, a Dkk4 transgene, like ablation of Eda, did not affect initial induction of MG germs, but abrogated the further growth of MGs; the phenotype mimicked that of Tabby mice, consistent with compromised or counteracted action of Eda in both cases.
Increased Dkk4 cleavage after MG induction releases Wnt/β-catenin activity
Levels of endogenous Dkk4 and its cleavage products from E15.5 to E18.5 eyelids were also consistent with a ‘jack-in-the-box’ status of Dkk4 during development. Dkk4 was mainly present in a full-length form at E15.5 in both WT and Dkk4Tg mice, while cleavage products appeared increasingly at later stages (Fig. 4A). Neither FL-Dkk4 nor CL-Dkk4 protein was detected in Tabby mice (Fig. 4A), consistent with the lack of Dkk4 mRNA in MGs of Tabby mice (Fig. S2). Quantification indicated that the CL-Dkk4 level increased 6.3-fold in WT and 8.6-fold in Dkk4Tg mice, at E18.5 compared with that at E15.5. Over the same period, FL-Dkk4 decreased 4.8-fold in WT and 16.1-fold in Dkk4Tg (Fig. 4B,C). Meanwhile, active β-catenin was continuously upregulated from E15.5 to E18.5 in WT with a 5.4-fold increase, but remained at a lower level in both Dkk4Tg and Tabby mice at all four time points (Fig. 4A,D).
Fig. 4.

Dynamic modulation of Dkk4 cleavage regulates Wnt action and affects MG formation. (A) Protein levels of FL-Dkk4, CL-Dkk4 and active β-catenin during MG development. Eyelids from WT, Dkk4Tg and Tabby (Ta) mice were separated at different stages and lysed for immunoblotting analysis. Antibodies against Dkk4 or active β-catenin were used. Actin was detected as a loading control. Experiments were performed three times. Intensity of each protein band in immunoblots was measured. (B-D) Level of each protein in WT at E15.5 was normalized to 1. Average protein level from WT, Dkk4Tg or Tabby at each time point is shown. (E) Effect of each Dkk4 isoform on MG growth in WT MG cultures. Eyelids from WT at E15.5 were collected and cultured with application of indicated CM for 48 h, and fixed for IHC staining of Lef1 (red) and K14 (green). (F) Length of MGs. (G) MG formation is augmented by the Dkk inhibitors WAY-262611 and IIIC3. Eyelids were separated from WT or Dkk4Tg mice and cultured in medium with 2 µM DMSO (control), 2 µM WAY-262611 or 10 µM IIIC3 for 48 h. Cultured tissues were fixed and double-stained with Lef1 and K14 antibodies. (H) Length of MGs from WT or Dkk4Tg. Error bars are mean±s.e.m. of at least 15 MGs. White arrows indicate MGs. Scale bars: 50 µm.
MG formation is inhibited by uncleaved Dkk4 and augmented by chemical inhibitors of the Dkk4-Lrp6 complex
As an unequivocal test of Dkk4 action during MG development, we evaluated the effects of uncleaved and cleaved Dkk4 directly on MG growth in organotypic eyelid cultures with added CM from control, WT-Dkk4, CL-Dkk4 or PM-Dkk4. Preparations incubated with control CM or CL-Dkk4 CM exhibited multiple layers of MG germ cells and DC structures with Lef1-positive cells. By contrast, eyelids cultured in the presence of added WT-Dkk4 CM or PM-Dkk4 CM showed only a single layer of Lef1-positive cells, with no apparent DC formation (Fig. 4E). Quantifying the length of MGs, we found that MGs cultured in control or in CL-Dkk4 CM showed an average length of 6.2 µm, whereas no growth of MG germs beyond the initial length of 0.4 µm was seen when WT-Dkk4 CM or PM-Dkk4 CM was applied (Fig. 4F). Thus, only intact Dkk4 inhibited MG formation during the early induction phase.
To assess the effect of ‘loss of function’ of Dkk4, we utilized Dkk inhibitors WAY-262611 and IIIC3, which had previously been shown to inhibit Dkk1 or Dkk2 (Li et al., 2012; Pelletier et al., 2009). Either 2 µM WAY-262611 or 10 µM IIIC3 fully inhibited the binding of Dkk4-AP to HEK293 cells expressing Lrp6, as visualized by AP staining (Fig. S4A). Furthermore, in Topflash assays, both compounds reversed WT-Dkk4 CM-mediated inhibition of Topflash activity (Fig. S4B). We applied the inhibitors to organotypic WT eyelid cultures, and each induced enlarged MG germs, with WAY-262611 displaying a stronger effect than IIIC3 (Fig. 4G, top row). Quantifying MG length, WAY-262611 and IIIC3 induced much longer MG structures, with an average length of 41 and 26 µm, respectively – that is, 6.6- and 4-fold greater than in control cultures (Fig. 4H). Strikingly, both compounds also restored germ growth in Dkk4Tg eyelid cultures, where Lef1-positive germ cells patently started to grow into the dermis (Fig. 4G, bottom row). The average length of MGs was 6.3 µm or 2.7 µm (for WAY-262611 or IIIC3, respectively) compared with only 0.4 µm in the Dkk4 transgenic eyelid culture itself (Fig. 4H).
The organotypic culture assays thus confirmed the inhibitory function of uncleaved Dkk4 for MG formation, consistent with the notion that proteolytic cleavage or inhibition of Dkk4 eliminates its Wnt-blocking action and facilitates further MG development.
Dkk4 and other Wnt pathway components are induced by Eda during early MG development
To elucidate how Dkk4 might block MG formation with a phenotype similar to Tabby, we carried out transcriptome profiling from wild-type and Tabby embryo eyelids at E14.5 and E15.5, representing pre-induction and early induction stages. Notably, mRNA expression of several genes in the Wnt pathway, including Lrp6, Dkk4, Wnt10b and Fzd10, was significantly downregulated in Tabby (Table 1). Other genes in the Wnt signaling pathway showed no appreciable change in expression (Table S1). Dkk4, Wnt10b and Fzd10, which had previously been suggested as potential targets of Eda activation in skin appendages (Cui et al., 2014; Fliniaux et al., 2008; Voutilainen et al., 2012; Zhang et al., 2009), were comparably downregulated in Tabby MGs. Of particular interest, the Dkk4 receptor Lrp6, newly identified here as an Eda target, was reduced by 44% at E14.5 and 32% at E15.5 in Tabby eyelids. In contrast, expression of a well characterized Eda target, Shh, was reduced by 31% at E14.5 and 54% at E15.5 in Tabby compared with WT (Table 1). qRT-PCR assays confirmed that Lrp6, Dkk4 and Wnt10b, like Shh, were all reduced in Tabby at both E14.5 and E15.5, with Fzd10 expression also significantly decreased at E15.5 (Fig. 5A).
Table 1.
Eda downstream targets in Wnt pathway identified at early MG developmental stages
Fig. 5.

Both Dkk4 and Lrp6 are direct targets of Eda signaling during MG induction. (A) qRT-PCR assays show downregulation of Eda, Shh, Dkk4, Lrp6, Fzd10 and Wnt10b in Tabby eyelids. ***P≤0.001, **P≤0.01 by Student's t-test. Data are mean±s.e.m. for triplicate samples. (B) Luciferase reporter assays of Lrp6 WT or mutant promoter activation. Luciferase reporter assays were performed in 293 cells to measure the activity of Lrp6 WT or mutant promoter with or without recombinant Eda-A1 application. Luciferase activity from reporters of empty luciferase vector are normalized to 1. Each data point represents the mean±s.e.m. for triplicate samples. ***P≤0.001 by Student's t-test. (C) Antibody against NF-κB1 pulls down DNA fragments of Lrp6 or Dkk4 promoters containing NF-κB binding sites. Chromatin DNA input in first lane, negative controls in second (beads only) and IgG antibody in third lane. Lrp6_1 and Lrp6_2 are DNA sequences containing the first and second NF-κB binding sites in the Lrp6 promoter, Lrp6_c is the coding region of Lrp6 lacking the NF-κB binding site. (D) Lrp6 expression at E15.5 and E16.5 was reduced in Tabby. IHC staining indicates Lrp6 in red and K14 in green in WT and Tabby. Arrows indicate MG germs.
Lrp6 transcription is directly activated by Eda–Edar–NF-κB signaling
To further test whether Lrp6 was a direct transcriptional target of Eda-Edar signaling through its known NF-κB mediator, we first analyzed the promoter region of Lrp6 for potential NF-κB responsive elements. Two putative conserved NF-κB binding sites, 159 and 915 base pair upstream of the ATG codon, were found. We then cloned a 1.4 kb region upstream of Lrp6 into a luciferase reporter vector. As a negative control, the same DNA fragment was also cloned with mutations in both putative NF-κB binding sites. Previous studies suggested that the Edar itself can activate NF-κB (Botchkarev and Fessing, 2005; Kumar et al., 2001). Luciferase assays in cultured Kera-308 cells revealed that transfection of an Edar-expressing plasmid led to a 10.6-fold induction of the Lrp6 promoter reporter construct compared with a plasmid with no insert (Fig. 5B). In direct comparisons, transfection with Edar had no effect on luciferase activity driven by the mutated construct (Fig. 5B). Further confirmation of the activity of Eda signaling in the transcription of Lrp6 was provided by treating Edar-expressing cells with recombinant Eda, which led to an increase in luciferase activity from the wild-type construct but not the mutated version (Fig. 5B).
Interaction between Lrp6 promoter elements and NF-κB was directly confirmed by chromatin immunoprecipitation (ChIP) assays. NF-κB1 antibody pulled down both DNA binding fragments in the Lrp6 promoter (Lrp6_1 and Lrp6_2) and in the Dkk4 promoter, but not the DNA fragments in the Lrp6 coding region (Lrp6_c) (negative control) (Fig. 5C). Consistently, IHC showed the endogenous expression of Lrp6 in WT MG germs and its sharp reduction in Tabby mice at both E15.5 and E16.5 (Fig. 5D).
Thus, Dkk4 and its receptor Lrp6 are both direct transcriptional targets of Eda signaling, with their expression turned on at the induction stage of MG development. The results confirm that Dkk4 exerts Wnt-inhibitory function though Lrp6, which can also be induced by Eda for later modulation of Wnt action in MG development.
Elevated Lrp6 levels augment Wnt activity correlated with Dkk4 cleavage in cell culture
To examine Lrp6 mediation of dynamic Dkk4-Wnt activity, we re-constructed Dkk4-Lrp6-Wnt signaling in cell culture. First, we generated a HEK293 cell line stably expressing Dkk4 protein, and then transfected them with Lrp6 CRISPR activation (Lrp6-AC) plasmids followed by stimulation with recombinant Wnt10b (Fig. 6A). As a transcriptional induction method, the transfected Lrp6-AC plasmids indeed induced Lrp6 upregulation in time-course cultures (Fig. S5A).
Fig. 6.

Elevated Lrp6 reverses Dkk4-mediated Wnt inhibition in cell culture. (A) Schematic of reconstructing Dkk4-Lrp6-Wnt10b signaling in 293 cells. (B) Protein levels of Dkk4-Flag, Lrp6 and β-catenin in cell cultures. Cultured medium or cell lysates at each time point were collected for immunoblotting analysis. Experiments were performed three times. Actin was detected as a loading control. (C-F) Intensity of protein bands in immunoblots. Level of each protein in 293 cells at 24 h was normalized to 1. Average protein level at each time point was calculated. Error bars are mean±s.e.m. **P≤0.01 by Student's t-test.
When cultured for up to 72 h, comparable to the findings in Dkk4Tg mice, Dkk4-expressing cells also exhibited progressive cleavage (Fig. 6B,C). In this system, after transfection with Lrp6-AC plasmids for 24 h, Lrp6 protein levels increased around 3-fold in both WT and Dkk4-expressing cells (Fig. 6B,D). As expected, active β-catenin levels were upregulated 3.2-fold in Lrp6-AC-transfected WT cells. By comparison, in Dkk4-expressing cells, active β-catenin remained at a lower level of ∼0.4-fold that in non-transfected control 24 h cultures, where most Dkk4 protein remained in the uncleaved form, but was prominently increased in 48 h and 72 h cultures, when cleaved Dkk4 gradually rose. In contrast to active β-catenin, total β-catenin levels did not obviously change (Fig. 6B,E,F). Thus, upregulation of Lrp6 reverses Dkk4-induced Wnt inhibition only at later stages when most Dkk4 has been inactively cleaved, suggesting the intrinsic balance of Lrp6 level and Dkk4 activity is essential for Wnt signaling regulation in vivo.
Lrp6 promotes MG growth and rescues the MG defect in both Dkk4Tg and Tabby mice
To clarify the function of Wnt co-receptor Lrp6 during MG development, we used a lentivirus transfection system to manipulate Lrp6 activity by introducing shRNA to lower its level or Lrp6-AC to increase its level. Knockdown and activation efficacy of Lrp6 shRNA and Lrp6-AC were verified in kera308 cell culture (Fig. S5B,C). We then carried out transfections in cultured eyelids from E15.5 WT mice and examined MG morphology thereafter. IHC images showed that after incubation with both shRNA and control lentiviral particles, most epidermal and some dermal cells were transfected as evidenced by GFP expression. In contrast to transduction with control particles, Lrp6 shRNA transfection arrested MG growth, with a single layer of pre-germ morphology maintained (Fig. 7A, left and middle panels). This result is similar to that observed in Dkk4Tg mice. Furthermore, Lrp6-AC lentiviral particles added to parallel cultures promoted MG growth, as evidenced by larger germ structures invading the dermis (Fig. 7A, right panels). The effects were significant: MGs transduced with control virus showed a mean length of 5.6 µm, whereas the length was only 0.6 µm when Lrp6 shRNA was transfected, and increased to 8.7 µm upon Lrp6-AC lentivirus application (Fig. 7B). We conclude that Lrp6 promotes MG germ growth during the early developmental phase.
Fig. 7.

Lrp6 promotes MG germ growth and its activation rescues the MG defect in both Dkk4Tg and Tabby mice. (A) Effects of Lrp6 shRNA and Lrp6-AC on MG growth in WT eyelid cultures. Eyelids from WT at E15.5 were cultured with application of control, Lrp6 shRNA and Lrp6-AC lentiviral particles for 48 h. (B) Length of MGs. (C) Rescue effect of Lrp6-AC for MG formation defect. Eyelids were separated from Dkk4Tg or Tabby mice and cultured in medium with LentiCon, Dkk4 RNAi or Lrp6-AC lentiviral particles for 48 h. Cultured tissues were fixed and stained with GFP antibody. (D) Length of MGs from Dkk4Tg or Tabby. Error bars are mean±s.e.m. from at least 15 MGs. ***P≤0.001, by Student's t-test for B and D. MG germs are indicated by dotted lines. Scale bars: 20 µm.
To test the role of Lrp6 as a ‘link’ between Dkk4 and Eda action, we asked whether Lrp6 induction could rescue the MG defect in Dkk4Tg or Tabby mice. Eyelids from Dkk4Tg or Tabby embryos were cultured in the presence of control or Lrp6-AC lentiviral particles. Consistent with the data in Fig. 3, eyelids from Dkk4Tg or Tabby cultured with control virus showed only a single layer of MG pre-germs, with an average length of 0.4 µm. By contrast, Lrp6-AC restored MG germ growth to an average length of 5.8 µm in Dkk4Tg eyelid cultures and to 4.8 µm in Tabby eyelid culture (Fig. 7C,D). Unlike the potency of Lrp6, knockdown of Dkk4, which remains at a very low level in Tabby (Fig. 4A,D), did not rescue MG germ growth in Tabby (Fig. 7C,D). The similar MG defect was thus probably due to Lrp6 inhibition through Dkk4 binding in Dkk4Tg or blockage of Eda-mediated Lrp6 expression in the Eda-deficient Tabby mice.
DISCUSSION
Differential molecular function of Dkk1 and Dkk4
Given that Wnts have well-established fundamental roles in embryonic development, it is not surprising that Wnt inhibitor Dkks can participate in the control of formation of bone, limb, skin and eye (Cruciat and Niehrs, 2013). Dkk1 has been studied most extensively, primarily because of its clear and strong inhibition of Wnt action. Our cell-based results indeed showed promiscuous Dkk1 inhibition of all canonical Wnts (Fig. 1A). We found, however, that the effects of Dkk4 were limited to some Wnts (including Wnt1, Wnt10a and Wnt10b, but not the well-known canonical Wnt3a). The more selective inhibition of Wnts by Dkk4 provides much more nuanced regulatory possibilities. Also, selectivity can be augmented by the somewhat weaker and possibly time-dependent action of Dkk4 provided by the subsequent inactivating cleavage described here, which can further explain the ‘weaker’ function of Dkk4 compared with Dkk1. The possibility that Dkk function is modulated by proteolytic processing, had not, however, been explored. Our cell-based assays and the animal model showed that mouse Dkk4, unlike Dkk1, can indeed be cleaved. Importantly, the cleavage at Asp88 abolished the ability of Dkk4 to bind Lrp6, thus leading to loss of its Wnt-inhibitory activity. Notably, the sequence at the cleavage site around Asp88 is highly conserved in vertebrate Dkk4, but absent in Dkk1 and other Dkks (data not shown). The protease responsible for the cleavage remains to be identified. Interestingly, a previous study showed that, in their cell-culture system, human Dkk4 possibly has multiple cleavage sites, among those only one around Ser134 has been described (Krupnik et al., 1999). Thus, whether different homologs of Dkk4 have divergent properties of proteolytic cleavage and subsequent functional regulation remain open questions.
The results indicate features of the differential activity of Dkk1 and Dkk4 on a variety of Wnts. The diversity of binding to the co-receptors Lrp5 and Lrp6 may be correlated with the considerable sequence divergence of Dkks. All of them include four homologous repeats, termed E1 to E4 from N- to C-terminus. But the first two repeats, Lrp6 E1 and E2, bind to Wnt variants, including Wnt1, Wnt2, Wnt2b, Wnt6, Wnt8a, Wnt9a, Wnt9b and Wnt10b (Gong et al., 2010), whereas Lrp6 E3 and E4 bind other Wnts, including Wnt3a (Bourhis et al., 2010). Recent crystal structure studies revealed that Dkk1 binds to both regions of Lrp6: its C-terminal domain binds to Lrp6 E3 and E4, whereas its N-terminal portion binds to Lrp6 E1 and E2. Binding at two sites may help to explain the greater antagonistic potency of Dkk1 against various Wnt ligands (Ahn et al., 2011; Cheng et al., 2011) compared with Dkk4, which is much smaller and may bind less strongly or even fail to bind to the Wnt3a ligand portion of Lrp6.
Involvement of Dkk4 in skin appendage development
In the development of hair follicles, another skin appendage affected by Eda, previous studies showed that endogenous Dkk1 was mainly expressed in the mesenchyme surrounding developing hair follicles (Andl et al., 2002), and might conceivably cooperate with Dkk4 to regulate hair follicle density (Sick et al., 2006). By contrast, we did not detect Dkk1 expression at early MG initiation stages, whereas Dkk4 was strongly expressed in epithelial cells around MG early germs (Fig. 3B).
To study the function of Dkk4 and the consequences of Dkk4 cleavage, we took advantage of MG formation as an animal model. It provides (1) an ‘all or nothing’ phenotype of MG organ formation when comparing WT with Dkk4Tg; and (2) specific expression of Wnt10a and Wnt10b (Fig. 3B), Wnt ligands that are both susceptible to inhibition by Dkk4. Phenotypic analysis of transgenic mice and organ cultures showed that WT-Dkk4 or PM-Dkk4 indeed inhibited Wnt action and blocked MG germ growth at an early stage. In contrast, the cleaved form CL-Dkk4 showed no inhibitory effect on MG growth (Fig. 4E,F), reflecting the loss of receptor binding of the cleaved form (Fig. 2A,B). Dkk4 has been shown to affect secondary hair follicle formation through the Shh pathway (Cui et al., 2010). Interestingly, Dkk4 has a powerful effect on MG development by totally blocked germ formation. The differential effect of Dkk4 might be due to the sensitivity of different organs to Dkk4, Eda or its downstream effectors such as Shh or Lrp6. Actually, epidermal knockout of Shh also affects MG development with a similar phenotype found in hair follicle development (data not shown). The molecular and cell properties from different skin appendages need to be further studied.
Lrp6, a Dkk4 receptor and also a Wnt co-receptor, is induced by Eda–Edar–NF-κB signaling during early phases of MG development (Fig. 5). Thus, Lrp6 turns out to be a link mediating Dkk4 and Eda action. Indeed, Lrp6 activation could reverse Dkk4-induced Wnt inhibition, although only at a later stage, when Dkk4 protein has been inactivated by cleavage (Fig. 6). Confirmative results in MG cultures showed that Lrp6 promoted MG formation during early development. Interestingly, elevation of Lrp6 rescued the MG defect not only in Dkk4Tg mice, but also in Tabby mice (Fig. 7). Interestingly, Lrp6 activation in Tabby did not induce the overgrowth of MG germs, as observed with Dkk4 antagonist treatment, suggesting that other Eda targets besides Lrp6 also play a role in MG development. We then analyzed the expression of all Wnt ligands and Fzds in Tabby mice. Among all the Wnts and Fzds, besides Lrp6, Wnt10b and Fzd10 were also downregulated in Tabby (Fig. 5A, Table S1). Thus, among Eda targets, Lrp6 is the important but not only one involved in MG development.
Overall, our results suggest that Dkk4, induced by Eda, limits Wnt activity through binding Lrp6 during the MG induction stage, and its effect is then weakened progressively by auto-cleavage, releasing augmented Wnt action for further MG development at later stages (Fig. 8). The modulation of MG formation clearly depends on the precise level of active Dkk4, whether determined by the time and rate of its formation, by the levels of inhibitors of its interaction with Lrp6, or by the extent of cleavage of the intact molecule. How these parameters are set by genetic and developmental variables requires further study and may also be relevant to the intricate refinement of Wnt action in other systems.
Fig. 8.
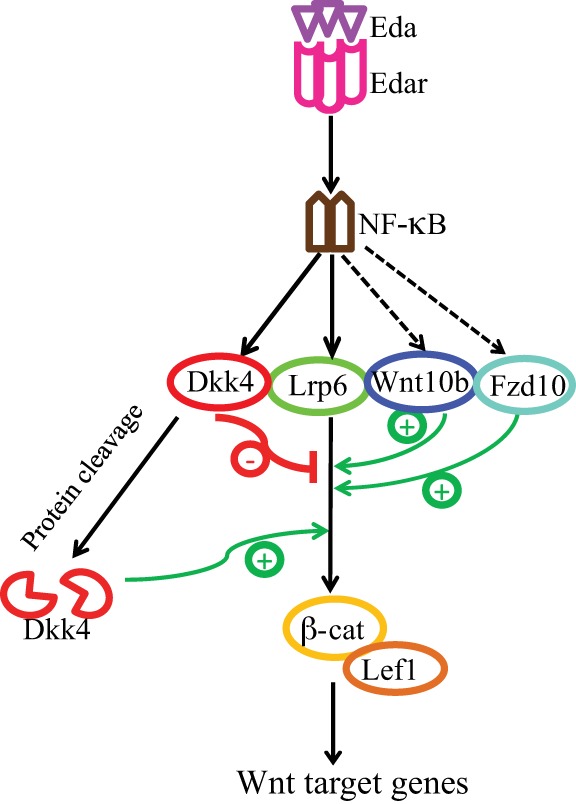
Schematic representation of Dkk4 dynamics and function in early meibomian gland development. At early stages of MG development, Eda/Edar signaling mediates NF-κB activation and thereby induces expression of Lrp6, negative Wnt modulator Dkk4, and positive Wnt effectors, including Wnt10b and Fzd10. Eda-induced Dkk4 then limits Wnt activity at the early phase, whereas subsequent Dkk4 inactivation via proteolytic protein cleavage facilitates MG growth at later stage.
MATERIALS AND METHODS
Conditioned medium collection and alkaline phosphatase cell surface binding assay
Dkk CM or Wnt CM was produced as previously described (Semenov et al., 2005). Briefly, HEK293 cells (2×108) were transfected with 100 µg WT-Dkk4, CL-DKK4, PM-Dkk4, WT-Dkk1 or various Wnt plasmids for 16 h, and then changed to culture in DMEM medium with 1% FBS. After 24 h, medium was concentrated 10-fold using Centricon Plus-10 filters (Millipore) for further use. Cell surface binding assays were performed as described (Mao et al., 2001) and stained with an AP staining kit (GenHunter).
Top/Fop luciferase reporter assay
To assess the transcriptional activity of β-catenin in Kera308 cells, we employed the 8× Top/Fop reporter system using a dual-luciferase kit (Promega). Kera308 cells (1×106/500 μl DMEM) were transiently transfected with 100 ng of constitutively active vector encoding Renilla luciferase (Promega) and 1 µg of Topflash (Addgene) or negative control Fopflash using the Nucleofector system (Lonza). After transfection for 24 h, cells were incubated with Dkk1 or Dkk4 CM with or without Dkk inhibitors for 2 h, followed by CM containing various individual Wnts for 24 h culture. Then firefly and Renilla luciferase activity was measured according to the manufacturer's instructions. The firefly luciferase activity was normalized to Renilla luciferase activity; the fold increase in Topflash activity compared with Fopflash was reported.
cDNAs, plasmids and antibodies
Mouse Dkk1 and Dkk4 cDNAs were purchased from ATCC and amplified by PCR and cloned into p3×FLAG-CMV-14 vector (Sigma). PM-Dkk4 (D88A)-expressing vector was obtained by using a site-directed mutagenesis kit (Agilent Technologies). CL-Dkk4 cDNA was cloned into p3×FLAG-CMV-13 vector (Sigma), which contained the leading signal peptide sequence. Plasmids expressing Lrp6, Topflash, Fopflash and all Wnts were from Addgene. hLrp6-AC plasmid was purchased from Santa Cruz. All antibodies used and suppliers are listed in Table S2.
Western blotting, immunoprecipitation, gel silver staining, mass spectral analysis and N-terminal Edman sequencing
Western blotting, immunoprecipitation, gel silver staining and mass spectral analyses were performed as previously described (Yan et al., 2010). Gel pieces containing FL-Dkk4 or CL-Dkk4 protein were cut out for mass spectral analysis (Alphalyse) or for N-terminal Edman sequencing (Alphalyse).
Mouse models, timed mating and genotyping
All research was conducted according to the guidelines of the Office of Animal Care and Use in the NIH Intramural Program, and all animal study protocols were approved by the NIA Animal Care and Use Committee (ACUC). Skin-specific Dkk4 transgenic mice (K14-Dkk4) were generated in our laboratory and have been reported previously (Cui et al., 2010). Tabby mice were purchased from The Jackson Laboratory. Two sets of timed mating were set up. K14-Dkk4Tg×C57BL/6J and WT C57BL/6J male mice×Tabby females crossed to obtain Dkk4Tg, Tabby homozygote and WT progeny. Eyelids for study and livers to provide DNA for genotyping were excised under dissection microscopy and were fixed, cultured or stored at 80°C until use. Genotyping for Dkk4Tg and Tabby was performed as previously described (Cui et al., 2014).
Histology, immunohistochemistry and in situ hybridization
For histological analyses, eyelids from mice at each time point were fixed in 10% formaldehyde and embedded in paraffin and 5 µm sections were then cut for H&E staining or immunofluorescence (antibodies employed are listed in Table S2). For in situ hybridization, 20 µm frozen sections were fixed in 4% paraformaldehyde as described previously (Cui et al., 2014). All probe cDNAs were amplified by PCR and subcloned into a pGEM-T vector (Promega). Images were then analyzed by DeltaVision microscopy.
Gene expression profiling and real-time RT-PCR
Eyelids from WT and Tabby embryos at E14.5 and E15.5 were collected under a dissection microscope, snap-frozen in dry ice and stored at −80°C for RNA isolation and microarray gene expression profiling as described (Cui et al., 2014). Frozen whole eyelid skin samples were separated into groups for each of the genotypes at various time points for biological duplicates, to assess crucial genes, including Eda and Shh. Hybridization data have been deposited in the Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE87742. Quantitative RT-PCR was performed using SYBR Green PCR master mix (Applied Biosystems) and all primers are listed in Table S3.
Promoter analysis, promoter luciferase reporter assay, and chromatin immunoprecipitation
Promoter analysis of Lrp6 used a computational program (http://ecrbrowser.dcode.org). A 1411 bp section of the Lrp6 promoter region, containing two putative NF-κB binding sites, was cloned into the pGL4.14 luciferase vector (Promega). The same Lrp6 promoter fragment with three nucleotides mutated at each NF-κB binding site was also cloned using a site-directed mutagenesis kit (Agilent Technologies). Promoter luciferase reporter assays were performed as previously described (Chen et al., 2011). Chromatin was extracted from WT eyelids at E15.5 and immunoprecipitated with a Chromatin-IP kit (Cell Signaling Technology) following the supplier's protocol. Treated DNA was amplified by PCR. All primers used are listed in Table S3.
Eyelid organotypic culture and MG length measurement
Eyelid skin from embryos at E15.5 was separated on ice under dissection microscopy. After two washes with cold PBS, eyelid skin was placed on a 0.4 µm Millicell culture insert (Millipore) and cultured with 10% FBS in full DMEM, or in 50% culture medium plus 50% conditioned medium of various types. In some cultures, the Dkk inhibitor WAY-262611/IIIC3 (Santa Cruz) or lentiviral particles (1×107 GC/ml) expressing LentiCon, shRNA or CRISPR activation motifs (Santa Cruz) were added to medium. After 48 h, eyelids were fixed in 10% formaldehyde and followed our immunohistochemistry protocol for paraffin-embedded tissue sections. The longest MG germs from serial sections were measured, and the distance between epidermal basal cell edge and the outer most edge of germs was calculated. A total of 10-15 MG germs were measured and quantified from each cultured eyelid tissue.
Acknowledgements
The authors thank Ramaiah Nagaraja, Weidong Wang, Zhijiang Yan for critical reading; Marc Michel, Yong Qian and Alexei Sharov for technical help.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
J.S. and D.S. developed the ideas and designed the experiments; J.S., Y.P. and Y.C. performed the experiments; J.S. and D.S. analyzed the data; J.S. and D.S. wrote the manuscript.
Funding
This work was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health. Deposited in PMC for release after 12 months.
Data availability
Hybridization data are deposited in Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE87742.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.143909.supplemental
References
- Ahn V. E., Chu M. L.-H., Choi H.-J., Tran D., Abo A. and Weis W. I. (2011). Structural basis of Wnt signaling inhibition by Dickkopf binding to LRP5/6. Dev. Cell 21, 862-873. 10.1016/j.devcel.2011.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andl T., Reddy S. T., Gaddapara T. and Millar S. E. (2002). WNT signals are required for the initiation of hair follicle development. Dev. Cell 2, 643-653. 10.1016/S1534-5807(02)00167-3 [DOI] [PubMed] [Google Scholar]
- Bafico A., Liu G., Yaniv A., Gazit A. and Aaronson S. A. (2001). Novel mechanism of Wnt signalling inhibition mediated by Dickkopf-1 interaction with LRP6/Arrow. Nat. Cell Biol. 3, 683-686. 10.1038/35083081 [DOI] [PubMed] [Google Scholar]
- Bazzi H., Fantauzzo K. A., Richardson G. D., Jahoda C. A. B. and Christiano A. M. (2007). The Wnt inhibitor, Dickkopf 4, is induced by canonical Wnt signaling during ectodermal appendage morphogenesis. Dev. Biol. 305, 498-507. 10.1016/j.ydbio.2007.02.035 [DOI] [PubMed] [Google Scholar]
- Botchkarev V. A. and Fessing M. Y. (2005). Edar signaling in the control of hair follicle development. J. Invest. Derm. Symp. Proc. 10, 247-251. 10.1111/j.1087-0024.2005.10129.x [DOI] [PubMed] [Google Scholar]
- Botchkarev V. A., Botchkareva N. V., Roth W., Nakamura M., Chen L.-H., Herzog W., Lindner G., McMahon J. A., Peters C., Lauster R. et al. (1999). Noggin is a mesenchymally derived stimulator of hair-follicle induction. Nat. Cell Biol. 1, 158-164. 10.1038/11078 [DOI] [PubMed] [Google Scholar]
- Bourhis E., Tam C., Franke Y., Bazan J. F., Ernst J., Hwang J., Costa M., Cochran A. G. and Hannoush R. N. (2010). Reconstitution of a Frizzled8·Wnt3a·LRP6 signaling complex reveals multiple Wnt and Dkk1 binding sites on LRP6. J. Biol. Chem. 285, 9172-9179. 10.1074/jbc.M109.092130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-H., Jiang T.-X., Lin C.-M., Burrus L. W., Chuong C.-M. and Widelitz R. (2004). Distinct Wnt members regulate the hierarchical morphogenesis of skin regions (spinal tract) and individual feathers. Mech. Dev. 121, 157-171. 10.1016/j.mod.2003.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Moradin A., Schlessinger D. and Nagaraja R. (2011). RXRalpha and LXR activate two promoters in placenta- and tumor-specific expression of PLAC1. Placenta 32, 877-884. 10.1016/j.placenta.2011.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z., Biechele T., Wei Z., Morrone S., Moon R. T., Wang L. and Xu W. (2011). Crystal structures of the extracellular domain of LRP6 and its complex with DKK1. Nat. Struct. Mol. Biol. 18, 1204-1210. 10.1038/nsmb.2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. (2006). Wnt/beta-catenin signaling in development and disease. Cell 127, 469-480. 10.1016/j.cell.2006.10.018 [DOI] [PubMed] [Google Scholar]
- Cruciat C. M. and Niehrs C. (2013). Secreted and transmembrane Wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 5, pii: a015081. 10.1101/cshperspect.a015081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C.-Y., Smith J. A., Schlessinger D. and Chan C.-C. (2005). X-linked anhidrotic ectodermal dysplasia disruption yields a mouse model for ocular surface disease and resultant blindness. Am. J. Pathol. 167, 89-95. 10.1016/S0002-9440(10)62956-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C. Y., Kunisada M., Piao Y., Childress V., Ko M. S. H. and Schlessinger D. (2010). Dkk4 and Eda regulate distinctive developmental mechanisms for subtypes of mouse hair. PLoS ONE 5, e10009 10.1371/journal.pone.0010009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C.-Y., Yin M., Sima J., Childress V., Michel M., Piao Y. and Schlessinger D. (2014). Involvement of Wnt, Eda and Shh at defined stages of sweat gland development. Development 141, 3752-3760. 10.1242/dev.109231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell R. R. and Watt F. M. (2015). Understanding fibroblast heterogeneity in the skin. Trends Cell Biol. 25, 92-99. 10.1016/j.tcb.2014.10.001 [DOI] [PubMed] [Google Scholar]
- Fliniaux I., Mikkola M. L., Lefebvre S. and Thesleff I. (2008). Identification of dkk4 as a target of Eda-A1/Edar pathway reveals an unexpected role of ectodysplasin as inhibitor of Wnt signalling in ectodermal placodes. Dev. Biol. 320, 60-71. 10.1016/j.ydbio.2008.04.023 [DOI] [PubMed] [Google Scholar]
- Fuchs E. (2007). Scratching the surface of skin development. Nature 445, 834-842. 10.1038/nature05659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka A., Wu W., Delius H., Monaghan A. P., Blumenstock C. and Niehrs C. (1998). Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 391, 357-362. 10.1038/34848 [DOI] [PubMed] [Google Scholar]
- Gong Y., Bourhis E., Chiu C., Stawicki S., DeAlmeida V. I., Liu B. Y., Phamluong K., Cao T. C., Carano R. A. D., Ernst J. A. et al. (2010). Wnt isoform-specific interactions with coreceptor specify inhibition or potentiation of signaling by LRP6 antibodies. PLoS ONE 5, e12682 10.1371/journal.pone.0012682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanskaya O., Glinka A., del Barco Barrantes I., Stannek P., Niehrs C. and Wu W. (2004). R-Spondin2 is a secreted activator of Wnt/beta-catenin signaling and is required for Xenopus myogenesis. Dev. Cell 7, 525-534. 10.1016/j.devcel.2004.07.019 [DOI] [PubMed] [Google Scholar]
- Krupnik V. E., Sharp J. D., Jiang C., Robison K., Chickering T. W., Amaravadi L., Brown D. E., Guyot D., Mays G., Leiby K. et al. (1999). Functional and structural diversity of the human Dickkopf gene family. Gene 238, 301-313. 10.1016/S0378-1119(99)00365-0 [DOI] [PubMed] [Google Scholar]
- Kumar A., Eby M. T., Sinha S., Jasmin A. and Chaudhary P. M. (2001). The ectodermal dysplasia receptor activates the nuclear factor-kappaB, JNK, and cell death pathways and binds to ectodysplasin A. J. Biol. Chem. 276, 2668-2677. 10.1074/jbc.M008356200 [DOI] [PubMed] [Google Scholar]
- Li X., Shan J., Chang W., Kim I., Bao J., Lee H.-J., Zhang X., Samuel V. T., Shulman G. I., Liu D. et al. (2012). Chemical and genetic evidence for the involvement of Wnt antagonist Dickkopf2 in regulation of glucose metabolism (vol 109, pg 11402, 2012). Proc. Natl. Acad. Sci. USA 109, 17141 10.1073/pnas.1205015109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien W.-H. and Fuchs E. (2014). Wnt some lose some: transcriptional governance of stem cells by Wnt/beta-catenin signaling. Gene Dev. 28, 1517-1532. 10.1101/gad.244772.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim X. and Nusse R. (2013). Wnt signaling in skin development, homeostasis, and disease. Cold Spring Harb. Perspect. Biol. 5, pii: a008029. 10.1101/cshperspect.a008029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan C. Y. and Nusse R. (2004). The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781-810. 10.1146/annurev.cellbio.20.010403.113126 [DOI] [PubMed] [Google Scholar]
- MacDonald B. T., Tamai K. and He X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9-26. 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao B. and Niehrs C. (2003). Kremen2 modulates Dickkopf2 activity during Wnt/LRP6 signaling. Gene 302, 179-183. 10.1016/S0378-1119(02)01106-X [DOI] [PubMed] [Google Scholar]
- Mao B., Wu W., Li Y., Hoppe D., Stannek P., Glinka A. and Niehrs C. (2001). LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 411, 321-325. 10.1038/35077108 [DOI] [PubMed] [Google Scholar]
- Mikkola M. L. (2008). TNF superfamily in skin appendage development. Cytokine Growth Factor Rev. 19, 219-230. 10.1016/j.cytogfr.2008.04.008 [DOI] [PubMed] [Google Scholar]
- Niehrs C. (2006). Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene 25, 7469-7481. 10.1038/sj.onc.1210054 [DOI] [PubMed] [Google Scholar]
- Pelletier J. C., Lundquist J. T., Gilbert A. M., Alon N., Bex F. J., Bhat B. M., Bursavich M. G., Coleburn V. E., Felix L. A., Green D. M. et al. (2009). (1-(4-(Naphthalen-2-yl)pyrimidin-2-yl)piperidin-4-yl)methanamine: a wingless beta-catenin agonist that increases bone formation rate. J. Med. Chem. 52, 6962-6965. 10.1021/jm9014197 [DOI] [PubMed] [Google Scholar]
- Semenov M. V., Tamai K., Brott B. K., Kuhl M., Sokol S. and He X. (2001). Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr. Biol. 11, 951-961. 10.1016/S0960-9822(01)00290-1 [DOI] [PubMed] [Google Scholar]
- Semenov M., Tamai K. and He X. (2005). SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 280, 26770-26775. 10.1074/jbc.M504308200 [DOI] [PubMed] [Google Scholar]
- Sick S., Reinker S., Timmer J. and Schlake T. (2006). WNT and DKK determine hair follicle spacing through a reaction-diffusion mechanism. Science 314, 1447-1450. 10.1126/science.1130088 [DOI] [PubMed] [Google Scholar]
- Veeman M. T., Slusarski D. C., Kaykas A., Louie S. H. and Moon R. T. (2003). Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr. Biol. 13, 680-685. 10.1016/S0960-9822(03)00240-9 [DOI] [PubMed] [Google Scholar]
- Voutilainen M., Lindfors P. H., Lefebvre S., Ahtiainen L., Fliniaux I., Rysti E., Murtoniemi M., Schneider P., Schmidt-Ullrich R. and Mikkola M. L. (2012). Ectodysplasin regulates hormone-independent mammary ductal morphogenesis via NF-kappaB. Proc. Natl. Acad. Sci. USA 109, 5744-5749. 10.1073/pnas.1110627109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widelitz R. B. (2008). Wnt signaling in skin organogenesis. Organogenesis 4, 123-133. 10.4161/org.4.2.5859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Wang Y., Dabdoub A., Smallwood P. M., Williams J., Woods C., Kelley M. W., Jiang L., Tasman W., Zhang K. et al. (2004). Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell 116, 883-895. 10.1016/S0092-8674(04)00216-8 [DOI] [PubMed] [Google Scholar]
- Yan Z., Delannoy M., Ling C., Daee D., Osman F., Muniandy P. A., Shen X., Oostra A. B., Du H., Steltenpool J. et al. (2010). A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol. Cell 37, 865-878. 10.1016/j.molcel.2010.01.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Tomann P., Andl T., Gallant N. M., Huelsken J., Jerchow B., Birchmeier W., Paus R., Piccolo S., Mikkola M. L. et al. (2009). Reciprocal requirements for EDA/EDAR/NF-kappaB and Wnt/beta-catenin signaling pathways in hair follicle induction. Dev. Cell 17, 49-61. 10.1016/j.devcel.2009.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]