

Abstract
Holoprosencephaly (HPE) is defined as the incomplete separation of the two cerebral hemispheres. The pathology of HPE is variable and, based on the severity of the defect, HPE is divided into alobar, semilobar, and lobar. Using a novel hypomorphic Six3 allele, we demonstrate in mice that variability in Six3 dosage results in different HPE phenotypes. Furthermore, we show that whereas the semilobar phenotype results from severe downregulation of Shh expression in the rostral diencephalon ventral midline, the alobar phenotype is caused by downregulation of Foxg1 expression in the anterior neural ectoderm. Consistent with these results, in vivo activation of the Shh signaling pathway rescued the semilobar phenotype but not the alobar phenotype. Our findings show that variations in Six3 dosage result in different forms of HPE.
KEY WORDS: Six3, Holoprosencephaly, Transcription factor, Gene dosage, Forebrain, Mouse
Summary: Analysis of a hypomorphic Six3 allele reveals that variability in Six3 dosage is sufficient to generate mice exhibiting a spectrum of holoprosencephaly forms in a non-strain-specific manner.
INTRODUCTION
Incomplete separation of the cerebral hemispheres during embryonic development causes holoprosencephaly (HPE). Depending on its severity, HPE is classified as alobar, semilobar, or lobar, with alobar being the most severe form. Mutations in any of at least nine genes involved in the Shh signaling pathway can cause congenital HPE in humans (Geng and Oliver, 2009; Roessler and Muenke, 2010). Examples include SHH (ligand), PTCH1 (receptor), GLI2 (transcription factor activated by SHH) and SIX3 (transcription factor that activates SHH). However, the Shh signaling pathway accounts for only 17% of familial HPE (Cohen, 2006). Furthermore, by studying patients with HPE and their family members, it was determined that the level of HPE penetrance and the severity of the disease are highly variable. Mutations in SIX3, which account for ∼1.3% of HPE cases, exemplify this phenomenon (Cohen, 2006). SIX3 mutations are associated with defects ranging from alobar HPE (cyclopia) to microforms of HPE (e.g. single medial incisor) (Lacbawan et al., 2009; Muenke and Cohen, 2000). Also, HPE patients with normal parents that carry the same mutation have been reported (Ribeiro et al., 2006).
We previously showed that Six3 directly activates the expression of Shh in the rostral diencephalon ventral midline (RDVM) of mice (Geng et al., 2008; Jeong et al., 2008). In turn, Shh maintains the expression of Six3 in the RDVM. This Six3-Shh feedback loop is necessary to initiate and maintain the expression of a few crucial players (e.g. Fgf8, Bmp4, Shh and Nkx2.1) in the telencephalon to regulate the specification of the ventral telencephalon and the separation of the cerebral hemispheres (Geng et al., 2008). The Six3-Shh feedback loop is compromised in Six3 heterozygous mice in a background-specific manner, leading to the development of semilobar HPE (Geng et al., 2008). However, alobar HPE rarely develops in Six3+/– mice. How alobar HPE develops in some patients with mutations in SIX3 is also unknown.
Gene dosage has been associated with variations in disease severity. For example, Pax6 deletion results in the absence of eyes; however, Pax6 haploinsufficiency leads to defects in the anterior segment of the eye that results in small eye in mice and aniridia in humans (Davis et al., 2009; Davis-Silberman et al., 2005; Schedl et al., 1996). Similarly, functional inactivation of Six3 in mice results in the absence of the forebrain and eyes, whereas removal of one allele leads to defects in the specification of the ventral telencephalon, and sometimes causes semilobar HPE (Geng et al., 2008; Lagutin et al., 2003). Six3 dosage also plays a role in determining telencephalic versus eye fate and eye patterning in medaka fish (Beccari et al., 2012; Carl et al., 2002). Furthermore, the dosage of various signaling factors also affects the pathogenesis of HPE (Mercier et al., 2013; Storm et al., 2006). Based on these results, we speculated that reduced levels of Six3 (below the 50% expected of Six3+/– mice) might cause a more severe form of HPE.
To test this hypothesis, we took advantage of a novel mouse model (Six3+/neo) that when crossed with other Six3 mutant strains expresses reduced amounts (less than 50%) of Six3. Interestingly, we found that Six3neo/neo and Six3neo/– embryos exhibit semilobar and alobar HPE, respectively, in a strain-independent manner. We also report that in Six3neo/neo embryos, the previously identified Six3-Shh feedback loop is affected in the RDVM. By contrast, expression of the transcription factor Foxg1 was severely reduced in the anterior neural ectoderm (ANE) of Six3neo/– embryos, a consequence of the direct regulation of Foxg1 by Six3. Together, these results provide valuable information on how variability in Six3 levels is directly responsible for the severity of HPE phenotypes.
RESULTS
Six3 haploinsufficiency causes semilobar HPE
To help us identify novel Six3 partners and downstream targets using ChIP-Seq, we generated a novel mouse strain (Six3+/Avi) in which one allele of Six3 was replaced by Avi-Six3 (Fig. S1A,B). The Avi tag is a 15-amino-acid peptide that can be biotinylated by biotin ligase (de Boer et al., 2003). Addition of the Avi tag does not interfere with Six3 activity, as Six3Avi/Avi mice are indistinguishable from wild-type littermates (data not shown).
During the generation of the Six3+/Avi strain, we inserted a neomycin/thymidine kinase (Neo/TK) selection cassette flanked by two loxP sites before exon 1, in a region that is not evolutionarily conserved (Six3+/neo; Fig. S1A). Previous studies have shown that the insertion of a Neo/TK cassette in the targeted allele may affect the transcription of a downstream gene, resulting in a hypomorphic allele (White et al., 1997). We demonstrated previously that haploinsufficiency of Six3 causes semilobar HPE-like phenotypes in Six3 heterozygous (Six3+/– or Six3+/ki) embryos (Geng et al., 2008). Therefore, we speculated that the newly generated Six3neo/neo embryos might also exhibit HPE-like phenotypes.
To evaluate this possibility, we performed a detailed morphological analysis of Six3neo/neo embryos. As revealed by scanning electron microscopy (SEM) of embryonic day (E) 10.5 embryos, the telencephalic vesicles and the medial nasal prominences were well separated in wild-type embryos (Fig. 1A). However, in Six3neo/neo embryos, the telencephalic vesicles and the medial nasal prominences appeared smaller and were not separated (Fig. 1E). Coronal sections of postnatal day (P) 0 brains showed that the mutant brains lacked the cartilage nasal septum (compare Fig. 1B and F), the septum, and the corpus callosum (compare Fig. 1C and G). No obvious defects were identified in the posterior separation of the two cerebral hemispheres, and dorsal midline structures such as the hippocampus were present in the mutant brains, although the lateral ventricles appeared enlarged and the cerebral cortex was thinner (compare Fig. 1D and H). These phenotypes recapitulated those observed in Six3 heterozygous embryos and resembled the clinical manifestations of patients with semilobar HPE.
Fig. 1.

Changes in Six3 levels lead to different types of HPE-like phenotypes in mice. Scanning electron microscopy analysis was performed on E10.5 wild-type (A), Six3neo/neo (E) and Six3neo/– (I) embryos. The telencephalic vesicles are pseudocolored in magenta, medial nasal prominences (MNPs) in yellow, and lateral nasal prominences (LNPs) in green. (A) In control embryos, both the telencephalic vesicles and the MNPs are well separated (n=3). (E) In Six3neo/neo embryos, the LNPs are well formed; however, a single telencephalic vesicle is present and the MNP is not separated (n=2). (I) In Six3neo/– embryos, the single telencephalic vesicle is small, the MNPs are absent and the LNPs are not separated (n=2). Coronal sections [rostral (R) to caudal (C)] of P0 control (B-D), Six3neo/neo (F-H) and Six3neo/– (J-L) pups stained with Hematoxylin and Eosin. At the most rostral level, the cartilage nasal septum is seen in controls (B, arrow), but this structure is severely defective in Six3neo/neo pups (F, arrow). The nasal structures are absent in Six3neo/– pups (J, arrow). At the mid level, the corpus callosum (C,G,K, red arrows) and the septum (C,G,K, black arrows) are absent in Six3neo/neo and Six3neo/– embryos. More caudally, the cerebral hemispheres are well formed and separated by the diencephalon and the hippocampus in wild-type and in Six3neo/neo pups (D,H, arrows). However, the hippocampus is relatively enlarged and misplaced on the surface of the brain (L, arrow). n=3 control; n=4 Six3neo/neo; n=3 Six3neo/−.
HPE is normally associated with dorsoventral patterning defects of the telencephalon. Using different telencephalic markers we performed a detailed molecular characterization of the Six3neo/neo telencephalon. As revealed by serial coronal sections of E12.5 embryos, the severity of the dorsoventral patterning defects decreased along the rostrocaudal axis of the telencephalon. At the most rostral level, two well-separated telencephalic vesicles were seen in control embryos (Fig. S2). However, only one telencephalic vesicle was present in the mutant brain (Fig. S2). The dorsal telencephalic markers Ngn2 (Neurog2) and Pax6, and the ventral telencephalic markers Mash1 (Ascl1) and Dlx2, were expressed in the dorsal and ventral regions, respectively, of the telencephalon of control embryos (Fig. S2A,C,E,G). However, in the mutant telencephalon the expression of Ngn2 and Pax6 expanded ventrally to surround the vesicle at the expense of Mash1 and Dlx2 expression (Fig. S2B,D,F,H).
At the mid-caudal level, expression of the dorsal telencephalic marker Ngn2 was restricted to the dorsal telencephalon in control and mutant brains (Fig. 2A,B). Ventrally, four ganglionic eminences [two lateral ganglionic eminences (LGEs) and two medial ganglionic eminences (MGEs)] were present in the control telencephalon (Fig. 2D,G,J,M). However, as indicated by the expression of Dlx2 and Ebf1, only one ganglionic eminence with LGE identity was present in the mutant brain (Fig. 2E,H,K,N). Caudally, the two hemispheres of the mutant telencephalon were separated by the diencephalon, and the dorsal midline structures (i.e. the hippocampus, cortical hem and choroid plexus) were well formed (Fig. S2I-P). Similar dorsoventral patterning defects of the telencephalon have been observed in Six3+/ki;Shh+/– embryos (Geng et al., 2008).
Fig. 2.

Activation of the Shh signaling pathway rescues dorsoventral patterning defects in the Six3neo/neo telencephalon. E12.5 wild-type, Six3neo/neo and Six3neo/neo;Ptch1+/– embryos were coronally sectioned and analyzed for possible alterations in dorsoventral patterning of the telencephalon. Ngn2 expression is restricted to the dorsal telencephalon (DT) in control (A), Six3neo/neo (B) and Six3neo/neo;Ptch1+/– (C) embryos. (D) Dlx2, a marker for the ventral telencephalon, is expressed in both the lateral ganglionic eminence (LGE) and medial ganglionic eminence (MGE) of control embryos. A single Dlx2-positive lobe is seen in Six3neo/neo embryos (E). Normal separation of the ganglionic eminences is restored in Six3neo/neo;Ptch1+/– embryos (F). Ebf1 expression labels the LGEs in control embryos (G). The single ganglionic eminence in Six3neo/neo embryos is positive for Ebf1 (H). Proper separation of the LGE lobes is rescued in Six3neo/neo;Ptch1+/– embryos (I). Nkx2.1 is expressed in the two MGEs of control embryos (J) but is absent in the Six3neo/neo embryos (K). Normal expression of Nkx2.1 is restored in Six3neo/neo;Ptch1+/– embryos (L). n=4 control; n=5 Six3neo/neo; n=4 Six3neo/neo;Ptch1+/−. Schematic representation of the various telencephalic domains in control (M), Six3neo/neo (N) and Six3neo/neo; Ptch1+/− (O) embryos.
Six3+/– embryos are grossly normal in an outbred background (NMRI) and exhibit HPE phenotypes only at low penetrance (16%) in mixed backgrounds (129/Sv;C57BL/6). The penetrance increases to 85% when animals are backcrossed into the C57BL/6 background for five generations (Geng et al., 2008). However, Six3neo/neo mice were maintained in an outbred background (NMRI) and exhibited HPE phenotypes with 100% penetrance. The patterning of the forebrain is very sensitive to the amount of Six3; thus, we reasoned that the dosage of Six3 might contribute to this difference in background dependency. To test this possibility we determined the transcriptional activity of the targeted allele in Six3+/neo embryos by real-time PCR analysis at the 0-somite stage, a stage immediately after the specification of the ANE. Using this approach, we determined that 21.5% of total Six3 transcripts were transcribed from the Avi-targeted allele, and thus 78.5% were wild-type Six3. This indicates that the transcriptional activity of the targeted allele was 27% of that of the wild-type allele (Fig. S3A,B). These results are consistent with a previous report showing that the presence of the Neo/TK cassette affects the transcriptional activity of the targeted allele (White et al., 1997).
Elevated Shh signaling rescues semilobar HPE phenotypes
Six3 directly regulates Shh expression in the RDVM (Geng et al., 2008; Jeong et al., 2008). As the amount of functional Six3 decreases in Six3+/– and Six3+/ki embryos, it fails to activate Shh expression in the ventral forebrain and subsequently leads to HPE-like phenotypes. Supporting our previous findings, Shh expression in the ventral forebrain was severely downregulated in Six3neo/neo embryos (Fig. S4A,B, arrows); however, Shh expression in the prechordal plate was comparable to that of control embryos (Fig. S4A,B, arrowheads). Fgf8 expression in the commissural plate was also dramatically downregulated (Fig. S4D,E, arrow).
We previously showed that Shh signaling from the prechordal plate interacts with Six3 to activate its expression in the ventral forebrain (Geng et al., 2008). In fact, we determined that deleting one allele of Shh in Six3+/ki embryos dramatically increases the percentage of embryos that exhibit HPE-like phenotypes (Geng et al., 2008). From those results, we hypothesized that the HPE phenotypes observed in Six3 haploinsufficient embryos might be rescued by elevating Shh signaling levels. To test this we generated Six3neo/neo;Ptch1+/– embryos. Ptch1 is a receptor for Shh and acts as a negative regulator of the Shh signaling pathway by maintaining inactive Smo in the absence of Shh ligand. Ptch1 null embryos have open, overgrown neural tubes and die during embryogenesis (Goodrich et al., 1997). Ptch1 heterozygotes are grossly normal but ∼10% larger than their littermates. They are also prone to tumorigenesis due to elevated Shh signaling (Goodrich et al., 1997). Importantly, Ptch1 heterozygosity protects Cdon mutant mice with reduced Shh levels from alcohol-induced HPE (Hong and Krauss, 2013). As shown in Fig. 2, deletion of one allele of Ptch1 was sufficient to completely rescue the semilobar HPE phenotype of Six3neo/neo embryos (Fig. 2C,F,I,L,O). These results confirm our previous conclusion that Six3 cooperates with Shh in regulating the dorsoventral patterning of the telencephalon, and demonstrate that haploinsufficiency of Six3 can be compensated by increasing Shh signaling.
Further reduction of Six3 dosage causes alobar HPE
Six3 dosage is crucial for patterning of the forebrain; therefore, we examined the outcome of further reducing the amount of this transcription factor. We took advantage of Six3neo/– embryos, which, according to our calculations, should express Six3 mRNA at ∼13.5% of normal levels. We found that Six3neo/– embryos had the most severe phenotype, with only one small telencephalic vesicle and no medial nasal prominences (Fig. 1I). Coronal sections of P0 heads revealed that the nasal structures were completely missing in the mutant pups (Fig. 1J). The mutant brain consisted of only a small monoventricular cerebrum that lacked the interhemispheric fissure and a clear distinction between dorsal and ventral structures (Fig. 1K). Caudally, no separation was observed between the two cerebral hemispheres, probably because the dorsal midline structures were proportionally enlarged and formed on the surface of the brain instead of folding inside (Fig. 1L). Most aspects of the phenotypes resembled clinical manifestations of human alobar HPE. Therefore, we concluded that further reducing Six3 levels in Six3neo/– embryos was sufficient to promote alobar HPE-like phenotypes.
To further investigate the dorsoventral patterning defects of the telencephalon, we performed a detailed molecular characterization of coronal sections obtained from E14.5 Six3neo/– forebrains. At the most rostral level, in contrast to the interhemispheric fissure and the two lateral ventricles seen in control embryos, only one small cerebral vesicle was present in the mutant brain (Fig. 3A-J). The dorsal telencephalic marker Ngn2 and the ventral telencephalic markers Gad67 (subpallium marker), Ebf1 and Nkx2.1 were all expressed in the control dorsal and ventral telencephalon, respectively (Fig. 3A,C,E,G). However, the ventral expansion of Ngn2 and the absence of Gad67 (Gad1), Ebf1 and Nkx2.1 indicated the dorsalization of the mutant telencephalon (Fig. 3B,D,F,H). At the caudal level in control embryos, Ngn2 was restricted to the dorsal telencephalon and excluded from the caudal ganglionic eminences (CGEs) (Fig. 3K). The interhemispheric fissure was clearly formed by the invagination of the dorsal midline of the telencephalon, which was marked by Wnt8b expression and which develops into the hippocampus (labeled by Prox1 expression) and the choroid plexus (labeled by TTR expression) (Fig. 3M,O,Q,S). However, in the mutant embryos, Ngn2 expression was expanded ventrally and the CGEs were absent (Fig. 3L). The interhemispheric fissure was also absent, the dorsal midline of the telencephalon failed to invaginate, and the hippocampus and choroid plexus formed on the surface of the telencephalon (Fig. 3N,P,R,T).
Fig. 3.

Morphogenesis and dorsoventral patterning are defective in the Six3neo/– telencephalon. E14.5 control and Six3neo/– brains were coronally sectioned and in situ hybridization was performed for various markers at the rostral (A-H) and caudal (K-R) levels. At the rostral level, two well-separated cerebral vesicles are present in control embryos (A). Ngn2 expression is also restricted to the ventricle of the cerebral cortex (CC) (A), and Gad67, Ebf1 and Nkx2.1 are expressed in the ventral region (C,E,G). However, a single vesicle is observed in Six3neo/– embryos, and it is much smaller than that of the control telencephalon. In these mutant embryos, Ngn2 expression also expands ventrally to cover the entire ventricle (B) at the expense of Gad67, Ebf1 and Nkx2.1, which are no longer expressed in this region (D,F,H). These results suggest that the forebrain of Six3neo/– embryos is completely dorsalized, as schematically summarized (I,J). (K) In control embryos at the caudal level, Ngn2 is expressed along the ventricle of the CC and the dorsal thalamus, but excluded from the caudal ganglionic eminence (CGE). (L) In Six3neo/– embryos, Ngn2 expression is abnormally expanded ventrally. In control (M) and Six3neo/– (N) embryos, Prox1 expression is seen in the dentate gyrus neuroepithelium (DNE) of the hippocampal region (black arrows), and the dorsal thalamus (red arrows). Wnt8b expression labels the fimbria neuroepithelium (FNE) of the hippocampal region (O,P, black arrows) and eminentia thalami (EmT) (O,P, red arrows). Ttr is expressed in the choroid plexus (CP) (Q,R, arrows). The DNE, FNE and CP are localized to the surface of the brain (N,P,R, respectively). (S,T) Summary of the expression analysis results. Di, diencephalon; ST, septum. n=3 control; n=5 Six3neo/−.
In summary, these results further demonstrate that Six3neo/– embryos exhibit alobar HPE-like phenotypes. They also conclusively show that Six3 dosage is crucial for the pathogenesis of HPE. By fine-tuning the level of Six3 expression we generated mouse models of alobar and semilobar HPE.
Elevated Shh signaling fails to rescue the alobar HPE-like phenotype
The alobar HPE-like phenotypes of Six3neo/– embryos were reminiscent of those exhibited by Shh null embryos (Chiang et al., 1996; Ribeiro et al., 2006). To evaluate whether an increase in Shh activity would at least partially rescue the alobar HPE-like phenotype, similar to what we observed for the semilobar HPE phenotype, we generated Six3neo/–;Ptch1+/– embryos. Compared with the brain of Six3neo/– embryos, that of Six3neo/–;Ptch1+/– embryos appeared to have a longer diencephalon (Fig. S5). As revealed by serial coronal sections of E12.5 embryos, at the most anterior end of the mutant brains, the two brain vesicles (Fig. 4B, black arrows) are abnormally separated in the middle by the abnormally expanded diencephalon (Fig. 4B, red arrow).
Fig. 4.

Elevated Shh signaling fails to rescue brain defects in Six3neo/– embryos. Coronal sections of E12.5 control and Six3neo/–;Ptch1+/– forebrains. In controls, the dorsal telencephalic marker Ngn2 is excluded from the caudal ganglionic eminence (CGE) (A,C, arrows), which is identified by Dlx2 expression (C). By contrast, Ngn2 is expressed throughout the telencephalic neural epithelium in Six3neo/–;Ptch1+/– embryos (B, arrows). Rostral-expanded diencephalon separates the telencephalic vesicles (B, red arrow). Wnt8b labels the dorsal midline (DM) of the telencephalon and the eminentia thalami (EmT) in control and mutant samples (E,F, arrows). The presence of the EmT is confirmed by the expression of Lim1 (G,H, black arrows). Lim1 is also a marker for the zona limitans intrathalamica (ZLI), the ventral thalamus (VTh) and the hypothalamus (HTh). Compared with that in wild-type embryos (G, red arrows), the region of Lim1 expression is slightly smaller and located more ventrally in Six3neo/–;Ptch1+/– embryos (H, red arrows). Similarly, Dlx2, another marker for the VTh and HTh, is expressed more ventrally and in a smaller area in the mutant embryos (D, red arrows) than in controls (C, red arrows). The presence of the HTh is marked by Nkx2.1 expression in control and mutant embryos (I,J). Ngn2 and Gbx2 are expressed in the dorsal thalamus (DTh), and their expression domain is enlarged in the mutant brain (B,L) compared with that in controls (A,K). (M,N) Summary of the expression analysis results. DT, dorsal telencephalon; PT, pretectum. n=3 control; n=3 Six3neo/−;Ptch1+/−.
In order to confirm the identity of the mutant brain, we analyzed the expression of a panel of telencephalic and diencephalic markers. Similar to the telencephalon of Six3neo/– embryos, that of Six3neo/–;Ptch1+/– embryos was completely dorsalized, as indicated by the ventral expansion of Ngn2 expression and the absence of CGEs, as indicated by the lack of Dlx2 expression in the telencephalon (Fig. 4A-D). Similar to what was observed in Six3neo/− embryos, in Six3neo/−;Ptch1+/− embryos the dorsal midline of the telencephalon failed to invaginate and remained on the surface of the telencephalon (Fig. 4E,F). The identity of the enlarged diencephalon in the mutant brain was confirmed by expression of the dorsal thalamic markers Ngn2 and Gbx2, the ventral thalamic markers Dlx and Lim1 (Lhx1), and the hypothalamic marker Nkx2.1 (Fig. 4). Within the mutant diencephalon, the dorsal thalamus was enlarged and the ventral thalamus was slightly reduced (compare Fig. 4A,C,G,K with B,D,H,L). This observation is consistent with the previously proposed role of Shh in promoting the growth of the diencephalon (Ishibashi and McMahon, 2002; Kiecker and Lumsden, 2004). The hypothalamus is present in both control and mutant forebrain (Fig. 4I,J). A schematic representation of the various domains of the forebrain in control and Six3neo/−;Ptch1+/− embryos is included (Fig. 4M,N).
From these results we conclude that elevated Shh signaling failed to rescue the alobar HPE-like phenotype of Six3neo/– embryos. However, we could not distinguish whether this failure was caused by the level of Shh activity being too low to compensate for the loss of Six3 in Six3neo/–;Ptch1+/– embryos, or because haploinsufficiency of Six3 causes alobar HPE in an Shh-independent manner.
Six3 directly regulates Foxg1 expression in the anterior neuroectoderm independently of Shh signaling
Wnt, Shh and Fgf signaling play key roles during forebrain patterning (Ohkubo et al., 2002). Recent studies in zebrafish have shown that foxg1 coordinates the activity of the Shh and Wnt/β-catenin signaling pathways during dorsoventral patterning of the telencephalon (Danesin et al., 2009; Sylvester et al., 2013). Briefly, Shh activates foxg1 expression in the ventral forebrain and, in turn, Foxg1 is cell-autonomously required for the specification of the ventral telencephalon; Foxg1 also restricts the dorsal telencephalon by repressing wnt8b expression (Danesin et al., 2009). Carlin et al. (2012) showed that, in zebrafish, six3 cooperates with Hh signaling to promote Foxg1 expression during dorsoventral patterning of the telencephalon. Furthermore, Beccari et al. (2012) showed that, in medaka fish, Six3 directly activates Foxg1 to specify telencephalic fate.
To investigate the molecular mechanisms of Six3 haploinsufficiency-promoted alobar HPE, we compared the expression of Wnt8b, Shh, Fgf8 and Foxg1 in Six3neo/neo (semilobar) and Six3neo/– (alobar) mouse embryos. Shh expression in the midline of the ventral forebrain and Fgf8 expression in the commissural plate were dramatically downregulated in Six3neo/neo and Six3neo/– embryos (Fig. S4, arrows). In E8.5 controls and Six3neo/neo embryos, Wnt8b expression was restricted to the boundary between the forebrain and midbrain (Fig. 5A,B). However, in Six3neo/– embryos, Wnt8b expression was expanded to the rostral end of the anterior neural plate (Fig. 5C). The area of Foxg1 expression in E9.0 Six3neo/neo embryos appeared reduced, most likely because the mutant telencephalon is smaller than that in controls (compare Fig. 5D with E). In Six3neo/– embryos, Foxg1 expression in the telencephalon was dramatically downregulated (Fig. 5F). This downregulation was detected as early as the 7-somite stage (Fig. 6A,B). However, this downregulation is transient. In control embryos at E10.5, Foxg1 is weakly expressed in the dorsal telencephalon but strongly expressed in the ventral telencephalon (Fig. 6C). At this stage, weak Foxg1 expression was also detected in the monotelencephalic vesicle of Six3neo/– embryos (Fig. 6D). This weak Foxg1 expression in the mutant brain is consistent with the low level of Foxg1 expression in the dorsal telencephalon of control embryos and most likely further confirms the dorsal telencephalic identity of the mutant telencephalon. At later stages, Foxg1 expression is strong in the dorsal telencephalon and weak in the ventral telencephalon of control embryos (Fig. 6E,G). At these stages in the Six3neo/– telencephalon, Foxg1 expression also increases gradually in the dorsal telencephalon (Fig. 6F,H, Fig. S6). Similar to the results reported in zebrafish (Carlin et al., 2012), Six3 appears to transiently regulate Foxg1 expression during the early stages of mouse embryonic development.
Fig. 5.

Six3 regulates Foxg1 and Wnt8b expression in a dosage-dependent manner. (A-C) Frontal view of E8.5 control (A), Six3neo/neo (B) and Six3neo/– (C) embryos showing Wnt8b expression. Wnt8b labels the forebrain and midbrain boundary in control and Six3neo/neo embryos (A,B, arrowheads). By contrast, Wnt8b expression expands into the rostral end of the anterior neural plate in Six3neo/– embryos (C, arrowheads). (D-F) Lateral view of E9.0 control (D), Six3neo/neo (E) and Six3neo/– (F) embryos showing Foxg1 expression. Foxg1 is expressed in the telencephalon of control and Six3neo/neo embryos (D,E, arrowhead). By contrast, Foxg1 expression is dramatically downregulated in Six3neo/– embryos (F, arrowhead). n=3 for A-C,E; n=4 for D,F.
Fig. 6.
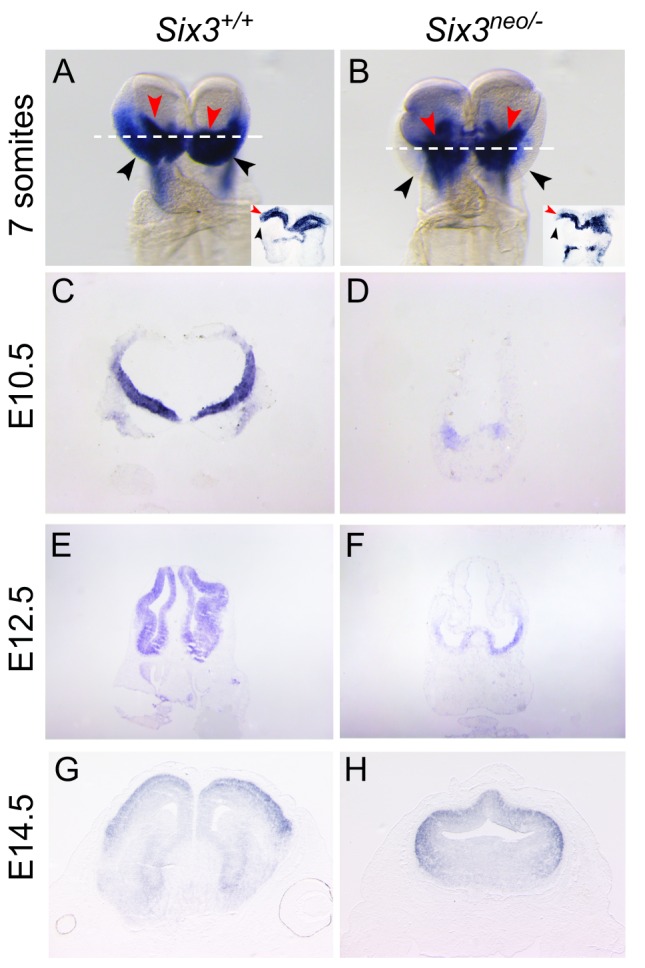
Foxg1 expression is downregulated in Six3 mutant embryos. Foxg1 expression during development (A-H). Frontal view of 7-somite embryos (A,B). Foxg1 is expressed in the anterior neuroectoderm (ANE) (black arrowheads) and the surface ectoderm beneath the neural plate (red arrowheads) of control embryos (A). Foxg1 expression in the ANE is significantly reduced in Six3neo/– embryos (black arrowheads); however, Foxg1 levels in the surface ectoderm are comparable to that of controls (B, red arrowheads). Insets illustrate the transverse sections of the stained embryos at the level indicated by the white dotted line. (C-F) Foxg1 expression in the frontal telencephalon at E10.5 and E12.5 is lower in Six3neo/– embryos than in Six3+/+ littermates. (G,H) Foxg1 expression levels appear normal in coronal sections of telencephalon of E14.5 Six3neo/– embryos.
How does Six3 regulate Foxg1 expression? One possibility is that, as suggested by the zebrafish studies, Six3 and Shh cooperate to regulate Foxg1 expression (Carlin et al., 2012); namely, Six3 gives the anterior neural plate the competence to respond to Shh signaling from the prechordal plate and activates Foxg1 expression. Alternatively, Six3 regulates Foxg1 expression independently of Shh, either directly, as in medaka fish, or through an intermediate player (Beccari et al., 2012). To distinguish between these two possibilities, we performed a detailed molecular analysis of early stage Shh null embryos.
At E8.5, Six3 was expressed at comparable levels in the anterior neural plate of control and Shh null embryos (Fig. 7A,B). Consistent with the role of Six3 in restricting Wnt1 expression, Wnt1 was expressed at the boundary of the forebrain and midbrain in control and Shh null embryos (Fig. 7C,D). Therefore, the anterior-posterior patterning of the neural tube is not defective in Shh null embryos, and Six3 expression in the ANE is not regulated by Shh. Surprisingly, Foxg1 expression in the ANE was also unaffected in Shh null embryos, although the midline of the ANE did not form properly (Fig. 7E,F). Consistent with the role of Foxg1 in restricting Wnt8b expression, Wnt8b expression was retained at the boundary of the forebrain and midbrain in controls and Shh null embryos (Fig. 7G,H). Thus, unlike in zebrafish, Six3 appears to regulate Foxg1 expression independently of Shh during patterning of the mouse forebrain.
Fig. 7.
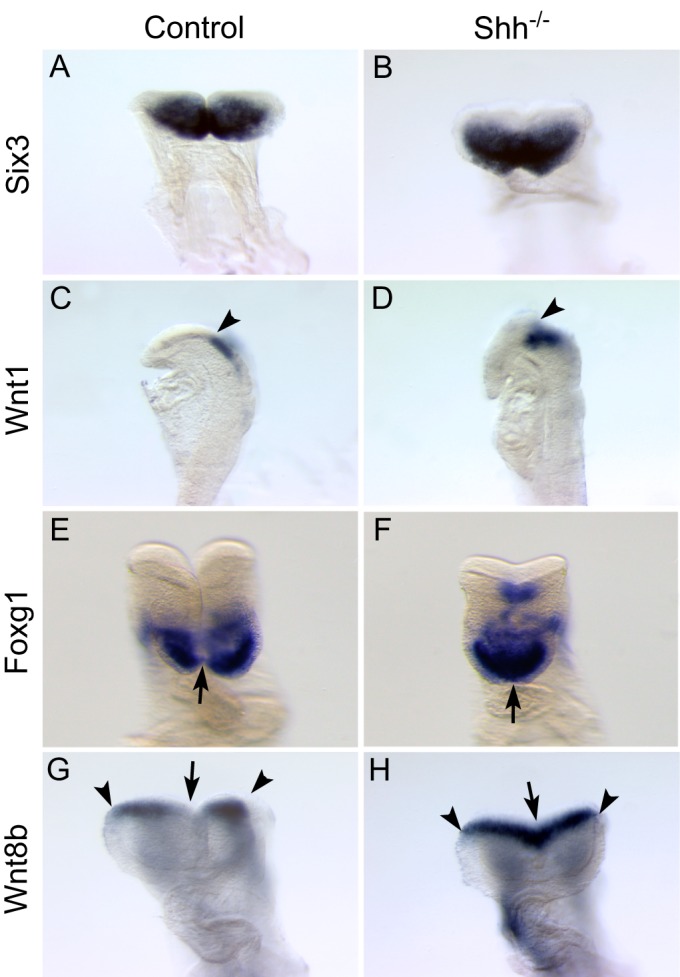
Shh is not necessary to regulate Foxg1 expression in the telencephalon. E8.5 control (A,C,E,G) and Shh–/– (B,D,F,H) embryos were analyzed for the expression of Six3 (A,B) and its target genes Wnt1 (C,D), Foxg1 (E,F) and Wnt8b (G,H). Although the midline remains fused in Shh–/– embryos (F,H, arrows), no obvious changes in gene expression are observed. Six3 and Foxg1 are expressed in the ANE of Shh–/– embryos (B,F) at a comparable level to that of control embryos (A,E). Consistently, Wnt1 and Wnt8b expression is restricted to the forebrain/midbrain boundary of control (C,G, arrowheads) and mutant (D,H, arrowheads) embryos. A, n=4; B, n=3.
In medaka fish, the Six3 homolog six3.2 directly regulates Foxg1 expression (Beccari et al., 2012). To evaluate whether this regulation is evolutionarily conserved in mammals, we performed ChIP analysis in E8.5-E9 (5- to 16-somite) mouse embryos. Computational analysis identified a putative Six3 binding site 1.5 kb upstream of the 5′ UTR, in a highly conserved region (Fig. 8A). qPCR analysis of Six3-immunoprecipitated Foxg1 DNA fragments revealed a 3-fold enrichment in the head versus trunk chromatin (Fig. 8B). No significant enrichment was observed in a similar analysis using the Gapdh promoter as a negative control (Fig. 8B). To evaluate the functional relevance of Six3 binding to the Foxg1 promoter, we cloned a 90 bp Foxg1 promoter element containing a Six3 binding site into the pGL4.10-basic vector (Foxg1-luc) and this luciferase reporter construct was then co-transfected together with a plasmid expressing Six3 (pCAB-Six3). We found that Six3 activates the luciferase reporter in a dosage-dependent manner (Fig. 8C). To further confirm that Six3 functions as a transcriptional activator and activates Foxg1 expression directly, we co-transfected Foxg1-luc together with plasmids that express wild-type (pCAB-Six3), an activator form (pCAB-Six3VP16) or a repressor form (pCAB-Six3EnR) of Six3 (Liu et al., 2006). As expected, Six3VP16 enhances the activation of Foxg1-luc more than 3.5-fold compared with wild type (Fig. 8D). Together, these results suggest that Six3 directly activates early Foxg1 expression in mammals.
Fig. 8.

Six3 directly regulates Foxg1 expression. (A) Alignment of the Foxg1 DNA sequences in different vertebrates reveals several conserved regions. The putative Six3 binding site identified in one conserved region 1.5 kb upstream of the 5′ UTR is indicated by the arrow and nucleotide sequence in red. (B) DNA enrichment after Six3 immunoprecipitation of Foxg1 DNA isolated from heads and trunks of 5- to 16-somite staged embryos. Gapdh promoter primers were used as a negative control. All samples were normalized with the input. (C,D) Six3 binding to the Foxg1 enhancer element as shown by activation of luciferase (Foxg1-Luc). (C) Luciferase activity was augmented significantly (***P<0.0001, ANOVA) as the concentration of the Six3 coding vector was increased. (D) The activator form of Six3 (Six3-VP16) induced a more significant increase (3.6-fold, **P<0.001, t-test) in luciferase activity than the normal reporter. No changes in luciferase activity were observed in the presence of a repressor form of Six3 (Six3-enR). Error bars indicate s.d.
DISCUSSION
Here we describe two new mouse models of HPE: Six3neo/neo embryos, which exhibit semilobar HPE-like phenotypes; and Six3neo/– embryos, which exhibit alobar HPE-like phenotypes. Consistent with our previous results, Six3 and Shh cooperated in the pathogenesis of semilobar HPE. We also showed that increasing Shh signaling is sufficient to rescue the semilobar HPE-like phenotype; however, it failed to rescue the alobar HPE-like phenotypes. In the latter mutant embryos, Foxg1 expression was severely reduced in the ANE; thus, we determined that Six3 directly regulates Foxg1 expression in that region of the developing brain.
An Shh-independent pathway regulates dorsoventral patterning of the mammalian telencephalon
Similar to Six3neo/– embryos, Shh null embryos exhibit alobar HPE-like phenotypes, and the expression of ventral telencephalic markers is substantially reduced (Chiang et al., 1996). Gli3 represses Shh signaling and is required for the development of the dorsal telencephalon (Tole et al., 2000). Interestingly, dorsoventral patterning of the telencephalon is mostly restored in Shh–/–;Gli3–/– embryos (Rallu et al., 2002). These results suggest that Shh and Gli3 regulate dorsoventral patterning of the telencephalon through mutual inhibition. They also argue that an Shh-independent pathway contributes to dorsoventral patterning of the telencephalon. In this study, we showed that Foxg1 expression in mammals is independent of Shh signaling and is directly regulated by Six3. Previous studies have shown that Foxg1 is cell-autonomously required for ventral telencephalon development (Manuel et al., 2010; Martynoga et al., 2005). Therefore, we propose that Six3 regulation of Foxg1 mediates dorsoventral patterning of the telencephalon in an Shh-independent pathway. The Fgf signaling pathway has also been suggested to promote ventral telencephalic specification independently of Shh (Gutin et al., 2006). Mutations in FGF8 and FGFR1 have been identified in human patients with HPE (McCabe et al., 2011; Simonis et al., 2013). Fgf signaling is both necessary and sufficient for Foxg1 expression in the ANE (Paek et al., 2009; Shimamura and Rubenstein, 1997). However, Fgf8 activates Foxg1 expression only in the ANE region where Six3 is normally expressed. In the posterior neural ectoderm, Fgf8 activates En2 expression instead (Crossley et al., 1996; Shimamura and Rubenstein, 1997).
Taken together, these results raise the possibility that in mammals Six3 co-operates with Fgf signaling to activate Foxg1 expression in the ANE and, subsequently, to specify the ventral telencephalon. Recently, Aguiar et al. (2014) showed that inhibition of Bmp signaling, via Bmp inhibitors secreted by the facial neural crest (FNC), is required for Foxg1 expression in the chicken telencephalon (Aguiar et al., 2014). It will be interesting to determine whether BMP inhibitors originating in the FNC are also required for Foxg1 expression in mammals.
Six3 dosage during mammalian forebrain development
Six3 functions as a transcriptional activator and as a repressor. It plays multiple roles at different time points during the development of the vertebrate forebrain (Fig. 9A). Six3 expression in the ANE starts as early as E6.5 and regulates forebrain patterning by directly repressing Wnt1 expression (Lagutin et al., 2003). Here we show that Six3 directly activates Foxg1 expression in the ANE. Foxg1 controls the growth of the telencephalon by regulating the expression of Fgf8 and Bmp4 (Martynoga et al., 2005). Six3 and Foxg1 also restrict dorsal telencephalon by directly repressing Wnt8b (Danesin et al., 2009; Carlin et al., 2012; Liu et al., 2010), while Six3 cooperation with Shh signaling from the prechordal plate activates Shh expression in the RDVM (Geng et al., 2008). Shh signaling from the RDVM subsequently regulates the formation of three signaling centers along the midline of the telencephalon, ensuring the proper patterning and morphogenesis of the telencephalon (Geng et al., 2008).
Fig. 9.

Model for the dosage effect of Six3 during normal and pathological forebrain development. (A) Six3 serves multiple roles during mammalian forebrain development, as a transcriptional activator and repressor of major signaling pathways. (B) Dosage effect of Six3 in the pathogenesis of HPE. Depending on the amount of functional Six3 in the embryo, the brain phenotype can range from normal, semilobar HPE, alobar HPE to aprosencephaly/atelencephaly.
Our results indicate that the downstream targets of Six3 have different sensitivities to Six3 dosage (Fig. 9B). Among these targets, Shh is the most sensitive to reduced levels of Six3, a result consistent with our observation that Six3 expression is enriched in the RDVM and colocalizes with Shh expression (Geng et al., 2008). When Six3 activity is 100% (Six3+/+) or 50% (Six3+/–, Six3+/ki or Six3+/ki;Shh+/–) in an outbred background, the telencephalon develops normally. However, when Six3 activity is reduced to 27% (Six3neo/neo) in an outbred background, or to 50% (Six3+/–, Six3+/ki or Six3+/ki;Shh+/–) in an inbred background, haploinsufficiency of Six3 fails to activate Shh expression in the RDVM, resulting in semilobar HPE (Geng et al., 2008). This form of HPE can be rescued by elevating Shh signaling (Six3neo/neo;Ptch1+/–). However, when Six3 levels are reduced even further (∼13.5% of total Six3 mRNA in Six3neo/–), Six3 activity is not sufficient to activate Foxg1 expression in the ANE. As a consequence of the reduced Six3 and Foxg1 levels, Wnt8b expression is expanded ventrally and alobar HPE eventually develops in the mutant embryos. When Six3 activity is absent (Six3–/–) or almost absent (Six3ki/ki), Wnt1 expression expands anteriorly and the telencephalon of the mutant embryos fails to form. In Six3ki/ki embryos, the diencephalon is properly developed (atelencephaly); however, in Six3 null embryos the rostral diencephalon is truncated (aprosencephaly) (Geng et al., 2008; Lagutin et al., 2003; Lavado et al., 2008). When the level of Six3 expression is more than 50% but less than 100%, it most likely produces the milder lobar HPE phenotype. However, patients with lobar HPE often exhibit no obvious craniofacial defects; therefore, we might not have identified mice with that form of HPE.
We want to emphasize that the dosage of Six3 that we described here was determined according to Six3 mRNA levels. We suspect that a corresponding reduction occurs at the protein level as well. However, we have not been able to quantify the protein owing to the limited number of Six3-expressing cells in early somite stage embryos.
Foxg1 as a candidate gene for screening HPE-associated mutations
Although the severe downregulation of Foxg1 contributes to the alobar HPE phenotypes observed in Six3neo/– embryos, Foxg1 null embryos failed to fully recapitulate the alobar HPE-like phenotypes of Six3neo/– embryos (Xuan et al., 1995). Similar to Six3neo/– embryos, the ventral telencephalon in Foxg1 null embryos is not specified, and the dorsal midline structures (i.e. hippocampus and choroid plexus) are enlarged and fail to fold inwards (Martynoga et al., 2005; Muzio and Mallamaci, 2005; Xuan et al., 1995). However, the cerebral hemispheres are well separated in Foxg1 null mice (Xuan et al., 1995). Together, the alobar HPE phenotypes of Six3neo/– embryos are most likely caused by the combination of Foxg1 downregulation in the anterior neural plate and Shh reduction in the RDVM. Therefore, mutations in Foxg1 alone are unlikely to result in HPE (Santen et al., 2012). Screening additional FOXG1 mutations in cases of familial HPE might shed additional light on the pathological variability of HPE.
Pathological variability of HPE
Pathological variability is a feature of HPE. Ming and Muenke (2002) proposed that intrafamiliar variability in human HPE is due to multiple genetic hits. Our results strongly support this proposal. In the case of familial HPE associated with SIX3 mutations, additional mutations in SHH, PTCH1 or FOXG1, or mutations in the SIX3 regulatory region will lead to different forms of HPE. Therefore, identifying such additional mutations could define the pathologic variability of HPE. HPE is a multifactorial disease. Besides genetic modifiers, environmental factors also contribute to the phenotype both in human patients and animal models (Ming and Muenke, 2002). Important work has been done to determine how environmental exposure to teratogens affects HPE phenotypes in mouse models predisposed to HPE (Billington et al., 2015; Hong and Krauss, 2012; Lipinski et al., 2010). In summary, our study reveals some of the molecular mechanisms behind the pathological variability of HPE. We conclude that the dosage of Six3 is critical for the pathogenesis of various forms of HPE.
MATERIALS AND METHODS
Mice
NMRI mice were purchased from the Jackson Laboratory. Ptch1+/– mice were provided by Dr Peter McKinnon (St Jude Children's Research Hospital). Six3+/– mice have been reported previously (Lagutin et al., 2003). To generate the Six3+/neo mice, we generated a targeting vector containing an Avi tag in frame with Six3 (de Boer et al., 2003). An extrinsic HindIII site was also introduced into the targeting vector for genotyping convenience. A Neo/TK selection cassette flanked by two loxP sites was inserted at the SacI site upstream of Six3 exon 1. W9.5 129/Sv embryonic stem cells were electroporated with this targeting vector and selected by standard procedures. Positive clones were used to generate chimeras by blastocyst injection. All animal experiments were approved by the St. Jude Children's Research Hospital Animal Care and Use Committee.
Scanning electron microscopy
E11.5 embryos were fixed in 2% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) overnight at 4°C. The embryos were washed and postfixed in 1% osmium tetroxide in 0.1 M cacodylate buffer for 2 h and subsequently dehydrated in a graded ethanol series. The embryos were further dehydrated using a Tousimis Samdri 790 critical point dryer. Dry embryos were sputter-coated with Au/Pd particles (EMS 550X Sputter Coater) and observed under a scanning electron microscope (FEI XL30 ESEM).
Quantitative analysis of Six3 transcripts
The yolk sac of a 0-somite staged embryo was collected for genotyping. Embryos were lysed in TRIzol reagent (Invitrogen). After genotyping, only Six3+/neo samples were processed for RNA extraction. The cDNA was synthesized using an Advantage RT-for-PCR Kit (Clontech) and processed for TaqMan-based real-time PCR (Applied Biosystems). To distinguish Avi-Six3 transcripts from wild-type Six3 transcripts, two sets of primers and probes were designed. One set detected Six3 and Avi-Six3 transcripts: probe, 5′-CAAACTTCGCCGATTCTCACCACTGCT-3′; forward primer, 5′-TCTCTATTCCTCCCACTTCTTGTTG-3′; and reverse primer, 5′-GCCGCTACTCGCCAGAAGTA-3′. The other set was designed to detect only Avi-Six3 transcripts (i.e. the forward primer was specific for the Avi tag sequence inserted into the Six3 locus): probe, 5-CAAGCTTGGCATGGTATTCCGCTCC-3′; forward primer, 5′-ATCGAGTGGCATGAGAACCTGTA-3′; reverse primer, 5′-ACAAGAAGTGGGAGGAATAGAGATCT-3′. Both probes were labeled with fluorescein (FAM) at the 5′-end and with BHQ1 dark quencher at the 3′-end. For normalization, β-actin was used as an internal control: probe, 5′-CCCTCCATCGTGCACCGCAA-3′; forward primer, 5′-CAGCAAGCAGGAGTACGATGAG-3′; reverse primer, 5′-CAGTAACAGTCCGCCTAGAAGCA-3′. The β-actin probe was labeled with tetrachlorofluorescein at the 5′-end and BHQ1 dark quencher at the 3′-end.
In situ hybridization and immunohistochemistry
For whole-mount in situ hybridization, embryos were fixed in 4% paraformaldehyde (PFA) at 4°C overnight and processed as previously described (Lagutin et al., 2003). In situ hybridization of tissue sections was performed as previously described (Lavado et al., 2008).
For immunohistochemistry, embryos were collected at the indicated embryonic stages and fixed at room temperature in 4% PFA for 15 min to 1 h depending on the stage. After sucrose cryoprotection, they were embedded in OCT (Tissue-tek, Sakura) and sectioned at 10 µm. DAB immunostaining was performed using rabbit anti-Prox1 (AngioBio, cat. no. 11002; 1:500) as the primary antibody, donkey anti-rabbit biotin-conjugated secondary antibody (Jackson Laboratories, cat. no. 711-065-152; 1:250) and diaminobenzidine as a substrate (ABC Kit, Vector Laboratories). Foxg1 immunostaining was performed after blocking for 2 h in 5% BSA, 3% fetal bovine serum and 0.1% Triton X-100 using rabbit anti-Foxg1 (Takara-Clontech, cat. no. M227; 1:100) as the primary antibody. Donkey anti-rabbit Cy3-conjugated secondary antibody (Jackson Laboratories, cat. no. 711-165-152; 1:400) was used for this staining.
Chromatin immunoprecipitation (ChIP)
Briefly, 5- to 16-somite staged embryos were dissected into heads and posterior trunks and fixed separately in 16% formaldehyde for 8 min at room temperature. Fixed tissues were washed once in PBS containing 0.1 M glycine for 5 min. Subsequently, they were washed three times in PBS for 5 min each at room temperature. Washed tissues were lysed separately in lysis buffer (LowCell# ChIP Kit, Diagenode). Chromatin was sonicated for 18 min in a Diagenode Bioruptor. ChIP was performed using the LowCell# ChIP Kit with rabbit anti-Six3 antibody (Geng et al., 2008); rabbit anti-IgG antibody (Santa Cruz, Diagenode, cat. no. C15400001; 1 µl) was used as the control. The chromatin was reverse crosslinked, purified using the iPure Kit (Diagenode), and processed for SYBR Green real-time PCR analysis (Life Technologies). Primers used to amplify the Six3 binding site in the Foxg1 regulatory region were 5′-TCGAAATGATTCTGTTTCTT-3′ and 5′-CTGATTGCATGCTCATAA-3′. Primers used to amplify Gapdh were 5′-ACCAGGGAGGGCTGCAGTCC-3′ and 5′-TCAGTTCGGAGCCCACACGC-3′. ChIP was repeated four times, with an average 3.3-fold enrichment in the head versus trunk samples.
Luciferase assay
The 293T HEK cell line was maintained in DMEM with 10% fetal bovine serum. 24 h prior to transfection, 0.5-1×105 cells/well were seeded into a 24-well plate. Cells were transfected at 70% confluency with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instruction. 100 ng Foxg1-luc, 1 ng pGL4.76 renilla luciferase (as an internal control) and 10, 100, 250 or 500 ng pCAB-Six3 were co-transfected. Cells were harvested 48 h after transfection. Dual-Glo Luciferase Assay (Promega) was performed on the cell lysates according to the manufacturer's protocol. To validate that Six3 functions as a transcriptional activator, 100 ng Foxg1-luc, 1 ng pGL4.76 renilla luciferase and 250 ng pCAB-Six3, pCAB-Six3VP16 or pCAB-Six3EnR were co-transfected and analyzed as described previously (Liu et al., 2006).
Acknowledgements
We thank L. Boykins for assistance with scanning electron microscopy studies, Y. Lee for technical assistance with real-time PCR analysis, and Angela McArthur for scientific editing of the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
X.G. and G.O. designed the project and co-wrote the manuscript; X.G. and S.A. performed most of the experiments; HJ.G. contributed to the luciferase assay experiments; and O.L. generated the different Six3 strains.
Funding
This work was supported a National Institutes of Health grant [EY12162 to G.O.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.132142.supplemental
References
- Aguiar D. P., Sghari S. and Creuzet S. (2014). The facial neural crest controls fore- and midbrain patterning by regulating Foxg1 expression through Smad1 activity. Development 141, 2494-2505. 10.1242/dev.101790 [DOI] [PubMed] [Google Scholar]
- Beccari L., Conte I., Cisneros E. and Bovolenta P. (2012). Sox2-mediated differential activation of Six3.2 contributes to forebrain patterning. Development 139, 151-164. 10.1242/dev.067660 [DOI] [PubMed] [Google Scholar]
- Billington C. J. Jr, Schmidt B., Marcucio R. S., Hallgrimsson B., Gopalakrishnan R. and Petryk A. (2015). Impact of retinoic acid exposure on midfacial shape variation and manifestation of holoprosencephaly in Twsg1 mutant mice. Dis. Model. Mech. 8, 139-146. 10.1242/dmm.018275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carl M., Loosli F. and Wittbrodt J. (2002). Six3 inactivation reveals its essential role for the formation and patterning of the vertebrate eye. Development 129, 4057-4063. [DOI] [PubMed] [Google Scholar]
- Carlin D., Sepich D., Grover V. K., Cooper M. K., Solnica-Krezel L. and Inbal A. (2012). Six3 cooperates with Hedgehog signaling to specify ventral telencephalon by promoting early expression of Foxg1a and repressing Wnt signaling. Development 139, 2614-2624. 10.1242/dev.076018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C., Litingtung Y., Lee E., Young K. E., Corden J. L., Westphal H. and Beachy P. A. (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407-413. 10.1038/383407a0 [DOI] [PubMed] [Google Scholar]
- Cohen M. M., Jr (2006). Holoprosencephaly: clinical, anatomic, and molecular dimensions. Birth Defects Res. A Clin. Mol. Teratol 76, 658-673. 10.1002/bdra.20295 [DOI] [PubMed] [Google Scholar]
- Crossley P. H., Martinez S. and Martin G. R. (1996). Midbrain development induced by FGF8 in the chick embryo. Nature 380, 66-68. 10.1038/380066a0 [DOI] [PubMed] [Google Scholar]
- Danesin C., Peres J. N., Johansson M., Snowden V., Cording A., Papalopulu N. and Houart C. (2009). Integration of telencephalic Wnt and hedgehog signaling center activities by Foxg1. Dev. Cell 16, 576-587. 10.1016/j.devcel.2009.03.007 [DOI] [PubMed] [Google Scholar]
- Davis N., Yoffe C., Raviv S., Antes R., Berger J., Holzmann S., Stoykova A., Overbeek P. A., Tamm E. R. and Ashery-Padan R. (2009). Pax6 dosage requirements in iris and ciliary body differentiation. Dev. Biol. 333, 132-142. 10.1016/j.ydbio.2009.06.023 [DOI] [PubMed] [Google Scholar]
- Davis-Silberman N., Kalich T., Oron-Karni V., Marquardt T., Kroeber M., Tamm E. R. and Ashery-Padan R. (2005). Genetic dissection of Pax6 dosage requirements in the developing mouse eye. Hum. Mol. Genet. 14, 2265-2276. 10.1093/hmg/ddi231 [DOI] [PubMed] [Google Scholar]
- de Boer E., Rodriguez P., Bonte E., Krijgsveld J., Katsantoni E., Heck A., Grosveld F. and Strouboulis J. (2003). Efficient biotinylation and single-step purification of tagged transcription factors in mammalian cells and transgenic mice. Proc. Natl. Acad. Sci. USA 100, 7480-7485. 10.1073/pnas.1332608100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X. and Oliver G. (2009). Pathogenesis of holoprosencephaly. J. Clin. Invest. 119, 1403-1413. 10.1172/JCI38937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X., Speirs C., Lagutin O., Inbal A., Liu W., Solnica-Krezel L., Jeong Y., Epstein D. J. and Oliver G. (2008). Haploinsufficiency of Six3 fails to activate Sonic hedgehog expression in the ventral forebrain and causes holoprosencephaly. Dev. Cell 15, 236-247. 10.1016/j.devcel.2008.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich L. V., Milenković L., Higgins K. M. and Scott M. P. (1997). Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109-1113. 10.1126/science.277.5329.1109 [DOI] [PubMed] [Google Scholar]
- Gutin G., Fernandes M., Palazzolo L., Paek H., Yu K., Ornitz D. M., McConnell S. K. and Hébert J. M. (2006). FGF signalling generates ventral telencephalic cells independently of SHH. Development 133, 2937-2946. 10.1242/dev.02465 [DOI] [PubMed] [Google Scholar]
- Hong M. and Krauss R. S. (2012). Cdon mutation and fetal ethanol exposure synergize to produce midline signaling defects and holoprosencephaly spectrum disorders in mice. PLoS Genet. 8, e1002999 10.1371/journal.pgen.1002999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M. and Krauss R. S. (2013). Rescue of holoprosencephaly in fetal alcohol-exposed Cdon mutant mice by reduced gene dosage of Ptch1. PLoS ONE 8, e79269 10.1371/journal.pone.0079269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi M. and McMahon A. P. (2002). A sonic hedgehog-dependent signaling relay regulates growth of diencephalic and mesencephalic primordia in the early mouse embryo. Development 129, 4807-4819. [DOI] [PubMed] [Google Scholar]
- Jeong Y., Leskow F. C., El-Jaick K., Roessler E., Muenke M., Yocum A., Dubourg C., Li X., Geng X., Oliver G. et al. (2008). Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat. Genet. 40, 1348-1353. 10.1038/ng.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecker C. and Lumsden A. (2004). Hedgehog signaling from the ZLI regulates diencephalic regional identity. Nat. Neurosci. 7, 1242-1249. 10.1038/nn1338 [DOI] [PubMed] [Google Scholar]
- Lacbawan F., Solomon B. D., Roessler E., El-Jaick K., Domene S., Velez J. I., Zhou N., Hadley D., Balog J. Z., Long R. et al. (2009). Clinical spectrum of SIX3-associated mutations in holoprosencephaly: correlation between genotype, phenotype and function. J. Med. Genet. 46, 389-398. 10.1136/jmg.2008.063818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagutin O. V., Zhu C. C., Kobayashi D., Topczewski J., Shimamura K., Puelles L., Russell H. R. C., McKinnon P. J., Solnica-Krezel L. and Oliver G. (2003). Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes Dev. 17, 368-379. 10.1101/gad.1059403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavado A., Lagutin O. V. and Oliver G. (2008). Six3 inactivation causes progressive caudalization and aberrant patterning of the mammalian diencephalon. Development 135, 441-450. 10.1242/dev.010082 [DOI] [PubMed] [Google Scholar]
- Lipinski R. J., Godin E. A., O'Leary-Moore S. K., Parnell S. E. and Sulik K. K. (2010). Genesis of teratogen-induced holoprosencephaly in mice. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 29-42. 10.1002/ajmg.c.30239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Lagutin O. V., Mende M., Streit A. and Oliver G. (2006). Six3 activation of Pax6 expression is essential for mammalian lens induction and specification. EMBO J. 25, 5383-5395. 10.1038/sj.emboj.7601398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Lagutin O., Swindell E., Jamrich M. and Oliver G. (2010). Neuroretina specification in mouse embryos requires Six3-mediated suppression of Wnt8b in the anterior neural plate. J. Clin. Invest. 120, 3568-3577. 10.1172/JCI43219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuel M., Martynoga B., Yu T., West J. D., Mason J. O. and Price D. J. (2010). The transcription factor Foxg1 regulates the competence of telencephalic cells to adopt subpallial fates in mice. Development 137, 487-497. 10.1242/dev.039800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynoga B., Morrison H., Price D. J. and Mason J. O. (2005). Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev. Biol. 283, 113-127. 10.1016/j.ydbio.2005.04.005 [DOI] [PubMed] [Google Scholar]
- McCabe M. J., Gaston-Massuet C., Tziaferi V., Gregory L. C., Alatzoglou K. S., Signore M., Puelles E., Gerrelli D., Farooqi I. S., Raza J. et al. (2011). Novel FGF8 mutations associated with recessive holoprosencephaly, craniofacial defects, and hypothalamo-pituitary dysfunction. J. Clin. Endocrinol. Metab. 96, E1709-E1718. 10.1210/jc.2011-0454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier S., David V., Ratie L., Gicquel I., Odent S. and Dupe V. (2013). NODAL and SHH dose-dependent double inhibition promotes an HPE-like phenotype in chick embryos. Dis. Model. Mech. 6, 537-543. 10.1242/dmm.010132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming J. E. and Muenke M. (2002). Multiple hits during early embryonic development: digenic diseases and holoprosencephaly. Am. J. Hum. Genet. 71, 1017-1032. 10.1086/344412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenke M. and Cohen M. M. Jr (2000). Genetic approaches to understanding brain development: holoprosencephaly as a model. Ment. Retard. Dev. Disabil. Res. Rev. 6, 15-21. 10.1002/(SICI)1098-2779(2000)6:1<15::AID-MRDD3>3.0.CO;2-8 [DOI] [PubMed] [Google Scholar]
- Muzio L. and Mallamaci A. (2005). Foxg1 confines Cajal-Retzius neuronogenesis and hippocampal morphogenesis to the dorsomedial pallium. J. Neurosci. 25, 4435-4441. 10.1523/JNEUROSCI.4804-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo Y., Chiang C. and Rubenstein J. L. R. (2002). Coordinate regulation and synergistic actions of BMP4, SHH and FGF8 in the rostral prosencephalon regulate morphogenesis of the telencephalic and optic vesicles. Neuroscience 111, 1-17. 10.1016/S0306-4522(01)00616-9 [DOI] [PubMed] [Google Scholar]
- Paek H., Gutin G. and Hebert J. M. (2009). FGF signaling is strictly required to maintain early telencephalic precursor cell survival. Development 136, 2457-2465. 10.1242/dev.032656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rallu M., Machold R., Gaiano N., Corbin J. G., McMahon A. P. and Fishell G. (2002). Dorsoventral patterning is established in the telencephalon of mutants lacking both Gli3 and Hedgehog signaling. Development 129, 4963-4974. [DOI] [PubMed] [Google Scholar]
- Ribeiro L. A., El-Jaick K. B., Muenke M. and Richieri-Costa A. (2006). SIX3 mutations with holoprosencephaly. Am. J. Med. Genet. A 140, 2577-2583. 10.1002/ajmg.a.31377 [DOI] [PubMed] [Google Scholar]
- Roessler E. and Muenke M. (2010). The molecular genetics of holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 52-61. 10.1002/ajmg.c.30236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen G. W. E., Sun Y., Gijsbers A. C. J., Carre A., Holvoet M., Haeringen A., Lesnik Oberstein S. A. J., Tomoda A., Mabe H., Polak M. et al. (2012). Further delineation of the phenotype of chromosome 14q13 deletions: (positional) involvement of FOXG1 appears the main determinant of phenotype severity, with no evidence for a holoprosencephaly locus. J. Med. Genet. 49, 366-372. 10.1136/jmedgenet-2011-100721 [DOI] [PubMed] [Google Scholar]
- Schedl A., Ross A., Lee M., Engelkamp D., Rashbass P., van Heyningen V. and Hastie N. D. (1996). Influence of PAX6 gene dosage on development: overexpression causes severe eye abnormalities. Cell 86, 71-82. 10.1016/S0092-8674(00)80078-1 [DOI] [PubMed] [Google Scholar]
- Shimamura K. and Rubenstein J. L. (1997). Inductive interactions direct early regionalization of the mouse forebrain. Development 124, 2709-2718. [DOI] [PubMed] [Google Scholar]
- Simonis N., Migeotte I., Lambert N., Perazzolo C., de Silva D. C., Dimitrov B., Heinrichs C., Janssens S., Kerr B., Mortier G. et al. (2013). FGFR1 mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly. J. Med. Genet. 50, 585-592. 10.1136/jmedgenet-2013-101603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm E. E., Garel S., Borello U., Hebert J. M., Martinez S., McConnell S. K., Martin G. R. and Rubenstein J. L. (2006). Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development 133, 1831-1844. 10.1242/dev.02324 [DOI] [PubMed] [Google Scholar]
- Sylvester J. B., Rich C. A., Yi C., Peres J. N., Houart C. and Streelman J. T. (2013). Competing signals drive telencephalon diversity. Nat. Commun. 4, 1745 10.1038/ncomms2753 [DOI] [PubMed] [Google Scholar]
- Tole S., Ragsdale C. W. and Grove E. A. (2000). Dorsoventral patterning of the telencephalon is disrupted in the mouse mutant extra-toes(J). Dev. Biol. 217, 254-265. 10.1006/dbio.1999.9509 [DOI] [PubMed] [Google Scholar]
- White J. K., Auerbach W., Duyao M. P., Vonsattel J.-P., Gusella J. F., Joyner A. L. and MacDonald M. E. (1997). Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nat. Genet. 17, 404-410. 10.1038/ng1297-404 [DOI] [PubMed] [Google Scholar]
- Xuan S., Baptista C. A., Balas G., Tao W., Soares V. C. and Lai E. (1995). Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron 14, 1141-1152. 10.1016/0896-6273(95)90262-7 [DOI] [PubMed] [Google Scholar]