

Abstract
Ebola viruses and Marburg viruses, members of the filovirus family, are zoonotic pathogens that cause severe disease in people. The Ebola virus epidemic in West Africa, which was first recognized in early 2014, highlights the threat posed by these deadly viruses. Filovirus disease is characterized by uncontrolled virus replication and the activation of damaging host pathways. Underlying these phenomena is the potent suppression of host innate antiviral responses, particularly the type I interferon (IFN) response, which allows high levels of replication. Here we review the mechanisms deployed by filoviruses to block host innate immunity and discuss aspects of virus replication that promote disease.
Introduction
In March 2014, cases of a severe, frequently lethal illness with symptoms that included fever, severe diarrhea and vomiting were reported to be occurring in the West African country of Guinea1. The causative pathogen was determined to be Ebola virus (EBOV), a member of the filovirus family of enveloped, negative-strand RNA viruses noted for their ability to cause outbreaks of severe, often fatal, viral hemorrhagic fevers2,3. Analysis of viral genome sequences and epidemiological investigation of the viruses from Guinea suggested an index case in a 2-year old child in December of 2013 with subsequent transmission of the virus to the prefectures of Guéckédou, Macenta, and Kissidougou1. The outbreak subsequently expanded into neighbouring Sierra Leone and Liberia. Consequent in-depth analysis of 78 EBOV sequences from Sierra Leone, obtained by using deep sequencing approaches, helped trace the spread of EBOV from Guinea and shed further light on transmission dynamics and genetic variation as the virus spread in the human population through mid-June 20144. After a period of exponential spread in West Africa, the transmission of Ebola infections has generally decreased, but has not ended. As of May 10, 2015 the WHO reported a total of 26,759 confirmed, probable and suspected cases of EBOV disease and 11,080 deaths across 9 countries: Guinea, Liberia, Sierra Leone, United States, Mali, Nigeria, Senegal, Spain, and the United Kingdom, although the vast majority of cases have been in West Africa5. This outbreak of Ebola virus disease (EVD) is unique in that it is by far the largest outbreak on record and the first outbreak of EVD in West Africa (see Box 1 for description of past filovirus outbreaks).
Box 1. Filovirus outbreaks – a historical perspective.
The first documented outbreak caused by filoviruses occurred in 1967 in Marburg, Germany when animal staff and lab workers were infected while harvesting tissues from Cercopithecus aethips monkeys. These animals were imported from Uganda to produce kidney cell cultures needed for the production of live poliomyelitis vaccine141,142. In efforts to identify and characterize the infectious agent, plasma from infected guinea pigs were formalinized and analyzed by electron microscopy. The causative agent was identified as a novel virus and named Marburg virus (MARV)142. This pathogen is now classified in the Marburgvirus genus of the filovirus family. In 1976, another outbreak of hemorrhagic fever occurred in northern Zaire, now the Democratic Republic of the Congo (DRC), resulting in 318 cases with an 88% lethality rate. The causative agent of this outbreak was a second filovirus, Ebola virus (EBOV)143. Ebolavirus represents a second genus of the filovirus family. In the same year, a second outbreak of EBOV hemorrhagic fever also occurred in Sudan causing 284 cases with a 53% lethality rate144. This second outbreak was caused by a distinct virus, Sudan ebolavirus (SUDV). In the years since these early outbreaks, small sporadic outbreaks of MARV infection have occurred in Africa with the largest epidemics occurring in 1998 in the DRC, with 141 cases and an 82% lethality rate, and in 2004 in Angola, with 252 cases and a 90% lethality rate145,146. Subsequent outbreaks of EBOV revealed the presence of three additional species with varying pathogenicity in humans. In 1989 Reston ebolavirus (RESTV) was isolated from cynomolgus macaques in the Philippines and was found to be non-pathogenic in humans147. In 1994, the Ivory Coast ebolavirus (now called Tai Forrest ebolavirus; TAFV) was discovered from an infected patient in the Tai Forest reserve in Cote d’Ivoire148. Bundibugyo ebolavirus (BDBV) was discovered in 2007 in Uganda where it caused 149 cases and a 25% lethality rate. Most human cases of Ebola virus disease (EVD) are due to the emergence and re-emergence of EBOV in rural areas of Gabon, Republic of Congo, and DRC and of SUDV in Sudan and Uganda. Until 2014, there have been 1,383 EBOV and 779 SUDV cases, with fatality rates ranging from 50–90% and 40–65%, respectively8. The 2014 West Africa outbreak is unique in that it is the largest outbreak on record to date. The most recently identified filovirus, Lloviu virus (LLOV), which is classified in a distinct filovirus genus Cuevavirus, was identified in bats in Spain7. LLOV has yet to be cultured, but a near full-length viral genomic sequence has been determined from RNA in bat tissue. The pathogenic potential of LLOV is uncertain.
The virus family Filoviridae is divided into three genera, Ebolavirus, Marburgvirus and Cuevavirus6. Within the ebolavirus genus are five species: Zaire ebolavirus (EBOV), Sudan ebolavirus (SUDV), Bundibugyo ebolavirus (BDBV), Tai Forrest ebolavirus (TAFV) and Reston ebolavirus (RESTV). The marburgvirus genus consists of a single species, Marburg marburgvirus (MARV). The genome of another filovirus, Lloviu virus, which is the only member of the Cuevavirus genus, was demonstrated to be present in bats in Northern Spain, but this virus has not yet been cultured7. EBOV is responsible for the current West Africa outbreak and along with SUDV, BDBV and MARV has caused past outbreaks of human disease; TAFV has only been associated with a single, non-fatal human infection8. RESTV appears to originate from the Philippines and has been isolated from non-human primates and pigs; infected non-human primates have been exported from the Philippines to other countries8. Humans exposed to RESTV have not become ill, suggesting that RESTV is significantly attenuated in humans compared to other filoviruses9. Whether Lloviu virus is hazardous to humans is not yet known. As is discussed below, filovirus pathogenesis is best characterized for the EBOV species, particularly in non-human primates. Current models suggest that potent suppression of host innate immune responses by filoviruses promotes high levels of systemic virus replication and that this triggers potent inflammatory responses that lead to severe manifestations of disease such as high fever, vascular leakage and bleeding.
The filoviruses are suspected to use bats as reservoir hosts, although current evidence is stronger for MARV than for EBOV (Box 2). As might be expected for a reservoir host, filoviruses do not appear to be overtly pathogenic in bats, despite the high virulence of these viruses in humans and non-human primates10,11. The absence of fatal disease following filovirus infection is not a property unique to bats. For example, experimentally-infected guinea pigs and mice do not die of filovirus infection, although serial passage of EBOV or MARV in these species can select for viruses that are lethal to these animals12–15. The factors that determine whether or not a filovirus is pathogenic in a particular host are not well-defined, although the type I interferon (IFN) response plays a role in containing infections in mice16.
Box 2. Bats as filovirus reservoir hosts.
Elucidating the reservoir host that harbors filoviruses has long been of interest. The identification of the reservoir might suggest mechanisms that drive the emergence of the viruses into human populations and strategies to avoid human infections. Because it is presumed that the reservoir host does not suffer the extreme mortality seen in humans and non-human primates, studies of virus-reservoir interaction could also provide unique insight into filovirus pathogenesis. However, it is only recently that conclusive evidence has been developed implicating select bat species as natural hosts of Marburg virus.
Circumstances surrounding several Marburg virus outbreaks suggested bats as a possible source of the virus for human infection. For example, bats were one of several animals to which the presumed index case of a small Marburg virus outbreak in 1975 had been exposed149. In several cases, human infections occurred in individuals, including miners and tourists, who had entered caves containing significant bat populations146,150–152. Rousettus aegyptiacus were repeatedly found in these caves153. More direct evidence of Marburg virus infection of bats was the detection of Marburg virus nucleic acid and anti-Marburg virus antibodies in R. aegyptiacus153. However, the conclusive evidence was the isolation of live virus from R. aegyptiacus151. Follow up studies, focused on Python Cave in Uganda where two tourists had become infected previously, found evidence of a seasonal pattern of infections within the bat population154. Consistent with the expected outcome, where infection of a reservoir host should not be consistently lethal, experimental inoculations of filoviruses into bats has not yielded signs of overt disease10,11. Interestingly, current data suggests that Marburg virus infections of R. aegyptiacus are not persistent, but instead are cleared after a period of replication. The animals develop a transient viremia, but mount a detectable antibody response to the infection and the virus is cleared. However, oral shedding as well as replication in multiple bat tissues has been detected. These data suggest potential routes that might lead to spread of infections among bats or to other species, including humans, although transmission between experimentally-infected bats has not been demonstrated10,155.
The evidence for bats as Ebola virus reservoirs is currently based on serology and the presence of Ebola virus-derived RNAs in tissues and sera from bats in both Africa and Asia (see10). The near complete genome of Lloviu virus was characterized from bats in Northern Spain7. The isolation of infectious virus from bats, particularly from healthy bats, would provide stronger evidence that bats are reservoir hosts for Ebola virus and Lloviu virus. Nonetheless, the available data suggest that filoviruses may have a broader geographic distribution than is generally appreciated.
With the identification of bats as being susceptible to Marburg virus infection, but resistant to disease, an understanding of the mechanisms of disease resistance is needed. It will be of significant interest to determine how interactions of filoviruses with the bat innate or adaptive immune responses somehow contribute to disease outcome.
An overview of the filovirus genome and replication cycle is provided in Figure 13. Filoviruses possess single-stranded, negative-sense RNA genomes of approximately 19 kilobases. They have seven distinct genes, each of which functions as a separate transcriptional unit (F). These encode seven viral structural proteins known as nucleoprotein (NP), viral protein (VP) 35, VP40, glycoprotein (GP), VP30, VP24, and the large protein (L). For the ebolaviruses, the GP gene encodes multiple different proteins due to transcriptional editing by the viral RNA-dependent RNA polymerase (RDRP) where non-template encoded “As” are added co-transcriptionally to the nascent mRNA. The unedited transcript encodes sGP, while editing results in translational frameshifting such that the membrane-bound GP and a second secreted GP are produced from edited RNAs3,17,18. Additional editing events recently described for both EBOV and MARV mRNAs may further diversify the proteins translated from filovirus mRNAs19.
Figure 1. Filovirus genome structure and life cycle.
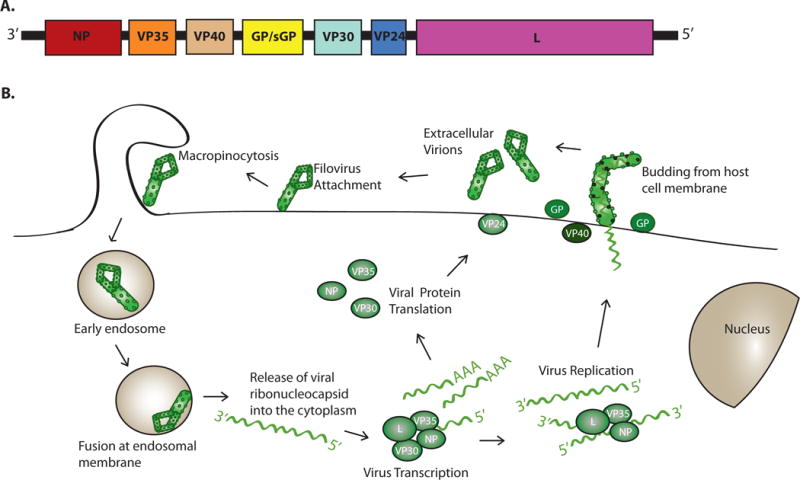
A. The genome of EBOV is depicted. The genome is depicted in the 3′ to 5′ orientation to indicate that the genomic RNA is negative-sense. The genes are named after the proteins encoded by each. They are NP, nucleoprotein; VP35, viral protein 35; VP40, viral protein 40; GP/sGP, glycoprotein/soluble glycoprotein; VP30, viral protein 30; VP24, viral protein 24; L, Large protein (the viral polymerase). Note that Marburg virus does not encode a sGP protein. Filoviruses have unusually long non-coding regions at the 5′ and 3′ end of their mRNAs. The genome regions corresponding to these non-protein coding sequences are not drawn to scale. B. The general life cycle of a filovirus is displayed. First, the filovirus attaches to the cell membrane via its surface GP protein. Virus is taken up by a process known as macropinocytosis38,156–158. Upon acidification of the endosome, cathepsins B and L (cellular proteases) cleave GP33. This allows GP to interact with host protein NPC-1, an interaction which is a prerequisite for fusion of viral and endosomal membranes34,35. Host endosomal calcium channels called two-pore channels (TPCs) play a critical role in the endosomal trafficking of incoming viral particles to the site of fusion159. GP mediates fusion of the viral and the endosomal membrane, releasing the viral ribonucleocapsid into the cytoplasm where the negative strand RNA genome undergoes transcription and replication160. Production of 5′-capped, 3′-polyadenylated mRNAs from individual viral genes occurs and genome replication follows, in which the genomic RNA template is copied into a full-length complement. The full-length complement serves as template for synthesis of additional negative-sense genome. The RNA synthesis reactions require the NP, VP35 and L proteins. Initiation of transcription for Ebola virus also requires the VP30 protein, whereas the replication reactions do not require VP30. Viral proteins, including NP, VP35, VP40, GP, VP30, VP24, and L are translated from the viral mRNAs. New viral particles are assembled at the plasma membrane. The VP40 protein functions as the viral matrix protein and directs budding of particles from the cell surface161–163. GP, a type I transmembrane protein, is incorporated into the budding particles, as are viral nucleocapsids containing the viral genome164.
With the exception of EBOV sGP, each viral protein is present in the enveloped viral particle. GP is a type I transmembrane protein and is the only viral protein exposed on the surface of viral particles. GP mediates entry into host cells, acting as the attachment and fusion protein. Within the cytoplasm, viral RNA synthesis occurs on the uncapped, non-polyadenylated template viral RNA, which is associated with the NP protein. NP, VP35, VP30 and L proteins are all required for viral RNA synthesis. L is the enzymatic component of the RDRP complex and in addition to RDRP activity possesses methyltransferase and guanyltransferase activities needed to 5′ cap viral mRNAs. VP35 acts as a viral polymerase cofactor, interacting with both L and NP, to deliver L to the NP-encapsidated template. VP30 is required for viral transcription (mRNA synthesis). The phosphorylation state of VP30 may also regulate the transcriptase and replicase functions of the viral polymerase complex. Tubular nucleocapsids form in the infected cell cytoplasm, and transfection-based studies indicate that these consist of RNA, NP, VP35 and VP24. The nucleocapsids are delivered to sites of viral assembly and release at the plasma membrane. Virus release occurs by budding and involves interactions of VP40 with components of the cellular ESCRT pathway. The VP40 protein is sufficient to bud on its own but budding efficiency is enhanced by other viral proteins, including GP, which reaches the plasma membrane. As is discussed in detail below, features that contribute to the severity of filovirus infections in humans and non-human primates include robust systemic virus replication and excessive inflammatory responses. This leads to the manifestations of filoviral disease, including high fever, vascular leakage and coagulopathy2. Underlying the uncontrolled replication is a potent suppression of innate antiviral defenses by filovirus gene products20. Below we discuss how several filoviral proteins counteract host defenses. Examples include the EBOV and MARV virus VP35s (eVP35 and mVP35, respectively), EBOV VP24 (eVP24) and MARV VP40 (mVP40) that disable specific innate immune signaling pathways and the GP protein that counteracts the antiviral effects of the cellular protein tetherin21–25. We also describe how these viruses have evolved mechanisms to take advantage of host pathways to increase the life of infected cells and promote optimal levels of viral protein translation.
Filovirus pathogenesis
Human infections are acquired by direct contact with infected bodily fluid and likely enter the human body via breaks in the skin or through mucosal surfaces26. Studies using nonhuman primates, the gold standard animal model for studying EBOV disease, have provided significant insight into the pathogenesis of filovirus infections. Immunohistochemistry and in-situ hybridization demonstrate that filoviruses can infect many cell types including macrophages, monocytes, dendritic cells (DCs), Kuppfer cells in the liver, fibroblasts, hepatocytes, adrenal gland tissue, endothelial cells and epithelial cells27,28. Time-course studies of infection with EBOV show that it preferentially and initially replicates in macrophages and DCs28. This conclusion is consistent with histopathology studies performed on human tissues29. A definitive basis for the preferential infection of these cells is lacking. However, Ebola virus has been demonstrated to effectively infect macrophages and DCs following their differentiation from monocytes30–32. Differentiation correlates with the upregulation of cellular cathepsins and the protein Niemann-Pick C1 (NPC1), factors critical for GP-mediated membrane fusion32–36. Further, specificity for macrophages and DCs may be promoted through the interaction of virus with cell surface attachment receptors including dendritic-cell-specific ICAM-3-grabbing non-integrin (DC-SIGN) and phosphatidylserine binding proteins27,28,37–40. Infection of these mobile cells allows the transport of the virus via lymphatics to lymph nodes and via blood to the liver and spleen, where the virus can infect resident tissue macrophages, including Kuppfer cells and DCs27,28. In vivo, filoviruses trigger the release of proinflammatory cytokines and chemokines, such as IL-1β, IL-6, IL-8, IL-10, MCP-1, MIP-1α, MIP-1β and TNFα, in addition to reactive oxygen species and nitric oxide41–46. This likely reflects production of these factors by infected macrophages43,47–49. Cytokines MIP-1α and MCP-1 can recruit additional macrophages to areas of infection, allowing EBOV to infect more target cells28. Infection of DCs inhibits their maturation and prevents antigen presentation to T cells. In vitro studies have shown that EBOV infection inhibits upregulation of co-stimulatory molecules CD40, CD80, CD86 and MHC class II50–52. These DC-suppressing functions have been linked to suppression of innate immune signaling pathways by viral proteins (see below)50,52–54. Exactly to what extent inhibition of DCs influences adaptive immune responses remains to be seen as EBOV-infected mice and people who survive infection have been demonstrated to develop adaptive immune responses to viral antigens55,56.
In addition to disrupting the innate immune branch, EBOV infection has consequences for B and T cell responses. Lymphopenia has been detected following infection with EBOV and MARV57. For EBOV this occurs most notably among the CD4 T cells, CD8 T cells and NK cell populations in mouse models (2–3 dpi) and in nonhuman primates (4 dpi)58,59. Similarly, EBOV infection results in the loss of human CD4+ and CD8+ T cells in vitro 4 days post infection60. Apoptosis likely contributes to loss of lymphocytes, and several mechanisms have been proposed to explain this phenomenon43,46,57,61–65. The consequences of lymphopenia for pathogenesis are not fully elucidated, and in a mouse model, prevention of lymphocyte apoptosis did not prevent death in EBOV-infected mice62. However, loss of CD4+ T cells, which are required for isotype class switching, could result in the observed absence of EBOV specific IgM and IgG antibodies in case fatalities61. Another way in which EBOV may modulate B cell responses is via the sGP protein, which may redirect GP-specific antibody responses to non-neutralizing epitopes66.
Hemorrhage and intravascular coagulation are features of severe EBOV infection. EBOV-induced paralysis of an effective host response may facilitate viral dissemination to hepatocytes, adrenal cortical cells and endothelial cells of connective tissue28. Hepatocellular necrosis may result in the decreased synthesis of coagulation proteins while adrenocortical infection and necrosis may negatively affect blood pressure homeostasis, potentially leading to hemorrhaging28. The increased vascular permeability and vascular leakage that typically lead to hemorrhagic fever could be attributed to the large release of TNFα from infected macrophages, which can increase endothelial permeability43. Similarly, the release of nitric oxide, which is an important effector molecule in the homeostasis of the cardiovascular system, may result in loss of vascular smooth-muscle tone and hypotension44,67.
More directly, studies in nonhuman primates show that coagulation abnormalities are initiated early during EBOV infection. A dramatic decrease in plasma levels of anticoagulant protein C occurred early in the coagulation cascade (~2 dpi). This was followed by an increase of both tissue plasminogen activator (tPA), which is involved in dissolving blood clots, and fibrin-degradation products (D-dimers) at day 5 post-infection68. In addition, EBOV infection of macrophages leads to the upregulation of tissue factor (TF) on the surface of monocytes and macrophages and the release of membrane microparticles that express TF. These likely promote the over-activation of the extrinsic pathway of coagulation and may promote disseminated intravascular coagulation68. It is postulated that expression levels of TF may be further upregulated by proinflammatory cytokines such as IL-6 that are abundant during acute EBOV, thereby exacerbating the intravascular coagulation phenotype69. Thrombocytopenia and prolonged pro-thrombin time are also indicators of dysregulation of blood coagulation. Fibrinolysis during filovirus infection may manifest as petechiae, ecchymoses, mucosal hemorrhages, and congestion68,70. The compounding effects of the increase of pro-coagulation factors and decrease of anti-coagulant factor Protein C could plausibly explain the activation of coagulation and hemorrhagic features characteristic of filovirus infection. Towards the terminal stage of infection and after the onset of hemorrhagic abnormalities, EBOV replicates in endothelial cells27. However, while endothelial cells are thought to play a role in pathogenesis, the molecular mechanisms that affect endothelial function are not yet fully understood27,71–74.
The lethality of filovirus infection is likely due, in part, to the ability to replicate largely unimpeded by the host innate antiviral responses. This innate immune evasion probably facilitates the excessive virus replication that activates the damaging host responses outlined above. Underlying this robust growth are multiple mechanisms that allow filoviruses to counteract the host innate antiviral defenses, particularly IFN responses20. The type I IFNs (IFNβ and multiple IFNαs) constitute the major components of the innate immune response to viral infections75. IFNα/β expression can be triggered via signaling from pattern recognition receptors, including the RIG-I-like receptors (RLRs), which include RIG-I, MDA5 and LGP2, and select toll-like receptors (TLRs). The RLRs are cytoplasmic RNA helicases that detect viral RNAs and promote IFN-α/β responses. Of these, the RLRs RIG-I and MDA5 appear most relevant to filovirus infection. Activated by “immunostimulatory RNAs,” such as RNAs with 5′-triphosphates and double-stranded RNAs that may be produced during filovirus replication, RIG-I and MDA5 activate the Ser/Thr kinases TBK-1 and IKKε that phosphorylate IRF-3 and IRF-7. This promotes their nuclear accumulation and stimulates IFN-α/β gene expression. When expressed, IFNαβ is secreted from cells and signals via a heterodimeric receptor, the IFN-α/β receptor. This activates a JAK-STAT signaling pathway and induces expression of numerous genes, triggering an antiviral state76.
EBOV and MARV infections effectively counter this critical antiviral defense system, blocking both the production of and the cellular responses to exogenously added IFNs25,47,77–79 (Figure 2). That this inhibition is important is suggested by the facts that EBOV genomic RNA can activate RIG-I, activation of RIG-I signaling before EBOV infection substantially reduced virus growth and recombinant viruses unable to block IFN responses are attenuated both in cell culture and in experimentally-infected animals (discussed further below)16,80–83. The mechanisms by which IFNα may exert its anti-filoviral effects have not been systematically studied, but several proteins encoded by IFN-stimulated genes (ISGs) exert anti-filoviral effects in cell culture. For example, interferon-induced transmembrane (IFITM) proteins inhibit filovirus entry into host cells and ISG15 is reported to impair budding by VP4084,85. Nonetheless, while IFNs can control infection in rodent models, filoviral infections are not controlled by host defenses in non-human primates and death of the host frequently ensues14,16,86,87.
Figure 2. Subversion of IFN induction.
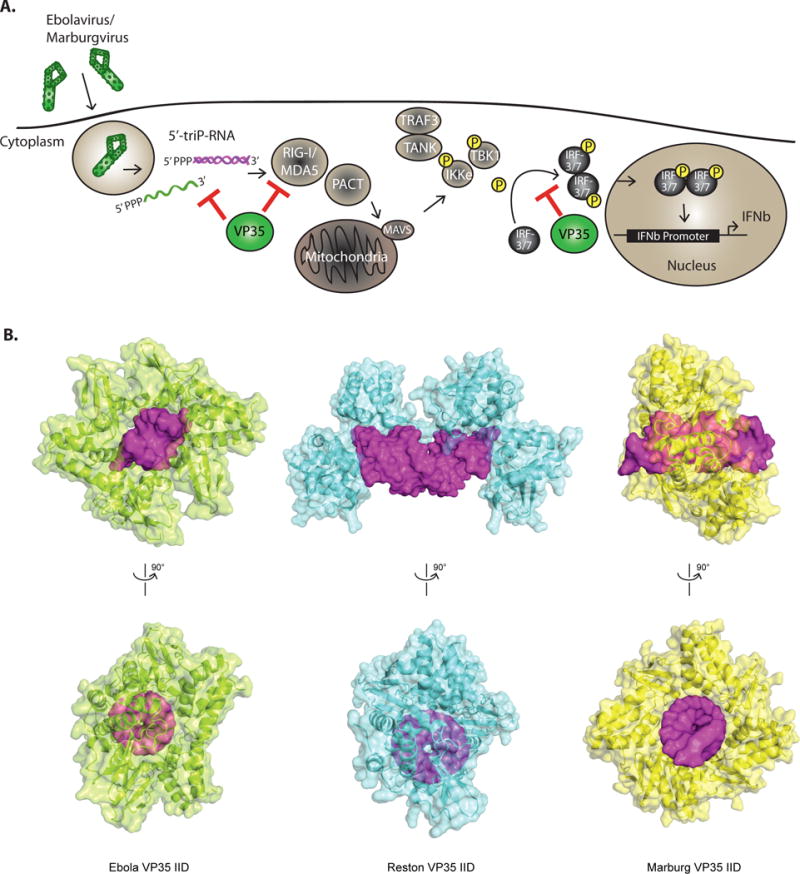
A. Schematic of the IFN production pathway and mechanisms of filoviral evasion. The image depicts how EBOV VP35 (eVP35) and MARV VP35 (mVP35) proteins antagonize signaling pathways that lead to IFN-α/β gene expression. RIG-I-like receptors (RLRs) which include RIG-I and MDA5, detect products of viral replication, RNAs such as cytoplasmic dsRNAs or RNAs with 5′-triphosphates. Activation of RLRs is facilitated by the protein PACT. Upon activation, RLRs signal through the mitochondria-associated protein MAVS to activate kinases IKKε and TBK1. These phosphorylate interferon regulatory factor (IRF)-3 or 7 then accumulate in the nucleus and promote IFN-α/β gene expression. VP35 proteins can bind to dsRNAs and to PACT, preventing RLR activation. In addition, VP35s interact with and act as decoy substrates for the kinases IKKε and TBK1. B. Structural analysis of filoviral VP35 protein interferon inhibitory domain (IID) binding to dsRNAs reveals differences among the dsRNA recognition mode for EBOV, RESTV, and MARV VP35 proteins. (Left) The structure of eVP35 bound to 8 bp dsRNA (PDB 3L25). (Middle) rVP35 bound to 12bp (PDB 4LG2). (Right) mVP35 IID bound to 12 bp dsRNA (PDB 4GHL). dsRNA shown in magenta.
VP35 has multiple roles in suppressing cytosolic sensing
EBOVs and MARVs very effectively impair production and this is largely mediated by the VP35 protein. VP35 is multifunctional, serving not only as an antagonist of the innate immune response, but also functioning as a non-enzymatic cofactor for the viral RNA-dependent RNA polymerase and as a viral structural protein. The VP35 proteins of EBOV and MARV block signaling through RIG-I-like receptors and prevent the phosphorylation of IRF-3 and IRF-7, thereby short-circuiting the IFN response88. These inhibitory activities likely reflect inhibition at both upstream and downstream steps in the RLR signaling cascades. Downstream targets include the kinases IKKε and TBK1; VP35 has been demonstrated to interact with and act as a decoy substrate for these kinases such that IRF-3 interaction with the kinase is blocked and VP35 is phosphorylated instead89. VP35 is also reported to interact with IRF-7, SUMO conjugating enzyme Ubc9 and SUMO E3 protein ligase PIAS1 to promote SUMOylation of IRF-7, a modification that impairs IRF-7-dependent transcriptional responses90,91. Inhibitory activities upstream of RIG-I are connected to the dsRNA binding capacity of VP35, which resides in a carboxy-terminal domain referred to as the interferon inhibitory domain (IID)92. While wild-type VP35 (wtVP35) can bind dsRNA and suppress RLR-mediated activation of IFN responses induced by Sendai virus infection or by transfected dsRNAs, VP35 point mutants that lack dsRNA binding activity are greatly reduced in suppressive activity53,93–95. These data suggest a model where VP35 sequesters RLR-activating dsRNAs. Supporting this view, purified wild-type VP35, but not VP35 dsRNA binding mutants, can prevent dsRNA activation of RIG-I ATPase activity in vitro96.
VP35 was also demonstrated to interact with the cellular protein PACT, a multifunctional protein that was originally described as a protein activator of the IFN-induced antiviral kinase, PKR97. PACT has also been shown to interact with and facilitate the activation of RIG-I98; VP35 blocks this interaction, thereby preventing PACT activation of RIG-I both in cells and in vitro. Interestingly, the interaction of VP35 with PACT can occur in the absence of dsRNA, but requires the same basic patch that mediates interaction with dsRNA96. These basic amino acid residues are capable of participating in either protein-dsRNA or protein-protein interactions. In the X-ray crystal structure of VP35 complexed with dsRNA, these residues were found to contact the RNA phosphodiester backbone and to facilitate VP35-VP35 contacts95,99. VP35 point mutants lacking dsRNA binding activity failed to interact with PACT and failed to block PACT-mediated activation of RIG-I. Demonstrating the relevance of PACT for EBOV infection, PACT over-expression overcomes the inhibitory effects of VP35 and triggers an IFN-response in wild-type EBOV infected cells; on the other hand, the IFN response induced by EBOV carrying a mutated VP35 is decreased in PACT knockdown cells96. In sum, current models suggest that VP35 can inhibit RLR responses both by the sequestration of immunostimulatory dsRNAs and through interaction with PACT. These inhibitory mechanisms rely on the VP35 IID and contribute to the potent suppression of the IFNα/β response in filovirus-infected cells.
In addition to suppressing the production of IFNα/β, VP35 inhibition of RLR signaling suppresses maturation and function of DCs. As noted above, DCs are important targets of filovirus infection in vivo and are productively infected28,50–52. The role of DCs is to stimulate adaptive immune responses upon encountering pathogen100, which requires that the DCs undergo a “maturation” process triggered by innate immune signaling pathways. However, in vitro infection of DCs with filoviruses results in aberrant DC maturation50–52. In contrast, EBOVs with mutant VP35s do induce DC maturation. Further, the expression of eVP35 in DCs recapitulates the suppressive effects of wild-type EBOV infection, with the inhibitory effects being mediated in large part by VP35 RLR inhibitory activity50,52–54,101. Therefore, the impact of VP35 RLR suppression likely extends beyond IFNα/β production and also impairs activation of adaptive immune responses.
Structural determinants of VP35 binding to dsRNA
The dsRNA binding activity of both EBOV and MARV VP35 has been explained by structural and biophysical methods. Overall, VP35 contains two identifiable domains, an N-terminal oligomerization domain and a C-terminal “interferon inhibitory domain (IID).” VP35 residues 220–340 were defined as the minimal region important for dsRNA binding and IFN inhibition using a combination of limited proteolysis and nuclear magnetic resonance (NMR)-based studies92. Consistent with this finding, the C-terminal IID is sufficient for suppression of IFN production, but the addition of the oligomerization domain enhances inhibition102. The structure of the EBOV VP35 IID, which has an alpha-helical subdomain and a beta-sheet subdomain, contains two basic regions. These regions, termed the first basic patch (FBP) and the central basic patch (CBP), are functionally important, with the latter playing a role in dsRNA binding and IFN inhibition92,95,103. Comparison of EBOV, RESTV and MARV VP35 IIDs reveal remarkable similarities in the overall structure as well as the highly conserved molecular surfaces23,24,95,103. Moreover, evaluation of the RESTV and MARV IIDs confirms that the CBP is conserved, whereas EBOV has the most extensive charge on the CBP95,104. In contrast, the FBP residues, which are conserved among ebolaviruses, appear to be important for virus replication, but not for dsRNA binding or IFN inhibition105.
Despite the structural and functional similarities described above, the EBOV and MARV IIDs also show differences in their biochemical and structural characteristics. For example, in vitro EBOV VP35 (eVP35) can bind dsRNA with >8 bp (Figure 2B, left) and RESTV VP35 (rVP35) can bind dsRNA with >12 bp (Figure 2B, middle), whereas MARV VP35 (mVP35) binds to dsRNA with at least 12 bp23 (Figure 2B, right); for dsRNAs >12 bps, eVP35 binds with greater affinity than mVP35. Data also suggests that eVP35 and mVP35 show significant differences in how they bind dsRNA: eVP35 displays two different binding modes, recognizing both blunt ends of dsRNA and the dsRNA backbone, whereas mVP35 IID can only recognize the dsRNA backbone (Figure 2B). Because the 5′-triphosphate plays a critical role in RIG-I activation, the ability of eVP35 to mask dsRNA ends may provide eVP35 with an increased capacity to block IFN production.
Subversion of IFN-induced signaling
In addition to preventing IFNα/β induction, EBOVs and MARV block the cellular response to IFNs. As a consequence, IFN-induced gene expression is substantially impaired in EBOV- and MARV-infected cells78,79. In uninfected cells, IFNα/β addition activates a Jak-STAT signaling pathway in which receptor-associated Jak family kinases Jak1 and Tyk2 are activated. These kinases phosphorylate STAT1 and STAT2 (PY-STAT), which dimerize via phosphotyrosine-SH2 interactions106. The phosphorylation/dimerization reveals a non-conventional nuclear localization signal (ncNLS) on STAT1 that can interact with any of the three members of the NPI-1 subfamily of karyopherin alpha (KPNA) proteins, which comprises KPNA1, 5 and 6107. The interaction of PY-STAT1 with KPNAs mediates the nuclear import of the STAT protein dimers107.
EBOV VP24 interferes with STAT nuclear entry
In EBOV-infected cells, IFNα/β trigger STAT phosphorylation; however, PY-STAT1 nuclear entry is impaired by eVP2422. eVP24 interacts with the same NPI-1 subfamily of KPNAs that are bound by PY-STAT1 and mapping studies suggest that eVP24 occupies the PY-STAT1 binding site on KPNA, thereby acting as a competitive inhibitor of PY-STAT1108. Structural and biochemical studies support this competition model of eVP24 binding to KPNA. eVP24 forms a pyramid-shaped tertiary structure with a series of α-helices resting on a β-sheet pedestal109,110 (Figure 3AB, left). The KPNA proteins possess an N-terminal importin beta binding domain, ten armadillo (ARM) repeats and a C-terminal domain. Whereas proteins with classical, basic nuclear localization signals (cNLSs) bind to the central ARM repeats of KPNAs, PY-STAT interacts via its ncNLS with C-terminal regions of the NPI-1 KPNAs including ARMs 8–10. A recent study generated a stable KPNA construct (KPNA5C), which contains ARMs 7–10111. The structure of KPNA5C in complex with eVP24 (Figure 3B, right) identified three regions within eVP24, termed clusters 1, 2, and 3, that interact with KPNA5C ARMs 8, 9, and 10, respectively. Moreover, residues within KPNA5C identified as critical for eVP24 binding were also important for STAT1-KPNA binding, directly correlating binding with inhibitory activity. Thus, eVP24 targets well-conserved, functionally significant interactions to prevent the host response111. This same study also revealed that the KPNA binding site of monopartite cNLSs is largely unperturbed by eVP24 binding. Therefore, eVP24 may preserve nuclear import of such cargo. However, bipartite cNLSs as well as non-classical NLSs may be affected by eVP24-KPNA binding111,112. Further work will be required to determine if eVP24 mainly affects STAT1 or if import of other cargo is also affected.
Figure 3. Subversion of IFN-induced signaling and of IFN-induced protein tetherin.
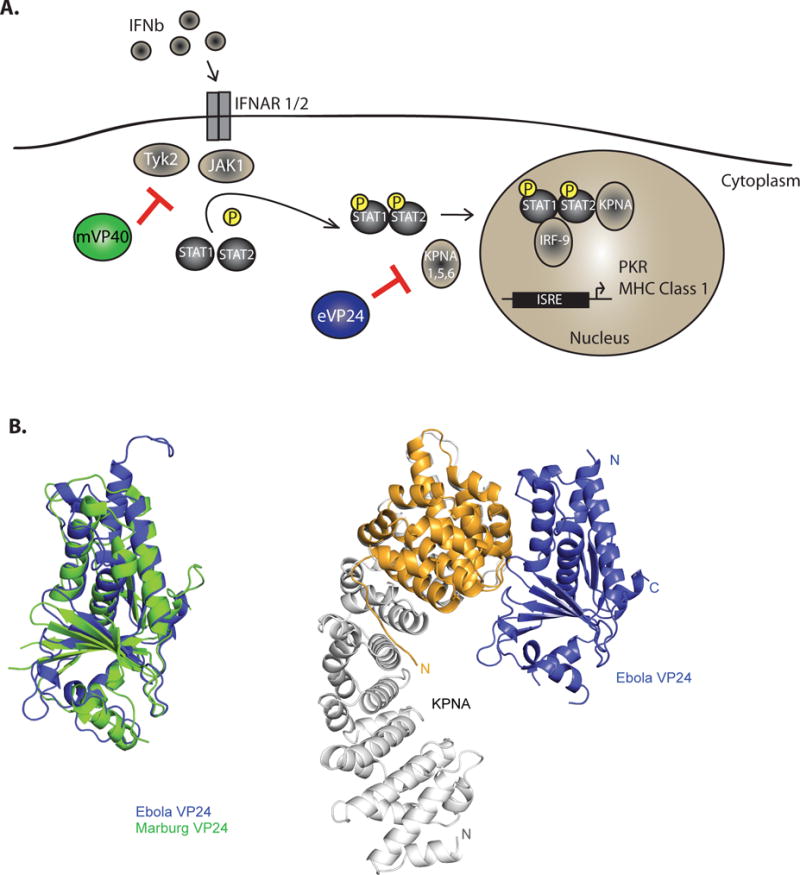
A. Schematic of the IFN response pathway and mechanisms of filoviral evasion. IFN-α/β binds the extracellular domains of the heterodimeric IFN alpha receptor composed of subunits IFNAR1 and IFNAR2. This activates receptor-associated tyrosine kinases Jak1 and Tyk2. These phosphorylate STAT1 and STAT2 causing them to heterodimerize, associate with karyopherin alpha (KPNA)-1, 5 and 6 and be transported into the nucleus to induce interferon stimulating genes, including the antiviral kinase PKR and major histocompatibility complex 1 MARV VP40 (mVP40) inhibits the IFN induced activation of tyrosine kinase JAK1. EBOV VP24 (eVP24) binds KPNA1, 5 and 6 to block KPNA-STAT1 interactions. Both mechanisms inhibit IFN-induced gene expression. B. EBOV VP24 uses a unique non-classical nuclear localization site to interact with KPNA-1, 5 or 6. (Left) EBOV VP24 (PDB 4M0Q) and SUDV VP24 (PDB 3VNF) show a similar pyramidal structure110. (Right) eVP24 proteins bind the KPNA C-terminus (PDB 4U2X). C. The IFN-inducible protein tetherin can restrict budding of the filovirus VP40 protein (left). However, filovirus GP counteracts the antiviral effects of tetherin, allowing Ebola and Marburg viruses to be released in the present of tetherin (right).
mVP40 blocks JAK1 signaling
In contrast to eVP24, MARV VP24 (mVP24) does not bind to KPNAs or significantly alter IFN-induced gene expression25. The inhibition of IFN-signaling in MARV infected cells is instead mediated by mVP40, which blocks the activation and function of Jak125 (Figure 3A). This recapitulates the phenotype of Jak1 knockout cells in that Jak family kinase and STAT protein tyrosine phosphorylation events were absent after addition of either IFNα/β or IFNγ113. Over-expression studies also suggest that mVP40 acts by specifically inhibiting Jak1; whereas over-expression of JAK1 triggers JAK1 autophosphorylation and STAT1 and STAT2 phosphorylation, mVP40 coexpression prevents these phosphorylation events. In contrast, when Tyk2 was over-expressed, phosphorylation of Tyk2, STAT1 and STAT2 was induced and expression of mVP40 had little to no impact on these phosphorylation events25. Although Jak1 function is inhibited by mVP40, the underlying mechanism remains to be defined and direct interaction of mVP40 with Jak1 has not been demonstrated. Thus, in contrast to EBOV, MARV infection abrogates IFN signaling by blocking the IFNα/β induced tyrosine phosphorylation Jak1, STAT1 and STAT2.
Inhibition of IFN-induced antiviral proteins
In addition to blocking cellular signaling pathways, mechanisms that counteract select IFN-induced antiviral proteins, including PKR and tetherin, have been described. PKR is a well-characterized IFN-induced, dsRNA-activated kinase with antiviral activity and VP35 has been shown to block its activity114,115. Although PKR is dsRNA-activated, some VP35 mutants unable to bind to dsRNA retain the capacity to inhibit PKR, suggesting a dsRNA-independent mechanism of action. Although the mechanism of inhibition remains to be elucidated, it is intriguing that VP35 interactor PACT is an activator of PKR97,116. The impact of PKR inhibition on EBOV replication also remains to be defined. That this function is biologically-significant is suggested by the facts that PKR phosphorylation and phosphorylation of the PKR substrate eIF-2α were not detected in EBOV-infected 293 cells and by the observation that the status of eIF-2α phosphorylation regulates replication in cells that are persistently infected with EBOV in vitro115,117.
Tetherin, also known as BST-2 and CD317, is an IFN-inducible protein that restricts the budding and release of some enveloped viruses by tethering the particles to the cell surface118,119. The release of virus-like particles (VLPs), which occurs upon over-expression of either the EBOV or MARV VP40 matrix proteins, is likewise blocked by tetherin120,121 (Figure 3C). In contrast to the inhibition of budding seen when VP40 was expressed outside the context of virus infection, particle release from EBOV-infected cells was relatively unaffected by tetherin because filoviral GPs counteract the anti-budding effects of tetherin122–124 (Figure 3C). The molecular basis for GP antagonism of tetherin remains ambiguous, but GP does not alter tetherin levels, decrease tetherin levels at the cell surface or prevent tetherin association with lipid rafts124–126.
Subversion of other host pathways
There is accumulating evidence that filoviruses not only block innate immune responses, but that they also adapt to their host to sustain replication and subvert translational regulation mechanisms of host cells. Filoviruses are unusual compared to many other non-segmented, negative-strand RNA viruses in that they produce mRNAs with long 5′ and 3′ untranslated regions (UTRs). The 5′UTRs of the EBOV VP35, VP30, VP24 and L mRNAs contain upstream AUGs (uAUGs), and the uAUGs of EBOV VP30, VP24 and L mRNAs mark the beginning of short upstream open reading frames (uORFs)127. When assessed in reporter gene assays under normal conditions, the uORF in the L mRNA was found to suppress expression of the primary L ORF by approximately 10-fold. This suppression required the uAUG and reflects decreased translation initiation at the primary ORF. During cellular stress, translation initiation factor eIF-2α is phosphorylated, leading to a general decrease in translation (Figure 4). This suppression can serve as a mechanism to allow cells to recover from stresses or, in the case of virus infections, serve as a means to impair virus replication. However, translation of some cellular mRNAs that contain uORFs is increased under these conditions128. Therefore, the impact of the L 5′UTR and L uORF were compared in cells that were either not stressed or that were stressed by addition of thapsigargin, which triggers eIF-2α phosphorylation. The results demonstrated that the L mRNA uORF can sustain translation at the primary ORF when eIF-2α is phosphorylated, such as in the presence of thapsigargin, by increasing translation initiation efficiency at the primary AUG. This suggests a model whereby an uORF helps EBOV maintain expression of its polymerase L under conditions of stress and eIF-2α phosphorylation, such as might occur when the innate immune response leads to PKR activation (Figure 4). Furthermore, a virus mutant lacking the L uAUG site was impaired in replication by about 10-fold relative to wild-type virus and showed increased sensitivity to stress induced by thapsigargin, demonstrating that the L uORF is important for EBOV replication127.
Figure 4. Subversion of host translation mechanisms to support virus replication.
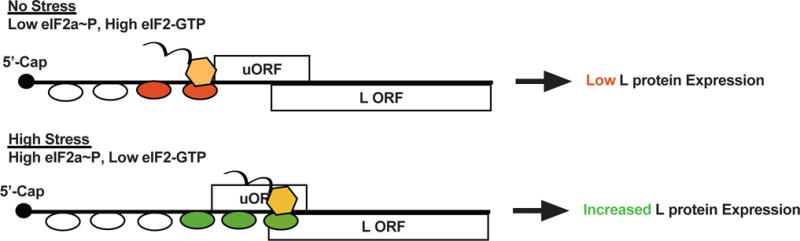
It has been observed that several of the 5′-capped viral EBOV mRNAs possess upstream open reading frames (uORFs) in their 5′ untranslated regions (UTRs). Under lowstress conditions, the uORF present in the mRNA encoding the L protein attenuates L translation initiation. This is because scanning ribosomes (depicted as ovals) initiate efficiently at the uORF AUG instead. However, under stress, such as occurs due to activation of innate immune responses, eIF2α phosphorylation (eIF2α~P) increases and translation initiation at the uORF becomes less efficient. This allows the scanning ribosome to bypass the uORF AUG and increases initiation at the L AUG, helping to sustain L expression despite the increase in eIF2α phosphorylation.
In another example, mVP24 has been demonstrated to interact with and modulate the function of cellular protein kelch-like ech-associated protein 1 (Keap1)110,129,130. Keap1 normally directs Cul3 ubiquitin ligase to target proteins such as nuclear factor (erythroid-derived 2)-like 2 (Nrf2), a transcription factor that binds to and activates promoters possessing antioxidant response element (ARE) sequences. Such genes encode proteins that allow cells to recover from oxidative stress and other stresses. Under homeostatic conditions, Nrf2 is constitutively targeted for degradation due to ubiquitination directed by Keap1. However, when a cell experiences stress, Keap1-Nrf2 interaction can be disrupted, stabilizing Nrf2 and promoting ARE gene expression131. mVP24 interacts with the Kelch domain of Keap1 via an acidic motif present within a loop (called the “K-loop”). This loop possesses the amino acid residues “GE” preceded by acidic residues, which is reminiscent of interacting motifs on other Keap1 binding partners, including Nrf2. The specificity of mVP24 for the Keap1 KELCH domain is derived, in large part, from the extended beta-strands that can protrude from the body of mVP24 and presumably can reach into the deep binding pocket within the KELCH structure110,132. Functionally, mVP24 interaction with Keap1 disrupts Keap1-Nrf2 interaction and activates ARE gene expression110. Consistent with the binding data, eVP24 did not upregulate ARE genes, but chimeric VP24s possessing the mVP24 K-loop did induce upregulation. A comparison of MARV- and EBOV-infected cells demonstrated an upregulation of ARE gene expression MARV infection of macrophage-like THP1 cells and the absence of a response in EBOV-infected cells, consistent with the observation that mVP24 interacts with Keap1 but eVP24 does not. We hypothesize that turning on antioxidant responses could prolong the life of virus-infected cells.
Impact of immune evasion on pathogenesis
Among the described filoviral immune evasion functions, those most directly linked to pathogenesis are those that directly block innate immune signaling pathways. Recombinant EBOVs possessing mutated VP35s that are unable to bind dsRNA or PACT trigger nuclear accumulation of IRF-3 and induce robust IFNα/β responses, whereas wild-type EBOV does not. In vivo, these mutant viruses exhibited reduced replication and were non-lethal at the doses tested, providing compelling evidence that VP35 IFN-antagonist function is a critical determinant of EBOV disease in vivo81,82.
Studies also implicate the IFN signaling inhibitors eVP24 and mVP40 as virulence determinants in rodent models. Although EBOV does not display virulence in mice or guinea pigs, the virus can be made lethal by serial passage in these animals12,133. The adapted viruses acquire amino acid changes in eVP24 and several other genes12,133. In the guinea pig-adapted EBOV, changes in VP24 were sufficient to confer lethality134 and for mouse-adapted EBOV, a mutation in VP24 and a mutation in NP together make the virus lethal to mice135. It is presently unclear to what extent the adaptive changes are related to eVP24 IFN-antagonist activities, although the mouse-adapted EBOV was more resistant to the antiviral effects of IFNs in murine macrophages than was the parental, non-adapted virus135.
Like EBOV, MARV does not cause lethal illness in guinea pigs or mice unless experimentally-adapted to these species15,136. Adaptation of the Ravn strain of MARV (RAVV) to mice led to changes across the viral genome; among these were several amino acid changes in VP40137. When the inhibitory activities of the adapted and parental mVP40 variants toward IFN signaling were compared, the parental mVP40 was impaired in inhibiting IFN responses in mouse cell lines relative to human cell lines. In contrast, the mouse-adapted mVP40 effectively inhibited IFN responses in both human and mouse cell lines138,139. Based on these data, it appears that the inhibitory function of VP40 is connected to virulence in mice138,139. Interestingly, the mouse-adapted mVP40 virus exhibits budding defects in human cells, and the defect was mapped to the same residues in mVP40 implicated in enhanced inhibition of the mouse IFN response. This correlates with an increased sensitivity to restriction by human, but not mouse, tetherin140. Therefore, there appear to be functional costs associated with increased IFN-antagonism in mice.
Conclusions and Future Directions
As discussed above, the mechanisms by which EBOV and MARV counteract host defenses are becoming increasingly clear. The VP35 protein antagonizes several aspects of host defense by blocking the cytoplasmic sensors RIG-I and MDA5, by inhibiting the antiviral kinase PKR and by impairing DC maturation. The eVP24 protein blocks cellular responses to IFNs by selectively blocking the nuclear import of phospho-STAT1; the mVP40 protein counters IFN signaling through inhibition of Jak1 and the filovirus GPs counter the antiviral effects of tetherin. In addition, mechanisms whereby EBOV and MARVs capitalize on specific cellular functions, such as stress-regulated translation or antioxidant responses, to promote virus replication are also coming into focus. There is also strong evidence that these functions contribute to the severe infections that make filoviruses notable.
Nonetheless, fundamental questions remain to be asked. For example, it is unclear how these functions relate to specific mechanisms that drive disease, such as inflammation, vascular leakage and coagulopathy and how they relate to the relative virulence of different filovirus species. Differences in the efficiencies of innate immune antagonism, which might arise due to differences in how viral proteins function and/or in expression levels, could contribute to different degrees of virulence seen between various filoviruses, but such studies are yet to be performed. Because non-human primate models of filovirus infection are considered the gold-standard for pathogenesis studies, it will be important to define the contribution of innate immune antagonism and other proposed virulence determinants in these models. Given that filoviruses are zoonotic pathogens that are apparently not pathogenic in their presumed reservoir hosts, it will also be important to understand how filoviral immune evasion functions differ in bats versus humans. If the responses of different hosts diverge, it will be important to understand the mechanistic basis for such differences. Finally, it remains to be determined whether these functions can be exploited for therapeutic development. The West Africa Ebola epidemic has reinforced the need for effective medical interventions, yet approved therapeutics are lacking. Given the critical role of VP35 for virulence, drugs that either disrupt or bypass this function to restore RLR signaling in infected cells would be expected to have therapeutic benefit. Similarly, drugs that counteract eVP24 or mVP40 IFN-antagonist functions might enhance the benefit of naturally-produced IFNs or synergize with IFN administered therapeutically. Studies to identify inhibitors of other functions, such as the mechanisms regulating L translation and antioxidant responses are also warranted, as these would be useful as molecular probes for these functions and as possible leads for antiviral development. The need for better therapeutics and for improved understanding of filoviral disease should prompt continued delineation of filovirus innate immune evasion functions.
Acknowledgments
The authors thank Christine Schwall for critical reading of this manuscript. This work was supported by NIH grant U19 AI109945 to C.F.B (Basler- PI), I.M., and G.K.A., R01 AI059536 and U19 AI109664 to C.F.B. and U19 AI070489 (Holtzman- PI) and R01 AI081914 to G.K.A.
Glossary
- Cathepsins B and L
endosomal cysteine proteases that cleave EBOV GP during viral entry
- humoral responses
antibody responses
- isotype class switching
a recombination process that alters the isotype of an antibody (e.g. from IgM to IgG)
- lymphopenia
low levels of lymphocytes in blood
- micropinocytosis
an actin-dependent form of endocytosis that results in the formation of large fluid-filled vacuoles
- necrosis
a non-apoptotic form of cell death
- negative-sense RNA viruses
viruses possessing single-stranded RNA genomes that are negative-sense (i.e. complementary to the viral mRNAs)
- phosphatidylserine
a phospholipid found on the surface of apoptotic bodies and on viral membranes that can promote intracellular uptake via interaction with specific cell surface receptors
- pro-thrombin time
a blood test that measures clotting time
- thrombocytopenia
low levels of platelets in the blood
References
- 1.Baize S, et al. Emergence of Zaire Ebola Virus Disease in Guinea – Preliminary Report. N Engl J Med. 2014;371:1418–1425. doi: 10.1056/NEJMoa1404505. [DOI] [PubMed] [Google Scholar]
- 2.Feldmann H, Geisbert TW. Ebola haemorrhagic fever. Lancet. 2010;377:849–862. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanchez A, Geisbert TW, Feldmann H. In: Fields Virology. Knipe DM, Howley PM, et al., editors. Lippincott Williams and Wilkins; 2007. pp. 1410–1448. [Google Scholar]
- 4.Gire SK, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345:1369–1372. doi: 10.1126/science.1259657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO. Ebola Situation Report – 4 February 2015. 2015 [Google Scholar]
- 6.Kuhn JH, et al. Virus nomenclature below the species level: a standardized nomenclature for filovirus strains and variants rescued from cDNA. Arch Virol. 2013;159:1229–1237. doi: 10.1007/s00705-013-1877-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Negredo A, et al. Discovery of an ebolavirus-like filovirus in europe. PLoS pathogens. 2011;7:e1002304. doi: 10.1371/journal.ppat.1002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.CDC. Outbreaks Chronology: Ebola Virus Disease. 2015 < http://www.cdc.gov/vhf/ebola/outbreaks/history/chronology.html>.
- 9.Miranda ME, Miranda NL. Reston ebolavirus in humans and animals in the Philippines: a review. J Infect Dis. 2011;204(Suppl 3):S757–760. doi: 10.1093/infdis/jir296. [DOI] [PubMed] [Google Scholar]
- 10.Amman BR, et al. Oral shedding of marburg virus in experimentally infected egyptian fruit bats (rousettus aegyptiacus) J Wildl Dis. 2014;51:113–124. doi: 10.7589/2014-08-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paweska JT, et al. Virological and serological findings in Rousettus aegyptiacus experimentally inoculated with vero cells-adapted hogan strain of Marburg virus. PloS one. 2012;7:e45479. doi: 10.1371/journal.pone.0045479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volchkov VE, Chepurnov AA, Volchkova VA, Ternovoj VA, Klenk HD. Molecular characterization of guinea pig-adapted variants of Ebola virus. Virology. 2000;277:147–155. doi: 10.1006/viro.2000.0572. [DOI] [PubMed] [Google Scholar]
- 13.Lofts LL, Ibrahim MS, Negley DL, Hevey MC, Schmaljohn AL. Genomic differences between guinea pig lethal and nonlethal Marburg virus variants. J Infect Dis. 2007;196(Suppl 2):S305–312. doi: 10.1086/520585. [DOI] [PubMed] [Google Scholar]
- 14.Bray M, Davis K, Geisbert T, Schmaljohn C, Huggins J. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J Infect Dis. 1999;179(Suppl 1):S248–258. doi: 10.1086/514292. [DOI] [PubMed] [Google Scholar]
- 15.Warfield KL, et al. Development and characterization of a mouse model for Marburg hemorrhagic fever. J Virol. 2009;83:6404–6415. doi: 10.1128/JVI.00126-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bray M. The role of the Type I interferon response in the resistance of mice to filovirus infection. J Gen Virol. 2001;82:1365–1373. doi: 10.1099/0022-1317-82-6-1365. [DOI] [PubMed] [Google Scholar]
- 17.Mehedi M, et al. A new Ebola virus nonstructural glycoprotein expressed through RNA editing. J Virol. 2011;85:5406–5414. doi: 10.1128/JVI.02190-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanchez A, Trappier SG, Mahy BW, Peters CJ, Nichol ST. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc Natl Acad Sci U S A. 1996;93:3602–3607. doi: 10.1073/pnas.93.8.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shabman RS, et al. Deep sequencing identifies noncanonical editing of Ebola and Marburg virus RNAs in infected cells. MBio. 5:e02011. doi: 10.1128/mBio.02011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basler CF, Amarasinghe GK. Evasion of interferon responses by Ebola and Marburg viruses. J Interferon Cytokine Res. 2009;29:511–520. doi: 10.1089/jir.2009.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basler CF, et al. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:12289–12294. doi: 10.1073/pnas.220398297. This was the first demonstration that the Ebola virus VP35 protein blocks interferon responses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reid SP, et al. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol. 2006;80:5156–5167. doi: 10.1128/JVI.02349-05. This was the first demonstration that the Ebola virus VP24 protein blocks interferon-induced signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramanan P, et al. Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc Natl Acad Sci U S A. 2012;109:20661–20666. doi: 10.1073/pnas.1213559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bale S, et al. Marburg virus VP35 can both fully coat the backbone and cap the ends of dsRNA for interferon antagonism. PLoS pathogens. 2012;8:e1002916. doi: 10.1371/journal.ppat.1002916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valmas C, et al. Marburg virus evades interferon responses by a mechanism distinct from ebola virus. PLoS Pathog. 2010;6:e1000721. doi: 10.1371/journal.ppat.1000721. This study demonstrated that the Marburg virus VP40 protein blocks interferon-induced signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dowell SF, et al. Transmission of Ebola hemorrhagic fever: a study of risk factors in family members, Kikwit, Democratic Republic of the Congo, 1995. Commission de Lutte contre les Epidemies a Kikwit. The Journal of infectious diseases. 1999;179(Suppl 1):S87–91. doi: 10.1086/514284. [DOI] [PubMed] [Google Scholar]
- 27.Geisbert TW, et al. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. The American journal of pathology. 2003;163:2371–2382. doi: 10.1016/S0002-9440(10)63592-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geisbert TW, et al. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am J Pathol. 2003;163:2347–2370. doi: 10.1016/S0002-9440(10)63591-2. This study demonstrated macrophages and dendritic cells as major targets of Ebola virus throughout the course of infection in non-human primates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaki SR, et al. A novel immunohistochemical assay for the detection of Ebola virus in skin: implications for diagnosis, spread, and surveillance of Ebola hemorrhagic fever. Commission de Lutte contre les Epidemies a Kikwit. J Infect Dis. 1999;179(Suppl 1):S36–47. doi: 10.1086/514319. [DOI] [PubMed] [Google Scholar]
- 30.Yonezawa A, Cavrois M, Greene WC. Studies of ebola virus glycoprotein-mediated entry and fusion by using pseudotyped human immunodeficiency virus type 1 virions: involvement of cytoskeletal proteins and enhancement by tumor necrosis factor alpha. J Virol. 2005;79:918–926. doi: 10.1128/JVI.79.2.918-926.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dube D, et al. Cell adhesion promotes Ebola virus envelope glycoprotein-mediated binding and infection. J Virol. 2008;82:7238–7242. doi: 10.1128/JVI.00425-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez O, et al. Ebola virus exploits a monocyte differentiation program to promote its entry. J Virol. 2013;87:3801–3814. doi: 10.1128/JVI.02695-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carette JE, et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cote M, et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature. 2011;477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schornberg KL, et al. Alpha5beta1-integrin controls ebolavirus entry by regulating endosomal cathepsins. Proc Natl Acad Sci U S A. 2009;106:8003–8008. doi: 10.1073/pnas.0807578106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moller-Tank S, Maury W. Phosphatidylserine receptors: enhancers of enveloped virus entry and infection. Virology. 2014:468–470. doi: 10.1016/j.virol.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hunt CL, Kolokoltsov AA, Davey RA, Maury W. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J Virol. 2011;85:334–347. doi: 10.1128/JVI.01278-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimojima M, et al. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J Virol. 2006;80:10109–10116. doi: 10.1128/JVI.01157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simmons G, et al. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology. 2003;305:115–123. doi: 10.1006/viro.2002.1730. [DOI] [PubMed] [Google Scholar]
- 41.Baize S, et al. Inflammatory responses in Ebola virus-infected patients. Clin Exp Immunol. 2002;128:163–168. doi: 10.1046/j.1365-2249.2002.01800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ebihara H, et al. Host response dynamics following lethal infection of rhesus macaques with Zaire ebolavirus. J Infect Dis. 2011;204(Suppl 3):S991–999. doi: 10.1093/infdis/jir336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hensley LE, Young HA, Jahrling PB, Geisbert TW. Proinflammatory response during Ebola virus infection of primate models: possible involvement of the tumor necrosis factor receptor superfamily. Immunol Lett. 2002;80:169–179. doi: 10.1016/s0165-2478(01)00327-3. [DOI] [PubMed] [Google Scholar]
- 44.Sanchez A, et al. Analysis of human peripheral blood samples from fatal and nonfatal cases of Ebola (Sudan) hemorrhagic fever: cellular responses, virus load, and nitric oxide levels. J Virol. 2004;78:10370–10377. doi: 10.1128/JVI.78.19.10370-10377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Villinger F, et al. Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. The Journal of infectious diseases. 1999;179(Suppl 1):S188–191. doi: 10.1086/514283. [DOI] [PubMed] [Google Scholar]
- 46.Wauquier N, Becquart P, Padilla C, Baize S, Leroy EM. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Negl Trop Dis. 2010;4 doi: 10.1371/journal.pntd.0000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupta M, Mahanty S, Ahmed R, Rollin PE. Monocyte-derived human macrophages and peripheral blood mononuclear cells infected with ebola virus secrete MIP-1alpha and TNF-alpha and inhibit poly-IC-induced IFN-alpha in vitro. Virology. 2001;284:20–25. doi: 10.1006/viro.2001.0836. [DOI] [PubMed] [Google Scholar]
- 48.Stroher U, et al. Infection and activation of monocytes by Marburg and Ebola viruses. J Virol. 2001;75:11025–11033. doi: 10.1128/JVI.75.22.11025-11033.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wahl-Jensen V, et al. Ebola virion attachment and entry into human macrophages profoundly effects early cellular gene expression. PLoS Negl Trop Dis. 2011;5:e1359. doi: 10.1371/journal.pntd.0001359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lubaki NM, et al. The lack of maturation of Ebola virus-infected dendritic cells results from the cooperative effect of at least two viral domains. J Virol. 2013;87:7471–7485. doi: 10.1128/JVI.03316-12. This study demonstrates that Ebola viruses with mutated VP35s are greatly impaired in suppression of dendritic cell maturation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mahanty S, et al. Cutting edge: impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J Immunol. 2003;170:2797–2801. doi: 10.4049/jimmunol.170.6.2797. This is one of the first in vitro studies demonstrating suppression of dendritic cell maturation by filovirus infection. [DOI] [PubMed] [Google Scholar]
- 52.Bosio CM, et al. Ebola and Marburg viruses replicate in monocyte-derived dendritic cells without inducing the production of cytokines and full maturation. J Infect Dis. 2003;188:1630–1638. doi: 10.1086/379199. This is one of the first in vitro studies demonstrating suppression of dendritic cell maturation by filovirus infection. [DOI] [PubMed] [Google Scholar]
- 53.Yen B, Mulder LC, Martinez O, Basler CF. Molecular Basis for Ebola Virus VP35 Suppression of Human Dendritic Cell Maturation. J Virol. 2014;88:12500–12510. doi: 10.1128/JVI.02163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin H, et al. The VP35 Protein of Ebola Virus Impairs Dendritic Cell Maturation Induced by Virus and Lipopolysaccharide. J Gen Virol. 2010;91(Pt 2):352–361. doi: 10.1099/vir.0.017343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bradfute SB, Warfield KL, Bavari S. Functional CD8+ T cell responses in lethal Ebola virus infection. J Immunol. 2008;180:4058–4066. doi: 10.4049/jimmunol.180.6.4058. doi:180/6/4058 [pii] [DOI] [PubMed] [Google Scholar]
- 56.McElroy AK, et al. Human Ebola virus infection results in substantial immune activation. Proc Natl Acad Sci U S A. 2015;112:4719–4724. doi: 10.1073/pnas.1502619112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Geisbert TW, et al. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab Invest. 2000;80:171–186. doi: 10.1038/labinvest.3780021. [DOI] [PubMed] [Google Scholar]
- 58.Bradfute SB, et al. Lymphocyte death in a mouse model of ebola virus infection. J Infect Dis. 2007;196(Suppl 2):S296–304. doi: 10.1086/520602. [DOI] [PubMed] [Google Scholar]
- 59.Reed DS, Hensley LE, Geisbert JB, Jahrling PB, Geisbert TW. Depletion of peripheral blood T lymphocytes and NK cells during the course of ebola hemorrhagic Fever in cynomolgus macaques. Viral immunology. 2004;17:390–400. doi: 10.1089/vim.2004.17.390. [DOI] [PubMed] [Google Scholar]
- 60.Gupta M, Spiropoulou C, Rollin PE. Ebola virus infection of human PBMCs causes massive death of macrophages, CD4 and CD8 T cell sub-populations in vitro. Virology. 2007;364:45–54. doi: 10.1016/j.virol.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 61.Baize S, et al. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat Med. 1999;5:423–426. doi: 10.1038/7422. [DOI] [PubMed] [Google Scholar]
- 62.Bradfute SB, et al. Mechanisms and consequences of ebolavirus-induced lymphocyte apoptosis. J Immunol. 2010;184:327–335. doi: 10.4049/jimmunol.0901231. [DOI] [PubMed] [Google Scholar]
- 63.Rath PC, Aggarwal BB. TNF-induced signaling in apoptosis. Journal of clinical immunology. 1999;19:350–364. doi: 10.1023/a:1020546615229. [DOI] [PubMed] [Google Scholar]
- 64.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis : an international journal on programmed cell death. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- 65.Snyder CM, Shroff EH, Liu J, Chandel NS. Nitric oxide induces cell death by regulating anti-apoptotic BCL-2 family members. PloS one. 2009;4:e7059. doi: 10.1371/journal.pone.0007059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohan GS, Li W, Ye L, Compans RW, Yang C. Antigenic subversion: a novel mechanism of host immune evasion by Ebola virus. PLoS pathogens. 2012;8:e1003065. doi: 10.1371/journal.ppat.1003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kang ES, et al. Hypotension during hemodialysis: role for nitric oxide. The American journal of the medical sciences. 1997;313:138–146. doi: 10.1097/00000441-199703000-00003. [DOI] [PubMed] [Google Scholar]
- 68.Geisbert TW, et al. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J Infect Dis. 2003;188:1618–1629. doi: 10.1086/379724. doi:JID30697 [pii] [DOI] [PubMed] [Google Scholar]
- 69.Neumann FJ, et al. Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arteriosclerosis, thrombosis, and vascular biology. 1997;17:3399–3405. doi: 10.1161/01.atv.17.12.3399. [DOI] [PubMed] [Google Scholar]
- 70.Bray M, Hatfill S, Hensley L, Huggins JW. Haematological, biochemical and coagulation changes in mice, guinea-pigs and monkeys infected with a mouse-adapted variant of Ebola Zaire virus. J Comp Pathol. 2001;125:243–253. doi: 10.1053/jcpa.2001.0503. [DOI] [PubMed] [Google Scholar]
- 71.Feldmann H, et al. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J Virol. 1996;70:2208–2214. doi: 10.1128/jvi.70.4.2208-2214.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ray RB, et al. Ebola virus glycoprotein-mediated anoikis of primary human cardiac microvascular endothelial cells. Virology. 2004;321:181–188. doi: 10.1016/j.virol.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 73.Wahl-Jensen VM, et al. Effects of Ebola virus glycoproteins on endothelial cell activation and barrier function. J Virol. 2005;79:10442–10450. doi: 10.1128/JVI.79.16.10442-10450.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang ZY, et al. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat Med. 2000;6:886–889. doi: 10.1038/78645. [DOI] [PubMed] [Google Scholar]
- 75.Schneider WM, Chevillotte MD, Rice CM. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Immunology. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schoggins JW. Interferon-stimulated genes: roles in viral pathogenesis. Current opinion in virology. 2014;6:40–46. doi: 10.1016/j.coviro.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harcourt BH, Sanchez A, Offermann MK. Ebola virus inhibits induction of genes by double-stranded RNA in endothelial cells. Virology. 1998;252:179–188. doi: 10.1006/viro.1998.9446. This is the first study to demonstrate that Ebola virus infection blocks cellular production of interferon. [DOI] [PubMed] [Google Scholar]
- 78.Harcourt BH, Sanchez A, Offermann MK. Ebola virus selectively inhibits responses to interferons, but not to interleukin-1beta, in endothelial cells. J Virol. 1999;73:3491–3496. doi: 10.1128/jvi.73.4.3491-3496.1999. This is the first study to demonstrate that Ebola virus infection blocks cellular responses to interferons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kash JC, et al. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J Virol. 2006;80:3009–3020. doi: 10.1128/JVI.80.6.3009-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spiropoulou CF, et al. RIG-I activation inhibits ebolavirus replication. Virology. 2009;392:11–15. doi: 10.1016/j.virol.2009.06.032. [DOI] [PubMed] [Google Scholar]
- 81.Hartman AL, et al. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of ebola virus. J Virol. 2008;82:2699–2704. doi: 10.1128/JVI.02344-07. This study demonstrates that disruption of VP35 interferon-antagonist function attenuates Ebola virus replication in a mouse model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Prins KC, et al. Mutations abrogating VP35 interaction with double-stranded RNA render Ebola virus avirulent in guinea pigs. J Virol. 2010;84:3004–3015. doi: 10.1128/JVI.02459-09. This study demonstrates that disruption of VP35 interferon-antagonist function renders Ebola virus non-lethal in a guinea pig model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mahanty S, et al. Protection from lethal infection is determined by innate immune responses in a mouse model of Ebola virus infection. Virology. 2003;312:415–424. doi: 10.1016/s0042-6822(03)00233-2. doi:S0042682203002332 [pii] [DOI] [PubMed] [Google Scholar]
- 84.Huang IC, et al. Distinct Patterns of IFITM-Mediated Restriction of Filoviruses, SARS Coronavirus, and Influenza A Virus. PLoS Pathog. 2011;7:e1001258. doi: 10.1371/journal.ppat.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Okumura A, Pitha PM, Harty RN. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc Natl Acad Sci U S A. 2008;105:3974–3979. doi: 10.1073/pnas.0710629105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bray M, Raymond JL, Geisbert T, Baker RO. 3-Deazaneplanocin A induces massively increased interferon-alpha production in Ebola virus-infected mice. Antiviral Res. 2002;55:151–159. doi: 10.1016/s0166-3542(02)00018-9. [DOI] [PubMed] [Google Scholar]
- 87.Jahrling PB, et al. Evaluation of immune globulin and recombinant interferon-alpha2b for treatment of experimental Ebola virus infections. J Infect Dis. 1999;179(Suppl 1):S224–234. doi: 10.1086/514310. [DOI] [PubMed] [Google Scholar]
- 88.Basler CF, et al. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol. 2003;77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prins KC, Cardenas WB, Basler CF. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol. 2009;83:3069–3077. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang TH, et al. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 2009;5:e1000493. doi: 10.1371/journal.ppat.1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kubota T, et al. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J Biol Chem. 2008;283:25660–25670. doi: 10.1074/jbc.M804479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leung DW, et al. Structure of the Ebola VP35 interferon inhibitory domain. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:411–416. doi: 10.1073/pnas.0807854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cardenas WB, et al. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol. 2006;80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hartman AL, Towner JS, Nichol ST. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology. 2004;328:177–184. doi: 10.1016/j.virol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 95.Leung DW, et al. Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nature structural & molecular biology. 2010;17:165–172. doi: 10.1038/nsmb.1765. This study describes the structural basis for VP35 interferon-antagonist and dsRNA binding activities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Luthra P, et al. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe. 2013;14:74–84. doi: 10.1016/j.chom.2013.06.010. This study demonstrates that Ebola virus VP35 interaction with cellular protein PACT contributes to its suppression of interferon production. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Patel RC, Sen GC. PACT a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998;17:4379–4390. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kok KH, et al. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe. 2011;9:299–309. doi: 10.1016/j.chom.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 99.Bale S, et al. Ebolavirus VP35 coats the backbone of double-stranded RNA for interferon antagonism. J Virol. 2013;87:10385–10388. doi: 10.1128/JVI.01452-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–258. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 101.Leung LW, et al. Ebolavirus VP35 suppresses IFN production from conventional but not plasmacytoid dendritic cells. Immunol Cell Biol. 2011;89:792–802. doi: 10.1038/icb.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Reid SP, Cardenas WB, Basler CF. Homo-oligomerization facilitates the interferon-antagonist activity of the ebolavirus VP35 protein. Virology. 2005;341:179–189. doi: 10.1016/j.virol.2005.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kimberlin CR, et al. Ebolavirus VP35 uses a bimodal strategy to bind dsRNA for innate immune suppression. Proc Natl Acad Sci U S A. 2009;107:314–319. doi: 10.1073/pnas.0910547107. This study describes the structural basis for VP35 interferon-antagonist and dsRNA binding activities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Leung DW, et al. Structural and functional characterization of Reston Ebola VP35 Interferon Inhibitory Domain. J Mol Biol. 2010;399:347–357. doi: 10.1016/j.jmb.2010.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Prins KC, et al. Basic residues within the ebolavirus VP35 protein are required for its viral polymerase cofactor function. J Virol. 2010;84:10581–10591. doi: 10.1128/JVI.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 107.McBride KM, Reich NC. The ins and outs of STAT1 nuclear transport. Sci STKE. 2003;2003:RE13. doi: 10.1126/stke.2003.195.re13. [DOI] [PubMed] [Google Scholar]
- 108.Reid SP, Valmas C, Martinez O, Sanchez FM, Basler CF. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J Virol. 2007;81:13469–13477. doi: 10.1128/JVI.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang AP, et al. The ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog. 2012;8:e1002550. doi: 10.1371/journal.ppat.1002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Edwards MR, et al. The Marburg virus VP24 protein interacts with Keap1 to activate the cytoprotective antioxidant response pathway. Cell Rep. 2014;6:1017–1025. doi: 10.1016/j.celrep.2014.01.043. This study explains the molecular basis by which Marburg virus VP24 interacts with cellular protein Keap1 to activate the antioxidant response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu W, et al. Ebola Virus VP24 Targets a Unique NLS Binding Site on Karyopherin Alpha 5 to Selectively Compete with Nuclear Import of Phosphorylated STAT1. Cell Host Microbe. 2014;16:187–200. doi: 10.1016/j.chom.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shabman RS, Gulcicek EE, Stone KL, Basler CF. The Ebola virus VP24 protein prevents hnRNP C1/C2 binding to karyopherin alpha1 and partially alters its nuclear import. J Infect Dis. 2011;204(Suppl 3):S904–910. doi: 10.1093/infdis/jir323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rodig SJ, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93:373–383. doi: 10.1016/s0092-8674(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 114.Feng Z, Cerveny M, Yan Z, He B. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J Virol. 2007;81:182–192. doi: 10.1128/JVI.01006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schumann M, Gantke T, Muhlberger E. Ebola virus VP35 antagonizes PKR activity through its C-terminal interferon inhibitory domain. J Virol. 2009;83:8993–8997. doi: 10.1128/JVI.00523-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Peters GA, Hartmann R, Qin J, Sen GC. Modular structure of PACT: distinct domains for binding and activating PKR. Mol Cell Biol. 2001;21:1908–1920. doi: 10.1128/MCB.21.6.1908-1920.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Strong JE, et al. Stimulation of Ebola virus production from persistent infection through activation of the Ras/MAPK pathway. Proc Natl Acad Sci U S A. 2008;105:17982–17987. doi: 10.1073/pnas.0809698105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 119.Van Damme N, et al. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jouvenet N, et al. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol. 2009;83:1837–1844. doi: 10.1128/JVI.02211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Neil SJ, Sandrin V, Sundquist WI, Bieniasz PD. An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe. 2007;2:193–203. doi: 10.1016/j.chom.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Radoshitzky SR, et al. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J Virol. 2010;84:10569–10580. doi: 10.1128/JVI.00103-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A. 2009;106:2886–2891. doi: 10.1073/pnas.0811014106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kuhl A, et al. The Ebola virus glycoprotein and HIV-1 Vpu employ different strategies to counteract the antiviral factor tetherin. J Infect Dis. 2011;204(Suppl 3):S850–860. doi: 10.1093/infdis/jir378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lopez LA, et al. Anti-tetherin activities of HIV-1 Vpu and Ebola virus glycoprotein do not involve removal of tetherin from lipid rafts. J Virol. 2012;86:5467–5480. doi: 10.1128/JVI.06280-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lopez LA, et al. Ebola virus glycoprotein counteracts BST-2/Tetherin restriction in a sequence-independent manner that does not require tetherin surface removal. J Virol. 2010;84:7243–7255. doi: 10.1128/JVI.02636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shabman RS, et al. An upstream open reading frame modulates ebola virus polymerase translation and virus replication. PLoS Pathog. 2013;9:e1003147. doi: 10.1371/journal.ppat.1003147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 129.Pichlmair A, et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature. 2012;487:486–490. doi: 10.1038/nature11289. [DOI] [PubMed] [Google Scholar]
- 130.Page A, et al. Marburgvirus hijacks nrf2-dependent pathway by targeting nrf2-negative regulator keap1. Cell Rep. 2014;6:1026–1036. doi: 10.1016/j.celrep.2014.02.027. This study describes the interaction of Marburg virus VP24 with cellular protein Keap1 and demonstrates a role for the host antioxidant response in Marburg virus pathogenesis in a mouse model. [DOI] [PubMed] [Google Scholar]
- 131.Copple IM. The keap1-nrf2 cell defense pathway – a promising therapeutic target? Adv Pharmacol. 2012;63:43–79. doi: 10.1016/B978-0-12-398339-8.00002-1. [DOI] [PubMed] [Google Scholar]
- 132.Zhang AP, Bornholdt ZA, Abelson DM, Saphire EO. Crystal structure of Marburg virus VP24. J Virol. 2014;88:5859–5863. doi: 10.1128/JVI.03565-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bray M, Davis K, Geisbert T, Schmaljohn C, Huggins J. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J Infect Dis. 1998;178:651–661. doi: 10.1086/515386. [DOI] [PubMed] [Google Scholar]
- 134.Mateo M, et al. VP24 is a molecular determinant of Ebola virus virulence in guinea pigs. J Infect Dis. 2011;204(Suppl 3):S1011–1020. doi: 10.1093/infdis/jir338. [DOI] [PubMed] [Google Scholar]
- 135.Ebihara H, et al. Molecular determinants of Ebola virus virulence in mice. PLoS Pathog. 2006;2:e73. doi: 10.1371/journal.ppat.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Warfield KL, et al. Development of a model for marburgvirus based on severe-combined immunodeficiency mice. Virol J. 2007;4:108. doi: 10.1186/1743-422X-4-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lofts LL, Wells JB, Bavari S, Warfield KL. Key genomic changes necessary for an in vivo lethal mouse marburgvirus variant selection process. J Virol. 2011;85:3905–3917. doi: 10.1128/JVI.02372-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Valmas C, Basler CF. Marburg Virus VP40 Antagonizes Interferon Signaling in a Species-Specific Manner. J Virol. 2011;85:4309–4317. doi: 10.1128/JVI.02575-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Feagins AR, Basler CF. Amino Acid Residue at Position 79 of Marburg Virus VP40 Confers Interferon Antagonism in Mouse Cells. J Infect Dis. 2015 doi: 10.1093/infdis/jiv010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Feagins AR, Basler CF. The VP40 protein of Marburg virus exhibits impaired budding and increased sensitivity to human tetherin following mouse adaptation. J Virol. 2014;88:14440–14450. doi: 10.1128/JVI.02069-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Feldmann H, Slenczka W, Klenk HD. Emerging and reemerging of filoviruses. Archives of virology. Supplementum. 1996;11:77–100. doi: 10.1007/978-3-7091-7482-1_9. [DOI] [PubMed] [Google Scholar]
- 142.Slenczka W, Klenk HD. Forty years of marburg virus. The Journal of infectious diseases. 2007;196(Suppl 2):S131–135. doi: 10.1086/520551. [DOI] [PubMed] [Google Scholar]
- 143.Johnson KM, Lange JV, Webb PA, Murphy FA. Isolation and partial characterisation of a new virus causing acute haemorrhagic fever in Zaire. Lancet. 1977;1:569–571. doi: 10.1016/s0140-6736(77)92000-1. [DOI] [PubMed] [Google Scholar]
- 144.WHO. International Commission to Sudan: Ebola haemorrhagic fever in Sudan, 1976. Bull World Health Organ. 1978;56:247–270. [PMC free article] [PubMed] [Google Scholar]
- 145.Towner JS, et al. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J Virol. 2006;80:6497–6516. doi: 10.1128/JVI.00069-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bausch DG, et al. Marburg hemorrhagic fever associated with multiple genetic lineages of virus. The New England journal of medicine. 2006;355:909–919. doi: 10.1056/NEJMoa051465. [DOI] [PubMed] [Google Scholar]
- 147.Jahrling PB, et al. Preliminary report: isolation of Ebola virus from monkeys imported to USA. Lancet. 1990;335:502–505. doi: 10.1016/0140-6736(90)90737-p. [DOI] [PubMed] [Google Scholar]
- 148.Formenty P, et al. Human infection due to Ebola virus, subtype Cote d’Ivoire: clinical and biologic presentation. The Journal of infectious diseases. 1999;179(Suppl 1):S48–53. doi: 10.1086/514285. [DOI] [PubMed] [Google Scholar]
- 149.Conrad JL, et al. Epidemiologic investigation of Marburg virus disease, Southern Africa, 1975. Am J Trop Med Hyg. 1978;27:1210–1215. doi: 10.4269/ajtmh.1978.27.1210. [DOI] [PubMed] [Google Scholar]
- 150.Swanepoel R, et al. Studies of reservoir hosts for Marburg virus. Emerg Infect Dis. 2007;13:1847–1851. doi: 10.3201/eid1312.071115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Towner JS, et al. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009;5:e1000536. doi: 10.1371/journal.ppat.1000536. This study describes the first isolation of infectious Marburg virus from wild-caught bats. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Timen A, et al. Response to imported case of Marburg hemorrhagic fever, the Netherland. Emerg Infect Dis. 2009;15:1171–1175. doi: 10.3201/eid1508.090015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Towner JS, et al. Marburg virus infection detected in a common African bat. PLoS One. 2007;2:e764. doi: 10.1371/journal.pone.0000764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Amman BR, et al. Seasonal pulses of Marburg virus circulation in juvenile Rousettus aegyptiacus bats coincide with periods of increased risk of human infection. PLoS Pathog. 2012;8:e1002877. doi: 10.1371/journal.ppat.1002877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Paweska JT, et al. Virological and serological findings in Rousettus aegyptiacus experimentally inoculated with vero cells-adapted hogan strain of Marburg virus. PLoS One. 2012;7:e45479. doi: 10.1371/journal.pone.0045479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Aleksandrowicz P, et al. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J Infect Dis. 2011;204(Suppl 3):S957–967. doi: 10.1093/infdis/jir326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. Cellular entry of ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS pathogens. 2010;6:e1001110. doi: 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nanbo A, et al. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS pathogens. 2010;6:e1001121. doi: 10.1371/journal.ppat.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sakurai Y, et al. Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science. 2015;347:995–998. doi: 10.1126/science.1258758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Muhlberger E. Filovirus replication and transcription. Future Virol. 2007;2:205–215. doi: 10.2217/17460794.2.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jasenosky LD, Neumann G, Lukashevich I, Kawaoka Y. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J Virol. 2001;75:5205–5214. doi: 10.1128/JVI.75.11.5205-5214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Noda T, et al. Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. J Virol. 2002;76:4855–4865. doi: 10.1128/JVI.76.10.4855-4865.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Harty RN, Brown ME, Wang G, Huibregtse J, Hayes FP. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc Natl Acad Sci U S A. 2000;97:13871–13876. doi: 10.1073/pnas.250277297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hartlieb B, Weissenhorn W. Filovirus assembly and budding. Virology. 2006;344:64–70. doi: 10.1016/j.virol.2005.09.018. [DOI] [PubMed] [Google Scholar]