

Sustaining a healthy population of mitochondria in a neuron requires a balance of de novo biogenesis of new mitochondria or mitochondrial components and removal of damaged mitochondrial membrane, proteins, and DNA (1, 2). Recent evidence suggests that mitochondria must be actively transported between the cell body and axon (2). A new study by Davis et al. (3) offers a surprising alternative: Axons expel mitochondria to neighboring astrocytes for degradation. This unexpected finding might reshape how we think of mitochondrial disposal in the nervous system.
Axons enable long-distance communication in the nervous system by propagating signals to distant synapses at extraordinary lengths from the neuronal cell body. Axons within the spinal tract of the blue whale (perhaps the longest in nature) are estimated to exceed 30 m in length, and the longest human axons extend an average of 1 m from the base of the spine to the toes. By comparison, a neuronal cell body is less than 100 μm in diameter (4). Thus, long axons can represent the majority of a neuron’s volume and pose a major logistical challenge to ship nutrients, organelles, and other biomaterials essential for maintaining axon integrity, synaptic connectivity, and neuronal function (2, 4).
The active transport of organelles and vesicles, as well as the maintenance of membrane potential along the length of the axon, are energetically demanding tasks that require a constant supply of adenosine triphosphate from axonal mitochondria (5). As such, mitochondria must be both properly distributed and functioning efficiently to maintain neuronal energy homeostasis and neural activity (2, 6).
Most newly formed mitochondria are transported from the cell body down the length of the axon and often fuse with resident mitochondria along the way (2). Mitochondrial fusion and fission together constitute an important quality-control mechanism that is believed to help adjust mitochondrial length and vitality, replenish supplies of biomolecules, and dilute out defective mitochondrial components (1). Such maintenance is essential not only for proper bioenergetic function, but also to prevent the release of reactive oxygen species or molecules that trigger programmed cell death (apoptosis) from partially depolarized mitochondria (7, 8).
Ultimately, aged mitochondria are believed to become damaged irreparably, at which point cells discard them or fission off sections to be degraded through an autophagy-based process (mitophagy). Autophagosomes form around mitochondria, which then fuse with lysosomes, delivering digestive enzymes and accomplishing their degradation (1, 9). Neuronal autophagosomes likely mature by fusing with endosomes and lysosomes during retrograde transport from the axon to the cell body (10). How or where mitophagy is induced in the axon, or precisely how many mitochondria are eliminated by autophagy versus alternate mechanisms (axonal or otherwise), is not clear (11).
A study of retinal ganglion cells at the optic nerve head in a mouse revealed that membrane-bound vesicles that were shed from their axons were internalized by surrounding astrocytes (12). Davis et al. further analyzed this shedding event. Using scanning electron microscopy and cell-specific labeling, they identified mitochondria as one of the major constituents of the shed axonal evulsions. A clear continuum of events was observed whereby mitochondria clustered in axons near sites of astrocyte membranes, evulsions from axons were filled with mitochondria, and shed evulsions were internalized by astrocytes. The authors further determined that the mitochondria-rich evulsions were degraded in astrocytes, as the organelles co-localized with lysosomal markers and were surrounded by astrocyte membranes.
To trace the fate of these mitochondria-rich evulsions in astrocytes, Davis et al. targeted a red/green, acid-resistant/acid-sensitive fluorescent protein to neuronal mitochondria. In healthy mitochondria, red and green signals colocalize, whereas the green fluorescence is eliminated in an acidic lysosomal environment. The authors confirmed that mitochondria undergoing lysosomal degradation were not in axons but in surrounding glia (astrocytes). They also detected degraded neuronal mitochondrial DNA in astrocytes. The study provides compelling evidence that retinal ganglion cells can transfer mitochondria to astrocytes for destruction rather than sending the organelles down the axon to the cell body for recycling. In addition, Davis et al. show that a majority of axonal mitochondria in the optic nerve head undergo degradation in surrounding astrocytes. This process therefore represents a major route for mitochondrial disposal from these neurons.
Retinal ganglion cells are particularly energy-hungry cells and may demand a level of mitochondrial turnover that exceeds axonal transport capacity. Their axons project through a variety of target regions in the brain, are unmyelinated in the retina but myelinated for the remainder of their length, and experience stressors such as light, varying intraocular pressure, and poor oxygen supply (13). Such an environment could stand in contrast to the more protected regions of the brain and nerve tracts, and so this may be a specialization of these neurons. However, one could also imagine low levels of transcellular mitochondrial degradation being dismissed as artifactual in these tissues or going wholly unnoticed because they are only readily identified by electron microscopy or with a fluorescent marker. Indeed, this phenomenon appears to be more widely used in the nervous system, as Davis et al. report the identification of similar mitochondria-rich protrusions in the superficial layers of the cerebral cortex of young mice.
Davis et al. have named this process of transcellular degradation of mitochondria “transmitophagy.” But although cell-autonomous mitophagy is an autophagic event, it remains to be determined whether axonal mitochondria transferred to astrocytes are degraded by an autophagic process or a phagocytic event; both would result in the association of transferred material with lysosomal compartments. Key next steps include determining whether these particles are enclosed in a double membrane-bound vesicle, implying autophagy, or a single membrane-bound phagosome-like vesicle. Equally important is exploring whether this process is regulated by autophagic, phagocytic, or other signaling pathways.
The process of transmitophagy not only goes against dogma—cells don’t necessarily autonomously destroy all of their own mitochondria—but it raises many intriguing questions about the biology of mitochondria, axons, and astrocytes. For instance, how do mitochondria tag themselves for disposal, cluster at the appropriate axonal site, and load themselves into these evulsions? It is also not clear whether this transcellular degradation process is specific for mitochondria, or is used to dispose of other axonal organelles. How astrocytes discriminate between axonal evulsions and healthy axonal material, and ultimately drive uptake and disposal of the correct target is also a curiosity. What drives the formation of evulsions, loading, and pinching off is also an open question. The finding also raises the question of whether signals are sent from the axon to the astrocyte to ensure uptake.
Even if transmitophagy is rare in healthy neuronal populations, it will be necessary to determine whether each neuron has the intrinsic capability to use this mechanism to dispose of unwanted mitochondria, if we wish to understand neuronal physiology, and whether it is up-regulated in response to stress or disease. Interestingly, Davis et al. noted an increase of transmitophagy in retinal ganglion cells after exposure to rotenone, an inhibitor of mitochondrial respiratory chain complex I. Autophagosomes containing mitochondria reportedly accumulate in neurodegenerative disease models (9, 14). In these situations, transmitophagy may compensate for interruptions in axonal transport by intracellular aggregates or disruption of microtubule networks. Going forward, it will be exciting to explore a possible increase of transmitophagy as a neuroprotective target.
Eye on mitochondria.
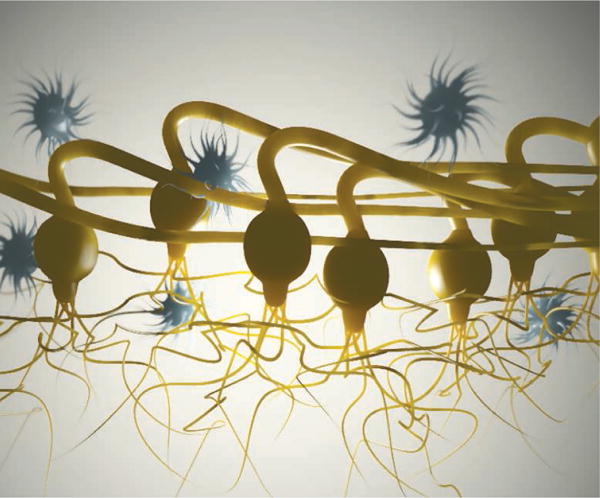
Retinal ganglion cells in the optic nerve head of the mouse are enwrapped by astrocytes and rapidly turn over mitochondria to meet high metabolic demands.
Transmitophagy.
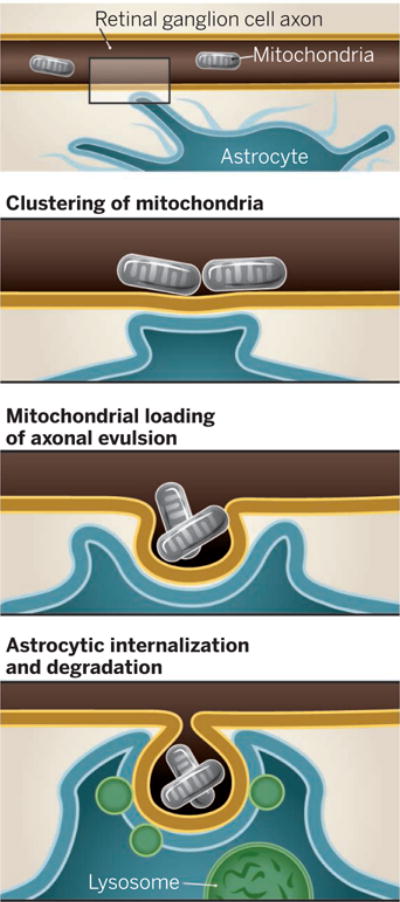
Mitochondria cluster in the axon of a retinal ganglion cell in the optic nerve head. This forms an evulsion that is internalized by a nearby astrocyte. Mitochondria are then degraded in lysosomes.
References
- 1.Youle RJ, van der Bliek AM. Science. 2012;337:1062. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sheng ZH. J Cell Biol. 2014;204:1087. doi: 10.1083/jcb.201312123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis CH, et al. Proc Natl Acad Sci USA. 2014;111:9633. [Google Scholar]
- 4.Smith DH. Prog Neurobiol. 2009;89:231. doi: 10.1016/j.pneurobio.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrison BM, Lee Y, Rothstein JD. Trends Cell Biol. 2013;23:644. doi: 10.1016/j.tcb.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sajic M, et al. PLOS Biol. 2013;11:e1001754. doi: 10.1371/journal.pbio.1001754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Court FA, Coleman MP. Trends Neurosci. 2012;35:364. doi: 10.1016/j.tins.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Weaver D, et al. Mol Cell. 2014;54:870. doi: 10.1016/j.molcel.2014.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maday S, Holzbaur ELF. Autophagy. 2012;8:858. doi: 10.4161/auto.20055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maday S, et al. J Cell Biol. 2012;196:407. doi: 10.1083/jcb.201106120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashrafi G, Schwarz TL. Cell Death Differ. 2013;20:31. doi: 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen JV, et al. Proc Natl Acad Sci USA. 2011;108:1176. [Google Scholar]
- 13.Yu DY, et al. Prog Retin Eye Res. 2013;36:217. doi: 10.1016/j.preteyeres.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Wang QJ, et al. J Neurosci. 2006;26:8057. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]