

Abstract
Adenosine derivatives developed to activate adenosine receptors (ARs) revealed µM activity at serotonin 5HT2B and 5HT2C receptors (5HTRs). We explored the SAR at 5HT2Rs and modeled receptor interactions in order to optimize affinity and simultaneously reduce AR affinity. Depending on N6 substitution, small 5′-alkylamide modification maintained 5HT2BR affinity, which was enhanced upon ribose substitution with rigid bicyclo[3.1.0]hexane (North (N)-methanocarba), e.g. N6-dicyclopropylmethyl 4′-CH2OH derivative 14 (Ki 11 nM). 5′-Methylamide 23 was 170-fold selective as antagonist for 5HT2BR vs. 5HT2CR. 5′-Methyl 25 and ethyl 26 esters potently antagonized 5HT2Rs with moderate selectivity in comparison to ARs; related 6-N,N-dimethylamino analogue 30 was 5HT2R-selective. 5′ position flexibility of substitution was indicated in 5HT2BR docking. Both 5′-ester and 5′-amide derivatives displayed in vivo t1/2 of 3–4 h. Thus, we used GPCR modeling to repurpose nucleoside scaffolds in favor of binding at nonpurine receptors, as novel 5HT2R antagonists, with potential for cardioprotection, liver protection or CNS activity.
Keywords: 5HT receptor, G protein-coupled receptor, scaffold repurposing, adenosine derivatives, molecular modeling, structure-activity relationship
Graphical abstract

Introduction
The process of de-risking potential clinical candidate molecules typically involves broad screening at hundreds of off-target activities, consisting of receptors, ion channels, transporters and enzymes.1 The structure-activity relationship (SAR) of such off-target activities can be analyzed and controlled to either suppress or enhance a secondary activity.2 Sometimes a secondary activity within a compound series can become the major focus of a drug discovery program and even lead to an approved drug, as was accomplished for a protease-activated receptor (PAR) 1 antagonist.3 Chemical scaffolds from already approved drugs have been repurposed for new clinical indications, such as kinases drugs that served as the basis of inhibitors of trypanosome replication.4 Numerous nucleoside derivatives are approved as pharmaceuticals, indicating that this is a general scaffold that is well-tolerated in vivo.5 Other scaffolds have been applied to multiple GPCR families, such as 1,4-dihydropyridines, which have been termed a privileged scaffold, because it can be molded to fit diverse sites.6,7
In the process of examining off-target interactions of nucleoside derivatives that were designed as potent ligands of the A3 adenosine receptor (AR), we found that occasionally µM affinity was observed at 5HT2B and 5HT2C serotonin receptors (5HT2Rs). A prototypical A3AR agonist IB-MECA 10 (Chart 1), containing a ribose 5′-N-methyluronamide group, showed binding Ki values at the 5HT2BR and 5HT2CR of 1.08 and 5.42 µM, respectively.8,9 Certain adenine (1, 2) and truncated adenosine derivatives (3, 4) also displayed significant 5HT2R affinity with Ki <5 µM.9 In the same study, we reported that the substitution of the ribose tetrahydrofuryl ring of adenosine derivatives with a fused bicyclo[3.1.0]hexyl moiety (termed (N)-methanocarba), which maintained a conformation preferred by the A3AR, was compatible with binding at the 5HT2Rs. Thus, an otherwise A3AR-selective agonist 35, which like 10 has an N6-3-halobenzyl group, bound with even greater affinity than riboside 10 at 5HT2Rs; the Ki values of 35 at 5HT2BR and 5HT2CR were 75 and 122 nM, respectively.9 Therefore, these findings provided an opportunity to define more clearly the interaction of nucleosides with 5HT2Rs and to shift the affinity in favor of this nonpurine receptor activity.
Chart 1.

Adenine (1, 2), nucleoside (10) and (N)-methanocarba nucleoside (3, 4, 31) derivatives that were previously reported to interact with the 5HT2BR (binding Ki values are shown in italics).9
Technology for the discovery of new GPCR ligands is now largely structure-based, with the determination of the X-ray structures of >170 GPCR-ligand complexes.10 The four ARs belong to the family of rhodopsin-like G protein-coupled receptors (GPCRs). Thirteen of the fourteen 5HTRs, including 5HT2BR and 5HT2CR, are also GPCRs and are located on a different, but closely related branch of rhodopsin-like GPCRs that includes various biogenic amine receptors. The interaction of 35 with the 5HT2BR and 5HT2CR was modeled based on a recently determined X-ray crystallographic structure of the 5HT2BR complex with agonist ergotamine.7 Paoletta et al. predicted the binding mode to feature a hydrophobic region deep in the binding site that could accommodate the substituted N6-benzyl group of 35.9 Furthermore, a truncated (N)-methanocarba-adenosine derivative containing an N6-dicyclopropylmethyl group 4 that acts as a moderately selective agonist of the A1AR was found to interact at µM concentrations with 5HT2Rs.9 By in silico docking of several adenine and adenosine derivatives to 5HT2Rs, self-consistent hypotheses for the orientation of the relevant nucleosides in this receptor family were proposed.9 Here, we have further explored the SAR of (N)-methanocarba-adenosine derivatives at 5HT2BR and 5HT2CR in an effort to optimize affinity at 5HTRs and simultaneously reduce AR affinity. Thus, we have incorporated and modified structural features of the nucleosides to repurpose the scaffold toward the 5HT-R family.
Selective AR ligands and selective 5HT-R ligands both have varied beneficial activities in disorders of the peripheral and central nervous system (CNS) and in other tissues.11,12 5HT2BR agonist activity is a desirable characteristic in selective serotonin reuptake inhibitors (SSRIs), but peripheral actions at 5HT2Rs might also have therapeutic potential. 5HT2BR antagonists are sought as agents potentially useful for treating chronic liver and heart disease.13,14 Activation of the 5HT2BR by serotonin agonists has been associated with valvulopathy and fibrosis in many organs,15–17 5HT2BR antagonists improved liver function in fibrotic disease models and were described as a “clinically safe” therapy for humans.13 In mouse models of coronary function, 5HT2BR antagonists reduced collagen deposits and the resultant right ventricular fibrosis (RVF).14 A 5HT2BR antagonist is also under development for irritable bowel syndrome.18 Therefore, the goal of this study is to discover selective 5HT2BR antagonists that might have in vivo activities indicative of the potential for disease treatment. This requires deselecting for 5HT2CR affinity, which is closely associated with 5HT2BR in the present structural series. The combination of potent 5HT2BR antagonist and A3AR agonist activities19 in a single molecule might also be beneficial or synergistic for liver protection or cardioprotection.
Results
We have utilized some of the previously reported analogues of adenosine (Table 1, 1–13, 35–41) that were characterized as either A1 or A3AR-selective agonists,20–27 to examine the effects of structural modification on the interaction with 5HT2Rs. The objectives were to use structural insights to synthesize novel nucleosides (14–34, 42) with enhanced 5HT2R affinity and at the same time progressively achieve selectivity over the ARs.
Table 1.
Structures and binding affinities of nucleoside derivatives at 5HT2Rs and at A1 and A3ARs, including newly synthesized (14–34, 42) derivatives and previously reported (1–13, 35–41) nucleosides. R3 = NHCH3 and X = N, unless noted. Human (h) ARs, unless noted (r = rat).
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| No. |
R1 | R2 | 5HT2AR % inhibition or Ki (µM)a |
5HT2BR % inhibition or Ki (µM)a |
5HT2CR % inhibition or Ki (µM)a |
A1AR, % inhibition or Ki (µM)b |
A2AAR, % inhibition or Ki (µM)b |
A3AR, % inhibition or Ki (µM)b |
| Adenine and truncated nucleoside derivatives | ||||||||
| 1b | 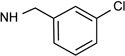 |
Cl | 26% | 2.16± 0.045 |
3.35± 0.34 |
45% | 37% | 0.165 |
| 2b | 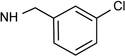 |
 |
<10% | 4.00± 1.36 |
11% | <10% | <10% | 0.120 |
| 3b | 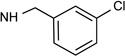 |
Cl | <10% | 2.39± 0.53 |
4.10± 1.62 |
1.6 | 4.52 | 4.9 |
| 4b | 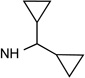 |
Cl | <10% | 0.675± 0.100 |
1.86± 0.22 |
0.048 | 3.95 | 0.47 |
| Riboside 5′-OH derivatives | ||||||||
| 5 | NH2 | Cl | 46% | <10% | <10% | 0.0028 | 0.063 (r)c | 0.087c |
| 6 | 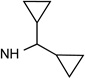 |
H | <10% | 38% | 11% | 0.0008 (r)b |
1.37 (r)c | 0.041c |
| Riboside 5′-N-alkylamide derivatives | ||||||||
| 7 | NH2 | H | <10% | <10% | <10% | 0.084 (r)c | 0.0668 (r)c |
0.072 (r)c |
|
8 R3 = EtNH |
NH2 | H | <10% | <10% | <10% | 0.0068 | 0.0103 (r)b |
0.016 |
|
9 R3 = cPrNH |
NH2 | H | <10% | <10% | <10% | 0.013 (r)c | 0.0134 (r)c |
1.6 (r)c |
| 10 | 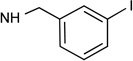 |
H | <10% | 1.08 | 5.42 | 0.70 | 6.20 | 0.0017c |
| 11 | 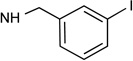 |
Cl | ~30% | 2.77± 0.56 |
~10 | 0.22 | 5.40 | 0.0014c |
| (N)-methanocarba 5′-OH derivatives | ||||||||
| 12 | NH2 | Cl | <10% | >10 | <10% | 0.273 (r)c | 1.91 (r)c | 0.085 (r)c |
| 13 | NHCH3 | Cl | <10% | 6.47± 1.24 |
6.4d | 1.47 (r)c |
<10% (r)c |
0.023c |
| 14 | 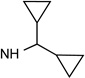 |
Cl | <10% | 0.0111± 0.0034 |
0.084± 0.024 |
0.039± 0.017 |
2.2d | 1.60 ±0.21 |
| 15 | NH- CH(CH2CH3)2 |
Cl | 12% | 0.476± 0.063 |
0.703± 0.058 |
0.112± 0.010 |
1.75± 0.19 |
1.2±0.4 |
| 16 | 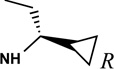 |
Cl | <10% | 0.186± 0.058 |
0.371± 0.043 |
0.076± 0.014 |
1.88± 0.20 |
0.16± 0.05 |
| 17 | 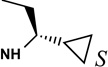 |
Cl | <10% | 0.073± 0.010 |
0.251± 0.023 |
0.117± 0.010 |
2.95± 0.62 |
1.81± 0.11 |
| 18 | 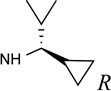 |
Cl | <10% | 0.163± 0.016 |
0.190± 0.031 |
0.103± 0.007 |
2.20± 0.31 |
1.24± 0.32 |
| 19 | 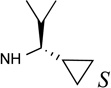 |
Cl | <10% | 0.163± 0.018 |
0.672± 0.202 |
0.104± 0.006 |
1.52± 0.12 |
3.45± 0.67 |
| 20 | 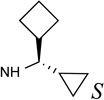 |
Cl | <10% | 0.034± 0.007 |
0.298± 0.064 |
0.298± 0.004 |
2.69± 0.37 |
3.24± 0.46 |
| 21 | 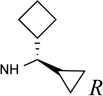 |
Cl | <10% | 0.077± 0.003 |
0.270± 0.026 |
0.144± 0.032 |
3.94± 0.32 |
3.47± 0.49 |
| 22 | N(CH3)2 | Cl | <10% | 4.80± 0.34 |
8.1±0.8 | 11±5% | <10% | 2.47± 0.44 |
| (N)-methanocarba 5′-ester, amide and carboxylic acid derivatives | ||||||||
| 23 | 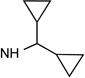 |
Cl | <10% | 0.023± 0.002 |
0.269± 0.007 |
0.110± 0.014 |
4.32± 0.87 |
0.034± 0.011 |
| 24 | N(CH3)2 | Cl | <10% | 2.10± 0.68 |
4.3d | 26% | <10% | 1.48± 0.62 |
|
25 R3=OCH3 |
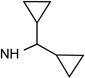 |
Cl | <10% | 0.019 ±0.001 |
0.117 ± 0.011 |
0.437± 0.021 |
33% | 0.766± 0.193 |
|
26 R3= O-CH2-CH3 |
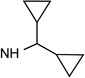 |
Cl | 19% | 0.015± 0.003 |
0.030± 0.002 |
0.360± 0.074 |
1.57± 0.18 |
0.236± 0.041 |
|
27 R3= O-(CH2)2-CH3 |
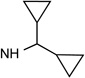 |
Cl | <10% | 0.045± 0.003 |
0.205± 0.073 |
0.440± 0.047 |
19±4% | 0.499± 0.242 |
|
28 R3= OH |
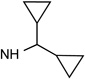 |
Cl | <10% | 1.48± 0.12 |
2.58± 1.34 |
ND | <10% | 5.98± 0.30 |
|
29 R3= O-CH2-CH3 |
N(CH3)2 | Cl | <10% | 0.671± 0.184 |
1.35± 0.19 |
<10% | 13±6% | 30±10% |
|
30 R3= OH |
N(CH3)2 | Cl | <10% | 4.70± 0.01 |
8.0d | ND | <10% | 1.08± 0.14 |
|
31e R3=NH(CH2)2-NH2 |
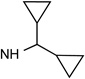 |
Cl | <10% | 0.630± 0.146 |
0.142± 0.001 |
2.58± 1.22 |
23±5% | 8.11± 1.27 |
|
32e R3=NH(CH2)3-NH2 |
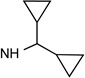 |
Cl | <10% | 1.12± 0.31 |
0.137± 0.019 |
45±4% | 26±5% | 55±1% |
|
33 R3=NH(CH2)2-NHAc |
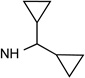 |
Cl | <10% | 0.332± 0.092 |
1.70± 0.90 |
0.397± 0.081 |
20±2% | 1.65± 0.93 |
|
34 R3=NH-(CH2)3NH-Ac |
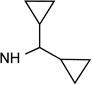 |
Cl | <10% | 0.534± 0.131 |
2.31± 0.64 |
46±1% | 26±0% | 1.97± 0.90 |
| 35c | 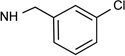 |
Cl | 35% | 0.075± 0.007 |
0.168± 0.047 |
0.26 | 2.30 | 0.00029 |
| (N)-methanocarba 5′-amide derivatives with C2-arylethynyl groups | ||||||||
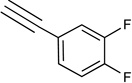
| 36c | NHCH3 | 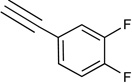 |
11% | <10% | <10% | <10% | <10% | 0.0017 |
| 37c | 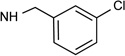 |
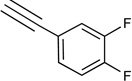 |
<10% | 2.58± 0.01 |
7.2d | <10% | 41% | 0.0035 |
| 38c | 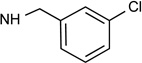 |
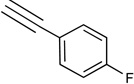 |
26% | 2.12± 0.14 |
8.9d | 20% | 42% | 0.0022 |
| 39c | NHCH3 | 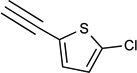 |
19% | <10% | 30% | <10% | 24% | 0.00070 |
| 40c | NHC2H5 | 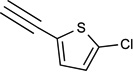 |
<10% | 13% | 38% | 28% | 12% | 0.0038 |
|
41c X=CH |
NHC2H5 | 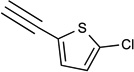 |
47% | 2.21± 0.30 |
<10% | 10% | <10% | 0.0017 |
| 42 | N(CH3)2 | 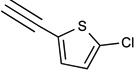 |
27% | <10% | 8.5d | 25±5% | 15±8% | 0.0235± 0.0106 |
Binding in membranes prepared from cells stably expressing one of three 5HT2 receptors: 5HT2AR, HEKT cells, binding with [3H]ketanserin (Kd 1.57 nM); 5HT2BR, stable HEK cells, binding with [3H]lysergic acid diethylamide 43 (Kd 1.04 nM); 5HT2CR, Flp IN HEK cells, binding with [3H]mesulergine 44 (Kd 2.92 nM). Values are expressed as the mean ± SEM. A percent in italics refers to inhibition of binding at 10 µM. Nonspecific binding was determined using 10 µM clozapine (5HT2AR), 49 (5HT2BR) or 50 (5HT2CR). Ki values were calculated as reported.41 All of the compounds displayed <50% inhibition at the human 5HT1A, 5HT1B, 5HT1D, 5HT1E, 5HT3 and 5HT5ARs, unless noted. Compound 3 bound weakly at human 5HT3 and compound 30 bound weakly at the human 5HT5AR (Supporting Information).
Affinity at the A1AR (binding with [3H]N6-phenylisopropyladenosine 45) and A3AR (binding with [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide 46) determined as reported,22,24 unless noted. Selected compounds were tested for binding inhibition ([3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine 47) at hA2AAR in HEK293 cell membranes. The binding affinity was expressed as Ki values (n = 2−4, unless noted). A percent in italics refers to inhibition of binding at 10 µM. Nonspecific AR binding was determined using adenosine-5′-N-ethyluronamide 48 (10 µM). Values are expressed as the mean ± SEM. Ki values were calculated as reported.53
n = 1.
ND – not determined.
The synthesis of the (N)-methanocarba nucleoside analogues was performed as shown in Schemes 1 and 2. The (N)-methanocarba sugar derivative 53 (Scheme1) was obtained from D-ribose by a known method.28 Mitsunobu condensation of compound 53 with 2,6-dichloropurine gave the nucleoside derivative 54, which underwent amination with various amines to give compounds 55–63. Treatment of TBAF solution with compounds (55–62) gave the desilylated compounds 64–71. The isopropylidine group of compounds (64–72) was deprotected with Dowex50 resin to give the nucleoside derivatives 14–21. Compound 63 was treated with 10% TFA solution at 70 °C to afford compound 22. The 4′-ethylester derivative 74 (Scheme 2), which obtained from L-ribose by a reported method,29 was converted to nucleoside derivative 75 by a Mitsunobu condensation with 2,6-dichloropurine. Amination of compound 75 with dicyclopropyl amine and N,N-dimethyl amine gave compounds 76 and 77, respectively, which upon acid hydrolysis provided the nucleoside derivatives 26 and 29. Hydrolysis of ethyl ester group of compounds 26 and 29 with 2N aqueous sodium hydroxide provided the carboxylic acid derivatives 28 and 30. Coupling of compound 28 with methanol and propanol in the presence of DCC and DMAP gave the ester derivatives 25 and 27. Treatment of compound 26 and 29 with 40% MeNH2 solution gave the 5′-methylamide nucleosides 23 and 24, respectively. Compound 26 was also treated with 1,2-diaminoethane and 1,3-diamino propane to afford the compound 31 and 32, respectively, which underwent acetylation upon treatment of acetic acid N-hydroxysuccinimide ester in the presence of triethylamine to provide acetyl derivatives 33 and 34.
Scheme 1.

Synthesis of N6-alkyl-(N)-methanocarba-adenosine (5′-OH) derivatives. Reagents: (a) 2,6-dichloropurine, Ph3P, DIAD, THF, rt, 69%; (b) RNH2, Et3N, MeOH, rt, 78–85%; (c) TBAF, THF, rt, 68–80%; (d) Dowex50, MeOH-H2O, 67–85%. All steps were performed at rt. Compound 22 was prepared from 63 without isolation of protected intermediate.
Scheme 2.

Synthesis of N6-alkyl-(N)-methanocarba-adenosine (4′-carbonyl) derivatives. Reagents and Condition: (a) 2,6-dichloropurine, Ph3P, DIAD, THF, rt; (b) RNH2, Et3N, MeOH, rt, 85–87%; (c) Dowex50, MeOH-H2O, rt, 86–88%; (d) 1N NaOH, MeOH, rt, 76–77%; (e) 40% MeNH2, MeOH, rt, 65–67%; (f) H2N(CH2)nNH2, MeOH, rt, 63–64%; (g) 2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl acetate, Et3N, DMF, rt, 58–59%; (h) ROH, DMAP, DCC, DMF, rt.
The binding assays at diverse receptors and transporters, other than ARs, were performed at the Psychoactive Drug Screening Program (PDSP) at the University of North Carolina under the direction of Bryan L. Roth.1 Initially a concentration of 10 µM was used to probe binding at diverse sites, and compounds that achieved >50% inhibition at this concentration were analyzed with full curves to determine Ki values. We have focused on the 5HT2BR and 5HT2CR, because none of the compounds tested showed significant interaction (>50% inhibition at 10 µM) in binding assays at other 5HTRs: 5HT1A, 5HT1B, 5HT1D, 5HT1E, 5HT4, 5HT6 and 5HT7. Affinity at the 5HT2AR, which is related in sequence to 5HT2BR and 5HT2CR, was also consistently low (Ki > 10 µM) or absent; when <50% inhibition was noted at any of the 5HT2Rs, the % values at are provided in the table. However, one compound displayed measurable affinity at the 5HT3R, e.g. truncated nucleoside 3, Ki 3.26 ± 0.54 µM (n = 3), and compound 30 bound weakly to the 5HT5AR.
Binding at the 5HT2BR and 5HT2CR was performed using standard high affinity radioligands, [3H]43 and [3H]44, respectively. As we reported previously, adenine derivatives 1 and 2, lacking N9 substitution, or with a 2′,3′-dihydroxy-bicyclo[3.1.0]hexyl substitution of ribose, i.e. 3 and 4, bound to the 5HT2BR with µM affinity.9 Appending the adenine C2 substitution with a rigid arylethynyl group in 2 maintained affinity at the 5HT2BR, but eliminated µM affinity at the 5HT2CR that was observed with compounds 1, 3 and 4. Ribonucleosides 5–9, known AR agonists that display varied AR selectivities, were found to be inactive at 5HT2Rs. Compound 5 is often taken to closely approximate the pharmacology of adenosine itself; thus, it is unlikely that endogenous adenosine would be interacting with 5HT2Rs in situ. Depending on N6-substitution, appending a 9-ribose moiety with a small alkyl amide modification at the 5′ position was tolerated at the 5HT2BR, as with compounds 10 and 11. Changing C2-H 10 to C2-Cl 11 in the ribose series did not have a major effect on the 5HT2R affinity; both of those A3AR-selective agonists are in current clinical trials.30
The SAR of (N)-methanocarba derivatives 12 – 42 was analyzed in detail. In the 4′-CH2OH series 12 – 22, a slight increase of 5HT2R affinity was observed with introduction of an N6-methyl group on the exocyclic amine (as in previously reported (N)-methanocarba nucleosides 13 vs. 12). Enhancement of 5HT2R affinity was observed upon introduction of a rigid (N)-methanocarba moiety, compared to ribose, in analogues containing a 4′-hydroxymethyl group (as in 12 vs. 5; 14 vs. 6, in which the C2 substitution also changes). Notably, N6-dicyclopropylmethyl 4′-CH2OH derivative 14 containing a (N)-methanocarba substitution displayed a potent Ki value of 11 nM at the 5HT2BR with slight (8-fold) selectivity vs. 5HT2CR (P < 0.05, unpaired t-test). Consequently, analogues of 14 with slight modifications of the N6-dicyclopropylmethyl group and defined stereochemistry (compounds 15–21) were evaluated in an attempt to tune the pharmacological properties. Cyclopropyl was changed to ethyl, isopropyl or cyclobutyl in an effort to enhance 5HT2BR affinity or selectivity. Thus, pairs of diastereomeric nucleosides differing in the chirality of the Cα group at N6 were compared. In comparing N6-dicyclopropylmethyl derivative 14 with R-isomers 16, 18, and 21 and with S-isomers 17, 19, and 20, there was no consistent preference for a particular stereoisomer, and all of these variations of 14 moderately reduced the 5HT2BR affinity. Also, the N6-achiral derivative 15 was most reduced in 5HT2R affinity compared to 14. Among these chiral N6 derivatives of 14, S-isomer 20 was the most potent at the 5HT2BR (Ki 34 nM), and R-isomer 18 was the most potent at the 5HT2CR (Ki 190 nM).
5′-Carbonyl group modifications had variable effects on A3AR affinity in comparison to the corresponding 4′-CH2OH. The 5′-methylamide derivative 23, related to N6-dicyclopropyl-methyl analogue 14, was 12-fold selective in binding for 5HT2BR vs. 5HT2CR (P < 0.05, unpaired t-test). However, the same 5′-methylamide in the (N)-methanocarba series generally resulted in small decreases in 5HT2BR affinity (Ki 23 nM, 23 vs. 11 nM, 14) and 5HT2CR affinity (Ki 269 nM, 23 vs. 84 nM, 14). 5′-Methyl ester 25 and 5′-ethyl ester 26 derivatives were particularly potent and selective at the 5HT2Rs (15–19 nM at 5HT2BR) with only intermediate affinity at the ARs (roughly µM). Compound 25 was 6-fold selective for the 5HT2BR compared to 5HT2CR (P < 0.05, unpaired t-test),
Representative inhibition curves are shown for 5′-methyl ester 25 (Figure 1), which bound to the 5HT2BR as potently as the reference compound 49. The corresponding 5′-propyl ester 27 and 5′-carboxylic acid derivative 28 were less potent at the 5HT2Rs than the methyl and ethyl esters.
Figure 1.


Representative binding curves at the (A) 5HT2BR and (B) 5HT2CR for compound 25 (black curves), in comparison to reference compounds (red curves): (A) 3,5-dihydro-5-methyl-N-3-pyridinylbenzo[1,2-b:4,5-b′]dipyrrole-1(2H)-carboxamide (49, SB206553) and (B) 6-[2-[4-[bis(4-fluorophenyl)methylene]-1-piperidinyl]ethyl]-7-methyl-5H-thiazolo[3,2-a]pyrimidin-5-one (50, ritanserin). Radioligands used were: 5HT2BR, binding with [3H]lysergic acid diethylamide 43; 5HT2CR, binding with [3H]mesulergine 44. Representative functional assays at the (C) 5HT2BR and (D) 5HT2CR for compounds 23, 25 and 26, in comparison to reference compound 5-HT applied to desensitize the response. 5-HT (3 nM, EC80) was used as an agonist in both C and D. Compound 26 (10 µM) did not display significant agonist or antagonist activity (>10%) at the 5HT2AR or agonist activity at 5HT2BR or 5HT2CR (data not shown). RFU, relative fluorescence units. Mean ± s.e.m. is shown (n = 3). The curves for all compounds were best fit using a monophasic isotherm with a variable Hill slope (F-test, P< 0.05 was considered as statistically significant).
For comparison, we include affinities at the human A1AR, A2AAR and A3AR, often from archival data (Table 1).21–26 Selected newly synthesized structures were assayed at 10 µM at the A2AAR, which is disfavored in binding of (N)-methanocarba adenosines in general. We did not include affinity at the human A2BAR, because we already established that in the (N)-methanocarba series, especially those containing compounds C2 substitution, the affinities tend to be much lower at that subtype than at the other AR subtypes.25 (N)-Methanocarba 4′-hydroxymethyl 14 and 5′-methylamide 23 derivatives were more potent at the A3AR than the corresponding 5′-alkyl esters 25 – 27. However, 14 was most potent at the A1AR with a Ki value of 39 nM. The affinity at the A1AR and A3AR of the diastereomeric nucleosides 16 – 21 was significantly reduced (typically by 20- to 30-fold) compared to 14. Similarly, in a previous modeling study of the 4′-truncated equivalents of compounds 14 – 21,22 any deviation from the N6-dicyclopropylmethyl group reduced both A1AR and A3AR affinity. Furthermore, 25 was significantly more potent at 5HT2BR than at A1AR (23-fold) and A3AR (40-fold) (P < 0.05, one-way ANOVA with post-hoc test). Functional assays of A1AR-mediated inhibition of cAMP formation22 showed that 14 and 26 were full agonists, with maximal efficacy (at 10 µM) of 104±4% and 89±3% of N6-cyclopentyladenosine (1 µM). In a functional assay of A3AR-mediated inhibition of cAMP formation (n=3),22 esters 25 and 26 were partial agonists (at 10 µM) with maximal efficacy of 41.4±2.8% and 26.3±6.1%, respectively, compared to 48 set at 100%, and amide 23 was a full agonist (96.7±1.0%).
All of the previous analogues mentioned up to this point contained either an exocyclic NH2 group or a monosubstituted NH. The effects of di-methyl substitution of the N6 group was examined in the following 2-Cl analogues: 4′-hydroxymethyl 22, 5′-methylamide 24, 5′-ethyl ester 29 and 5′-carboxylic acid 30 derivatives. The 4′-CH2OH analogue 22 displayed Ki values of 5–8 µM at 5HT2BR and 5HT2CR and ~2.5 µM at the A3AR. The corresponding 5′-CONH-Me analogue 24 displayed only slightly improved µM affinity at 5HT2BR, 5HT2CR and A3AR. However, the corresponding 5′-ethyl ester analogue 29 displayed Ki values of 671 nM at 5HT2BR and 1.35 µM at 5HT2CR with no measurable affinity at the A3AR. Thus, N6-dimethyl analogue 29 was 5HT2R-selective in binding assays. 5′-Carboxylate derivatives 28 and 30 displayed roughly µM affinity at the 5HT2BR, 5HT2CR and A3AR.
In order to reduce AR affinity while further enhancing 5HT2R affinity, we examined in detail the molecular modeling of these GPCRs. The docking of nucleoside derivatives at the 5HT2BR by Paoletta et al. proposed two hypothetical binding modes depending on substituent groups.9 The most general docking mode, applicable to all nucleosides examined, proposed that a nonpolar N6 group, i.e. a 3-chlorobenzyl group in 35, was anchored the analogues in a deep hydrophobic region.2 This mode would allow the ribose moiety access to the exofacial side of the receptor and therefore might provide greater flexibility of substitution. An alternative mode of 5HT2BR docking, which was very similar to the position of related adenosine derivatives in ARs with the ribose moiety facing downward in a deep hydrophilic region, was possible only in the absence of an extended C2 substituent. We proceeded initially assuming the first predicted binding mode (above), leading to the hypothesis that the more external ribose moiety (or (N)-methanocarba moiety) would have considerable freedom of substitution in binding to the 5HT2BR. In order to design analogues that might lack interaction with ARs, we referred to our previous studies of the modification of 5′-amide derivatives in the adenosine (9-riboside) series.31 It was noted that 5′-N-2-aminoethyl and 5′-N-3-aminopropyl amides in the ribose series were of greatly reduced AR affinity. Therefore, we prepared compounds 31 and 32, which are 5′-N-(aminoalkyl)amide derivatives containing the preferred N6-dicyclopropylmethyl group to test if the interaction with the 5HT2Rs would be maintained. In fact, these two compounds bound with moderate affinity at 5HT2BR and 5HT2CR, with a preference for the 5HT2CR (both displayed Ki values of ~140 nM). Moreover, as predicted by receptor docking modes, compounds 31 and 32 were selective for the 5HT2Rs in comparison to ARs. Therefore, we added two more analogues 33 and 34, which were the N-acetylation products of 31 and 32 and that displayed roughly µM or sub-µM affinity in 5HT2Rs and Ki values of ~1.7–2.0 µM at the A3AR. Evidently, there is a freedom of substitution at the 5HT2Rs of this extended functionalized chain attached at the 5′-CO position, consistent with modeling predictions that indicate the ribose moiety to be pointing toward the extracellular medium. However, in AR docking, based on the X-ray structures of nucleoside-bound A2AARs, the ribose binding subpocket and the region around the 5′ position are more sterically limited. Curiously, compound 33 displayed a Ki value of 0.40 µM at the A1AR, which was more potent than other members of the extended 5′-amide series.
Effects on 5HT2R affinity of extensions at the C2 position were compared. Compound 35 has been studied as a potent A3AR agonist in an inflammation model.30 Although it displayed substantial affinity at 5HT2Rs, 35 was nevertheless 260-fold selective for A3AR vs. 5HT2BR. Other (N)-methanocarba derivatives with extended C2 substituents 36–41, which were already reported as particularly potent and selective at the A3AR,20,22–26 displayed at best µM affinity at 5HT2Rs, e.g. 37, 38, and 41. Thus, the approach of C2 extension that we have taken in recent SAR studies to enhance A3AR selectivity generally precluded activity at 5HTRs. The dialkylation of the exocyclic amine of 42 slightly increased affinity at the 5HT2CR with an accompanying 34-fold reduction of A3AR affinity (39 vs. 42).
Functional assays indicated antagonist activity of 14 and 26 at 5HT2B/CRs, similar to our previous report with adenine/adenosine derivatives that are moderately potent 5HT2R ligands.9 Compounds 1 and 35 were previously found to be antagonists of the 5HT2BR and 5HT2CR, with functional IC50 values at 5HT2BR of 0.89 and 3.26 µM, respectively.9 Here, Ki values in fully antagonizing receptor activation by serotonin at 5HT2BR and 5HT2CR were determined (Figures 1C,D), respectively, for compounds (n=): 14, 22.3±4.0 nM (2) and 337±27 nM (2); 23, 4.4±2.6 nM (4) and 752±431 nM (3); 25, 3.2±1.9 nM (4) and 4.1±1.6 nM (4); 26, 1.8±0.7 nM (3) and 3.9±2.9 nM (3). Thus, 5′-methyamide 23 displayed 170-fold selectivity at 5HT2BR. Compounds 14, 23, 25, and 26 at 10 µM had no appreciable agonist or partial agonist activity at 5HT2BR and 5HT2CR and no activity as agonist or antagonist at 5HT2AR (Figure 2, Figure S6, Supporting Information). Therefore, within the 5HT2Rs, all four derivatives were potent antagonists at two subtypes, and 5HT2BR functional selectivity was observed for amide derivative 23 (Figure S7, Supporting information). The ligand efficiency (LE) in 5HT2BR binding for four compounds was: 14, 0.41; 23, 0.37; 25, 0.37; 26, 0.37.
Figure 2.

Representative functional assays at the (A, B) 5HT2AR, (C, D) 5HT2BR and (D, E) 5HT2CR for compound 14, in comparison to reference compound 5-HT applied to desensitize the response. 5-HT (3 nM, EC80) was used as agonist in B, D and F. RFU, relative fluorescence units.
Two representative N6-dicyclopropylmethyl derivatives 23 and 25 that differed only at the 5′ position were examined for ADME and toxicological parameters (Table 2, Supporting Information). 5′-Methylamide 23 and 5′-methyl ester 25 demonstrated ~100% stability in human plasma over 2 h, and 87% 23 and 94% 25 remained after 1 h exposure to human liver microsomes with cofactor present. Both compounds lacked inhibition of cytochrome P450 enzymes (Table 2) and were found to be relatively non-toxic to HepG2 liver cells (growth inhibition <30% at 30 µM). Their stability at pH 6.5 was high, with 97% of each compound remaining after 2 h in simulated intestinal fluid). However, the stability in simulated gastric fluid at pH 1.6 showed only ~50% of each compound remaining after 2 h. The reduced acid stability is likely common to compounds containing a N6-dicyclopropylmethyl group. 23 was moderately permeable in CACO-2 cells with evidence of efflux (A-to-B Papp = 4.73 × 10−6 cm/sec; efflux ratio = 11.7), while 25 was highly permeable with no significant efflux (A-to-B Papp = 18.5 × 10−6 cm/sec; efflux ratio = 1.67). The half-life following intravenous administration (1.0 mg/kg) in male Sprague-Dawley rats was 2.89±0.67 h for 23 and 3.56±0.32 h for 25 (mean ± SEM, n = 3). Thus, the ester group of 25 was not subject to rapid hydrolysis, probably due to the steric hindrance of the tertiary 4′-carbon. Compound 23 (C0 = 1070±30 ng/mL; Vdss = 3.16±0.41 L/kg) displayed better pharmacokinetic exposure than 25 (C0 = 349±7 ng/mL; Vdss = 4.62±0.18 L/kg).
Table 2.
In vitro stability parameters for 5′-methylamide 23 and 5′-methyl ester 25.a
 | ||
|---|---|---|
| Test Compound: | 23 | 25 |
| Stability in simulated fluids (t1/2, min): | ||
| Gastric (pH 1.6) | 132 | 125 |
| Intestinal (pH 6.5) | >480 | >480 |
| Stability in plasma (% remaining at 120 min): | ||
| Human | 104 | 95.3 |
| Rat | 88.7 | 70.9 |
| Mouse | 95.0 | 61.0 |
| CACO cell permeability: | ||
| Papp, A->B (10−6 cm/sec) | 4.73 | 18.47 |
| Papp, B->A (10−6 cm/sec) | 46.21 | 30.59 |
| Efflux ratio | 11.7 | 1.67 |
| Inhibition of 5 CYP isozymes: | ||
| IC50, µM | >30 | ≥20b |
| Stability in liver microsomes (t1/2, min): | ||
| Human d | 130 | >140 |
| Rat | >140 | >140 |
| Mouse | >145 | >140 |
| Cytotoxicity in HepG2 cells: | ||
| CC50, µM | >30 | >30 |
Procedures were described in Supporting Information and in Tosh et al. and Carlin et al.22,77
IC50 of both compounds at the following isozymes was >30 µM (except for 25 at the 2C9, which was 20.1 µM): A2, 2C9, 2C19, 2D6, 3A4; (average of n = 2).
CLint values (µL/min/mg protein) in human liver microsomes: 23, 1.75; 25, 0.77.
A few other off-target activities (Ki < 10 µM) at receptors and ion channels determined by the PDSP that are not already reported were detected (Figure S5, Supporting Information). For example, riboside derivative 9 showed only two such interactions (Ki at σ1 and σ2 receptors, 4.1 and 1.4 µM, respectively). Compounds 13 and 42 displayed Ki values at the σ2 receptor of 2.22±0.88 and 7.3 µM, respectively. Compound 14 inhibited binding at the dopamine transporter (Ki 4.8 µM) and compounds 26 and 27 at the translocator protein (TSPO, Ki 2.1 µM and 2.2 µM, respectively). There were no such interactions with other closely related GPCR families: muscarinic cholinergic, α- and β- adrenergic, dopaminergic or histaminergic receptors.
Molecular Modeling
We performed docking and MD simulations to explain the observed binding data by focusing on the h5HT2BR. The available X-ray structure of the receptor in complex with the biased agonist ergotamine (ERG) features signatures of both active and inactive states,32 thus entailing potential limitations in using this structure to elucidate the binding of antagonists. We therefore built a homology model for the native h5HT2BR using the ERG-h5HT2BR X-ray structure as template,32 docked a selected compound by means of induced fit docking (IFD), and refined the obtained binding pose with 30 ns of membrane MD simulations.
Induced Fit Docking
For our modeling analysis, we focused on the N6-dicyclopropylmethyl 5′-methylamide derivative 23, which is 12-fold selective in binding at the h5HT2BR in comparison to the h5HT2CR. The compound was docked into the h5HT2BR homology model by means of IFD (docking score −8.265 kcal/mol). As depicted in Figure 3A, the ligand lies perpendicular to the membrane plane with the pseudo-sugar moiety pointing toward the extracellular side in an orientation similar to the most general (ribose-up) binding mode previously described.9 A network of hydrogen bonds and extended hydrophobic contacts (Figure 3A) anchor the ligand to the upper part of TM2, TM3 and TM7, and to the downstream region of the second extracellular loop (EL2). The NH group of the amide moiety in 5′ and the secondary amino group (N6) act as H-bond donors to the backbone of Cys207EL2 of the conserved disulfide bridge and the sidechain of the conserved Asp1353.32 residue, respectively. The 2′ and 3′ OH groups of the pseudo-sugar moiety establish H-bond interactions with the sidechains of Gln3597.32, Glu3637.36, and Thr1142.64. The N6-dicyclopropylmethyl moiety engages in extended hydrophobic contacts with residues located in TM3 (Leu132 and Val136), TM6 (Trp337 and Phe340), TM7 (Val366 and Tyr370), and at the interface between EL2 and TM5 (Leu209 and Phe217). The methyl group of the 5′-methylamide points toward TM3 and establishes hydrophobic contacts with Trp121EL1, Trp1313.28, and the sidechain of Cys207EL2. The (N)-methanocarba ring faces the extracellular side and engages in hydrophobic contacts with Val208EL2.
Figure 3.

Hypothetical binding modes predicted by IFD (A) and following MD (B) simulation of N6-dicyclopropylmethyl 5′-methylamide (N)-methanocarba derivative 23 (cyan carbon atoms, sticks representation) at the h5HT2BR. Side chains of residues important for ligand recognition (grey carbon atoms) and water molecules (highlighted with different colors according to the ligand moiety they interact with) are reported as sticks. Residues establishing hydrophobic contacts with the ligand are depicted also as transparent surfaces. H-bonds and π-π stacking interactions are pictured as dashed yellow and cyan lines, respectively. Nonpolar hydrogen atoms are omitted. Helices are shown as ribbon structures numbered (from right to left) in A, TMs 1, 2, 3, 4, 5, 6 and 7, and in B, TMs 3, 4, 5, 6 and 7.
MD simulations
The above-described binding mode of 23 was subjected to 30 ns of all-atom MD simulations. The visualization of the trajectory (Video S1) along with the Interaction Energy (IE) profile (Lower right panel in Video S1 and Figure S1) showed that the interaction network did not persist when water molecules were allowed to diffuse into the membrane-embedded system. Indeed, after a few ns, a flip of the glycosidic bond (Video S1) triggered the ligand to move deeper in the binding side and to establish an extended network of H-bond interactions with the conserved Asp1353.32 and Tyr3707.43 through the interplay of water molecules. The pseudoglycosidic bond angle was −117.2° for the docked compound (anti) and 12.7° for the final structure after MD (syn). In the other two trajectories (data not shown), the flip of the glycosidic bond was assisted by an H-bond interaction between the side chain of Glu3637.36 and the 5′-methylamide moiety. As this residue occurs as Asn7.36 in the h5HT2CR, we believe that this transient interaction might assist the ligand while approaching the binding site and therefore might account for the h5HT2BR selectivity of the 5′-methylamide with respect to 5′-alkylester groups. The new placement of the ligand (Figure 3B) - with the adenine core lying almost parallel to the membrane plane and the N6-dicyclopropylmethyl moiety pointing toward TM5 and TM6 - persisted for the rest of the trajectory and was stabilized by extended water-mediate H-bond interactions. The binding mode proposed by the MD analysis (Figure 3B) features a π-π stacking interaction between the adenine core and Phe3406.51 and nine tightly bound water molecules (w1–w9) mediating the ligand-protein H-bond interactions: w1 (highlighted in yellow) anchors the 5′-carbonyl group to the sidechain of Gln3597.32 (acting as H-bond donor) and the backbone of Leu209EL2 (acting as H-bond acceptor); w2 and w3 (highlighted in magenta) connect the 2′ OH group of the pseudo-sugar moiety to conserved Asp1353.32 and Tyr3707.43; w4 and w5 (highlighted in green) bridge the backbone of Cys207 to the sidechain of the conserved Asp1353.32 through the interplay of the 3′ OH group; w6 (highlighted in purple) mediates the H-bond interaction between the sidechain of Ser1393.39 and the N6 amine; w7–w9 (highlighted in cyan) connect the N3 nitrogen atom of the adenine core to the sidechains of the conserved Asp1353.32 and Asn3446.55 residues. This putative binding mode agrees with the flexibility of substitution at the 5′ position of the pseudo-sugar moiety as well as with the intolerance of bulkier groups at the adenine C2 position (pointing toward TM6). Indeed, active compounds bearing different groups at the 5′ position (14: hydroxy; 25: methyl ester; 26: ethyl ester; 27: propyl ester, Figure 4A–D) as well as the N6-3-chlorobenzyl 5′-methylamide derivative 35 (data not shown) docked to this receptor conformation with good scores (Table S1) by maintaining the above describe interaction pattern and by retaining water molecules. Notably, as reported in Table S1, we were able to dock the compounds using also the XP scoring function, which is known to require a greater ligand-receptor shape complementarity, thus further emphasizing the feasibility of this interaction network.
Figure 4.

Hypothetical binding mode predicted by rigid docking at the h5HT2BR (including all water molecules) for N6-dicyclopropylmethyl (N)-methanocarba derivatives bearing different 5′-groups: A) 14 (5′-hydroxy, orange carbon atoms); B) 25 (5′-methyl ester, green carbon atoms); C) 26 (5′-ethyl ester, yellow carbon atoms, two alternative binding modes are reported); and D) 27 (5′-propyl ester, pink carbon atoms). Side chains of residues important for ligand recognition (grey carbon atoms) and water molecules (highlighted with different colors according to the ligand moiety they interact with) are represented as sticks. Residues establishing hydrophobic contacts with the ligand are also depicted as transparent surfaces. H-bonds and π-π stacking interactions are pictured as dashed yellow and cyan lines, respectively. Nonpolar hydrogen atoms, the sidechain of Asn3446.55 and w8 and w9 are omitted to aid visualization.
As emerged from this analysis, all of the ligand-receptor interactions, except the H-bond network mediated by w1 and a hydrophobic contact with Met2185.39 (transparent surface on the right in Figure 3B), involve highly conserved residues. Nonetheless, we believe that these two interactions might account for the greater affinity of 23 for the h5HT2BR. Indeed, the h5HT2CR features a Glu residue (which sidechain that cannot act as H-bond donor) in place of Gln7.32 and a shorter EL2. While the Glu7.32 side chain would not enable the H-bond network as mediated by w1 in the h5HT2BR, the shorter EL2 is expected to affect the three-dimensional arrangement of the downstream loop region as well as of the extracellular tip of TM5 – where Leu 209 and Met5.39 (occurring as Val5.39 in the h5HT2CR), respectively, are located.
Concerning the activity at the hA1 and hA3ARs exhibited by 5′-methylamide derivatives, we expect binding modes similar to the previously discussed poses of (N)-methanocarba adenosines21,33 envisaging the placement of the scaffold perpendicular to the membrane plane with the pseudo-sugar pointing toward the intracellular side. In this binding mode, the C2′ and C3′ OH groups interact with the sidechains of the conserved His7.43 and Ser7.42, respectively, and the NH group of the 5′-methylamide engages in an H-bond interaction with Thr3.36.
Analysis of Receptor Structures
The overlay between the starting docking structure and the MD-refined complex (Figure S2A, alignment base on alpha carbon atoms of TM domains) shows that the largest structural rearrangements in the protein occurred in TM5, TM6, and TM7. In the final 23-h5HT2BR structure (cyan ribbons in Figure S2A), the bulge in TM5 - that protruded into the binding site in the initial structure (magenta ribbons in Figure S2A) - was pushed outward by the N6-dicyclopropylmethyl moiety of the ligand, the intracellular tip of TM6 moved toward the TM bundle, and a disruption of the alpha helix structure in TM7 (residues 372 to 374) occurred. All these structural rearrangements are compatible with a transition from an active-like to an inactive protein structure, as emerged by the comparison between the β2-adrenergic active34 and inactive35 structures. Moreover, the average distances of the sidechains of Phe6.41 and Phe6.44 with respect to TM5 (d(C4-Phe6.44-CA-Pro5.50)= 12.8 Å; d(C4-Phe6.41-CA-Met5.54)= 9.8 Å) and the sidechain of Tyr7.53 oriented toward TM3 further suggest that the final receptor structure is in the inactive state.36,37 In addition, the average density of water molecules calculated during the trajectory (Figure S2B) highlighted the presence of the previously hypothesized36 hydrophobic connector that hampers the entrance of water molecules.
We also performed the check of the “inactive signatures”37 by comparing the results with MD simulations of the apo receptor. Surprisingly, the ionic lock between Arg3.50 and Glu6.30 persisted in the h5HT2BR-apo structure but not in the ligand-bound structure (Figure S3A, Supporting Information). Indeed, in the 23-h5HT2BR complex, Arg3.50 establishes a persistent H-bond interaction with the sidechain of Tyr7.53 of the conserved NPxxY motif (Figure S3B). In both structures, the salt bridge between Asp3.49 and Arg3.50 (conserved DRY motif, Figure S3C) is not present because Asp3.49 faces TM4 and establishes a salt bridge with Lys4.45 (Figure S3D). According to structure-based sequence alignments,38 Lys4.45 is a not conserved residue present only in the h5HT2AR, h5HT2BR, h5HT2CR and human histamine H3R. Indeed, other biogenic amine receptors as well as nucleotide receptors feature a positively charged residue (Lys or Arg) in the 4.41, 4.43 or 4.44 position (Table S2). Moreover, in the h5HT2BR structure experimentally determined through serial femtosecond crystallography,39 a salt bridge between Glu3.60 and the non-conserved Lys5.68 was found. This provided further evidence of the interplay between non-conserved Lys residues and negatively charged conserved residues as well as the existence of a slightly different interaction network in the h5HT2BR with respect to other class A GPCRs. However, it is still to be determined whether these differences can be ascribed to the biased signaling of the co-crystallized ligand, and hopefully future X-ray structures in complex with biased agonists will clarify this point.
Discussion
Rigid nucleosides also appear to be a privileged scaffold at diverse sites, in addition to purine receptors. In our repurposing of nucleosides to obtain novel 5HT2R ligands, we utilized a structure-based approach. Similar methods could be suitable for the repurposing of scaffolds to other GPCRs. The lead for our present study originated from a fortuitous hit in the off-target screening of known AR ligands, i.e. the 5HT2R interaction of 10, but it is conceivable to structurally modify a given scaffold to fit to a GPCR target of choice purely using computational methods and not be dependent on fortuitous empirical screening. By repurposing a nucleoside scaffold from one set of GPCRs, i.e. ARs, to interact selectively with 5HT2Rs as guided by structural information, we can create novel ligands with physicochemical characteristics that are different from known 5HT2R ligands. The rational design of new GPCR ligands aided by computational analysis of new X-ray structures is gaining momentum.19
Several compounds demonstrated moderate selectivity for the 5HT2Rs. Small alkyl esters 25 and 26 were the most potent in this series at 5HT2BR, with 25 being slightly selective in binding with respect to 5HT2CR. Both compounds were modestly selective in comparison to ARs, at which they display only µM affinity. The corresponding 6-N,N-dimethylamino analogue 29 was less potent but still 5HT2R-selective. An amino-propylamide 32 was moderately selective for 5HT2CR in comparison to ARs. Moderate binding selectivity for the 5HT2BR in comparison to 5HT2CR was achieved with compounds 14, 20, 23 and 41. In functional assays, 5′-amide 23, which also bound to the A3AR, was 5HT2BR-selective, while 14, 25 and 26 were nonselective. Several compounds in addition to 23 were mixed ligands at ARs and 5HT2Rs. For example, 14 was a mixed potent ligand at A1AR (agonist) and 5HT2Rs (antagonist). Compounds 16 and 21 were mixed potent ligands at A1AR/A3AR and A1AR (agonists), respectively, and at 5HT2BR (likely antagonists). Compound 24 was a mixed ligand at A3AR and 5HT2B/CRs. Thus, we have identified (N)-methanocarba nucleosides that favored 5HT2BR affinity (Figure S4, Supporting Information, and in some cases also 5HT2CR). The 5′-methyl or ethyl ester modification lowered affinity at the A1AR by roughly 4-fold and at A3AR by 10- to 20-fold. The effects of amide chain extension at the 5′ position were complex and resulted in only modest selectivity.
Thus, we can tune the mixture of AR and 5HT2R activities by structural manipulation to provide novel compounds displaying mixed activities within the two families of receptors or favoring activity at either family. The SAR herein determined is consistent with a previous prediction that the (N)-methanocarba nucleosides bind at the h5HT2Rs in an orientation different from the presumed binding mode at the ARs (i.e. with the pseudo-sugar portion projecting into the TM bundle). The specific preference for the N6-dicyclopropylmethyl group is consistent with a small pocket at the TM5/TM6 interface. The modeling analysis herein presented suggests that ligand-recognition at the h5HT2Rs might occur with the scaffold lying almost parallel to the membrane plane. In this position, the 5′ group points toward the EL2 and the extracellular tip of TM5, a region exhibiting structural diversity in the h5HT2Rs, thus revealing the potential for further derivatization at this position. In particular, the selection of suitable anchoring points among non-conserved residues might achieve selectivity at the desired h5HT2R subtype.
The design of selective ligands of the 5HT2BR is warranted based on recent biological findings. Selective antagonists might be useful for treating the heart or pulmonary hypertension.14,40 Their experimental use in liver protection has been demonstrated; 5HT2BR blockade reduces chronic fibrosis and enhances hepatic regeneration.14 A3AR is highly expressed in the liver, and its agonists are also of interest for the treatment of liver disease.20 Thus, a combined agent with both 5HT2BR antagonist and A3AR agonist activity might have a synergistic benefit in the liver. The application of 5HT2BR antagonists for treating irritable bowel syndrome was also proposed.2,18 However, one recent study comparing selective 5HTR antagonists failed to confirm that activity.41 A variety of approaches have contributed to recent efforts to discover selective 5HT2R agonists and antagonists from screening and computational analysis. For example, computational activity prediction and microfluidics-assisted synthesis enabled the discovery of new 5HT2BR antagonists.42 Other novel antagonists were discovered as a result of broad screening programs.43,44 Activity at the 5HT2AR, considered to be a mechanism of hallucinogenic activity of a subset of its agonists,45 is consistently low or absent throughout this compound series.
In general, 5HT2R modulation in the brain contributes to the activity of antidepressant drugs. Various antidepressant drugs raise levels of 5HT, which can activate the 5HT2BR and downregulate the 5HT2CR to contribute to the action of these drugs. Inhibition of the 5HT2CR, which forms functional GPCR heterodimers with the melatonin MT2R,46 has both antidepressant and anxiolytic effects. However, 5HT2BR activation has antinociceptive properties related to the association of this subtype with descending inhibitory pain pathways originating in the rostroventral medial medulla (RVM).47–49 The typically low penetration of nucleoside derivatives into the brain50 would be of benefit when applying 5HT2BR antagonists for peripheral conditions such as cardiac and hepatic fibrosis. This might be an advantage of the nucleoside 5HT2BR antagonists over brain penetrant heterocycles. We have not determined if the 5HT2R antagonist activity in this chemical series represents neutral antagonism or inverse agonism, which has a bearing on potential therapeutic applications because of receptor constitutive activity.51 It was also noted that AR agonists might facilitate passage of various solutes across the blood brain barrier,52 but if this activity is removed from the current analogues, then that would not be a factor.
In conclusion, we have transformed rigid carbocyclic nucleosides that are A3AR-selective agonists into moderately potent 5HT2BR and/or 5HT2CR antagonists using a structure-based approach. Although many of the compounds are nonselective between these two receptor subtypes, there is a tendency toward greater affinity at the 5HT2BR. We have analyzed the molecular recognition in this chemical series, aided by the X-ray structural determination of rhodopsin-like GPCRs and the availability of broad screening, to shift and optimize the binding of the same scaffold to a new family of receptors.
Experimental section
Chemical synthesis
Materials and instrumentation
All reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). 1H NMR spectra were obtained with a Bruker 400 spectrometer using CDCl3, CD3OD and DMSO as solvents. Chemical shifts are expressed in δ values (ppm) with tetramethylsilane (δ 0.00) for CDCl3 and water (δ 3.30) for CD3OD. NMR spectra were collected with a Bruker AV spectrometer equipped with a z-gradient [1H, 13C, 15N]-cryoprobe. TLC analysis was carried out on glass sheets precoated with silica gel F254 (0.2 mm) from Aldrich. The purity of final nucleoside derivatives was checked using a Hewlett–Packard 1100 HPLC equipped with a Zorbax SB-Aq 5 µm analytical column (50 × 4.6 mm; Agilent Technologies Inc., Palo Alto, CA). Mobile phase: linear gradient solvent system, 5 mM TBAP (tetrabutylammoniumdihydrogenphosphate)–CH3CN from 80:20 to 0:100 in 13 min; the flow rate was 0.5 mL/min. Peaks were detected by UV absorption with a diode array detector at 230, 254, and 280 nm. All derivatives tested for biological activity showed >95% purity by HPLC analysis (detection at 254 nm). Low-resolution mass spectrometry was performed with a JEOL SX102 spectrometer with 6-kV Xe atoms following desorption from a glycerol matrix or on an Agilent LC/MS 1100 MSD, with a Waters (Milford, MA) Atlantis C18 column. High resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine, unless noted. Observed mass accuracies are those expected based on known performance of the instrument as well as trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy. All of the monosubsituted alkyne and branched amine intermediates were purchased from Sigma-Aldrich (St. Louis, MO), Small Molecules, Inc. (Hoboken, NJ), Anichem (North Brunswick, NJ), PharmaBlock, Inc. (Sunnyvale, CA), Frontier Scientific (Logan, UT) and Tractus (Perrineville, NJ).
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((dicyclopropylmethyl)amino)-9H-purin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (14)
Dowex50 (27 mg) was added to a solution of compound 64 (49 mg, 0.11 mmol) in MeOH (2 mL) and water (1 mL) and the mixture stirred for 5 h at room temperature. After completion of reaction, the reaction mixture filtered and filtrate was evaporated under vacuum. The crude mixture was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 30:1) to give compound 14 (37 mg, 84%) as a colorless foamy powder. 1H NMR (CD3OD, 400 MHz) δ 8.44 (s, 1H), 4.80 (s, 1H), 4.78 (d, J = 6.4 Hz, 1H), 4.29 (d, J = 11.6 Hz, 1H), 3.88 (d, J = 6.4 Hz, 1H), 3.49 (br s, 1H), 1.64-1.61 (m, 1H), 1.55-1.53 (m, 1H), 1.18-1.09 (m, 2H), 0.78-0.74 (m, 1H), 0.60-0.55 (m, 2H), 0.47-0.39 (m, 6H). HRMS calculated for C19H25ClN5O3 (M + H)+: 406.1646; found 406.1653.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-(pentan-3-ylamino)-9H-purin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (15)
Compound 15 (85%) was prepared from compound 65 following the method for compound 14. 1H NMR (CD3OD, 400 MHz) δ 8.44 (s, 1H), 4.81 (s, 1H), 4.79 (d, J = 6.8 Hz, 1H), 4.29 (d, J = 11.6 Hz, 1H), 4.19 (br s, 1H), 3.89 (d, J = 6.8 Hz, 1H), 1.75-1.67 (m, 2H), 1.64-1.53 (m, 4H), 0.97 (t, J = 7.2 Hz, 6H), 0.78-0.75 (m, 1H). HRMS calculated for C17H25ClN5O3 (M + H)+: 382.1646; found 382.1647.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-(((1R)-cyclopropylpropyl)amino)-9H-purin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (16)
Compound 16 (73%) was prepared from compound 66 following the method for compound 14. 1H NMR (CD3OD, 400 MHz) δ 8.44 (s, 1H), 4.80 (s, 1H), 4.79 (d, J = 6.4 Hz, 1H), 4.29 (d, J = 11.6 Hz, 1H), 3.88 (d, J = 6.4 Hz, 1H), 3.66 (br s, 1H), 1.88-1.72 (m, 2H), 1.64-1.61 (m, 1H), 1.55 (t, J = 5.2 Hz, 1H), 1.02-0.99 (m, 4H), 0.78-0.74 (m, 1H), 0.61-0.56 (m, 1H), 0.47-0.41 (m, 2H), 0.36-0.33 (m, 1H). HRMS calculated for C18H25ClN5O3 (M + H)+: 394.1646; found 394.1647.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-((1-cyclopropylpropyl)amino)-9H-purin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (17)
Compound 17 (72%) was prepared from compound 67 following the method for compound 14. 1H NMR (CD3OD, 400 MHz) δ 8.44 (s, 1H), 4.80 (s, 1H), 4.79 (d, J = 6.4 Hz, 1H), 4.29 (d, J = 11.6 Hz, 1H), 3.88 (d, J = 6.4 Hz, 1H), 3.67 (br s, 1H), 1.88-1.70 (m, 2H), 1.63-1.61 (m, 1H), 1.56 (t, J = 5.2 Hz, 1H), 1.02-0.99 (m, 4H), 0.78-0.75 (m, 1H), 0.59-0.56 (m, 1H), 0.45-0.42 (m, 2H), 0.36-0.30 (m, 1H). HRMS calculated for C18H25ClN5O3 (M + H)+: 394.1646; found 394.1642.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-(((1R)-cyclopropyl-2-methylpropyl)amino)-9H-purin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (18)
Compound 18 (67%) was prepared from compound 68 following the method for compound 14. 1H NMR (CD3OD, 400 MHz) δ 8.44 (s, 1H), 4.80 (s, 1H), 4.79 (d, J = 6.4 Hz, 1H), 4.29 (d, J = 11.6 Hz, 1H), 3.89 (d, J = 6.4 Hz, 1H), 3.61 (br s, 1H), 2.09-2.02 (m, 1H), 1.64-1.62 (m, 1H), 1.54 (t, J = 5.2 Hz, 1H), 1.08-1.04 (m, 7H), 0.78-0.74 (m, 1H), 0.67-0.61 (m, 1H), 0.45-0.36 (m, 3H). HRMS calculated for C19H27ClN5O3 (M + H)+: 408.1802; found 408.1806.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-(((1S)-cyclopropyl-2-methylpropyl)amino)-9H-purin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (19)
Compound 19 (68%) was prepared from compound 69 following the method for compound 14. 1H NMR (CD3OD, 400 MHz) δ 8.44 (s, 1H), 4.80 (s, 1H), 4.79 (d, J = 6.4 Hz, 1H), 4.29 (d, J = 11.6 Hz, 1H), 3.89 (d, J = 6.4 Hz, 1H), 3.61 (br s, 1H), 2.08-2.02 (m, 1H), 1.64-1.61 (m, 1H), 1.55 (t, J = 5.2 Hz, 1H), 1.08-1.04 (m, 7H), 0.78-0.74 (m, 1H), 0.66-0.61 (m, 1H), 0.45-0.36 (m, 3H). HRMS calculated for C19H27ClN5O3 (M + H)+: 408.1802; found 408.1798.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-(((S)-cyclobutyl(cyclopropyl)methyl)amino)-9H-purin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (20)
Compound 20 (69%) was prepared from compound 70 following the method for compound 14. 1H NMR (CD3OD, 400 MHz) δ 8.44 (s, 1H), 4.80 (s, 1H), 4.79 (d, J = 6.4 Hz, 1H), 4.29 (d, J = 11.6 Hz, 1H), 3.89-3.82 (m, 2H), 2.73-2.65 (m, 1H), 2.12-2.07 (m, 1H), 2.01-1.87 (m, 4H), 1.85-1.80 (m, 1H), 1.64-1.61 (m, 1H), 1.54 (t, J = 5.2 Hz, 1H), 0.97-0.91 (m, 1H), 0.78-0.74 (m, 1H), 0.58-0.53 (m, 1H), 0.40-0.36 (m, 3H). HRMS calculated for C20H27ClN5O3 (M + H)+: 420.1796; found 420.1796.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-(((R)-cyclobutyl(cyclopropyl)methyl)amino)-9H-purin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (21)
Compound 21 (69%) was prepared from compound 71 following the method for compound 14. 1H NMR (CD3OD, 400 MHz) δ 8.44 (s, 1H), 4.80 (s, 1H), 4.79 (d, J = 6.4 Hz, 1H), 4.29 (d, J = 11.6 Hz, 1H), 3.89-3.82 (m, 2H), 2.71-2.67 (m, 1H), 2.12-2.07 (m, 1H), 2.01-1.87 (m, 4H), 1.83-1.80 (m, 1H), 1.64-1.61 (m, 1H), 1.54 (t, J = 5.2 Hz, 1H), 0.97-0.90 (m, 1H), 0.78-0.74 (m, 1H), 0.58-0.53 (m, 1H), 0.40-0.36 (m, 3H). HRMS calculated for C20H27ClN5O3 (M + H)+: 420.1802; found 420.1799.
(1R,2R,3S,4R,5S)-4-(2-Chloro-6-(dimethylamino)-9H-purin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (22)
10% TFA in water (1.5 mL) was added to a solution of compound 63 (16 mg, 0.02 mmol) in methanol (1.5 mL) and the mixture heated at 70 °C for 6 h. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel silica chromatography (CH2Cl2:MeOH = 30:1) to give compound 22 (6 mg, 68%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.44 (s, 1H), 4.81 (s, 1H), 4.77 (d, J = 6.4 Hz, 1H), 4.30 (d, J = 11.6 Hz, 1H), 3.85 (d, J = 6.8 Hz, 1H), 3.49 (br s, 6H), 1.64-1.61 (m, 1H), 1.55 (d, J = 5.2 Hz, 1H), 0.78-0.75 (m, 1H). HRMS calculated for C14H23ClN5O3 (M + H)+: 340.1176; found 340.1177.
(1S,2R,3S,4R,5S)-4-(2-Chloro-6-((dicyclopropylmethyl)amino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (23)
40% MeNH2 solution (2 mL) was added to a solution of compound 26 (41 mg, 0.09 mmol) in MeOH (2 mL) and the mixture stirred overnight at room temperature. Solvent was evaporated, and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH =20:1) to give compound 23 (25 mg, 65%) as asyrup. 1H NMR (CD3OD, 400 MHz) δ 8.04 (s, 1H), 5.07 (d, J = 6.4 Hz, 1H), 4.80 (s, 1H), 4.01 (d, J = 6.4 Hz, 1H), 3.49 (br s, 1H), 2.87 (s, 3H), 2.08-2.04 (m, 1H), 1.82 (t, J = 4.8 Hz, 1H), 1.40-1.36 (m, 1H), 1.18-1.09 (m, 2H), 0.60-0.55 (m, 2H), 0.47-0.39 (m, 6H). HRMS calculated for C20H26ClN6O3 (M + H)+: 433.1755; found 433.1771.
(1S,2R,3S,4R,5S)-4-(2-Chloro-6-(dimethylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (24)
Compound 24 (67%) was prepared from compound 29 following the method for compound 23. 1H NMR (CD3OD, 400 MHz) δ 7.96 (s, 1H), 5.05 (d, J = 6.4 Hz, 1H), 4.80 (s, 1H), 3.99 (d, J = 6.4 Hz, 1H), 3.49 (br s, 6H), 2.87 (s, 3H), 2.06-2.04 (m, 1H), 1.83 (t, J = 4.8 Hz, 1H), 1.40-1.36 (m, 1H). HRMS calculated for C15H20ClN6O3 (M + H)+: 367.1285; found 367.1290.
Methyl (1S,2R,3S,4R,5S)-4-(2-Chloro-6-((dicyclopropylmethyl)amino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (25)
DCC (28 mg, 0.13 mmol) and DMAP (2.76 mg, 0.13 mmol) were added to a solution of compound 28 (19 mg, 0.04 mmol) in DMF (0.7 mL). After stirring the reaction mixture for 10 min at room temperature, anhydrous MeOH (6.0 µL, 0.13 mmol) was added and the mixture stirred overnight at same condition. Solvent was evaporated, and the residue was purified on flash silica gel column chromatography (ethyl acetate:MeOH = 85:1) to give compound 25 (13 mg, 68%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 7.94 (s, 1H), 5.22 (d, J = 6.8 Hz, 1H), 4.78 (s, 1H), 4.07 (d, J = 6.4 Hz, 1H), 3.80 (s, 3H), 3.49 (br s, 1H), 2.19-2.15 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.62-1.59 (m, 1H), 1.15-1.11 (m, 2H), 0.63-0.53 (m, 2H), 0.45-0.42 (m, 6H). HRMS calculated for C20H25ClN5O4 (M + H)+: 434.1595; found 434.1603.
Ethyl (1S,2R,3S,4R,5S)-4-(2-chloro-6-((dicyclopropylmethyl)amino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (26)
Dowex50 (107 mg) was added to a solution of compound 76 (166 mg, 0.34 mmol) in MeOH (4 mL) and water (3 mL) and the mixture heated for 1 h at 65 °C. After completion of reaction, the reaction mixture was filtered and the filtrate was evaporated under vacuum. The crude mixture was purified on flash silica gel column chromatography (hexane:ethyl acetate = 1:2) to give compound 26 (131 mg, 86%) as a colorless foamy powder. 1H NMR (CD3OD, 400 MHz) δ 7.96 (s, 1H), 5.23 (d, J = 6.8 Hz, 1H), 4.77 (s, 1H), 4.28-4.22 (m, 2H), 4.12 (d, J = 6.8 Hz, 1H), 3.49 (br s, 1H), 2.18-2.15 (m, 1H), 1.87 (t, J = 5.2 Hz, 1H), 1.63-1.60 (m, 1H), 1.32 (t, J = 7.2 Hz, 3H), 1.18-1.09 (m, 2H), 0.61-0.54 (m, 2H), 0.47-0.42 (m, 6H). HRMS calculated for C21H27ClN5O4 (M + H)+: 448.1752; found 448.1745.
Propyl (1S,2R,3S,4R,5S)-4-(2-chloro-6-((dicyclopropylmethyl)amino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (27)
Compound 27 (66%) was prepared from compound 28 following the method for compound 25. 1H NMR (CD3OD, 400 MHz) δ 7.95 (s, 1H), 5.23 (d, J = 6.4 Hz, 1H), 4.77 (s, 1H), 4.16 (t, J = 6.8 Hz, 2H), 4.10 (d, J = 6.4 Hz, 1H), 3.49 (br s, 1H), 2.19-2.16 (m, 1H), 1.88 (t, J = 4.8 Hz, 1H), 1.78-1.69 (m, 2H), 1.64-1.60 (m, 1H), 1.17-1.09 (m, 2H), 0.99 (t, J = 7.2 Hz, 3H), 0.61-0.54 (2H), 0.47-0.41 (m, 6H). HRMS calculated for C22H29ClN5O4 (M + H)+: 462.1908; found 462.1903.
(1S,2R,3S,4R,5S)-4-(2-Chloro-6-((dicyclopropylmethyl)amino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylic acid (28)
2N NaOH solution (1.5 mL) was added to a solution of compound 26 (39 mg, 0.08 mmol) in methanol (1.5 mL) and the mixture stirred at room temperature for 1.5 h. The reaction mixture was neutralized with acetic acid and evaporated under vacuum. The crude product was purified on flash silica gel column chromatography (CH2Cl2:MeOH:TFA=20:1:0.1) to give compound 28 (27 mg, 76%) as colorless powder. 1H NMR (CD3OD, 400 MHz) δ 7.99 (s, 1H), 5.18 (d, J = 6.8 Hz, 1H), 4.79 (s, 1H), 4.10 (d, J = 6.8 Hz, 1H), 3.49 (br s, 1H), 2.19-2.16 (m, 1H), 1.90 (t, J = 5.2 Hz, 1H), 1.66-1.62 (m, 1H), 1.16-1.11 (m, 2H), 0.61-0.55 (m, 2H), 0.47-0.39 (m, 6H). HRMS calculated for C19H23ClN5O4 (M + H)+: 420.1439; found 420.1438.
Ethyl (1S,2R,3S,4R,5S)-4-(2-chloro-6-(dimethylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (29)
10% TFA in water (2.5 mL) was added to a solution of compound 77 (58 mg, 0.137 mmol) in methanol (2.5 mL) and the mixture heated at 70 °C for 3 h. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel silica chromatography (CH2Cl2:MeOH = 40:1) to give compound 29 (46 mg, 88%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 7.88 (s, 1H), 5.22 (d, J = 6.4 Hz, 1H), 4.78 (s, 1H), 4.28-4.22 (m, 2H), 4.08 (d, J = 6.4 Hz, 1H), 3.49 (br s, 6H), 2.17-2.14 (m, 1H), 1.88 (t, J = 5.2 Hz, 1H), 1.64-1.60 (m, 1H), 1.32 (t, J = 7.2 Hz, 3H). HRMS calculated for C16H21ClN5O4 (M + H)+: 382.1282; found 382.1283.
(1S,2R,3S,4R,5S)-4-(2-Chloro-6-(dimethylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo [3.1.0]hexane-1-carboxylic acid (30)
Compound 30 (77%) was prepared from compound 29 following the method for compound 28. 1H NMR (CD3OD, 400 MHz) δ 7.89 (s, 1H), 5.16 (d, J = 6.4 Hz, 1H), 4.79 (s, 1H), 4.08 (d, J = 6.4 Hz, 1H), 3.63 (br s, 6H), 2.18-2.15 (m, 1H), 1.91 (t, J = 5.2 Hz, 1H), 1.66-1.62 (m, 1H). HRMS calculated for C14H17ClN5O4 (M + H)+: 354.0969; found 354.0969.
(1S,2R,3S,4R,5S)-N-(2-Aminoethyl)-4-(2-chloro-6-((dicyclopropylmethyl)amino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxamide (31)
Ethylenediamine (1 mL) was added to a solution of compound 26 (19 mg, 0.04 mmol) in methanol (0.3 mL) and the mixture stirred for 3 days at room temperature. Solvent was evaporated, and the residue was purified on flash silca gel column chromatography (CH2Cl2:MeOH:NH4OH = 9:1:0.1) to give compound 31 (12 mg, 63%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.07 (s, 1H), 5.13 (d, J = 6.4 Hz, 1H), 4.81 (s, 1H), 4.02 (d, J = 6.4 Hz, 1H), 3.45-3.39 (m, 2H), 2.84 (t, J = 6.4 Hz, 2H), 2.10-2.07 (m, 1H), 1.83 (t, J = 5.2 Hz, 1H), 1.42-1.38 (m, 1H), 1.18-1.09 (m, 2H), 0.60-0.55 (m, 2H), 0.46-0.39 (m, 6H). HRMS calculated for C21H29ClN7O3 (M + H)+: 462.2020; found 462.2021.
(1S,2R,3S,4R,5S)-N-(3-Aminopropyl)-4-(2-chloro-6-((dicyclopropylmethyl)amino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxamide (32)
Compound 32 (64%) was prepared from compound 29 following the method for compound 31. 1H NMR (CD3OD, 400 MHz) δ 8.04 (s, 1H), 5.13 (d, J = 6.4 Hz, 1H), 4.80 (s, 1H), 4.03 (d, J = 6.4 Hz, 1H), 3.47-3.38 (m, 2H), 2.82 (t, J = 7.2 Hz, 2H), 2.06-2.05 (m, 1H), 1.84-1.65 (m, 3H), 1.40-1.36 (m, 1H), 1.16-1.09 (m, 2H), 0.60-0.55 (m, 2H), 0.46-0.39 (m, 6H). HRMS calculated for C22H31ClN7O3 (M + H)+: 476.2177; found 476.2172.
(1S,2R,3S,4R,5S)-N-(2-Acetamidoethyl)-4-(2-chloro-6-((dicyclopropylmethyl)amino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxamide (33)
Triethylamine (3.2 µL, 0.011 mmol) was added to a solution of compound 31 (3.61 mg, 0.007 mmol) and acetic acid N-hydroxysuccinimide ester (1.77 mg, 0.011 mmol) in DMF (1 mL) and the mixture stirred at room temperature overnight. Solvent was evaporated, and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH=15:1) to give compound 33 (2.3 mg, 59%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.06 (s, 1H), 5.06 (d, J = 6.4 Hz, 1H), 4.81 (s, 1H), 4.01 (d, J = 6.4 Hz, 1H), 3.42-3.38 (m, 2H), 3.36-3.32 (m, 2H), 2.09-2.06 (m, 1H), 1.95 (s, 3H), 1.83 (t, J = 4.8 Hz, 1H), 1.40-1.37 (m, 1H), 1.17-1.09 (m, 2H), 0.60-0.56 (m, 2H), 0.55-0.44 (m, 6H). HRMS calculated for C23H31ClN7O4 (M + H)+: 504.2116; found 504.2126.
(1S,2R,3S,4R,5S)-N-(3-Acetamidopropyl)-4-(2-chloro-6-((dicyclopropylmethyl)amino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxamide (34)
Compound 34 (58%) was prepared from compound 32 following the method for compound 33. 1H NMR (CD3OD, 400 MHz) δ 8.09 (s, 1H), 5.06 (d, J = 6.4 Hz, 1H), 4.80 (s, 1H), 4.02 (d, J = 6.4 Hz, 1H), 3.51 (br s, 1H), 3.27-3.23 (m, 4H), 2.09-2.06 (m, 1H), 1.96 (s, 3H), 1.84 (t, J = 5.2 Hz, 1H), 1.76 (t, J = 6.8 Hz, 2H), 1.41-1.37 (m, 1H), 1.18-1.09 (m, 2H), 0.60-0.55 (m, 2H), 0.47-0.39 (m, 6H). HRMS calculated for C24H33ClN7O4 (M + H)+: 518.2283; found 518.2289.
9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2,6-dichloro-9H-purine (54)
DIAD (0.17 mL, 0.89 mmol) was added to a solution of 2,6-dichloropurine (170 mg, 0.89 mmol) and triphenylphosphine (235 mg, 0.89 mmol) in THF (4 mL) at 0 °C and the mixture was stirred for 20 min at room temperature. A solution of compound 53 (197 mg, 0.49 mmol) in THF (2 mL) was added into the reaction mixture and the mixture stirred overnight at room temperature. Solvent was evaporated, and the residue was purified on flash silica gel column chromatography (hexane:ethyl acetate=5:1) to give compound 54 (189 mg, 69%) as colorless foamy powder. 1H NMR (CDCl3, 400 MHz) δ 8.42 (s, 1H), 7.63-7.61 (m, 4H), 7.42-7.31 (m, 6H), 5.28 (d, J = 6.4 Hz, 1H), 5.09 (s, 1H), 4.58 (d, J = 7.2 Hz, 1H), 4.29 (d, J = 10.8 Hz, 1H), 3.70 (d, J = 10.8 Hz, 1H), 1.59-1.58 (m, 1H), 1.55 (s, 3H), 1.25 (s, 3H), 1.17-1.15 (s, 1H), 1.11 (s, 9H), 1.07-1.04 (m, 1H). HRMS calculated for C31H35Cl2SiN4O3 (M + H)+: 609.1856; found 609.1857.
9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-chloro-N-(dicyclopropylmethyl)-9H-purin-6-amine (55)
2,2-Dicyclopropyl-methylamine (80 mg, 0.54 mmol) and triethylamine (0.25 mL, 1.8 mmol) were added to a solution of compound 54 (110, 0.18 mmol) in methanol (4 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum, and the residue was purified on flash column chromatography (hexane:ethyl acetate = 4:1) to give the desired product 55 (105 mg, 85%) as a syrup. 1H NMR (CDCl3, 400 MHz) δ 8.06 (s, 1H), 7.66-7.62 (m, 4H), 7.44-7.33 (m, 6H), 5.29 (d, J= 6.4 Hz, 1H), 5.02 (s, 1H), 4.58 (d, J = 7.2 Hz, 1H), 4.30 (d, J = 10.8 Hz, 1H), 3.63-3.60 (m, 2H), 1.61-1.58 (m, 1H), 1.54 (s, 3H), 1.29-1.25 (m, 4H), 1.08 (s, 9H), 1.07-1.03 (m, 2H), 0.98-0.94 (m, 1H), 0.60-0.44 (m, 8H). HRMS calculated for C38H47ClSiN5O3 (M + H)+: 684.3131; found 684.3132.
9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-chloro-N-(pentan-3-yl)-9H-purin-6-amine (56)
Compound 56 (81%) was prepared from compound 54 following the method for compound 55. 1H NMR (CD3OD, 400 MHz) δ 8.26 (s, 1H), 7.66-7.64 (m, 4H), 7.43-7.31 (m, 6H), 5.35 (d, J= 6.8 Hz, 1H), 4.95 (s, 1H), 4.71 (d, J= 6.8 Hz, 1H), 4.23 (d, J= 10.4 Hz, 1H), 3.76 (d, J= 10.4 Hz, 1H), 1.76-1.69 (m, 2H), 1.63-1.56 (m, 3H), 1.52 (s, 3H), 1.26 (s, 3H), 1.14-1.11 (m, 1H), 1.09 (s, 9H), 1.02-0.95 (m, 8H). HRMS calculated for C36H47ClSiN5O3 (M + H)+: 660.3137; found 660.3135.
9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-chloro-N-((1R)-cyclopropylpropyl)-9H-purin-6-amine (57)
Compound 57 (83%) was prepared from compound 54 following the method for compound 55. 1H NMR (CD3OD, 400 MHz) δ 8.25 (s, 1H), 7.65-7.63 (m, 4H), 7.44-7.33 (m, 6H), 5.34 (d, J = 7.0 Hz, 1H), 4.94 (s, 1H), 4.70 (d, J = 7.0 Hz, 1H), 4.22 (d, J= 10.4 Hz, 1H), 3.77 (d, J = 10.4 Hz, 1H), 3.69 (br s, 1H), 1.90-1.72 (m, 2H), 1.61-1.60 (m, 1H), 1.52 (s, 3H), 1.26 (s, 3H), 1.13-1.10 (m, 1H), 1.08 (s, 9H), 1.06-1.02 (m, 4H), 0.98-0.95 (m, 1H), 0.63-0.56 (m, 1H), 0.48-0.40 (m, 2H), 0.37-0.32 (m, 1H). HRMS calculated for C37H47ClSiN5O3 (M + H)+: 672.3137; found 672.3127.
9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-chloro-N-((1S)-cyclopropylpropyl)-9H-purin-6-amine (58)
Compound 58 (82%) was prepared from compound 54 following the method for compound 55. 1H NMR (CD3OD, 400 MHz) δ 8.25 (s, 1H), 7.66-7.64 (m, 4H), 7.43-7.33 (m, 6H), 5.34 (d, J = 7.0 Hz, 1H), 4.94 (s, 1H), 4.70 (d, J= 7.0 Hz, 1H), 4.22 (d, J = 10.4 Hz, 1H), 3.77 (d, J= 10.4 Hz, 1H), 3.68 (br s, 1H), 1.88-1.72 (m, 2H), 1.62-1.60 (m, 1H), 1.52 (s, 3H), 1.26 (s, 3H), 1.13-1.08 (m, 1H), 1.08 (s, 9H), 1.04-1.00 (m, 4H), 0.99-0.95 (m, 1H), 0.62-0.57 (m, 1H), 0.48-0.44 (m, 2H), 0.38-0.35 (m, 1H). HRMS calculated for C37H47ClSiN5O3 (M + H)+: 672.3137; found 672.3136.
9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-chloro-N-((1R)-cyclopropyl-2-methylpropyl)-9H-purin-6-amine (59)
Compound 59 (81%) was prepared from compound 54 following the method for compound 55. 1H NMR (CD3OD, 400 MHz) δ 8.26 (s, 1H), 7.66- 7.64 (m, 4H), 7.42-7.31 (m, 6H), 5.33 (d, J = 6.8 Hz, 1H), 4.94 (s, 1H), 4.71 (d, J = 6.8 Hz, 1H), 4.23 (d, J = 10.8 Hz, 1H), 3.75 (d, J = 10.8 Hz, 1H), 3.64 (t, J = 6.4 Hz, 1H), 2.09-2.02 (m, 1H), 1.62-1.59 (m, 1H), 1.52 (s, 3H), 1.26 (s, 3H), 1.16-1.02 (m, 16H), 1.01-0.90 (m, 2H), 0.66-0.61 (m, 1H), 0.46-0.37 (m, 3H). HRMS calculated for C38H49ClSiN5O3 (M + H)+: 686.3293; found 686.3293.
9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-chloro-N-((1S)-cyclopropyl-2-methylpropyl)-9H-purin-6-amine (60)
Compound 60 (81%) was prepared from compound 54 following the method for compound 56. 1H NMR (CD3OD, 400 MHz) δ 8.27 (s, 1H), 7.66- 7.64 (m, 4H), 7.43-7.32 (m, 6H), 5.34 (d, J = 6.8 Hz, 1H), 4.95 (s, 1H), 4.71 (d, J = 6.8 Hz, 1H), 4.23 (d, J = 10.8 Hz, 1H), 3.74 (d, J = 10.8 Hz, 1H), 3.64 (t, J = 6.4 Hz, 1H), 2.11-2.02 (m, 1H), 1.63-1.60 (m, 1H), 1.52 (s, 3H), 1.26 (s, 3H), 1.15-1.05 (m, 16H), 0.99-0.93 (m, 2H), 0.67-0.63 (m, 1H), 0.46-0.40 (m, 3H). HRMS calculated for C38H49ClSiN5O3 (M + H)+: 686.3293; found 686.3298.
9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-chloro-N-((S)-cyclobutyl(cyclopropyl)methyl)-9H-purin-6-amine (61)
Compound 61 (78%) was prepared from compound 54 following the method for compound 55. 1H NMR (CD3OD, 400 MHz) δ 8.25 (s, 1H), 7.66- 7.64 (m, 4H), 7.43-7.31 (m, 6H), 5.33 (d, J = 6.8 Hz, 1H), 4.94 (s, 1H), 4.71 (d, J = 6.8 Hz, 1H), 4.23 (d, J = 10.4 Hz, 1H), 3.87 (br s, 1H), 3.77 (d, J = 10.4 Hz, 1H), 2.73-2.69 (m, 1H), 2.17-2.08 (m, 1H), 2.01-1.84 (m, 5H), 1.61-1.60 (m, 1H), 1.52 (s, 3H), 1.26 (s, 3H), 1.14-1.07 (m, 10H), 0.99-0.93 (m, 2H), 0.59-0.56 (m, 1H), 0.38-0.37 (m, 3H). HRMS calculated for C39H49ClSiN5O3 (M + H)+: 698.3293; found 698.3303.
9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-chloro-N-((R)-cyclobutyl(cyclopropyl)methyl)-9H-purin-6-amine (62)
Compound 62 (79%) was prepared from compound 54 following the method for compound 55. 1H NMR (CD3OD, 400 MHz) δ 8.25 (s, 1H), 7.66- 7.64 (m, 4H), 7.45-7.31 (m, 6H), 5.34 (d, J = 6.8 Hz, 1H), 4.94 (s, 1H), 4.71 (d, J = 6.8 Hz, 1H), 4.23 (d, J = 10.4 Hz, 1H), 3.86 (br s, 1H), 3.77 (d, J = 10.4 Hz, 1H), 2.72-2.68 (m, 1H), 2.12-2.08 (m, 1H), 1.99-1.84 (m, 5H), 1.62-1.61 (m, 1H), 1.52 (s, 3H), 1.26 (s, 3H), 1.16-1.04 (m, 10H), 0.99-0.93 (m, 2H), 0.57-0.55 (m, 1H), 0.39-0.37 (m, 3H). HRMS calculated for C39H49ClSiN5O3 (M + H)+: 698.3293; found 698.3281.
9-((3aR,3bR,4aS,5R,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)methyl)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-chloro-N,N-dimethyl-9H-purin-6-amine (63)
Compound 63 (84%) was prepared from compound 54 following the method for compound 55. 1H NMR (CD3OD, 400 MHz) δ 8.11 (s, 1H), 7.66-7.62 (m, 4H), 7.41-7.30 (m, 6H), 5.32 (d, J = 6.8 Hz, 1H), 4.91 (s, 1H), 4.72 (d, J = 6.8 Hz, 1H), 4.17 (d, J = 10.8 Hz, 1H), 3.86 (d, J = 10.8 Hz, 1H), 3.49 (br s, 6H), 1.58-1.52 (m, 4H), 1.26 (s, 3H), 1.11-1.06 (m, 10H), 0.99-0.95 (m, 1H). HRMS calculated for C33H41ClSiN5O3 (M + H)+: 618.2667; found 618.2675.
((3aR,3bR,4aS,5R,5aS)-5-(2-Chloro-6-(pentan-3-ylamino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-3b(3aH)-yl)methanol (65)
TBAF (0.08 mL, 1 M solution in THF) was added to a solution of compound 56 (39 mg, 0.06 mmol) in THF (2 mL) and the mixture stirred for 1 h at room temperature. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (hexane:ethyl acetate = 1:1) to give compound 65 (16.8 mg, 68%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.21 (s, 1H), 5.37 (d, J = 6.4 Hz, 1H), 4.95 (s, 1H), 4.70 (d, J = 6.8 Hz, 1H), 4.21-4.19 (m, 1H), 4.01 (d, J = 11.6 Hz, 1H), 3.62 (d, J = 11.6 Hz, 1H), 1.75-1.68 (m, 3H), 1.62-1.55 (m, 2H), 1.52 (s, 3H), 1.26 (s, 3H), 1.15 (t, J = 5.2 Hz, 1H), 1.00-0.93 (m, 7H). HRMS calculated for C20H29ClN5O3 (M + H)+: 422.1959; found 422.1954.
((3aR,3bR,4aS,5R,5aS)-5-(2-Chloro-6-(((1R)-cyclopropylpropyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-3b(3aH)-yl)methanol (66)
Compound 66 (78%) was prepared from compound 57 following the method for compound 65. 1H NMR (CD3OD, 400 MHz) δ 8.21 (s, 1H), 5.39 (d, J = 6.8 Hz, 1H), 4.95 (s, 1H), 4.70 (d, J = 6.4 Hz, 1H), 4.00 (d, J = 11.6 Hz, 1H), 3.67-3.65 (br m, 1H), 3.62 (d, J = 11.6 Hz, 1H), 1.88-1.81 (m, 1H), 1.77-1.68 (m, 2H), 1.52 (s, 3H), 1.26 (s, 3H), 1.15 (t, J = 5.2 Hz, 1H), 1.04-0.97 (m, 5H), 0.61-0.56 (m, 1H), 0.47-0.40 (m, 2H), 0.36-0.33 (m, 1H). HRMS calculated for C21H29ClN5O3 (M + H)+: 434.1959; found 434.1962.
((3aR,3bR,4aS,5R,5aS)-5-(2-Chloro-6-(((1S)-cyclopropylpropyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-3b(3aH)-yl)methanol (67)
Compound 67 (79%) was prepared from compound 58 following the method for compound 65. 1H NMR (CD3OD, 400 MHz) δ 8.21 (s, 1H), 5.39 (d, J = 6.8 Hz, 1H), 4.95 (s, 1H), 4.70 (d, J = 6.4 Hz, 1H), 4.01 (d, J = 11.6 Hz, 1H), 3.67-3.65 (br m, 1H), 3.62 (d, J = 11.6 Hz, 1H), 1.88-1.81 (m, 1H), 1.75-1.68 (m, 2H), 1.52 (s, 3H), 1.26 (s, 3H), 1.14 (t, J = 5.2 Hz, 1H), 1.03-0.95 (m, 5H), 0.62-0.55 (m, 1H), 0.44-0.41 (m, 2H), 0.36-0.33 (m, 1H). HRMS calculated for C21H29ClN5O3 (M + H)+: 434.1959; found 434.1952.
((3aR,3bR,4aS,5R,5aS)-5-(2-Chloro-6-(((1R)-cyclopropyl-2-methylpropyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-3b(3aH)-yl)methanol (68)
Compound 68 (80%) was prepared from compound 59 following the method for compound 65. 1H NMR (CD3OD, 400 MHz) δ 8.21 (s, 1H), 5.39 (d, J = 6.4 Hz, 1H), 4.95 (s, 1H), 4.70 (d, J = 6.8 Hz, 1H), 4.00 (d, J = 11.6 Hz, 1H), 3.62-3.59 (m, 2H), 2.08-2.02 (m, 1H), 1.72-1.68 (m, 1H), 1.52 (s, 3H), 1.26 (s, 3H), 1.15 (t, J = 5.2 Hz, 1H), 1.08-1.03 (m, 7H), 0.99-0.97 (m, 1H), 0.65-0.62 (m, 1H), 0.43-0.36 (m, 3H). HRMS calculated for C22H31ClN5O3 (M + H)+: 448.2115; found 448.2112.
((3aR,3bR,4aS,5R,5aS)-5-(2-Chloro-6-(((1S)-cyclopropyl-2-methylpropyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-3b(3aH)-yl)methanol (69)
Compound 69 (79%) was prepared from compound 60 following the method for compound 65. 1H NMR (CD3OD, 400 MHz) δ 8.21 (s, 1H), 5.38 (d, J = 6.4 Hz, 1H), 4.95 (s, 1H), 4.70 (d, J = 6.8 Hz, 1H), 4.00 (d, J = 11.6 Hz, 1H), 3.62-3.59 (m, 2H), 2.08-2.03 (m, 1H), 1.72-1.68 (m, 1H), 1.52 (s, 3H), 1.26 (s, 3H), 1.14 (t, J = 5.2 Hz, 1H), 1.08-1.04 (m, 7H), 0.99-0.95 (m, 1H), 0.66-0.61 (m, 1H), 0.42-0.36 (m, 3H). HRMS calculated for C22H31ClN5O3 (M + H)+: 448.2115; found 448.2115.
((3aR,3bR,4aS,5R,5aS)-5-(2-Chloro-6-(((S)-cyclobutyl(cyclopropyl)methyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-3b(3aH)-yl)methanol (70)
Compound 70 (77%) was prepared from compound 61 following the method for compound 65. 1H NMR (CD3OD, 400 MHz) δ 8.21 (s, 1H), 5.39 (d, J = 6.4 Hz, 1H), 4.95 (s, 1H), 4.68 (d, J = 6.4 Hz, 1H), 4.01 (d, J = 11.6 Hz, 1H), 3.86-3.82 (m, 1H), 3.62 (d, J = 11.6, Hz, 1H), 2.71-2.63 (m, 1H), 2.12-2.09 (m, 1H), 2.02-1.86 (m, 3H), 1.83-1.80 (m, 1H), 1.73-1.68 (m, 1H), 1.52 (s, 3H), 1.26 (s, 3H), 1.15 (t, J = 5.2 Hz, 1H), 0.99-0.86 (m, 3H), 0.58-0.53 (m, 1H), 0.40-0.36 (m, 3H). HRMS calculated for C23H31ClN5O3 (M + H)+: 460.2115; found 460.2109.
((3aR,3bR,4aS,5R,5aS)-5-(2-Chloro-6-(((R)-cyclobutyl(cyclopropyl)methyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-3b(3aH)-yl)methanol (71)
Compound 71 (78%) was prepared from compound 62 following the method for compound 65. 1H NMR (CD3OD, 400 MHz) δ 8.21 (s, 1H), 5.39 (d, J = 6.4 Hz, 1H), 4.95 (s, 1H), 4.70 (d, J = 6.4 Hz, 1H), 4.01 (d, J = 11.6 Hz, 1H), 3.87-3.84 (m, 1H), 3.62 (d, J = 11.6, Hz, 1H), 2.71-2.67 (m, 1H), 2.12-2.09 (m, 1H), 2.02-1.87(m, 3H), 1.83-1.80 (m, 1H), 1.71-1.68 (m, 1H), 1.52 (s, 3H), 1.26 (s, 3H), 1.15 (t, J = 5.2 Hz, 1H), 0.99-0.86 (m, 3H), 0.58-0.53 (m, 1H), 0.40-0.36 (m, 3H). HRMS calculated for C23H31ClN5O3 (M + H)+: 460.2115; found 460.2107.
Ethyl (3aR,3bS,4aS,5R,5aS)-5-(2-chloro-6-((dicyclopropylmethyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (76)
2,2-Dicyclopropyl-methylamine (244 mg, 1.94 mmol) and triethylamine (0.53 mL, 3.9 mmol) were added to a solution of compound 75 (161, 0.39 mmol) in methanol (5 mL) and the mixture stirred at room temperature overnight. The reaction mixture was evaporated under vacuum, and the residue was purified on flash column chromatography (hexane:ethyl acetate = 3:1) to give the desired product 76 (166 mg, 87%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 8.03 (s, 1H), 5.86 (d, J = 6.8 Hz, 1H), 4.96 (s, 1H), 4.80 (d, J = 6.8 Hz, 1H), 4.28-4.18 (m, 2H), 3.45 (br s, 1H), 2.27-2.23 (m, 1H), 1.68-1.64 (m, 1H), 1.54-1.51 (m, 4H), 1.33 (t, J = 7.2 Hz, 3H), 1.34 (s, 3H), 1.17-1.10 (m, 2H), 0.60-0.54 (m, 2H), 0.46-0.38 (m, 6H). HRMS calculated for C24H31ClN5O4 (M + H)+: 488.2065; found 488.2065.
Ethyl (3aR,3bS,4aS,5R,5aS)-5-(2-chloro-6-(dimethylamino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (77)
Compound 77 (85%) was prepared from compound 75 following the method for compound 76. 1H NMR (CD3OD, 400 MHz) δ 7.95 (s, 1H), 5.87 (d, J = 6.4 Hz, 1H), 4.94 (s, 1H), 4.83 (d, J = 6.4 Hz, 1H), 4.28-4.19 (m, 2H), 3.49 (br s, 6H), 2.25-2.21 (m, 1H), 1.67-1.63 (m, 1H), 1.53-1.55 (m, 4H), 1.34 (t, J = 7.2 Hz, 3H), 1.29 (s, 3H). HRMS calculated for C19H25ClN5O4 (M + H)+: 422.1595; found 422.1591.
Binding to ARs, 5HT2Rs and to other diverse sites
Binding assays at ARs was performed as described.15,25 Ki determinations using 96-well plates and binding profiles in a broad screen of receptors and channels were generously provided by the National Institute of Mental Health's Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. For experimental details please refer to the PDSP web site http://pdsp.med.unc.edu/ and click on "Binding Assay" or "Functional Assay" on the menu bar. The cell line used for preparing membranes for h5HT2AR binding is transiently transfected HEKT cells, grown in DMEM medium containing 10% serum. Stable HEK cells were used for preparing membranes for h5HT2BR binding and grown in medium containing 2 µg/mL Puromycin. Flp-In HEK cells were used for preparing membranes for h5HT2CR binding and grown in DMEM medium containing 100 µg/mL Hygromycin B. Radioligands used and their measured 5HT2R affinities from saturation experiments are given in Table 1. The binding buffer consisted of 50 mM Tris.HCl, 10 mM MgCl2, 0.1 mM EDTA at pH 7.4 and at RT, and the wash buffer was cold 50 mM Tris.HCl at pH 7.4. Nonspecific binding was determined as noted in Table 1. Untransfected HEK-293 cells do not express specific [3H]43 binding.54 Where we use a nonspecific radioligand, [3H]43, and an antagonist of broad specificity (10 µM clozapine for 5HT2A) to define nonspecific binding, only the receptor of interest is detected as specific binding because it is overexpressed in HEK cells. Although only a single 5HT receptor was overexpressed in the HEK cells, it is possible that [3H]43 binds to endogenous serotonin or non-serotonin receptors with affinities within the nanomolar range in these cells. Results were analyzed in Prism 5.0 by nonlinear least-squares regression to obtain Ki values. Statistical significance of the difference was assessed using unpaired t-test or One-Way Analysis of Variance (ANOVA) followed by post-hoc test where appropriate.
Functional assays at 5HT2Rs
In functional assays, the 5HT2AR, 5HT2BR and 5HT2CR were expressed in Flp-In HEK cells. Cells were plated into poly-L-lysine coated 96-well black clear bottom cell culture plates with DMEM supplemented with 1% dialyzed FBS and cultured overnight before assays. Activity an agonist or antagonist (using 5-HT as reference agonist at the EC80) was determined in calcium mobilization experiments using a FLIPRTETRA (Molecular Devices, Sunnyvale, CA). Plates were loaded with the Fluo-4 Direct Calcium dye (prepared in 20 mM HEPES, 1× HBSS, 2.5 mM Probenecid, pH 7.4) and incubated for 60 min at 37°C, followed by a 10 min incubation at room temperature in the dark. The FLIPR then collected 10 readings (1 read per second) first as a baseline before addition of solutions of the test compound. For agonist activity, the fluorescence intensity was recorded for 2 min after test compound addition. To measure antagonist activity, test compound stocks were prepared at 4× of the final concentration and added as above for potential effects on basal levels for 2 min first, followed by a 5 to 10 min incubation before addition of reference agonist 5-HT at a final concentration equivalent to the EC80 to measure remaining agonist activity.
Maximal fluorescence intensity (RFU) was recorded within a min after drug addition. IC50 values obtained were converted to Ki values using the Cheng-Prusoff equation.53 The FLIPR calcium assay does not differentiate between inverse agonist and neutral antagonist activity. In control HEK293 cells, there was no endogenous response to 5-HT, using either calcium transients in functional assays or specific radioligand binding assays (results not shown). Comparison between experimental curves was performed using F-test (P < 0.05 was considered as statistically significant).
Preclinical testing
Parameters in Table 2 were determined by GVK BIO Sciences Pvt Ltd Hyderabad, India.
Molecular Modeling
Computational facilities
Ligand geometry optimization, homology modeling and molecular docking simulations were carried out using a 6 Intel® Xeon® E5-1650 v3 CPU workstation. Membrane MD simulations were run in part on an in-house cluster composed of two NVIDIA® 970 GTX and one NVIDIA® 980Ti GTX, and in part by exploiting the computational resources (four NVIDIA® Tesla® K20X) of the NIH HPC Biowulf cluster (http://biowulf.nih.gov).
Ligand Preparation
Selected compounds were built using Maestro55 and subjected to gas phase geometry optimization with the Jaguar 8.9 quantum chemistry package56 using density functional theory (DFT) with the B3LYP hybrid functional and the 6-31G** basis set. Frequency calculations were performed to ensure the structures represented minima on the potential energy surfaces.
Homology modeling
The h5HT2BR homology model was built upon the X-ray structure of the ERG-h5HT2BR complex.11 The template structure was retrieved from the RCSB PDB database57 (http://www.rcsb.org, PDB ID: 4IB4), and the amino acid sequence of the target receptor was retrieved from the Universal Protein Resource58 (UniProt IDs: P41595). The template was pre-processed as follows: after manual removal of the apocytochrome b562 RIL (BRIL) fusion protein and co-crystallized ligand, water, and lipid molecules, ionization states of protein sidechains and hydrogen positions were assigned with the Protein Preparazion Wizard tool.59 Prime 4.160 was used to build homology models (ClustalW alignment method and knowledge-based building method) and to reconstruct and refine missing loop domains except IL3 (Serial loop sampling, default level of accuracy). N-terminal and C-terminal portions were not modeled if their lengths exceeded those of the template. The C- and N-termini of the protein and the boundaries of missing IL3 were capped with acetyl and methylamino groups, respectively.
Induced Fit Docking
Compound 23 was docked to the homology model by means of the IFD procedure based on Glide search algorithm61 using the Standard Protocol (SP) and OPLS3 force field.62 The centroid of Asp3.33 residue was selected as center of the Glide grid (inner box side = 10 Å; outer box side = auto). The ligand was initially docked rigidly into the receptor by applying a scaling factor of 0.5 to both ligand and protein van der Waals (vdW) radii. Up to 20 poses were collected, and the sidechains of residues within 5 Å of the ligand were refined with Prime.60 The ligand were re-docked into the newly generated receptor conformations with Glide61 by generating up to 10 poses using the SP scoring function and reverting the vdW radii scaling factors to their default values.
Molecular dynamics
The best obtained docking pose of 23 and the homology model of the apo receptor were subjected to 30 ns of membrane MD simulations. The structures were embedded in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid bilayer (80×80 Å wide, generated through the VMD63 Membrane Plugin tool) according to the orientation suggested by the “Orientations of Proteins in Membranes (OPM)” server.64 Overlapping lipids (within 0.6 Å) were removed upon protein insertion and the systems were solvated with TIP3P65 water and neutralized by Na+/Cl− counter-ions (final concentration 0.154 M). MD simulations with periodic boundaries conditions were carried out with the ACEMD program66 using the CHARMM3667,68/CGenFF(3.0.1)69,70 force fields for lipid and protein, and ligand atoms, respectively. Ligand parameters were obtained by analogy through the ParamChem service with no further optimization. After parameters validation, constrains were applied to the dihedral angles of the (N)-methanocarba ring to avoid unfeasible south ring puckering. The systems were equilibrated through a stepwise procedure: in the first stage, after 2500 cycles of conjugate-gradient minimization aimed at reducing steric clashes arising from the manual setup of the system, 10 ns of MD simulation were performed in the NPT ensemble, restraining protein (and ligand) atoms by a force constant of 1 kcal/mol·Å2. In the second stage, once water molecules diffused inside the protein cavity and the lipid bilayer reached equilibrium, the constraints on the ligand atoms were maintained whereas those on protein atoms were removed except for alpha carbon atoms (force constant = 0.5 kcal/mol·Å2) for other 10 ns. During the equilibration procedure, the temperature was maintained at 310 K using a Langevin thermostat with a low damping constant of 1 ps−1, and the pressure was maintained at 1 atm using a Berendensen barostat. Bond lengths involving hydrogen atoms were constrained using the M-SHAKE71 algorithm with an integration timestep of 2 fs. The equilibrated systems were then subjected to 30 ns of unrestrained MD simulations (NVT ensemble, damping constant of 0.1 ps−1). Long-range Coulomb interactions were handled using the particle mesh Ewald summation method (PME)72 with grid size rounded to the approximate integer value of cell wall dimensions. A non-bonded cutoff distance of 9 Å with a switching distance of 7.5 Å was used. The biophysical validity of the membrane-protein systems was assessed by measuring the average area per lipid headgroup (APL, Table S3) using Grid-MAT-MD.73 The simulation on the 23-h5HT2BR were run in triplicate and selection of a representative trajectory was based upon the cumulative total Ligand-Protein interaction energy (IEtot) expressed as the sum of van der Waals (IEvdW) and electrostatic (IEele) contribution and computed at frames extracted every 50 ps with the mdenergy function implemented in VMD60 exploiting NAMD 2.10.74 From the IEtot values so obtained, we computed the cumulative sum of ligand–protein IEtot (hereby denoted as “dynamic scoring function”, DSFtot) as follows:
and derived the slope values by fitting a linear function as previously reported.75 The replica that returned the highest absolute slope value (Supporting Information, Table S1) was selected. IE versus simulation time graphs were generated with Gnuplot.76
Docking of compounds 14, 25, 26, 27 and 35
The selected compounds were docked to the final 23-h5HT2B MD refined structure including all the selected water molecules (w1–w9) with Glide61 by generating up to 5 poses using both the SP and XP scoring functions. The centroid of 23 was selected as center of the Glide grid (inner box side = 10 Å; outer box side = 30 Å).
Supplementary Material
Acknowledgments
We thank Dr. John Lloyd and Dr. Noel Whittaker (NIDDK) for mass spectral determinations. This research was supported by the National Institutes of Health (Intramural Research Program of the NIDDK and R01HL077707). We thank Dr. Bryan L. Roth and Dr. X. P. Huang (Univ. North Carolina at Chapel Hill) and National Institute of Mental Health's Psychoactive Drug Screening Program (Contract # HHSN-271-2008-00025-C) for screening data.
Abbreviations
- AR
adenosine receptor
- cAMP
adenosine 3′,5′-cyclic monophosphate
- CHO
Chinese hamster ovary
- CNS
central nervous system
- CYP
cytochrome P450
- DMEM
Dulbecco’s modified Eagle medium
- DMF
N,N-dimethylformamide
- EL
extracellular loop
- GPCR
G protein-coupled receptor
- HEPES
2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- HEK
human embryonic kidney
- HRMS
high resolution mass spectroscopy
- 5HT
5-hydroxytryptamine
- IFD
induced fit docking
- NMR
nuclear magnetic resonance
- PBS
phosphate buffered saline
- PDSP
Psychoactive Drug Screening Program
- RFU
relative fluorescence unit
- SAR
structure-affinity relationship
- TBAP
tetrabutylammonium dihydrogenphosphate
- TEA
triethylamine
- TM
transmembrane helix
- tPSA
total polar surface area
- TSPO
translocator protein
- MW
molecular weight
Footnotes
Supporting Information Available:
NMR and mass spectra of selected synthesized compounds, results of PDSP screening, MD trajectory Video, additional modeling Figures S1, S2 and S3, Table S1, S2 and S3, 3D coordinates of the starting docking and MD-refined 23-h5HT2BR complexes, Figure S4 (affinity correlation plot), Figures S5 and S6 (biological data), in vitro ADME measurements. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, Simeons FR, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Gilbert IH, Roth BL, Hopkins AL. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobson KA, Costanzi S, Paoletta S. Computational studies to predict or explain GPCR polypharmacology. Trends Pharmacol. Sci. 2014;35:658–663. doi: 10.1016/j.tips.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, Boykow G, Hsieh Y, Palamanda J, Agans-Fantuzzi J, Kurowski S, Graziano M, Chintala M. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J. Med. Chem. 2008;51:3061–3064. doi: 10.1021/jm800180e. [DOI] [PubMed] [Google Scholar]
- 4.Patel G, Karver CE, Behera R, Guyett PJ, Sullenberger C, Edwards P, Roncal NE, Mensa-Wilmot K, Pollastri MP. Kinase scaffold repurposing for neglected disease drug discovery: discovery of an efficacious, lapatinib-derived lead compound for trypanosomiasis. J. Med. Chem. 2013;56:3820–3832. doi: 10.1021/jm400349k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jordheim LP, Durantel D, Zoulim F, Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discovery. 2013;12:447–464. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- 6.Triggle D. 1,4-Dihydropyridines as calcium channel ligands and privileged structures. Cell. Mol. Neurobiol. 2003;23:293–303. doi: 10.1023/A:1023632419813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welsch ME, Snyder SA, Stockwell BR. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallo-Rodriguez C, Ji X-D, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu Q-L, Olah ME, van Galen PJM, Stiles GL, Jacobson KA. Structure-activity relationships of N6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. J. Med. Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paoletta S, Tosh DK, Salvemini D, Jacobson KA. Structural probing of off-target G protein-coupled receptor activities within a series of adenosine/adenine congeners. PLoS ONE. 2014;9:e97858. doi: 10.1371/journal.pone.0097858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh E, Kumari P, Jaiman D, Shukla AK. Methodological advances: the unsung heroes of the GPCR structural revolution. Nat. Rev. Mol. Cell Biol. 2015;16:69–81. doi: 10.1038/nrm3933. [DOI] [PubMed] [Google Scholar]
- 11.Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets — what are the challenges? Nat. Rev. Drug Discovery. 2013;12:265–286. doi: 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diaz SL, Doly S, Narboux-Nême N, Fernández S, Mazot P, Banas SM, Boutourlinsky K, Moutkine I, Belmer A, Roumier A, Maroteaux L. 5-HT2B receptors are required for serotonin-selective antidepressant actions. Mol. Psychiatry. 2012;17:154–163. doi: 10.1038/mp.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebrahimkhani MR, Oakley F, Murphy LB, Mann J, Moles A, Perugorria MJ, Ellis E, Lakey AF, Burt AD, Douglass A, Wright MC, White SA, Jaffré F, Maroteaux L, Mann DA. Stimulating healthy tissue regeneration by targeting the 5-HT2B receptor in chronic liver disease. Nat. Med. (N. Y., NY, U. S.) 2011;17 doi: 10.1038/nm.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janssen W, Schymura Y, Novoyatleva T, Kojonazarov B, Boehm M, Wietelmann A, Luitel H, Murmann K, Krompiec DR, Tretyn A, Pullamsetti S, Weissmann N, Seeger W, Ghofrani HA, Schermuly RT. 5-HT2B Receptor antagonists inhibit fibrosis and protect from RV heart failure. BioMed Res. Int. 2015 doi: 10.1155/2015/438403. Article ID 438403 http://dx.doi.org/10.1155/2015/438403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roth BL. Drugs and valvular heart disease. N. Engl. J. Med. 2007;356:6–9. doi: 10.1056/NEJMp068265. [DOI] [PubMed] [Google Scholar]
- 16.Mann DA, Oakley F. Serotonin paracrine signaling in tissue fibrosis. Biochim. Biophys. Acta. 2013;1832:905–910. doi: 10.1016/j.bbadis.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elangbam CS, Job LE, Zadrozny LM, Barton JC, Yoon LW, Gates LD, Slocum N. 5-Hydroxytryptamine (5HT)-induced valvulopathy: compositional valvular alterations are associated with 5HT2B receptor and 5HT transporter transcript changes in Sprague-Dawley rats. Exp. Toxicol. Pathol. 2008;60:253–262. doi: 10.1016/j.etp.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Y, Ma J, Lin X, Huang XP, Wu K, Huang N. Structure-based discovery of novel and selective 5-hydroxytryptamine 2B receptor antagonists for the treatment of irritable bowel syndrome. J. Med. Chem. 2016;59:707–720. doi: 10.1021/acs.jmedchem.5b01631. [DOI] [PubMed] [Google Scholar]
- 19.Jazayeri A, Dias JM, Marshall FH. From G protein-coupled receptor structure resolution to rational drug design. J. Biol. Chem. 2015;290:19489–19495. doi: 10.1074/jbc.R115.668251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Structure-guided design of A3 adenosine receptor-selective nucleosides: Combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J. Med. Chem. 2012;55:4847–4860. doi: 10.1021/jm300396n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tosh DK, Paoletta S, Deflorian F, Phan K, Moss SM, Gao ZG, Jiang X, Jacobson KA. Structural sweet spot for A1 adenosine receptor activation by truncated (N)-methanocarba nucleosides: Receptor docking and potent anticonvulsant activity. J. Med. Chem. 2012;55:8075–8090. doi: 10.1021/jm300965a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tosh DK, Finley A, Paoletta S, Moss SM, Gao ZG, Gizewski E, Auchampach J, Salvemini D, Jacobson KA. In vivo phenotypic screening for treating chronic neuropathic pain: Modification of C2-arylethynyl group of conformationally constrained A3 adenosine receptor agonists. J. Med. Chem. 2014;57:9901–9914. doi: 10.1021/jm501021n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tosh DK, Paoletta S, Chen Z, Crane S, Lloyd J, Gao ZG, Gizewski E, Auchampach JA, Salvemini D, Jacobson KA. Structure-based design, synthesis by click chemistry and in vivo activity of highly selective A3 adenosine receptor agonists. Med. Chem. Comm. 2015;6:555–563. doi: 10.1039/C4MD00571F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tosh DK, Crane S, Chen Z, Paoletta S, Gao ZG, Gizewski E, Auchampach JA, Salvemini D, Jacobson KA. Rigidified A3 adenosine receptor agonists: 1-Deaza modification maintains high in vivo efficacy. ACS Med. Chem. Lett. 2015;6:804–808. doi: 10.1021/acsmedchemlett.5b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao ZG, Blaustein J, Gross AS, Melman N, Jacobson KA. N6-Substituted adenosine derivatives: Selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem. Pharmacol. 2003;65:1675–1684. doi: 10.1016/s0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee K, Ravi RG, Ji XD, Marquez VE, Jacobson KA. Ring-constrained (N)methanocarba-nucleosides as adenosine receptor agonists: Independent 5′-uronamide and 2′-deoxy modifications. Bioorg. Med. Chem. Lett. 2001;11:1333–1337. doi: 10.1016/s0960-894x(01)00213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melman A, Gao ZG, Kumar D, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Design of (N)-methanocarba adenosine 5′-uronamides as species-independent A3 receptor-selective agonists. Bioorg. Med. Chem. Lett. 2008;18:2813–2819. doi: 10.1016/j.bmcl.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tosh DK, Padia J, Salvemini D, Jacobson KA. Efficient, large-scale synthesis and preclinical studies of MRS5698, a highly selective A3 adenosine receptor agonist that protects against chronic neuropathic pain. Purinergic Signalling. 2015;11:371–387. doi: 10.1007/s11302-015-9459-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joshi BV, Melman A, Mackman RL, Jacobson KA. Synthesis of ethyl (1S,2R,3S,4S,5S)-2,3-O-(isopropylidene)-4-hydroxy-bicyclo[3.1.0]hexane-carboxylate from L-ribose: A versatile chiral synthon for preparation of adenosine and P2 receptor ligands. Nucleosides Nucleotides Nucleic Acids. 2008;27:279–291. doi: 10.1080/15257770701845253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA. Pharmacological and therapeutic effects of A3 adenosine receptor (A3AR) agonists. Drug Discovery Today. 2012;17:359–366. doi: 10.1016/j.drudis.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacobson KA, Ohno M, Duong HT, Kim SK, Tchilibon S, Cesnek M, Holy A, Gao ZG. A neoceptor approach to unraveling microscopic interactions between the human A2A adenosine receptor and its agonists. Chem. Biol. (Oxford, U. K.) 2005;12:237–247. doi: 10.1016/j.chembiol.2004.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, Liu W, Xu HE, Cherezov V, Roth BL, Stevens RC. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tosh DK, Paoletta S, Chen Z, Moss SM, Gao Z-G, Salvemini D, Jacobson KA. Extended N6 substitution of rigid C2-arylethynyl nucleosides for exploring the role of extracellular loops in ligand recognition at the A3 adenosine receptor. Bioorg. Med. Chem. Lett. 2014;24:3302–3306. doi: 10.1016/j.bmcl.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi H-J, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martí-Solano M, Sanz F, Pastor M, Selent J. A dynamic view of molecular switch behavior at serotonin receptors: Implications for Functional selectivity. PLoS ONE. 2014;9:e109312. doi: 10.1371/journal.pone.0109312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 38.Isberg V, de Graaf C, Bortolato A, Cherezov V, Katritch V, Marshall F, Mordalski S, Pin J-P, Stevens RC, Vriend G, Gloriam DE. Generic GPCR residue numbers - aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015;36:22–31. doi: 10.1016/j.tips.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu W, Wacker D, Gati C, Han GW, James D, Wang D, Nelson G, Weierstall U, Katritch V, Barty A, Zatsepin NA, Li D, Messerschmidt M, Boutet S, Williams GJ, Koglin JE, Seibert MM, Wang C, Shah ST, Basu S, Fromme R, Kupitz C, Rendek KN, Grotjohann I, Fromme P, Kirian RA, Beyerlein KR, White TA, Chapman HN, Caffrey M, Spence JC, Stevens RC, Cherezov V. Serial femtosecond crystallography of G protein-coupled receptors. Science. 2013;342:1521–1524. doi: 10.1126/science.1244142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.West JD, Carrier EJ, Bloodworth NC, Schroer AK, Chen P, Ryzhova LM, Gladson S, Shay S, Hutcheson JD, Merryman WD. Serotonin 2B receptor antagonism prevents heritable pulmonary arterial hypertension. PLoS One. 2016;11:e0148657. doi: 10.1371/journal.pone.0148657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morita H, Mochiki E, Takahashi N, Kawamura K, Watanabe A, Sutou T, Ogawa A, Yanai M, Ogata K, Fujii T, Ohno T, Tsutsumi S, Asao T, Kuwano H. Effects of 5-HT2B, 5-HT3 and 5-HT4 receptor antagonists on gastrointestinal motor activity in dogs. World J. Gastroenterol. 2013;19:6604–6612. doi: 10.3748/wjg.v19.i39.6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodrigues T, Hauser N, Reker D, Reutlinger M, Wunderlin T, Hamon J, Koch G, Schneider G. Multidimensional de novo design reveals 5-HT2B receptor-selective ligands. Angew. Chem. Int. Ed. 2015;54:1551–1555. doi: 10.1002/anie.201410201. [DOI] [PubMed] [Google Scholar]
- 43.Kwon YJ, Saubern S, Macdonald JM, Huang XP, Setola V, Roth BL. N-Tetrahydrothiochromenoisoxazole-1-carboxamides as selective antagonists of cloned human 5-HT2B. Bioorg. Med. Chem. Lett. 2010;20:5488–5490. doi: 10.1016/j.bmcl.2010.07.074. [DOI] [PubMed] [Google Scholar]
- 44.Moss N, Choi Y, Cogan D, Flegg A, Kahrs A, Loke P, Meyn O, Nagaraja R, Napier S, Parker A, Peterson JT, Ramsden P, Sarko C, Skow D, Tomlinson J, Tye H, Whitaker M. A new class of 5-HT2B antagonists possesses favorable potency, selectivity, and rat pharmacokinetic properties. Bioorg. Med. Chem. Lett. 2009;19:2206–2210. doi: 10.1016/j.bmcl.2009.02.126. [DOI] [PubMed] [Google Scholar]
- 45.González-Maeso J, Weisstaub NV, Zhou M, Chan P, Ivic L, Ang R, Lira A, Bradley-Moore M, Ge Y, Zhou Q, Sealfon SC, Gingrich JA. Hallucinogens recruit specific cortical 5-HT2A receptor-mediated signaling pathways to affect behavior. Neuron. 2007;53:439–452. doi: 10.1016/j.neuron.2007.01.008. (2007) [DOI] [PubMed] [Google Scholar]
- 46.Kamal M, Gbahou F, Guillaume JL, Daulat AM, Benleulmi-Chaachoua A, Luka M, Chen P, Kalbasi Anaraki D, Baroncini M, Mannoury la Cour C, Millan MJ, Prevot V, Delagrange P, Jockers R. Convergence of melatonin and serotonin (5-HT) signaling at MT2/5-HT2C receptor heteromers. J. Biol. Chem. 2015;290:11537–11546. doi: 10.1074/jbc.M114.559542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei F, Gu M, Chu YX. New tricks for an old slug: Descending serotonergic system in pain. Shengli Xuebao. 2012;64:520–530. [PubMed] [Google Scholar]
- 48.Urtikova N, Berson N, Van Steenwinckel J, Doly S, Truchetto J, Maroteaux L, Pohl M, Conrath M. Antinociceptive effect of peripheral serotonin 5-HT2B receptor activation on neuropathic pain. Pain. 2012;153:1320–1331. doi: 10.1016/j.pain.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 49.Janes K, Symons-Liguori AM, Jacobson KA, Salvemini D. Identification of A3 adenosine receptor agonists as novel non-narcotic analgesics. Br. J. Pharmacol. 2016;173:1253–1267. doi: 10.1111/bph.13446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schaddelee MP, Read KD, Cleypool CG, IJzerman AP, Danhof M, de Boer AG. Brain penetration of synthetic adenosine A1 receptor agonists in situ: role of the rENT1 nucleoside transporter and binding to blood constituents. Eur. J. Pharm. Sci. 2005;24:59–66. doi: 10.1016/j.ejps.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 51.Chanrion B, Mannoury la Cour C, Gavarini S, Seimandi M, Vincent L, Pujol JF, Bockaert J, Marin P, Millan MJ. Inverse agonist and neutral antagonist actions of antidepressants at recombinant and native 5-hydroxytryptamine2C receptors: differential modulation of cell surface expression and signal transduction. Mol. Pharmacol. 2008;73:748–757. doi: 10.1124/mol.107.041574. [DOI] [PubMed] [Google Scholar]
- 52.Carman AJ, Mills JH, Krenz A, Kim DG, Bynoe MS. Adenosine receptor signaling modulates permeability of the blood–brain barrier. J. Neurosci. 2011;31:13272–13280. doi: 10.1523/JNEUROSCI.3337-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng YC, Prusoff WH. Relationship between inhibition constant (K1) and concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic-reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 54.Roth BL, Craigo SC, Choudhary MS, Uluer A, Monsma FJ, Shen Y, Meltzer HY, Sibley DR. Binding of typical and atypical antipsychotic agents to 5-hydroxytryptamine-6 and 5-hydroxytryptamine-7 receptors. J. Pharmacol. Exp. Ther. 1994;268:1403–1410. [PubMed] [Google Scholar]
- 55.Maestro, version 10.3. New York, NY: Schrödinger, LLC; 2015. [Google Scholar]
- 56.Jaguar, version 8.9. New York, NY: Schrödinger, LLC; 2015. [Google Scholar]
- 57.Bernstein FC, Koetzle TF, Williams GJ, Meyer EE, Jr, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 58.The UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 60.Prime, version 4.1. New York, NY: Schrödinger, LLC; 2015. [Google Scholar]
- 61.Glide, version 6.8. New York, NY: Schrödinger, LLC; 2015. [Google Scholar]
- 62.Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016;12:281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 63.Humphrey W, Dalke A, Schulten K. VMD - Visual molecular dynamics. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 64.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–D376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 66.Harvey M, Giupponi G, De Fabritiis G. ACEMD: Accelerated molecular dynamics simulations in the microseconds timescale. J. Chem. Theory Comput. 2009;5:1632–1639. doi: 10.1021/ct9000685. [DOI] [PubMed] [Google Scholar]
- 67.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD., Jr Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi1 and chi2 dihedral angles. J. Chem. Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vanommeslaeghe K, MacKerell AD., Jr Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 2012;52:3144–3154. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vanommeslaeghe K, Raman EP, MacKerell AD., Jr Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012;52:3155–3168. doi: 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kräutler V, Van Gunsteren WF, Hünenberger PH. A Fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001;22:501–508. [Google Scholar]
- 72.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LGA. Smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 73.Allen WJ, Lemkul JA, Bevan DR. GridMAT-MD: A grid-based membrane analysis tool for use with molecular dynamics. J. Comput. Chem. 2009;30:1952–1958. doi: 10.1002/jcc.21172. [DOI] [PubMed] [Google Scholar]
- 74.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sabbadin D, Ciancetta A, Moro S. Bridging molecular docking to membrane molecular dynamics to investigate GPCR–ligand recognition: The human A2A adenosine receptor as a key study. J. Chem. Inf. Model. 2014;54:169–183. doi: 10.1021/ci400532b. [DOI] [PubMed] [Google Scholar]
- 76.Williams T, Kelley C. [accessed September 27, 2016];Gnuplot 4.6: An Interactive Plotting Program, version 4.6.6. 2014 [Online]; http://gnuplot.info. [Google Scholar]
- 77.Carlin JL, Tosh DK, Xiao C, Piñol RA, Chen Z, Salvemini D, Gavrilova O, Jacobson KA, Reitman ML. Peripheral adenosine A3 receptor activation causes regulated hypothermia in mice that is dependent on central histamine H1 receptors. J. Pharmacol. Exp. Ther. 2016;356:474–482. doi: 10.1124/jpet.115.229872. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.