

Summary
Objective
Perinatal nicotine exposure alters offspring lung structure and function; however, the underlying mechanisms remain incompletely understood. Whether epithelial– mesenchymal transition (EMT), a known contributor to pulmonary pathology, occurs following moderate perinatal nicotine exposure is not known.
Methods
Pregnant, pair-fed Sprague Dawley rat dams received either placebo (diluent) or nicotine [1 mg/kg, subcutaneously] once daily from embryonic day (e) 6 to postnatal day (PND) 21. Generation 1 (F1) and 3 (F3) offspring lungs were isolated at PND 21, and using Western analysis, q-RT-PCR and immunohistochemistry examined for evidence of EMT. To gain further supportive evidence for nicotine-induced EMT, embryonic day 19 primary rat lung alveolar type II (ATII) cells were cultured and treated with nicotine for 24 hr.
Results
Protein levels of α-smooth muscle actin, fibronectin, and calponin (myogenic differentiation markers) increased significantly. However, surfactant proteins B and C, and cholinephosphate cytidylyltransferase- α (epithelial cell markers), as well as the typical markers of EMT, E-cadherin, N-cadherin, and fibroblast specific protein (FSP)-1, in both F1 and F3 generation lungs, showed no significant change between the nicotine exposed and control dams. Immunostaining of lung sections and data from in vitro treated ATII cells strongly supported the Western data.
Conclusions
Enhanced myogenic molecular profile, without evidence of EMT, as evidenced by the absence of the loss of E-cadherin or gains in N-cadherin and FSP-1, suggest that perinatal nicotine exposure does not result in EMT, but it leads to myogenesis, which predominantly accounts for the lung phenotype seen in perinatally nicotine exposed rat offspring.
Keywords: epithelial, mesenchymal transition (EMT), smoking, pregnancy, asthma, lung phenotype
INTRODUCTION
Despite efforts at multiple levels, maternal smoking during pregnancy remains common.1 In addition, the recent increase in the use of electronic cigarettes by the women of the reproductive age group2 and the frequent use of nicotine replacement therapy during pregnancy3 render nicotine exposure of the developing fetus an even greater public health concern than it has been in the past. Although exposure of the developing fetus to nicotine affects many organ systems, the effects on the developing lung are particularly concerning, since we now know that the maternal nicotine exposure alters molecular programming of the developing lung, resulting in an asthmatic phenotype, not only in the exposed offspring, but also transgenerationally, that is, affecting at least two subsequent generations.4–6 The cellular and molecular mechanisms underlying the perinatal nicotine exposure-induced lung phenotype remain incompletely understood.7 Here, we determine whether epithelial–mesenchymal transition (EMT), an important contributor to various lung pathologies, plays any role in perinatal nicotine exposure-induced lung phenotype.
Epithelial–mesenchymal transition is a process by which epithelial cells transition to mesenchymal cells either in the process of organ development (physiologic EMT) or during tissue fibrosis, and/or tumor metastasis (pathologic EMT), as has been described in many organs and under a variety of experimental conditions.8 Epithelial and mesenchymal cells differ in both structural and functional phenotypes. For example, epithelial cells are closely connected to each other by tight junctions, gap junctions, and adherens junctions, have polarization of the actin cytoskeleton and an apico-basal polarity, and are bound by a lamina at their basal surface. In contrast, mesenchymal cells lack polarization, have a spindle-shaped morphology and interact with each other only through focal points.9 Furthermore, epithelial cells express high levels of E (epithelial)-cadherin, whereas mesenchymal cells express high levels of N (neural)-cadherin and other mesenchymal markers such as fibronectin and vimentin. Epithelial–mesenchymal transition entails profound changes in molecular, morphological, and phenotypic characteristics of a cell, where the transitioning cell sheds epithelial characteristics and acquires mesenchymal characteristics. Whether EMT contributes of pulmonary phenotype seen in perinatally nicotine exposed lungs has been only sparsely studied.10 Since cigarette smoke/nicotine has been shown to induce EMT in the adult airway/alveolar epithelial cells,11,12 we hypothesized that perinatal nicotine exposure results in EMT and this, at least in part, contributes to the resultant pulmonary phenotype seen in the exposed offspring. Using a well-published rat model of moderate perinatal nicotine exposure induced lung damage,4,6,13 we determined key molecular markers characterizing EMT in the offspring lung under control and perinatal nicotine exposed conditions. Our data demonstrate that perinatally nicotine exposed lungs had an unequivocal evidence for increased myogenesis, but not EMT, rendering it unlikely that EMT plays a significant role in the pathogenesis of perinatal nicotine-induced lung phenotype.
METHODS
The Animal Model
Time-mated, first-time pregnant, pair-fed Sprague Dawley rat dams (F0 generation) received either placebo (diluent) or nicotine [1 mg/kg, administered subcutaneously] in 100 μl volumes daily from embryonic day (e) 6 to postnatal day (PND) 21. This dose of nicotine has been used previously by us and others to mimic daily nicotine exposure to the fetus by the moderately heavy pregnant smoker.4–7,14 Following delivery at term F1 generation pups breast fed ad libitum. At PND 1 and 21 (~age 5 in human years), rat lungs were collected and probed for molecular markers indicative of EMT by Western analysis [surfactant protein B (SP-B), SP-C, CTP: cholinephosphate cytidylyltransferase (CCT)-α, E-cadherin, N-cadherin, and fibroblast specific protein-1 (FSP-1)], immunofluorescence staining for αSMA, calponin, SP-B, SP-C, and FSP-1, and mRNA analysis (E-cadherin, Twist, and Snail), performed according to our previously described methods.4,6,13,14 In order to explore if perinatal nicotine exposure-induced effects on EMT are also transmitted transgenerationally, following our previously described methods,6 generation 3 (F3) pups were generated and PND 21 lungs processed for markers of EMT (E-cadherin, N-Cadherin, and FSP-1). All studies were performed following NIH guidelines after approval by the Institutional Animal Care and Use Committee (project numbers: 12127 and 20280).
Immunoblot Analysis
At sacrifice, lungs were flash-frozen in liquid nitrogen, and then homogenized and sonicated in four volumes of ice-cold lysis buffer containing 50 mM β-glycerophosphate (pH 7.4), 150 mM NaCl, 1.5 mM EGTA, 1 mM EDTA, 1% Triton X-100, 100 mM NaF, 2 mM Na3VO4, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 2 μg/ml pepstatin A. After centrifugation at 13,200g for 15 min at 4°C, the supernatant was used for Western blot analysis for the relevant proteins. The total protein concentration of the supernatant was measured by the Bradford method, using bovine serum albumin as the protein standard. Aliquots of the supernatant, each containing 50 μg of protein, were separated by SDS–Polyacrylamide gel electrophoresis, and transferred to nitrocellulose membranes. Nonspecific binding sites were blocked with Tris-buffered saline (TBS) containing 5% nonfat dry powdered milk (w/v) for 1 hr at room temperature. After a brief rinse with TBS containing 0.1% tween 20 (TBST), the protein blots were incubated in 1:250 diluted anti-fibronectin polyclonal antibody (Santa Cruz sc-9068), 1:350 diluted anti-PPARg polyclonal antibody (Santa Cruz sc-7196), 1:3,000 diluted anti-calponin monoclonal antibody (Sigma–Aldrich, St. Louis, MO), 1:350 diluted anti-SP-C polyclonal antibody (Santa Cruz sc-13979), 1:200 diluted anti-SP-B polyclonal antibody (Santa Cruz sc-13978), 1;200 diluted anti-E-cadherin (Santa Cruz sc-8426), 1;200 anti-N-cadherin polyclonal antibody (Santa Cruz sc-7039), 1:800 diluted, anti-FSP 1 (Millipore Cat. ABF32, Billerica, MA), and 1:10,000 diluted anti-GAPDH monoclonal antibody (MILLIPORE MAB-374) overnight at 4°C. After three more washes in TBST, the blots were exposed to X-ray film using SuperSignal West Pico Chemiluminescent Substrate (Pierce Biotechnology, Rockford, IL), and developed. The relative densities of the protein bands were determined with UN-SCAN-IT software (Silk Scientific Inc., Orem, UT), and normalized to that of GAPDH.
Quantitative RT-PCR
Total RNA was isolated from rat lung tissues or cultured cells using Qiagen RNAeasy plus mini kit (Quigen Applied Biosystems, Foster City, CA, catalog #74134); the extracted RNA was quantitated by absorbance using a nanodrop spectrophotometer (Nanodrop Instruments, Wilmington, DE) and processed for q-RT-PCR according to our previously described methods.15 All PCR primers were obtained from Sigma–Aldrich, which included Snail: 5′-CACACTGGTGAGAAGCCTTT-3′ (forward) and 5′-CAGCCAGACTCTTGGTGTTT-3′ (reverse); Twist: 5′-TCGACTTCCTGTACCAGGTC-3′ (forward) and 5′-TCCATGTTCTCCTTCTCTGG-3′ (reverse); E-cadherin: 5′-AAAGCAGGAAGAAAACACCACTC-3′ (forward) and 5′-AAAGGGCACGCTATCAACATTAG-3′ (reverse); and GAPDH: 5′-CCATGACAACTTTGGTATCGTGG-3′ (forward) and 5′-TTGCTGTAGCCAAATTCGTTGTC-3′ (reverse).
Immunofluorescence Staining
For tissue immunofluorescence staining for the relevant proteins, rat lungs were inflated in situ with 4% paraformaldehyde in phosphate buffer at a standard inflation pressure of 20 cm H2O for 4 hr at 4°C. The lungs were subsequently transferred to PBS containing 30% sucrose (wt/vol) until equilibrated in the cold (4°C). After fixation, 5-μm paraffin sections were treated three times with Histo-Clear™ (National Diagnostics, Atlanta, GA) for 5 min, and then rehydrated by a sequential ethanol wash. Sections were then washed twice for 10 min with PBS, and blocked for 1 hr in PBS-5% normal goat serum-0.2% Triton X–100. Sections were incubated with primary antibodies for 1 hr at room temperature, and then with the appropriate secondary antibody for 1 hr, also at room temperature. Antibodies included calponin (primary antibody, 1:250 dilution, catalog# C2687, Sigma–Aldrich; secondary antibody, 1:100 dilution, Alexa Fluor, anti-mouse 488 green); α-SMA (primary antibody, 1:500, Sigma, catalog# A2547; secondary antibody, 1:250, Alexa Fluor, anti-mouse 488 green); SP-B (primary antibody 1:150 dilution, catalog# sc-13978, Santa Cruz; secondary antibody 1:100 dilution, Alexa Fluor, anti-rabbit 488 green); SP-C (primary antibody, 1:250, Santa Cruz, catalog# SC-13979; secondary antibody, 1:100, Alexa Fluor, anti-rabbit 488 green); and FSP-1 (primary antibody 1:250, Millipore, catalog# ABF32; secondary antibody, 1:150 anti-rabbit 594 red). Sections were washed with PBS and then mounted on glass slides with ProLong Gold antifade reagent with DAPI (Invitrogen, Carlsbad, CA) for visualization under a fluorescence microscope, following previously described methods.15
Alveolar Type II (ATII) Cell Isolation
Embryonic day 19 ATII cells were isolated and cultured according to previously described methods.13
Statistical Analysis
Statistical analysis was performed using a two-tailed Student’s t-test for comparison of the two groups (α = 0.05). Values are expressed as mean SEM.
RESULTS
In line with our previous observations4,6,13,14 at PND 21, we initially confirmed increased levels of myogenic proteins in perinatally nicotine exposed F1 lungs. Western analysis of PND 21 whole lung protein lysates showed significantly increased levels of α-SMA, fibronectin, and calponin in perinatally nicotine exposed versus control lungs (Fig. 1). To determine if EMT might be a contributing factor to increased myogenesis in the nicotine exposed lungs, we next determined protein levels of the established markers of EMT, that is, E-cadherin, N-cadherin, and FSP-1. As shown in Figure 2A, E-cadherin, N-cadherin, and FSP-1 protein levels were not significantly different between the control and nicotine exposed lungs, suggesting the absence of EMT in perinatally nicotine exposed F1 lungs at PND 21. Similarly, at PND 21, E-cadherin, N-cadherin, and FSP-1 protein levels of F3 lungs were also not different between the nicotine exposed (in F0 gestation) and control progeny (Fig. 2B). Since transgenerational transmission of EMT was ruled out, F3 lungs were not examined any further. In line with the data for the established markers of EMT, protein levels of the typical lung epithelial proteins, SP-B, SP-C, and CCT-α, were also not different between the control and nicotine exposed F1 lungs at PND21 (Fig. 2C). These Western analysis data were corroborated by co-immunostaining for one of the mesenchymal marker proteins such as the FSP-1, α-SMA, or calponin with one of the epithelial proteins, SP-B or SP-C. Fibroblast specific protein-1 staining co-localized with enhanced staining of mesenchymal proteins α-SMA and calponin, but not with epithelial proteins SP-B or SP-C (Fig. 3), indicating that the enhanced myogenic protein levels in nicotine exposed lungs was mesenchymal, and not epithelial in origin. To further confirm the absence of EMT following perinatal nicotine exposure, mRNA levels of key molecular intermediates that are known to drive EMT were examined by qRT-PCR. The mRNA expression of E-cadherin, Twist, and Snail, key mediators of EMT, were also not significantly different between the control and nicotine exposed groups (Fig. 4). The above data were also corroborated by the absence of any evidence of EMT in the lungs of nicotine exposed animals at an earlier time-point, that is, PND 1. In fact, contrary to what is expected in EMT, compared to the control group, the protein levels of E-cadherin significantly increased, those of N-cadherin significantly decreased, and FSP-1 levels were not different in perinatally nicotine exposed PND1 lungs (Fig. 5). Since the above outlined Western and mRNA data were obtained from whole lung tissue, we next examined for the occurrence of EMT following direct nicotine exposure to cultured primary ATII cells. Cultured embryonic e19 primary rat lung ATII cells were treated with nicotine (10−6 or 10−9 M) for 24 hr. Similar to the data obtained from the whole lung tissue, there was no evidence that direct nicotine exposure of fetal rat lung ATII cells led to EMT. In fact, contrary to what would be expected with EMT, E-cadherin protein levels increased and those of N-cadherin decreased (Fig. 6A), accompanying increases in SP-B, SP-C, and CCT-α protein levels, three key markers of lung ATII differentiation (Fig. 6B).
Fig. 1.
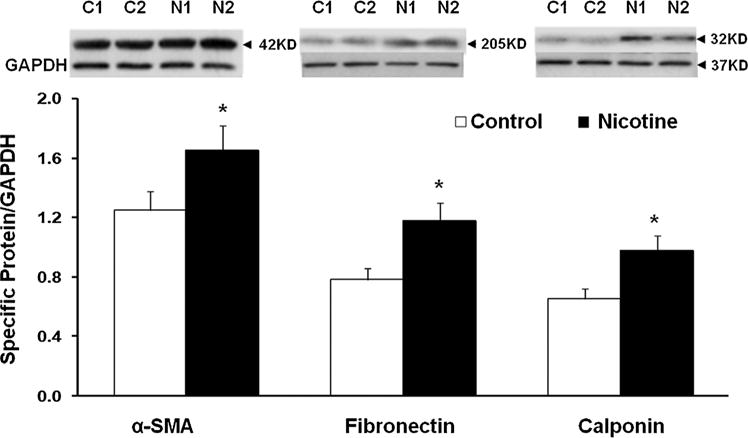
Effect of perinatal nicotine exposure on lung myogenic proteins α-smooth muscle actin (SMA), fibronectin, and calponin. Compared to controls (C1, C2), at postnatal day 21, whole lung protein levels of α-SMA, fibronectin, and calponin, normalized to GAPDH levels, increased significantly in perinatally nicotine (N1, N2) exposed animals. N = 6, *P < 0.05 versus control.
Fig. 2.
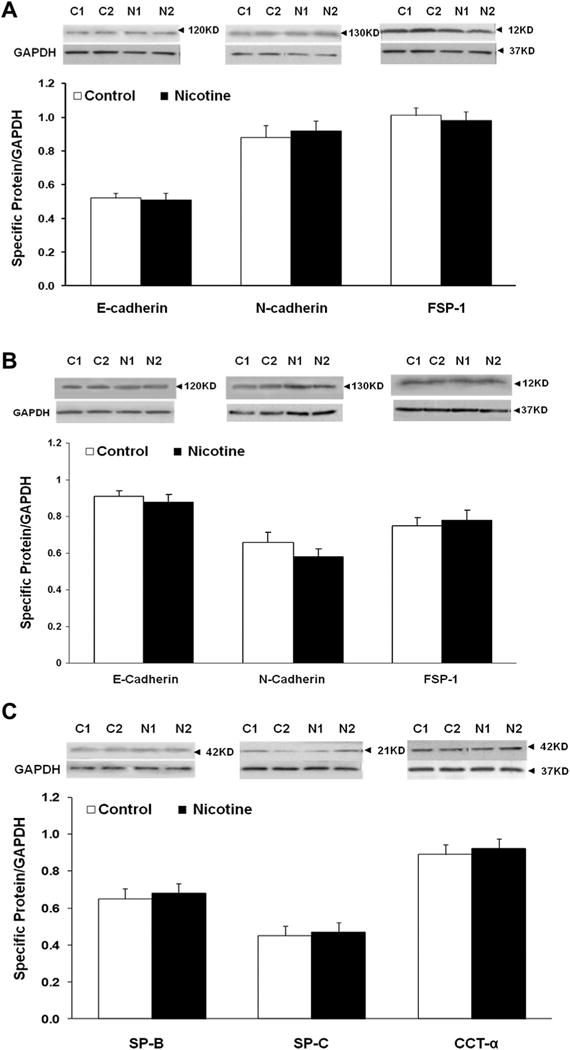
Effect of perinatal nicotine exposure on markers of epithelial–mesenchymal transition (E-cadherin, N-cadherin, and fibroblast-specific protein-1, FSP-1) and alveolar epithelial [surfactant protein B (SP-B), SP-C, and cholinephosphate cytidylyltransferase (CCT)-α] differentiation. E-cadherin, N-cadherin, and FSP-1 protein levels were not significantly different between the control (C1, C2) and nicotine (N1, N2) exposed F1 (A) and F3 (B) postnatal day 21 lungs. Similarly, protein levels of the SP-B, SP-C, and CCT-α, were also not different between the control and nicotine exposed lungs (C). N = 6, *P < 0.05 versus control.
Fig. 3.
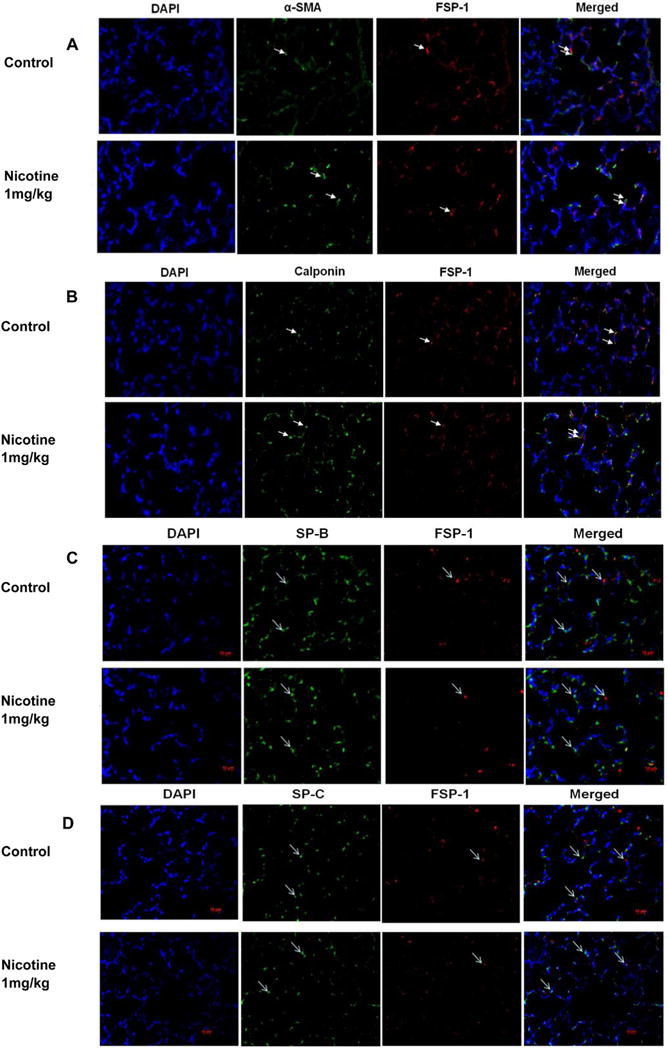
Double immunofluorescence for co-localization of mesenchymal myogenic [fibroblast-specific protein (FSP)-1, α-SMA, or calponin] and epithelial (surfactant protein B or C) marker proteins. Representative lung section stained for the co-localization of α-SMA and FSP-1 (A), calponin and FSP-1 (B), SP-B and FSP-1 (C), and SP-C and FSP-1 (D) are shown. Fibroblast specific protein-1 staining co-localized with enhanced staining of mesenchymal proteins α-SMA and calponin, but not with epithelial proteins SP-B or SP-C. Images were taken at 20×. White arrows indicate the specific signals. N = 4.
Fig. 4.
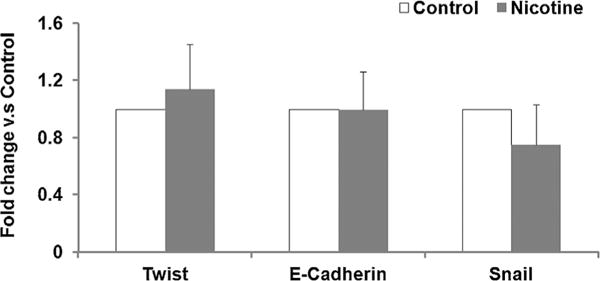
Effect of perinatal nicotine exposure on lung mRNA expression of EMT markers Twist, E-cadherin, and Snail. Total mRNA was extracted from PND21 control and perinatal nicotine exposed lungs and subjected to qRT-PCR. The mRNA expression of E-cadherin, Twist and Snail was not significantly different between the control and nicotine exposed groups. N = 6.
Fig. 5.
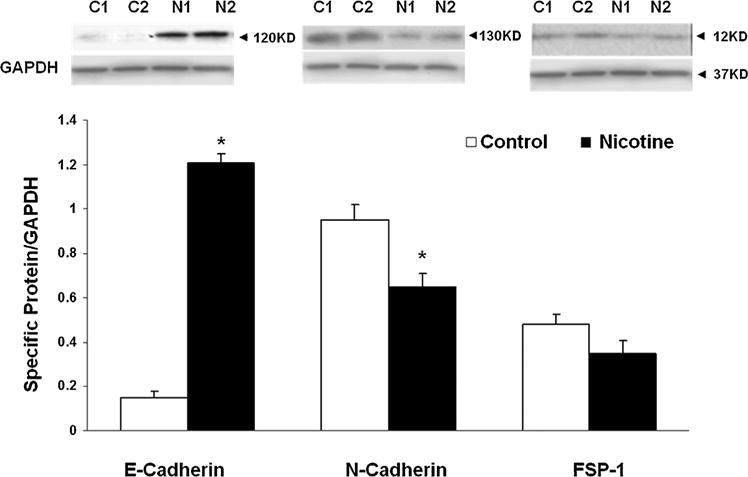
The effect of perinatal nicotine exposure on epithelial–mesenchymal differentiation [E-cadherin, N-cadherin, and fibroblast-specific protein (FSP-1)] markers in postnatal day 1 lungs. Normalized to GAPDH, compared to the controls, the protein levels of E-cadherin significantly increased, those of N-cadherin significantly decreased, and FSP-1 levels were not significantly different in perinatally nicotine exposed PND 1 lungs.
Fig. 6.
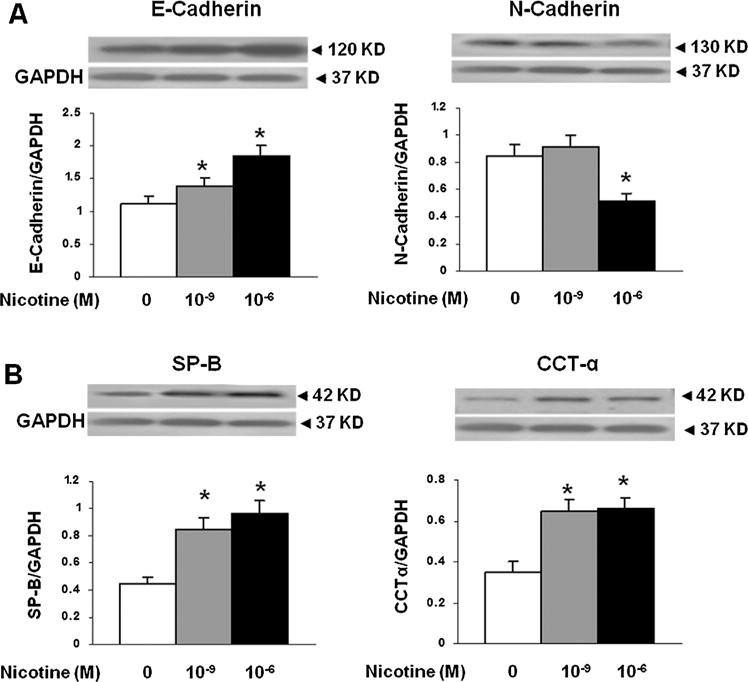
The effect of nicotine on epithelial-mesenchymal differentiation (E- and N-cadherins, surfactant protein-B [SP-B], and cholinephosphate cytidylyltransferase [CCT]-α) markers following direct exposure to cultured alveolar type II (ATII) cells. There was no evidence of EMT following direct nicotine exposure (10−6 or 10−9 M) for 24 hr to the cultured ATII. Contrary to what would be expected with EMT, E-cadherin protein levels increased and those of N-cadherin decreased (A), accompanying increases in SP-B, SP-C, and CCT-α protein levels (B). N = 3, *P < 0.05 versus control.
DISCUSSION
We tested the hypothesis that perinatal nicotine exposure induces EMT in the developing lung. Loss of E-cadherin is considered to be a fundamental event in EMT. Our Western analysis showed that perinatally nicotine exposed PND 21 lung tissue demonstrated increased myogenesis, but not EMT, as evidenced by the lack of a decrease in E-cadherin and increase in N-cadherin protein levels (Figs. 1–3). Protein levels of myogenic markers α-SMA, fibronectin, as well as calponin, which represent smooth muscle differentiation, increased significantly. Furthermore, key surfactant proteins expressed by the alveolar epithelium such as SP-B, SP-C, and CCT-α, a rate-limiting enzyme that regulates surfactant phospholipid synthesis, did not change significantly at PND 21. Co-immunostaining for epithelial and mesenchymal markers supported the Western data (Fig. 3). Furthermore, probing for the markers of EMT at an earlier time-point, that is, PND 1, also did not provide any support for nicotine-induced EMT (Fig. 5). Since there was no evidence of EMT in F1 lungs, it is not surprising that there was no transgenerational evidence of EMT as well. Markedly enhanced myogenic molecular profile, but no significant loss of E-cadherin or gain of N-cadherin and FSP-1, suggest that perinatal nicotine exposure does not result in EMT, but it leads to myogenesis, which predominantly accounts for the lung phenotype seen in perinatally nicotine exposed rat offspring.
The transdifferentiation of epithelial cells into motile mesenchymal cells is integral to the process of development, wound healing, and it also contributes pathologically to fibrosis and malignancy progression. Epithelial mesenchymal transition-inducing signaling pathways have many common intermediates, including the expression of EMT-associated genes. It is clear that each EMT event is regulated by a particular subset of EMT activators and repressors9 that result in the downregulation of E-cadherin and up-regulation of N-cadherin. Cadherins, a family of transmembrane glycoproteins involved in cell-to-cell interactions, are differentially expressed in various tissues. The best characterized cadherin, E-cadherin contains calcium binding extracellular domains responsible for interactions with other E-cadherin molecules on neighboring cells. The cytoplasmic domain of E-cadherin is linked to the actin cytoskeleton through cytoplasmic catenin proteins, thus, establishing a complex localized to adherens junctions. Although E-cadherin expression is regulated at multiple levels (epigenetic, transcriptional, and post-translational)16; at the transcriptional level, it is repressed by a number of nuclear factors (E-cadherin transcriptional repressors, EcTRs) such as ZEB 1 and 2, SNAI 1 (Snail) and 2 (Slug), E12/E47, and Twist1/2. The expression of EcTRs Snail, Slug, and Twist1/2 was not affected by perinatal nicotine exposure, likely explaining the lack of EMT following perinatal nicotine exposure in our model. The mechanism explaining the difference in EMT following nicotine exposure to the adult11,12 and perinatal pulmonary epithelial cells is not known. Differences in epigenetic or transcriptional profile, or proliferating capacity of these cells might explain the differential EMTresponse between these two developmental stages. In fact, following nicotine exposure, whether in vivo to the developing rat lung or in vitro to the cultured day 19 gestation ATII cells, contrary to what is expected in EMT, E-cadherin levels increased and those of N-cadherin decreased. Though the mechanism underlying this intriguing observation is unclear, it might be related to the well-documented increase in ATII cell proliferation induced by nicotine.7,13,17 However, it is also important to point out that in contrast to our data, Chen et al.,10 using a much higher dose of nicotine (6 mg/kg/day, delivered subcutaneously via a mini-osmotic pump, vs. 1 mg/kg/day, delivered subcutaneously without a mini-osmotic pump in our model) recently demonstrated EMT in prenatal and postnatal nicotine exposed rat lungs. This suggests that along with other factors nicotine-induced EMT is also regulated by the stage of lung development, and the dose and route of nicotine exposure.
CONCLUSIONS
Moderate maternal nicotine exposure during gestation and lactation does not cause EMT, but results in myogenesis, which contributes to the lung phenotype seen in perinatally exposed rat offspring.
Acknowledgments
This research was funded by grants from the NIH (HD51857, HL107118, HD071731, and HL127237) and the TRDRP (17RT-0170, and 23RT-0018). The authors had full access to all data and take responsibility for the integrity of the data and the accuracy of analysis.
Funding source: NIH, Numbers: HD51857, HL107118, HD071731, HL127237; TRDRP, Numbers: 17RT-0170, 23RT-0018.
Footnotes
Conflict of interest: None.
References
- 1.Tong VT, Dietz PM, Morrow B, D’Angelo DV, Farr SL, Rockhill KM, England LJ. Trends in smoking before, during, and after pregnancy-pregnancy risk assessment monitoring system, United States, 40 sites, 2000–2010. MMWR Surveill Summ. 2013;62:1–19. [PubMed] [Google Scholar]
- 2.Arrazola RA, Singh T, Corey CG, Husten CG, Neff LJ, Apelberg BJ, Bunnell RE, Choiniere CJ, King BA, Cox S, McAfee T, Caraballo RS. Tobacco use among middle and high school students—United States, 2011–2014. MMWR Morb Mortal Wkly Rep. 2015;64:381–385. [PMC free article] [PubMed] [Google Scholar]
- 3.Stead LF, Perera R, Bullen C, Mant D, Hartmann-Boyce J, Cahill K, Lancaster T. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2012;11:CD000146. doi: 10.1002/14651858.CD000146.pub4. [DOI] [PubMed] [Google Scholar]
- 4.Rehan VK, Liu J, Naeem E, Tian J, Sakurai R, Kwong K, Akbari O, Torday JS. Perinatal nicotine exposure induces asthma in second generation offspring. BMC Med. 2012;10:129. doi: 10.1186/1741-7015-10-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leslie FM. Multigenerational epigenetic effects of nicotine on lung function. BMC Med. 2013;11:27. doi: 10.1186/1741-7015-11-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rehan VK, Liu J, Sakurai R, Torday JS. Perinatal nicotine-induced transgenerational asthma. Am J Physiol Lung Cell Mol Physiol. 2013;305:L501–L507. doi: 10.1152/ajplung.00078.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rehan VK, Asotra K, Torday JS. The effects of smoking on the developing lung: insights from a biologic model for lung development, homeostasis, and repair. Lung. 2009;187:281–289. doi: 10.1007/s00408-009-9158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barriere G, Fici P, Gallerani G, Fabbri F, Rigaud M. Epithelial mesenchymal transition: a double-edged sword. Clin Transl Med. 2015;4:14. doi: 10.1186/s40169-015-0055-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 10.Chen CM, Chou HC, Huang LT. Maternal nicotine exposure induces epithelial-mesenchymal transition in rat offspring lungs. Neonatology. 2015;108:179–187. doi: 10.1159/000437012. [DOI] [PubMed] [Google Scholar]
- 11.Pillai S, Trevino J, Rawal B, Singh S, Kovacs M, Li X, Schell M, Haura E, Bepler G, Chellappan S. β-arrestin-1 mediates nicotine-induced metastasis through E2F1 target genes that modulate epithelial-mesenchymal transition. Cancer Res. 2015;75:1009–1020. doi: 10.1158/0008-5472.CAN-14-0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou W, Zou Y, Zhao Z, Li B, Ran P. Nicotine-induced epithelial-mesenchymal transition via Wnt/β-catenin signaling in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2013;304:L199–L209. doi: 10.1152/ajplung.00094.2012. [DOI] [PubMed] [Google Scholar]
- 13.Rehan VK, Wang Y, Sugano S, Santos J, Patel S, Sakurai R, Boros LG, Lee WP, Torday JS. In utero nicotine exposure alters fetal rat lung alveolar type II cell proliferation, differentiation, and metabolism. Am J Physiol Lung Cell Mol Physiol. 2007;292:L323–L333. doi: 10.1152/ajplung.00071.2006. [DOI] [PubMed] [Google Scholar]
- 14.Krebs M, Sakurai R, Torday JS, Rehan VK. Evidence for in vivo nicotine-induced alveolar interstitial fibroblast-to-myofibroblast transdifferentiation. Exp Lung Res. 2010;36:390–398. doi: 10.3109/01902141003714023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong M, Liu J, Sakurai R, Corre A, Anthony S, Rehan VK. Perinatal nicotine exposure suppresses PPARg epigenetically in lung alveolar interstitial fibroblasts. Mol Genet Metab. 2015;114:604–612. doi: 10.1016/j.ymgme.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peinado H, Olmeda D, Cano A. Snaik, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 17.Maritz GS, Thomas RA. Maternal nicotine exposure: response of type II pneumocytes of neonatal rat pups. Cell Biol Int. 1995;19:323–331. doi: 10.1006/cbir.1995.1075. [DOI] [PubMed] [Google Scholar]