

Abstract
Pluripotent stem cells (PSCs) proliferate rapidly with a characteristic cell cycle structure consisting of short G1- and G2- gap phases. This applies broadly to PSCs of peri-implantation stage embryos, cultures of embryonic stem cells, induced pluripotent stem cells and embryonal carcinoma cells. During the early stages of PSC differentiation however, cell division times increase as a consequence of cell cycle remodeling. Most notably, this is indicated by elongation of the G1-phase. Observations linking changes in the cell cycle with exit from pluripotency have raised questions about the role of cell cycle control in maintenance of the pluripotent state. Until recently however, this has been a difficult question to address because of limitations associated with experimental tools. Recent studies now show that pluripotency and cell cycle regulatory networks are intertwined and that cell cycle control mechanisms are an integral, mechanistic part of the PSC state. Studies in embryonal carcinoma, some 30 years ago, first suggested that pluripotent cells initiate differentiation when in the G1-phase. More recently, a molecular ‘priming’ mechanism has been proposed to explain these observations in human embryonic stem cells. Complexity in this area has been increased by the realization that pluripotent cells exist in multiple developmental states and that in addition to each having their own characteristic gene expression and epigenetic signatures, they potentially have alternate modes of cell cycle regulation. This review will summarize current knowledge in these areas and will highlight important aspects of interconnections between the cell cycle, self-renewal, pluripotency and cell fate decisions.
Keywords: Cell Cycle, Pluripotent Stem Cells, Differentiation, Embryonic Stem Cells
Introduction
Cultured pluripotent stem cells (PSCs) including embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs) and embryonal carcinoma cells (ECCs) are characterized by their ability to retain a broad differentiation potential following extended periods of time in culture. This latter property is known as ‘self-renewal’ and is maintained by cell cycle controls that maintain long-term proliferative capacity. These self-renewing populations are classified as being ‘pluripotent’ while they retain the ability to generate the three embryonic germ layers (ectoderm, endoderm and mesoderm) and, in principle, the ability to generate all lineages of the adult organism. Several core transcription factors are responsible for maintenance of pluripotency and self-renewal including SOX2, NANOG and OCT4 [1]. Together with MYC, these factors are capable of establishing the pluripotent state during what is commonly known as ‘reprogramming’ [2]. Their extensive proliferative capacity combined with wide-range differentiation potential places PSCs in a position of great interest because of their significant therapeutic utility. Before the full potential of PSCs can be exploited in areas such as regenerative medicine, drug discovery and tissue engineering, a thorough understanding of their biological properties is required. In this context, the goal of this review will be to investigate links between cell cycle controls, maintenance and establishment of pluripotency and then, cell fate decisions that PSCs make in response to developmental signals.
Note to the reader: Throughout this review there will be reference to mouse and human orthologs (genes, mRNAs and protein). Convention is that human components are usually specified in uppercase text (ie CDK2) while their mouse counterparts are specified in lower case text (ie Cdk2). For consistency and to prevent confusion in this review, orthologous gene names will be referred to in upper case, regardless of species.
Cell cycle regulation of pluripotent cells during early development
Although there are clear distinctions between fly, frog, fish, rodent and primate embryogenesis, several conserved themes should be noted. For example, pluripotent cells of all early embryos undergo rapid cell/nuclear divisions with cell cycles lacking fully-formed gap phases. This is particularly pronounced in flies, frogs and zebrafish where the early cell cycles are remarkably rapid and consist of alternating rounds of M-phase and interphase without discernable G1- and G2-gap phases [3]. Here, nuclear/cell divisions are synchronous but are later followed by slower, asynchronous cell divisions coinciding with progression through the mid-blastula transition (MBT) and the onset of zygotic gene activation [4, 5]. As cells transition through the MBT, the cell cycle acquires two identifiable gap phases for the first time during development.
In mouse development, zygotic transcription occurs from the second cell division and so does not involve a MBT as described for frogs, flies and fish. In mouse, a short G1-phase (1–2 hours) is identifiable at the second cell division [6] but the G2-phase at this time is unusually long (12–16 hours) and importantly, marks the beginning of zygotic genome activation [7, 8]. In the following divisions the duration of G2-phase is more comparable to that of G1 and cell division is primarily driven by de novo RNA and protein synthesis. As noted previously, this contrasts the situation in flies, fish and frogs where maternal pools of RNA and protein drive rapid cell division during the very early stages of development (pre-MBT). Throughout epiblast development, pluripotent cells in mice maintain a brief G1-phase and cycle rapidly compared to cells of the extra-embryonic tissues [9–11]. After the embryo undergoes gastrulation, several interesting cell cycle changes occur. For example, ‘giant cells’ of the trophoblast undergo endoreplication, resulting in a polyploid DNA content equal to about 500 haploid genomes [12, 13]. This involves an uncoupling of cell cycle events where multiple rounds of S-phase occur without an intervening M-phase. A second notable trend is that following gastrulation, descendants of pluripotent cells undergo dramatic changes in their cell cycle structure and consequently, their proliferation rate decelerates during the early stages of cell fate specification [11–13]. Although aspects of embryology differ between the species discussed above, they all show rapid nuclear/cell division cycles just before lineage commitment. These rapid cycles are therefore intimately linked to uncommitted embryonic cell types across wide evolutionary boundaries, suggesting some key relationship between cell cycle control and developmental status.
Pluripotent cells representing different stages of peri-implantation development
Pluripotency refers to the broad differentiation potential of a cell rather than a specific cell type or cell from a particular developmental stage. Because pluripotent cells exist throughout peri-implantation development [14], it is not surprising that different sub-types of PSCs exist. Different forms of pluripotent cells representing some of these different developmental stages have been isolated and cultured as stable populations, making them valuable tools to study early development. As a point of reference for this review, murine embryonic stem cells (mESCs) are classified as being ‘naïve’. These were the first PSCs to be characterized in the context of cell cycle control and are commonly maintained in the presence of leukemia inhibitory factor (LIF) and fetal calf serum (FCS). LIF/FCS-maintained mESCs are similar to PSCs of the pre-implantation epiblast (E3.5) but differ subtly at the epigenetic level [14]. More recently, a second early epiblast-like population has been described and is referred to as ‘ground-state’ PSCs [15]. These cells seem to represent an earlier stage of pluripotency than ‘naïve’ cells as indicated by their epigenetic and transcriptional signature. It should be noted that most work comparing the properties of ground-state and naïve cells has been performed in the mouse system and only limited information is available in human. The ground-state population in mouse is generated by treatment of LIF/FCS-maintained mESCs with two small molecule inhibitors (2i) that block MEK/ERK and GSK3 signaling.
Pluripotent cells reminiscent of primitive ectoderm in the embryonic epiblast have been isolated from mouse [16, 17] and human [18] sources and are referred to as ‘primed’ pluripotent cells. Human ESCs were the first primed PSCs to be characterized but following this, similar stem cells were isolated from early post-implantation stage mouse embryos and are known as ‘epiblast-like stem cells’ (EpiSCs). In mouse, conversion of naïve to the primed state can be achieved through 2i removal and FGF supplementation, demonstrating that different pluripotent states are inter-convertible and have developmental plasticity [14]. As will be seen later in this review, mechanisms of cell cycle control in naïve and primed cells may differ but the basic cell cycle structure is maintained, even across species [20–22].
The basic principals of cell cycle regulation in mammalian cells
Transition through the eukaryotic cell cycle is coordinated by phosphorylation and dephosphorylation of substrates required to promote events such as DNA replication (S-phase), chromosome segregation (mitosis) and cytokinesis. Each cell cycle phase has associated with it a specific, cell cycle regulated serine/threonine protein kinase that regulates substrates required for specific cell cycle events [23]. These protein kinases are known as cyclin-dependent protein kinases (CDKs) and they function in complexes with cyclin regulatory subunits which themselves accumulate and activate CDKs during specific windows of time during the cell cycle. Cyclin levels are rate-limiting for CDK activation and are regulated by transcriptional and post-transcriptional mechanisms such as ubiquitin-mediated proteolysis. In mitosis for example, cyclin B accumulates and complexes with CDK1 and then phosphorylates substrates required for spindle assembly, chromosome condensation and chromosome segregation. Importantly, the activity of this CDK-cyclin complex is restricted to the mitotic phase not only because of its requirement for chromosome segregation but also because its inactivation is required for cytokinesis. Another example relevant to this review is the role of G1-CDKs in promoting the G1 to S-phase transition. Here, growth factor-dependent signaling typically activates CDK4/6-cyclin D complexes and then, CDK2-cyclinA/E complexes that inactivate RB proteins by direct phosphorylation. RBp105 inactivation then allows E2F target genes to be derepressed in late G1. These genes are important because their activation status determines whether a cell will continue on into S-phase or, exit the cell cycle and enter a quiescent state (Go). This growth control mechanism, known as the Restriction (R)-point [31], represents a molecular switch that controls the decision made by cells in each cycle to either proliferate or enter Go (Fig. 1). Importantly, components of this pathway are frequently deregulated in tumor cells [32].
Figure 1. Kinetics and regulatory features of the cell cycle in ground-state, primed, and differentiated cells.
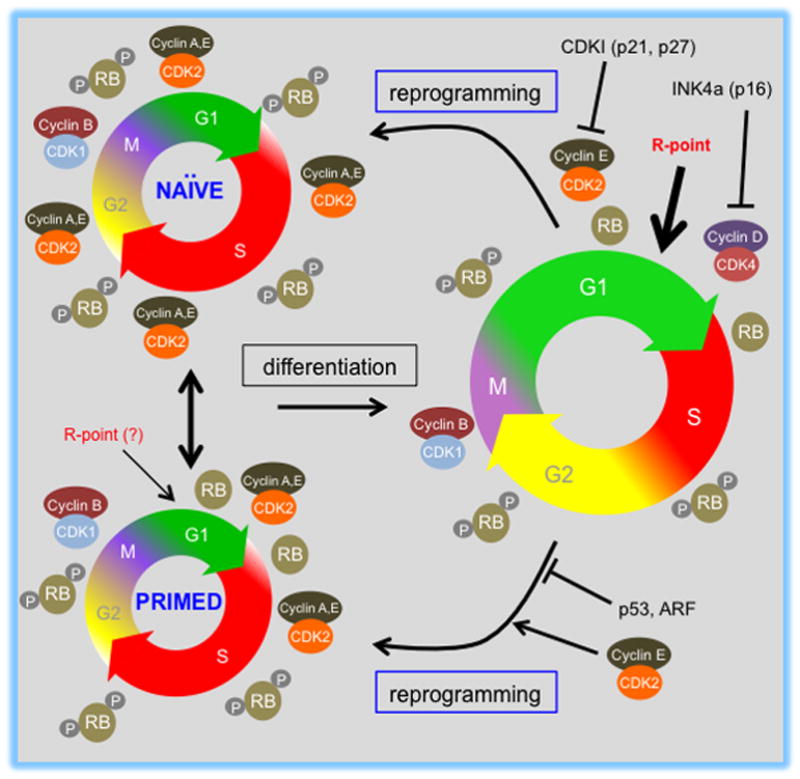
Cell cycle structures of pluripotent stem cells (PSC) and differentiated derivatives are shown. Phases are shown as an approximation of their relative length. Naïve (mESCs) and primed (hESCs, EpiSCs) pluripotent cells (left) proliferate rapidly and are characterized by a short G1 phase. mESCs represent the naïve pluripotent state and its respective cell cycle characteristics, while hESCs and mEpiSCs represent the primed pluripotent state. The potential for conversion between the naïve and primed states of pluripotency is indicated by the double-sided arrow. At the right, a representation of the cell cycle of a differentiated PSC-derivative. Note the differences in the molecular regulation of cell cycle events such as periodicity of CDK-cyclin complex assembly/activity, RB phosphorylation status and the presence or absence of an intact R-point. The relative activity of CDK-cyclin complexes and RB phosphorylation status is shown in relation to cell cycle position for each cell type. During differentiation, G1 length is extended and overall cell cycle duration increases. This is associated with up-regulation of D-type cyclin complexes, down-regulation of global CDK activity, establishment of cell cycle dependent CDK2-cyclin A and E activities and activation of a functional R-point in G1 cells. As cells differentiate, CDKIs are expressed and can negatively control CDK activity (p21, p27 and p16, for example). The figure also indicates that pluripotent cell cycle controls are restored upon reprogramming. The effects of cell cycle complexes (CDK2-cyclin E) and growth regulators (p53, ARF) on reprogramming are indicated.
The previous examples illustrate the need for tight, temporal regulation of CDK activity in the cell cycle. As will be seen, mESCs deviate from the typical mode of CDK regulation seen in most somatic cell types, particularly in relation to G1 control. Another class of molecules, the CDK inhibitors (CDKIs), bind to CDK-cyclin in ternary complexes and directly inhibit their activity [33]. CDKIs are upregulated in response to physiological cues such as DNA damage, growth factor deprivation, ageing etc. that serve to restrict cell division. The expression of these inhibitors is an important consideration when discussing the self-renewal capacity of PSCs. As will be discussed below, many key components of the cell cycle machinery are subject to a different mode of regulation in pluripotent stem cells compared to that typically seen in somatic cells.
Molecular regulation of the cell cycle in naïve PSCs
As described earlier for pluripotent cells in peri-implantation development, cultured naïve and primed PSCs divide rapidly and have a cell cycle structure composed of short gap phases (Figs. 1,2). Approximately 50–70% of the cell cycle is devoted to DNA replication (S-phase) and approximately 10–15% of time in G1-phase (Fig. 2). Although factors such as cell-cell contact, karyotype, cell line variation and culture conditions can impact cell cycle lengths [19], the cell cycle structure of PCSs is relatively consistent [20–22]. A key feature of PSCs in culture is that they have long-term proliferative capacity and do not undergo replicative senescence. There are questions however, about changes in the proliferative potential of PSCs over extended passage. For example, it has been suggested that PSCs have increased proliferative capacity and decreased differentiation potential after long periods of passaging [59–61]. This may be linked, in some cases at least, to the accumulation of genetic modifications and genomic instability that could impact the cell cycle machinery. It should also be noted that cell division rates change with cell density, making it difficult to precisely define generation times [45].
Figure 2. Cell cycle analysis of naïve and primed pluripotent cells.
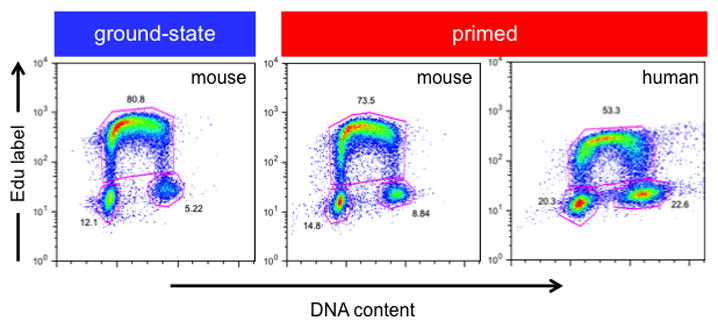
Cell cycle distribution of murine R1 naïve (2i) and primed (EpiSCs) cells and WA09 primed hPSCs. Cells were pulse-labeled with 5-ethynyl-2′-deoxyuridine (EdU, 10μM) to label actively replicating DNA shown on the Y-axis (S-phase cells), using the Click-iT Plus Edu Flow Cytometry Assay kit (Invitrogen). All cells were labeled for 15 mins. Cells were then fixed and stained with FxCycle Violet (Life Technologies; 30 mins, 1 μg/ml) to measure DNA content (X-axis). Cells were then analyzed using a Beckman Coulter CyAn ADP flow cytometer. Cells with low FxCycle Violet and low EdU fluorescence represent cells in G0/G1 phase. Cells with low to high FxCycle Violet and high EdU fluorescence represent cells in S-phase, while cells with high FxCycle Violet and low EdU fluorescence represent cells in G2/M-phase. The percentage of cells found in each respective fraction is indicated. Murine (naïve, primed) and human (primed) cells were cultured as described previously [15,43,62].
In mESCs, E- and A-type cyclins are constitutively expressed at high levels throughout the cell cycle, in contrast to the majority of somatic cell types where they are strictly cell cycle-regulated (Fig. 1). Correspondingly, CDK activities such as CDK2 are active throughout the cell cycle, explaining in part why mESCs transition quickly through G1- into S-phase. The mitotic cyclin CCNB1 (cyclin B1) is tightly cell cycle-regulated however, as is the activity of its catalytic partner, CDK1 [21, 24, 25]. Cyclin D1 is expressed at low levels in pluripotent cells of the ICM and naïve mESCs, while cyclin D2 is undetectable [21, 24, 26–28]. Cyclin D3 appears to be the only D-type cyclin expressed at robust levels in pluripotent cells during epiblast development and forms active complexes with CDK6 [24].
The following will discuss regulatory mechanisms that contribute to the truncated G1-phase in naïve PSCs beginning with CDK2. The consequences of constitutive CDK2 activity in mESCs are several-fold. First and perhaps most importantly, phospho-substrates such as the retinoblastoma tumor suppressor protein (RBp105) are held in a constitutively hyper-phosphorylated, inactive state due to the sustained activity of CDK2 throughout the cell cycle (Fig. 1). RB is therefore biochemically inactivated in mESCs indicating that R-point control is not intact. Consistent with this, knockout of all three pocket-protein family members (RBp105, RBL1p107, RBL2p130) has no major effect on mESC proliferation [29, 30], confirming that the RB growth control pathway is not required here. Because developmental defects occur in pocket protein triple knockout (TKO) cells, it seems that RB function only becomes critical as cells exit pluripotency and as they make cell fate decisions. It is interesting that TKO mouse embryonic fibroblasts (MEFs) share many properties that are characteristic of mESCs, indicating that the RB network is at the center of growth control for mESCs. For example, TKO MEFs progress through G1 at an accelerated rate and are not subject to contact inhibition. In addition, they fail to undergo replicative senescence and proliferate independently of growth factors [29, 30]. It can be argued then, that constitutive RB cell cycle kinases (ie CDK2) are responsible for RB inactivation in mESCs and that this is central to many properties associated with the naïve state.
Another consequence of constitutive CDK2 activity is the uncoupling of E2F target genes from cell cycle progression [21]. Activation of E2F target genes is a rate-limiting step for progression from G1 into S-phase and so when E2F activity is constitutive throughout the cell cycle, as a result of RB inactivation, cells progress through G1-phase more rapidly. Despite strong evidence showing that CDK2 is responsible for the short G1-phase in mESCs, details of how it acquires constitutive activity has yet to be established. This is clearly a key question that needs to be addressed in greater detail.
Another tier of cell cycle regulation that is absent in mESCs is the inhibition of CDK activity by CDKIs [21, 24, 25]. The absence of CDKIs in mESCs is an important contributor to the unrestrained activity of CDKs that contributes to inactivation of the RB pathway. CDKIs are also absent in peri-implantation development preceding gastrulation [24, 34], indicating that in vivo and in vitro pluripotency growth regulatory mechanisms are similar. MicroRNAs such as the miR-290 miRNA family also accelerate the progression of mESCs from G1- through to S-phase [35] by RB-dependent and independent mechanisms [36].
The cell cycle of ground-state PSCs has not been characterized in any great detail although mouse PSCs maintained in 2i media [15] have a cell cycle distribution that is comparable to their naïve counterparts (Fig. 2). It is anticipated that molecular mechanisms underpinning the rapid progression of ground-state PSCs through G1-phase will be similar to that in naïve cells but, more work needs to be done to resolve this question.
Cell cycles of primed pluripotent stem cells
The cell cycle of ‘primed’ hESCs has been characterized using a number of approaches including synchronization drugs [37–40], centrifugal elutriation [41] and fluorescence-based reporters [26, 37–39, 42–44]. The cell cycle length of hESCs is approximately 16–18 hours but changes with cell density [19, 42, 45]. As reported for other PSCs, hESCs have a short G1-phase and spend the majority of time in S-phase [39, 40, 42, 46] (Fig. 2). In contrast to naïve cells however, cyclin A levels are cell cycle regulated in hESCs suggesting that CDK2-cyclin A activity is also cell cycle-dependent. Primed hESCs also retain mitosis-specific cyclin B activity while cyclin E levels are invariant across the cell cycle [39–41, 47]. It is still critical however, to formally establish the activities of CDK complexes more directly by enzymatic assays.
The RB pathway seems to be active in G1-phase of hESCs [41, 46], although there is some confusion about which CDK-cyclin components are active (Fig. 1). For example, cyclin D and A-dependent mechanisms have been independently proposed to regulate RB in hESCs [37, 46]. It is also unclear which sites on RBp105 are regulated and how this impacts downstream targets such as E2F target genes. Overall, there is some confusion as to the function of R-point control in human primed cells and the possibility exists that different sub-populations exhibit slightly different modes of regulation. This is an area that requires further attention.
CDKIs such as KIP1p27 and CIP1p21 are maintained at low levels in hESCs by independent mechanisms [40]. p27 for example, is suppressed by elevated levels of the ubiquitin ligase SKP2 which presumably allows CDK2 activity to remain high in cycling hESCs [48]. p21 levels on the other hand, are maintained at low levels by microRNAs [49] and the epigenetic regulator JMJD5 [50]. Together this establishes conditions of elevated CDK activity that drives rapid cell division.
The mechanistic relationship between pluripotency and the cell cycle
The ubiquitous cell cycle structure described for PSCs suggests some mechanistic relationship with the pluripotent state. This question has been addressed by directly asking if a short G1-phase is important for maintenance of pluripotency. In primed hESCs a G1-S delay caused by CDK2 inhibition decreases the expression of pluripotency regulators such as OCT4 [46], indicating that the pluripotent state is destabilized by cell cycle perturbations. These observations also imply mechanistic connections between elevated CDK activity and the pluripotency network (Fig. 3). Other reports have described that a cell cycle arrest induced by ectopic expression of CDKIs [51] or knock-down of CDK2 [40] promotes the spontaneous differentiation of hESCs. Others however, have found that manipulating the length of G1 in mESCs has no impact on self-renewal [52]. It is possible that differences exist between primed and naïve PSCs but also that a critical threshold needs to be exceeded before lengthening of G1-phase impacts pluripotency. The possible mechanisms underpinning this will be discussed below. Apart from providing conditions where pluripotency is maintained, a short G1 phase and rapid cell division could be advantageous for the developing embryo, allowing it to expand in volume over a short developmental window. Another possibility is that up until gastrulation, regulation from environmental signals is not critical and it is only when intercellular signaling is required that an R-point and extended G1-phase are important. Both of these scenarios are likely to apply.
Figure 3. Regulatory networks impacting differentiation from G1-phase.
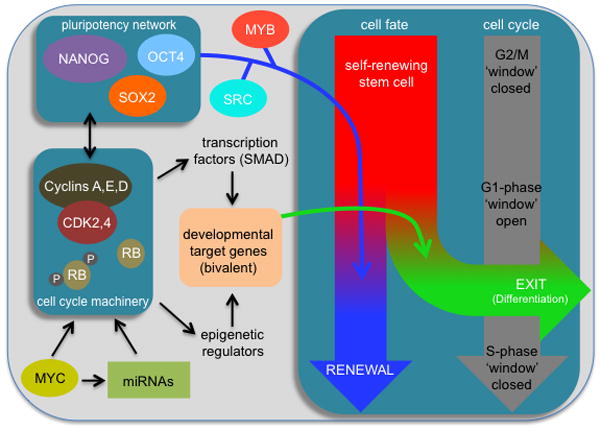
The G1-phase represents a ‘window of opportunity’ when pluripotent cells decide to self-renew or exit the pluripotent state. This involves interplay between the pluripotency network (OCT4, NANOG, SOX2) and cell cycle machinery (CDK-cyclin complexes, RB) that are impacted by other factors such as MYC and miRNAs. The pluripotency network, under self-renewing conditions, ensures maintenance of the pluripotent state. In response to the differentiation signaling, signal-regulated transcription factors (such as SMAD2,3) and epigenetic remodeling enzymes prime and then activate developmental genes in G1-phase. Activation of bivalent genes from G1-phase then initiates the exit from pluripotency and the differentiation program. Other factors such as MYB and SRC regulate this network. Impact that the pluripotency network has on the decision to ‘renew’ is indicated. Effects of developmental regulators, MYB and SRC on pluripotency ‘exit’ are also indicated. The G1-phase is shown as the ‘window’ in which these signals are integrated and where cell fate decisions are initially made. This window closes as cells transit through S-phase and remains closed until the subsequent G1.
Several reports have described an interplay between the pluripotency network and the cell cycle in PSCs. OCT4 and NANOG for example, have roles in controlling transition through the cell cycle [53–56]. In one study, NANOG was shown to directly regulate genes required for the G1 to S-phase transition, such as CDK6 and CDC25A, and to accelerate G1 progression [54]. A similar study in P19 ECCs showed that NANOG positively regulates multiple G1 regulators including CDK6, cyclin E and cyclin D [55]. A similar role has been proposed for OCT4, which is required for transition of mESCs through G1-phase and which regulates CIP1p21 [53]. Conversely, the cell cycle machinery has been implicated in regulation of the pluripotency network including targets such as OCT4, NANOG and SOX2 [57, 58]. For example, GEMININ regulates the expression of pluripotency regulators OCT4, NANOG and SOX2 [57] while CDK activity modulates pluripotency by directly phosphorylating SOX2 [58]. Although these are isolated examples, they suggest a mechanistic link between the cell cycle and pluripotency networks (Fig. 3). Clearly, more work needs to be done to validate this model.
Additional factors connecting the cell cycle to pluripotency and self-renewal
This section introduces additional regulators that are known to impact the cell cycle, and have potential links to pluripotency and self-renewal. Figure 3 shows how these factors may fit into a broad scheme.
(i) MYC
MYC family transcription factors have key roles in the maintenance [62–64] and establishment [2] of pluripotency. Moreover, MYC has established roles in control of cell fate in other multipotent cell types [65, 66] although a clear consensus as to how it functions has not been established [67]. MYC has well-characterized roles in control of the pluripotent cell cycle through positive and negative regulation of genes including those for G1 cyclins such as cyclin E, CDKIs and the DNA replication machinery (Fig. 3). In naïve PSCs, MYC maintains self-renewal through upregulation of the mir-17-92 miRNA cluster (Fig. 3) that targets cell cycle regulators including RBL2p130. MicroRNAs in this cluster have well-established roles in cell cycle control in cancer cells and have been implicated in regulation of RBL2p130, CIP1p21 and E2F1 [68, 69]. Here, miRNAs serve to decrease the stability and to block translation of cell cycle transcripts.
(ii) TP53p53
Several studies in hESCs indicate that TP53p53 antagonizes pluripotency through a cell cycle-dependent mechanism and promotes differentiation when its transcriptional activity increases. Under self-renewing conditions, p53 levels are low in the nucleus but in response to differentiation signals, p53 is stabilized resulting in lengthening of G1 phase and increased spontaneous differentiation [87]. This seems to be dependent on p53-dependent up-regulation of the CDK inhibitor, CIP1p21 and is consistent with other reports showing that delayed progression through G1 promotes exit from pluripotency [61,49]. Part of the p53 stabilization mechanism seems to involve its ability to form a complex with CDKN1Ap21 [70]. p53 also activates miR-34a and miR-145 that negatively regulate pluripotency genes such as OCT4, KLF4, LIN28A, and SOX2. Another study in mESCs screened for regulators of self-renewal using an shRNA library and identified the Aurora A (AURKA) mitotic kinase as having a role in stabilizing pluripotency by repressing TP53 [71]. In this scenario, TP53 transcription is repressed by AURKA, thereby maintaining lineage specific genes in a silent state. There are several unanswered questions about the link between p53, the cell cycle and pluripotency because TP53 does not seem to be essential for murine development [72].
(iii) SRC
The SRC non-receptor tyrosine kinase has well defined roles in cancer when deregulated and recently, was shown to regulate G1 progression in PSCs (Fig. 3). Inhibition of SRC blocks transition through G1-phase, indicating that SRC substrates are required for the G1-S transition in hESCs [73]. Importantly, SRC regulates the activity of RB1p105 by enhancing its phosphorylation and blocking genes required for entry into S-phase, although it is unclear if RB is a direct substrate of SRC. These studies once again however, identify RB as being a critical regulatory point for G1 control and maintenance of pluripotency.
(iv) MYBL2 (B-MYB)
B-MYB is an important but often overlooked regulator of pluripotency, but has well-defined roles as a regulator of the G1 to S-transition in somatic cell types. In the case of pluripotent cells, it is required for ICM formation during development and is essential for mESC generation and self-renewal [74]. Together with MYC and E2F1, B-MYB has been proposed to form part of a transcriptional network that regulates cell cycle target genes in mESCs [92]. Loss of MYBL2 activity causes cell cycle perturbations in PSCs and dramatic changes in global transcription patterns. Notably, cell cycle regulators such as CCNB1, CDC25C, CCND1, CDK6, PLK1, WEE1 and TP53 are downstream of B-MYB activity [75]. Together, this contributes to the short gap phases and rapid cell division associated with PSCs (Fig. 3).
Cell cycle remodeling is linked to early cell fate commitment
The unusual cell cycle structure of PSCs has been emphasized throughout this review, but the theme will now shift to events associated with early cell fate decisions. When PSCs undergo lineage commitment, proliferation rates decrease and organization of the cell cycle changes dramatically [20, 21, 76] (Fig. 2). Most notably, the length of G1 increases at around the time when pluripotency markers decline and when transcription of early differentiation genes such as T and GSC increase [21, 24, 25, 42]. This has generally been interpreted to indicate that cell cycle mechanisms are coupled to pluripotency and early cell fate decisions, consistent with the interplay between cell cycle and PSC regulatory networks discussed earlier in this review [53–58]. The mechanisms governing lineage decisions and cell cycle restructuring are thought to involve decreased CDK activity [21] and activation of the RB/E2F pathway [41, 46, 73] (Fig. 2). The question of whether cell fate commitment is reversible past the point of cell cycle restructuring has been recently investigated. This is an important question because it elaborates on earlier experiments showing that naïve cells in the early process of differentiation can reenter the pluripotent state if LIF signaling is restored within a critical time period [77]. Is cell cycle restructuring a boundary that defines commitment and irreversible exit from the pluripotent state? This question has been answered by showing that it is possible to restore the pluripotent state and its cell cycle structure after the cell cycle has remodeled during early differentiation [78]. Naïve cells can therefore tolerate G1 lengthening and a temporary decrease in pluripotency markers and still be competent to re-enter a stable stem cell state following the reestablishment of LIF signaling. This indicates continued plasticity even under conditions where the cell is preparing to permanently exit the PSC state. The same is likely to be true for primed PSCs.
As discussed earlier, global CDK activity decreases upon differentiation [21, 25]. This observation raises the question about whether this is a trigger to exit pluripotency or whether it is a consequence of early differentiation. Consistent with the idea that elevated CDK activity is required for self-renewal, knockdown of cyclin E delays progression from G1 to S-phase and triggers spontaneous differentiation [78]. Enforced CDK activity under differentiation conditions may therefore be anticipated to block or delay differentiation. This hypothesis has been addressed by showing that overexpressing cyclin E increases the resistance of ESCs to LIF deprivation [78]. Elevated cyclin E activity, probably in conjunction with CDK2, is therefore required for mESC self-renewal.
Changes in cell cycle structure during lineage commitment also appear to be controlled by the up-regulation of CDKIs such as the CIP/KIP and INK family members. The individual germ layers show differential patterns of CDKI expression, suggesting that there are lineage-specific mechanisms for CDK inhibition during early development [24, 25]. This may be related to different patterns of cyclin and CDK complex sub-types expressed in these lineages [24, 26, 28]. During hESC differentiation for example, p27 levels increase due to down-regulation of the SKP2 ubiquitin ligase subunit that targets p27 for degradation. Levels of SKP2 are high in hESCs but upon differentiation the activity of APC/CCDH1, a ubiquitin ligase that targets SKP2 for degradation, increases and remains active in differentiated cells [48]. How individual components of the cell cycle machinery are coordinated with cell fate decisions are now beginning to be investigated with some mechanistic links emerging [26]. For example, elevated levels of cyclin D1 destabilizes pluripotent cells and promotes neural differentiation [79]. In the future, it will be of interest to establish the roles of different cyclin-CDK complexes in alternate routes of cell fate commitment.
Primed pluripotent cells initiate cell fate decisions in the G1-phase
The idea that PSCs initiate cell fate decisions when they are in G1-phase was first described in ECCs by evaluating cell cycle-dependent colony forming activity [80, 81] and then later looking at the effects of retinoic acid as an induction signal [97]. More recently, this concept has been explored in greater detail in part because of recently developed tools. The cell cycle field has traditionally used synchronization drugs that introduce cell cycle perturbations and cytotoxicity to cultures. This problem can be addressed using centrifugal elutriation but the separation of cell cycle phases is quite limited using this approach. More recently, the fluorescence ubiquitin cell cycle indicator (FUCCI) system has been introduced which allows unperturbed cells to be isolated based on their cell cycle position using fluorescence activated cell sorting (FACS). Using the FUCCI system several groups have shown that naïve and primed PSCs initiate differentiation from G1 [42, 78]. These observations indicate that G1 represents a ‘window of opportunity’ where PSCs can more effectively respond to differentiation cues and are therefore competent for differentiation during this window of time (Fig. 3). Using different approaches, these observations have been confirmed by several other groups [26, 40, 41, 43, 46, 73]. Now the principal linking the cell cycle to developmental decisions has been clearly established and defining the mechanisms underpinning this is a high priority. Regulators of G1 progression such as SRC [73], RB [21, 24, 25, 27, 29, 30, 36, 41] and G1-specific CDK activities [26, 82] have been implicated in this general mechanism.
Using the FUCCI reporter, Paulkin and Vallier [27] showed that initiation of differentiation from G1 is determined by cell cycle-dependent loading of SMAD2,3 on developmental target genes (Fig. 3). These complexes are removed late in G1 by cyclin D activity, closing the window in which mesoderm and endoderm differentiation can initiate. This report makes the intriguing observation that early and late parts of G1 are involved in commitment to mesendoderm and ectoderm lineages, respectively. Consistent with this, developmental genes in hESCs are transiently transcribed in G1 under self-renewing conditions and fully activated in G1 when exposed to differentiation signals [42, 43] (Fig. 3). This coincides with epigenetic changes at developmental genes, including 5-hydroxy methylation [43] and increased H3K4 trimethylation at ‘bivalent’, developmental genes [82]. As human PSCs transition through G1, epigenetic changes are accompanied by the remodeling of chromosome architecture at developmental genes. All of these events require extrinsic signals that connect transcriptional networks to developmental genes in G1. Mechanisms driving cell fate decisions from G1 therefore require the convergence of signaling pathways with the cell cycle machinery which controls the activation of developmental genes through signal-regulated transcription factors. Another mechanism linking the cell cycle to cell fate decisions has been defined by Gonzales and co-workers [83] where S- and G2-phase regulators were shown to attenuate the propensity for differentiation. G1 cells however, do not exhibit this attenuation mechanism, explaining why they are competent to initiate cell fate decisions from this phase of the cell cycle.
The cell cycle in reprogramming to pluripotency
In 2006, Yamanaka and colleagues discovered that differentiated cells could acquire an embryonic-like, pluripotent state following the ectopic expression of four genes (MYC, OCT4, SOX2 and NANOG) [2]. Characteristics of cells reprogrammed to an iPSC state include long-term self-renewal, epigenetic and transcriptional changes, acquisition of broad-range differentiation potential, rapid cell division and a cell cycle structure with a short G1-phase [84, 85] (Fig. 2). Cell cycle remodeling has been shown to occur early during reprogramming, consistent with the idea that cell cycle regulation is important for establishment of pluripotency and not just for its maintenance [51]. Moreover, the ectopic expression of CDK-cyclin complexes, with known roles in G1-S progression, enhances reprogramming efficiency [51] (Fig. 1). These complexes potentially impact the rate of cell proliferation and proliferative capacity in addition to the suppression of factors that antagonize cell division such as RB, CDKIs and regulators of senescence such as TP53. Overall, these findings demonstrate a strong connection between cell cycle regulation and cell identity. Several studies have also established strong links between proliferative capacity and the ability to be reprogrammed. For example, reprogramming potential of primary fibroblasts decreases with serial passaging and progression towards replicative senescence [86]. Consistent with this, cells with low endogenous ARF and p53 levels reprogram with faster kinetics and greater efficiencies [86–88] (Fig. 1). Another study using a heterokaryon assay showed that mESCs in G2/M reprogram B lymphocytes and fibroblasts more efficiently than G1 mESCs [89]. Presumably, this indicates that biochemical activities in G2/M are critical for reprogramming in this assay. Whether this mechanism is completely analogous to cell cycle control mechanisms involved in iPSC generation is unclear. These findings however, clearly indicate that the cell cycle machinery is an important component of the mechanisms underpinning cellular reprogramming and that more work needs to be done to clarify this general area.
Conclusions
Throughout this review, mechanistic links between the cell cycle, maintenance of pluripotency and the onset of differentiation have been explored. On balance, the data clearly indicates that the distinct mode of cell cycle regulation associated with PSCs is directly linked to mechanisms of self-renewal. During differentiation, the cell cycle machinery undergoes dramatic changes and these appear to be driving cell fate decisions rather than responding to them. How the cell cycle machinery responds to specification cues is not understood at this time, however. Many other outstanding questions remain. For example, when does cell cycle remodeling occurs? Does it coincide with the initial decision in G1 to differentiate or, does this occur in subsequent cell divisions as pluripotency genes are down-regulated? Perhaps one of the most surprising observations to be reported is the cell cycle regulation of developmental genes at the transcriptional and epigenetic levels in self-renewing stem cells. This is completely unanticipated because lineage markers are generally thought to be repressed under self-renewing conditions. Instead, developmental genes marked by bivalent domains of H3K4 and H3K27 tri-methylation, are briefly activated in G1 indicating the existence of a priming mechanism where PSCs are competent for differentiation when they transition through this phase [43, 82]. During the early stages of differentiation, cells activate new CDK-cyclin complexes and CDKIs and the RB network becomes activated to impose a functional R-point, thereby coupling cell division to mitogenic signals. As mentioned previously, many of these cell cycle components are expressed in a germ-line and cell type specific pattern [21, 24, 25, 28, 48, 76, 90]. These patterns of regulation raise questions about roles for the cell cycle machinery in making specific cell fate decisions, perhaps by controlling developmentally regulated transcription factors. Different CDK-cyclin complexes for example, could target different subsets of developmental transcription factors required for progression down a specific lineage. As examples, cyclin D1 has been shown to regulate the activity of SMAD2,3 on developmental genes in G1-phase [27] and in neural cells, neurogenin 2 is subject to cell cycle dependent phosphorylation as part of its function in promoting neural differentiation [91].
The lineage-priming mechanism eluded to here is primarily based on data from hESCs and may also apply to naïve cells as they transition to the primed state but more broadly, it is unclear if this general mechanism applies to a wider range of multipotent cells such as hematopoietic and mesenchymal stem cells. There is evidence however, linking the G1 phase to cell fate decisions made by neural stem cells [92, 93]. The principles established using PSCs has led to a new paradigm linking the cell cycle to cell fate decisions. It will be of interest to determine roles for this in other aspects of cell fate transformation such as reprogramming and trans-differentiation.
Acknowledgments
This work was supported by a grant to SD from the National Institute for General Medical Sciences PO1 85354.
Footnotes
Ben Boward: Conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing
Tianming Wu: Collection and/or assembly of data, data analysis and interpretation
Stephen Dalton: Conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript
References
- 1.Chambers I, Tomlinson SR. The transcriptional foundation of pluripotency. DEVELOPMENT. 2009;136(14):2311–2322. doi: 10.1242/dev.024398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. CELL. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 3.Heasman J. Patterning the early Xenopus embryo. DEVELOPMENT. 2006;133(7):1205–1217. doi: 10.1242/dev.02304. [DOI] [PubMed] [Google Scholar]
- 4.Yarden A, Salomon D, Geiger B. Zebrafish cyclin D1 is differentially expressed during early embryogenesis. BIOCHIM BIOPHYS ACTA. 1995;1264(3):257–260. doi: 10.1016/0167-4781(95)00175-1. [DOI] [PubMed] [Google Scholar]
- 5.Kane DA, Kimmel CB. The zebrafish midblastula transition. DEVELOPMENT. 1993;119(2):447–456. doi: 10.1242/dev.119.2.447. [DOI] [PubMed] [Google Scholar]
- 6.Gamow EI, Prescott DM. The cell life cycle during early embryogenesis of the mouse. EXP CELL RES. 1970;59(1):117–123. doi: 10.1016/0014-4827(70)90630-0. [DOI] [PubMed] [Google Scholar]
- 7.Sikora-Polaczek M, Hupalowska A, Polanski Z, et al. The first mitosis of the mouse embryo is prolonged by transitional metaphase arrest. BIOL REPROD. 2006;74(4):734–743. doi: 10.1095/biolreprod.105.047092. [DOI] [PubMed] [Google Scholar]
- 8.Flach G, Johnson MH, Braude PR, et al. The transition from maternal to embryonic control in the 2-cell mouse embryo. EMBO J. 1982;1(6):681–686. doi: 10.1002/j.1460-2075.1982.tb01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mac Auley A, Werb Z, Mirkes PE. Characterization of the unusually rapid cell cycles during rat gastrulation. DEVELOPMENT. 1993;117(3):873–883. doi: 10.1242/dev.117.3.873. [DOI] [PubMed] [Google Scholar]
- 10.Snow MH, Bennett D. Gastrulation in the mouse: assessment of cell populations in the epiblast of tw18/tw18 embryos. J EMBRYOL EXP MORPHOL. 1978;47:39–52. [PubMed] [Google Scholar]
- 11.Lawson KA, Meneses JJ, Pedersen RA. Clonal analysis of epiblast fate during germ layer formation in the mouse embryo. DEVELOPMENT. 1991;113(3):891–911. doi: 10.1242/dev.113.3.891. [DOI] [PubMed] [Google Scholar]
- 12.Barlow P, Owen DA, Graham C. DNA synthesis in the preimplantation mouse embryo. J EMBRYOL EXP MORPHOL. 1972;27(2):431–445. [PubMed] [Google Scholar]
- 13.Varmuza S, Prideaux V, Kothary R, et al. Polytene chromosomes in mouse trophoblast giant cells. DEVELOPMENT. 1988;102(1):127–134. doi: 10.1242/dev.102.1.127. [DOI] [PubMed] [Google Scholar]
- 14.Nichols J, Smith A. Naive and primed pluripotent states. CELL STEM CELL. 2009;4(6):487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 15.Ying Q-L, Wray J, Nichols J, et al. The ground state of embryonic stem cell self-renewal. NATURE. 2008;453(7194):519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brons IGM, Smithers LE, Trotter MWB, et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. NATURE. 2007;448(7150):191–195. doi: 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- 17.Chenoweth JG, Tesar PJ. Isolation and maintenance of mouse epiblast stem cells. METHODS MOL BIOL. 2010;636(Chapter 2):25–44. doi: 10.1007/978-1-60761-691-7_2. [DOI] [PubMed] [Google Scholar]
- 18.Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. SCIENCE. 1998;282(5391):1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 19.Barbaric I, Biga V, Gokhale PJ, et al. Time-lapse analysis of human embryonic stem cells reveals multiple bottlenecks restricting colony formation and their relief upon culture adaptation. STEM CELL REPORTS. 2014;3(1):142–155. doi: 10.1016/j.stemcr.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fluckiger A-C, Marcy G, Marchand M, et al. Cell cycle features of primate embryonic stem cells. STEM CELLS. 2006;24(3):547–556. doi: 10.1634/stemcells.2005-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stead E, White J, Faast R, et al. Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. ONCOGENE. 2002;21(54):8320–8333. doi: 10.1038/sj.onc.1206015. [DOI] [PubMed] [Google Scholar]
- 22.Singh AM, Dalton S. The cell cycle and Myc intersect with mechanisms that regulate pluripotency and reprogramming. CELL STEM CELL. 2009;5(2):141–149. doi: 10.1016/j.stem.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. GENES & DEVELOPMENT. 2004;18(22):2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 24.Faast R, White J, Cartwright P, et al. Cdk6-cyclin D3 activity in murine ES cells is resistant to inhibition by p16(INK4a) ONCOGENE. 2004;23(2):491–502. doi: 10.1038/sj.onc.1207133. [DOI] [PubMed] [Google Scholar]
- 25.White J, Stead E, Faast R, et al. Developmental activation of the Rb-E2F pathway and establishment of cell cycle-regulated cyclin-dependent kinase activity during embryonic stem cell differentiation. MOLECULAR BIOLOGY OF THE CELL. 2005;16(4):2018–2027. doi: 10.1091/mbc.E04-12-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pauklin S, Vallier L. The cell-cycle state of stem cells determines cell fate propensity. CELL. 2013;155(1):135–147. doi: 10.1016/j.cell.2013.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savatier P, Huang S, Szekely L, et al. Contrasting patterns of retinoblastoma protein expression in mouse embryonic stem cells and embryonic fibroblasts. ONCOGENE. 1994;9(3):809–818. [PubMed] [Google Scholar]
- 28.Wianny F, Real FX, Mummery CL, et al. G1-phase regulators, cyclin D1, cyclin D2, and cyclin D3: up-regulation at gastrulation and dynamic expression during neurulation. DEV DYN. 1998;212(1):49–62. doi: 10.1002/(SICI)1097-0177(199805)212:1<49::AID-AJA5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 29.Dannenberg JH, van Rossum A, Schuijff L, et al. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. GENES & DEVELOPMENT. 2000;14(23):3051–3064. doi: 10.1101/gad.847700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sage J, Mulligan GJ, Attardi LD, et al. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. GENES & DEVELOPMENT. 2000;14(23):3037–3050. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pardee AB. A restriction point for control of normal animal cell proliferation. PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA. 1974;71(4):1286–1290. doi: 10.1073/pnas.71.4.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sherr CJ. Cancer cell cycles. SCIENCE. 1996;274(5293):1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 33.Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. DEV CELL. 2008;14(2):159–169. doi: 10.1016/j.devcel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 34.White J, Dalton S. Cell cycle control of embryonic stem cells. STEM CELL REVIEWS. 2005;1(2):131–138. doi: 10.1385/SCR:1:2:131. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Baskerville S, Shenoy A, et al. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. NATURE GENETICS. 2008;40(12):1478–1483. doi: 10.1038/ng.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Melton C, Li Y-P, et al. miR-294/miR-302 promotes proliferation, suppresses G1-S restriction point, and inhibits ESC differentiation through separable mechanisms. CELL REP. 2013;4(1):99–109. doi: 10.1016/j.celrep.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Becker KA, Ghule PN, Lian JB, et al. Cyclin D2 and the CDK substrate p220(NPAT) are required for self-renewal of human embryonic stem cells. JOURNAL OF CELLULAR PHYSIOLOGY. 2010;222(2):456–464. doi: 10.1002/jcp.21967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghule PN, Dominski Z, Yang XC, et al. Staged assembly of histone gene expression machinery at subnuclear foci in the abbreviated cell cycle of human embryonic stem cells. PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA. 2008;105(44):16964–16969. doi: 10.1073/pnas.0809273105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Becker KA, Ghule PN, Therrien JA, et al. Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. JOURNAL OF CELLULAR PHYSIOLOGY. 2006;209(3):883–893. doi: 10.1002/jcp.20776. [DOI] [PubMed] [Google Scholar]
- 40.Neganova I, Zhang X, Atkinson S, et al. Expression and functional analysis of G1 to S regulatory components reveals an important role for CDK2 in cell cycle regulation in human embryonic stem cells. ONCOGENE. 2009;28(1):20–30. doi: 10.1038/onc.2008.358. [DOI] [PubMed] [Google Scholar]
- 41.Sela Y, Molotski N, Golan S, et al. Human embryonic stem cells exhibit increased propensity to differentiate during the G1 phase prior to phosphorylation of retinoblastoma protein. STEM CELLS. 2012;30(6):1097–1108. doi: 10.1002/stem.1078. [DOI] [PubMed] [Google Scholar]
- 42.Calder A, Roth-Albin I, Bhatia S, et al. Lengthened G1 phase indicates differentiation status in human embryonic stem cells. STEM CELLS AND DEVELOPMENT. 2013;22(2):279–295. doi: 10.1089/scd.2012.0168. [DOI] [PubMed] [Google Scholar]
- 43.Singh AM, Chappell J, Trost R, et al. Cell-cycle control of developmentally regulated transcription factors accounts for heterogeneity in human pluripotent cells. STEM CELL REPORTS. 2013;1(6):532–544. doi: 10.1016/j.stemcr.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho RJ, Huang M, Campbell MJ, et al. Transcriptional regulation and function during the human cell cycle. NATURE GENETICS. 2001;27(1):48–54. doi: 10.1038/83751. [DOI] [PubMed] [Google Scholar]
- 45.Wu J, Fan Y, Tzanakakis ES. Increased culture density is linked to decelerated proliferation, prolonged G1 phase, and enhanced propensity for differentiation of self-renewing human pluripotent stem cells. STEM CELLS AND DEVELOPMENT. 2015;24(7):892–903. doi: 10.1089/scd.2014.0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Filipczyk AA, Laslett AL, Mummery C, et al. Differentiation is coupled to changes in the cell cycle regulatory apparatus of human embryonic stem cells. STEM CELL RESEARCH. 2007;1(1):45–60. doi: 10.1016/j.scr.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 47.Ghule PN, Becker KA, Harper JW, et al. Cell cycle dependent phosphorylation and subnuclear organization of the histone gene regulator p220(NPAT) in human embryonic stem cells. JOURNAL OF CELLULAR PHYSIOLOGY. 2007;213(1):9–17. doi: 10.1002/jcp.21119. [DOI] [PubMed] [Google Scholar]
- 48.Bar-On O, Shapira M, Skorecki K, et al. Regulation of APC/C (Cdh1) ubiquitin ligase in differentiation of human embryonic stem cells. CELL CYCLE. 2010;9(10):1986–1989. doi: 10.4161/cc.9.10.11727. [DOI] [PubMed] [Google Scholar]
- 49.Dolezalova D, Mraz M, Barta T, et al. MicroRNAs regulate p21(Waf1/Cip1) protein expression and the DNA damage response in human embryonic stem cells. STEM CELLS. 2012;30(7):1362–1372. doi: 10.1002/stem.1108. [DOI] [PubMed] [Google Scholar]
- 50.Zhu H, Hu S, Baker J. JMJD5 regulates cell cycle and pluripotency in human embryonic stem cells. STEM CELLS. 2014;32(8):2098–2110. doi: 10.1002/stem.1724. [DOI] [PubMed] [Google Scholar]
- 51.Ruiz S, Panopoulos AD, Herrerías A, et al. A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. CURRENT BIOLOGY: CB. 2011;21(1):45–52. doi: 10.1016/j.cub.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li VC, Ballabeni A, Kirschner MW. Gap 1 phase length and mouse embryonic stem cell self-renewal. PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA. 2012;109(31):12550–12555. doi: 10.1073/pnas.1206740109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee J, Go Y, Kang I, et al. Oct-4 controls cell-cycle progression of embryonic stem cells. THE BIOCHEMICAL JOURNAL. 2010;426(2):171–181. doi: 10.1042/BJ20091439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang X, Neganova I, Przyborski S, et al. A role for NANOG in G1 to S transition in human embryonic stem cells through direct binding of CDK6 and CDC25A. THE JOURNAL OF CELL BIOLOGY. 2009;184(1):67–82. doi: 10.1083/jcb.200801009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi S-C, Choi J-H, Park C-Y, et al. Nanog regulates molecules involved in stemness and cell cycle-signaling pathway for maintenance of pluripotency of P19 embryonal carcinoma stem cells. JOURNAL OF CELLULAR PHYSIOLOGY. 2012;227(11):3678–3692. doi: 10.1002/jcp.24076. [DOI] [PubMed] [Google Scholar]
- 56.Card DAG, Hebbar PB, Li L, et al. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. MOLECULAR AND CELLULAR BIOLOGY. 2008;28(20):6426–6438. doi: 10.1128/MCB.00359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang VS, Carter SA, Hyland SJ, et al. Geminin escapes degradation in G1 of mouse pluripotent cells and mediates the expression of Oct4, Sox2, and Nanog. CURRENT BIOLOGY: CB. 2011;21(8):692–699. doi: 10.1016/j.cub.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ouyang J, Yu W, Liu J, et al. Cyclin-dependent Kinase-mediated Sox2 Phosphorylation Enhances the Ability of Sox2 to Establish the Pluripotent State. THE JOURNAL OF BIOLOGICAL CHEMISTRY. 2015;290(37):22782–22794. doi: 10.1074/jbc.M115.658195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park YB, Kim YY, Oh SK, et al. Alterations of proliferative and differentiation potentials of human embryonic stem cells during long-term culture. EXP MOL MED. 2008;40(1):98–108. doi: 10.3858/emm.2008.40.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Draper JS, Smith K, Gokhale P, et al. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. NAT BIOTECHNOL. 2004;22(1):53–54. doi: 10.1038/nbt922. [DOI] [PubMed] [Google Scholar]
- 61.Mitalipova MM, Rao RR, Hoyer DM, et al. Preserving the genetic integrity of human embryonic stem cells. NAT BIOTECHNOL. 2005;23(1):19–20. doi: 10.1038/nbt0105-19. [DOI] [PubMed] [Google Scholar]
- 62.Smith KN, Singh AM, Dalton S. Myc represses primitive endoderm differentiation in pluripotent stem cells. CELL STEM CELL. 2010;7(3):343–354. doi: 10.1016/j.stem.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chappell J, Sun Y, Singh A, et al. MYC/MAX control ERK signaling and pluripotency by regulation of dual-specificity phosphatases 2 and 7. GENES & DEVELOPMENT. 2013;27(7):725–733. doi: 10.1101/gad.211300.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cartwright P, McLean C, Sheppard A, et al. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. DEVELOPMENT. 2005;132(5):885–896. doi: 10.1242/dev.01670. [DOI] [PubMed] [Google Scholar]
- 65.Wilson A, Murphy MJ, Oskarsson T, et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. GENES & DEVELOPMENT. 2004;18(22):2747–2763. doi: 10.1101/gad.313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Watt FM, Frye M, Benitah SA. MYC in mammalian epidermis: how can an oncogene stimulate differentiation? NAT. REV CANCER. 2008;8(3):234–242. doi: 10.1038/nrc2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kress TR, Sabò A, Amati B. MYC: connecting selective transcriptional control to global RNA production. NAT REV CANCER. 2015;15(10):593–607. doi: 10.1038/nrc3984. [DOI] [PubMed] [Google Scholar]
- 68.Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. CELL. 2008;133(2):217–222. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu Z, Wang C, Wang M, et al. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. THE JOURNAL OF CELL BIOLOGY. 2008;182(3):509–517. doi: 10.1083/jcb.200801079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jain AK, Allton K, Iacovino M, et al. p53 regulates cell cycle and microRNAs to promote differentiation of human embryonic stem cells. PLOS BIOLOGY. 2012;10(2):e1001268. doi: 10.1371/journal.pbio.1001268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee D-F, Su J, Ang Y-S, et al. Regulation of embryonic and induced pluripotency by aurora kinase-p53 signaling. CELL STEM CELL. 2012;11(2):179–194. doi: 10.1016/j.stem.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shigeta M, Ohtsuka S, Nishikawa-Torikai S, et al. Maintenance of pluripotency in mouse ES cells without Trp53. SCI REP. 2013;3:2944. doi: 10.1038/srep02944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chetty S, Engquist EN, Mehanna E, et al. A Src inhibitor regulates the cell cycle of human pluripotent stem cells and improves directed differentiation. THE JOURNAL OF CELL BIOLOGY. 2015;210(7):1257–1268. doi: 10.1083/jcb.201502035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tanaka Y, Patestos NP, Maekawa T, et al. B-myb is required for inner cell mass formation at an early stage of development. THE JOURNAL OF BIOLOGICAL CHEMISTRY. 1999;274(40):28067–28070. doi: 10.1074/jbc.274.40.28067. [DOI] [PubMed] [Google Scholar]
- 75.Zhan M, Riordon DR, Yan B, et al. The B-MYB transcriptional network guides cell cycle progression and fate decisions to sustain self-renewal and the identity of pluripotent stem cells. Wu Q, ed PLOS ONE. 2012;7(8):e42350. doi: 10.1371/journal.pone.0042350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Savatier P, Lapillonne H, van Grunsven LA, et al. Withdrawal of differentiation inhibitory activity/leukemia inhibitory factor up-regulates D-type cyclins and cyclin-dependent kinase inhibitors in mouse embryonic stem cells. ONCOGENE. 1996;12(2):309–322. [PubMed] [Google Scholar]
- 77.Boeuf H, Merienne K, Jacquot S, et al. The ribosomal S6 kinases, cAMP-responsive element-binding, and STAT3 proteins are regulated by different leukemia inhibitory factor signaling pathways in mouse embryonic stem cells. THE JOURNAL OF BIOLOGICAL CHEMISTRY. 2001;276(49):46204–46211. doi: 10.1074/jbc.M106718200. [DOI] [PubMed] [Google Scholar]
- 78.Coronado D, Godet M, Bourillot PY, et al. A short G1 phase is an intrinsic determinant of naive embryonic stem cell pluripotency. STEM CELL RESEARCH. 2013;10(1):118–131. doi: 10.1016/j.scr.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 79.Chen C-L, Wang L-J, Yan Y-T, et al. Cyclin D1 acts as a barrier to pluripotent reprogramming by promoting neural progenitor fate commitment. FEBS LETT. 2014;588(21):4008–4017. doi: 10.1016/j.febslet.2014.08.039. [DOI] [PubMed] [Google Scholar]
- 80.Wells RS. An in vitro assay for growth regulation of embryonal carcinoma by the blastocyst. CANCER RESEARCH. 1982;42(7):2736–2741. [PubMed] [Google Scholar]
- 81.Pierce GB, Aguilar D, Hood G, et al. Trophectoderm in control of murine embryonal carcinoma. CANCER RESEARCH. 1984;44(9):3987–3996. [PubMed] [Google Scholar]
- 82.Singh AM, Sun Y, Li L, et al. Cell-Cycle Control of Bivalent Epigenetic Domains Regulates the Exit from Pluripotency. STEM CELL REPORTS. 2015 doi: 10.1016/j.stemcr.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gonzales KAU, Liang H, Lim Y-S, et al. Deterministic Restriction on Pluripotent State Dissolution by Cell-Cycle Pathways. CELL. 2015;162(3):564–579. doi: 10.1016/j.cell.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 84.Mikkelsen TS, Hanna J, Zhang X, et al. Dissecting direct reprogramming through integrative genomic analysis. NATURE. 2008;454(7200):49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sridharan R, Tchieu J, Mason MJ, et al. Role of the murine reprogramming factors in the induction of pluripotency. CELL. 2009;136(2):364–377. doi: 10.1016/j.cell.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Utikal J, Polo JM, Stadtfeld M, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. NATURE. 2009;460(7259):1145–1148. doi: 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hong H, Takahashi K, Ichisaka T, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. NATURE. 2009;460(7259):1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kawamura T, Suzuki J, Wang YV, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. NATURE. 2009;460(7259):1140–1144. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tsubouchi T, Soza-Ried J, Brown K, et al. DNA synthesis is required for reprogramming mediated by stem cell fusion. CELL. 2013;152(4):873–883. doi: 10.1016/j.cell.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Becker KA, Stein JL, Lian JB, et al. Human embryonic stem cells are pre-mitotically committed to self-renewal and acquire a lengthened G1 phase upon lineage programming. JOURNAL OF CELLULAR PHYSIOLOGY. 2010;222(1):103–110. doi: 10.1002/jcp.21925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ali F, Hindley C, McDowell G, et al. Cell cycle-regulated multi-site phosphorylation of Neurogenin 2 coordinates cell cycling with differentiation during neurogenesis. DEVELOPMENT. 2011;138(19):4267–4277. doi: 10.1242/dev.067900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Salomoni P, Calegari F. Cell cycle control of mammalian neural stem cells: putting a speed limit on G1. TRENDS CELL BIOL. 2010;20(5):233–243. doi: 10.1016/j.tcb.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 93.Arai Y, Pulvers JN, Haffner C, et al. Neural stem and progenitor cells shorten S-phase on commitment to neuron production. NAT COMMUN. 2011;2(1):154. doi: 10.1038/ncomms1155. [DOI] [PMC free article] [PubMed] [Google Scholar]