

Abstract
Purpose of review
The most common type of ovarian cancer, high-grade serous ovarian carcinoma (HGSOC), was originally thought to develop from the ovarian surface epithelium. However, recent data suggest that the cells that undergo neoplastic transformation and give rise to the majority of HGSOC are from the fallopian tube. This development has impacted both translational research and clinical practice, revealing new opportunities for early detection, prevention, and treatment of ovarian cancer.
Recent findings
Genomic studies indicate that approximately 50% of HGSOC are characterized by mutations in genes involved in the homologous recombination pathway of DNA repair, especially BRCA1 and BRCA2. Clinical trials have demonstrated successful treatment of homologous recombination-defective cancers with poly-ribose polymerase inhibitors through synthetic lethality. Recently, amplification of CCNE1 was found to be another major factor in HGSOC tumorigenesis, accounting for approximately 20% of all cases. Interestingly, amplification of CCNE1 and mutation of homologous recombination repair genes are mutually exclusive in HGSOC.
Summary
The fallopian tube secretory cell is the cell of origin for the majority of ovarian cancers. Although it remains unclear what triggers neoplastic transformation of these cells, certain tumors exhibit loss of BRCA function or amplification of CCNE1. These alterations represent unique therapeutic opportunities in ovarian cancer.
Keywords: BRCA1, BRCA2, Cyclin E (CCNE1), fallopian tube epithelium, high-grade serous ovarian carcinoma
INTRODUCTION
Worldwide, approximately 240 000 women are diagnosed with ovarian cancer each year, and 140 200 are expected to succumb to the disease in 2016 [1,2]. This case-to-fatality ratio is nearly three times that of breast cancer, and makes ovarian cancer the most deadly gynecologic malignancy in developed countries. Patients with stage III or IV disease have a dismal 25% 5-year survival rate [2]. However, despite its aggressive clinical course, the American Cancer Society expects the number of ovarian cancer survivors to increase by 45 000 over the next decade [3].
Ovarian cancer is a nonspecific term for a variety of tumors that involve the ovary. Ovarian cancers can be classified into three large groups: epithelial, germ cell, and specialized stromal cell tumors. The vast majority of ovarian cancers are epithelial ovarian cancers (EOCs). EOC can be further subdivided into various histological subtypes that fall into two main groups: Type I and Type II tumors. Type I tumors include low-grade serous, mucinous, endometrioid, clear cell carcinomas and tend to grow more slowly, often from an identifiable precursor. In contrast, Type II tumors are characterized by high-grade and rapidly progressive disease. High-grade serous ovarian carcinoma (HGSOC) is the most common Type II tumor, accounting for almost 75% of all EOCs. Unfortunately, it is also one of the most aggressive. There are currently no robust methods for early detection of HGSOC. As a result, the majority of women are diagnosed when the cancer has already metastasized to other tissues, usually within the peritoneal cavity. The lack of specific symptoms, even when the disease has spread to the peritoneum, contributes to delayed diagnosis and poor survival rates.
The post-TCGA (The Cancer Genome Atlas) landscape for HGSOC is marked by surprisingly few recurrent somatic mutations [4]. Instead, this disease exhibits a complex genomic terrain, marked by copy number alterations that are so widespread that few other cancer types mirror its complexity. Intertumoral and intratumoral heterogeneity in HGSOC further decrease the likelihood of finding a single therapy that will prove beneficial for the majority of patients. Thus, HGSOC will require individualized therapy in which we unbraid a tumor's genomic profile to identify altered genes or pathways that offer an opportunity for therapeutic intervention.
Box 1.

no caption available
Pathogenesis of epithelial ovarian cancer
Originally, the ovary was thought to be the primary site of HGSOC tumorigenesis and the ovarian surface epithelium (OSE) represented the cell of origin. The ‘incessant ovulation’ hypothesis suggested that HGSOC developed because of repetitive injury to the OSE with each ovulatory cycle [5]. It was thought that this repetitive injury causes increased inflammation and changes in hormone levels, leading to DNA damage produced by oxidative stress [5]. Incessant ovulation, through a rupture and repair mechanism, along with the normal proliferation of the OSE, was thought to drive metaplastic changes toward a more Müllerian-type epithelium. If this Müllerian-type epithelium harbored unresolved DNA damage, it would represent a prime target for neoplastic transformation [6]. Although the OSE model could account for a number of important features associated with ovarian cancer, particularly Type I tumors, it fails to present a path toward understanding of Type II tumors. Perhaps most importantly, attempts to reproducibly identify precursor lesions for HGSOC in the OSE have been largely unsuccessful.
The cloning of the BRCA1 and BRCA2 genes quickly led to the practice of risk-reducing bilateral salpingo-oophorectomies in mutation carriers to reduce the risk of developing ovarian cancer [7▪]. These specimens afforded pathologists the opportunity to examine these tissues for occult cancers. Some of the earliest studies suggesting that the fallopian tube epithelium plays a much larger role in the development of ovarian cancer were reported by Piek et al.[8,9]. Subsequent studies confirmed the paradoxical observation that in the search for early ovarian cancers, most lesions were identified in the fallopian tube [10–15]. The development of a pathology protocol, called the SEE-FIM (Sectioning and Extensively Examining the FIMbriated end) protocol, to systematically evaluate the fallopian tubes of BRCA mutation carriers led to the reproducible identification of early serous carcinomas in the distal end of the fallopian tube. The vast majority of cases was localized to the fimbria and included serous tubal intraepithelial carcinoma (STIC) [16–18]. No intraepithelial or invasive serous carcinomas were identified in the ovaries of these samples [18,19]. Like the foci of invasive HGSOC, the STIC lesions were proliferative, as measured by Ki67 immunohistochemistry (IHC) and stained strongly for p53. More importantly, DNA sequencing revealed that the majority of STIC lesions harbor the same TP53 mutation as the concurrent HGSOC [20,21], indicative of their clonal nature.
Further examination of the fallopian tubes identified short stretches of benign-appearing secretory cells that stained strongly for p53 and γ-H2AX, a marker of DNA damage. These foci of p53-positive cells harbored TP53 mutations but were not proliferative [17]. These patches were called ‘p53 signatures’ based on the requisite p53 IHC necessary to identify the otherwise benign looking cells. Importantly, the ‘p53 signature’, the STIC lesion, and HGSOC from the same patient harbor the same TP53 mutation [17], implying a clonal relationship between the nonproliferative ‘p53 signature’, the intraepithelial lesion, and the invasive cancer (Fig. 1).
FIGURE 1.
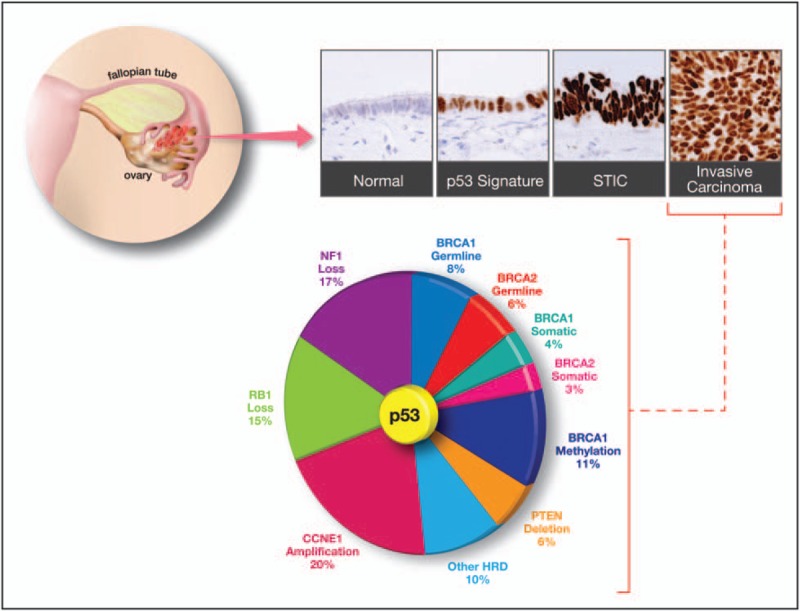
Pathological and genomic features of high-grade serous ovarian carcinomas (HGSOCs). The majority of HGSOCs emerge from the fallopian tube epithelium through a series of precursor lesions that target the secretory cell. Normal fallopian tube epithelium contains both secretory and ciliated cells and is typically immunonegative for p53. The benign ‘p53 signature’ is composed entirely of secretory cells that exhibit strong p53 expression and evidence of DNA damage but are not proliferative. With progression to a serous tubal intraepithelial carcinoma or ‘STIC’, there is acquisition of nuclear pleomorphism, mitoses, and loss of polarity. Invasive HGSOC shares all these properties and clinical symptoms typically emerge with advanced disease [22].
What percentage of HGSOCs arises from the fallopian tube? Studies that implement the SEE-FIM protocol report that approximately 50–60% of HGSOCs are associated with a STIC lesion in the fallopian tube (Table 1). A number of explanations have been offered to explain why the association between HGSOC and STIC is not higher. These include insufficient sampling of tissue blocks [50,51], interobserver variability [52–54], consumption of precursors by the invasive carcinoma, and the high frequency of p53-negative STIC lesions [55]. It is also possible that extrauterine Müllerian epithelium [56] or derivatives of the OSE harbor precursor lesions. However, until reproducible precursors are identified at these sites, their contributions remain unclear. Resolving whether all HGSOCs arise from the fallopian tube or other sites remains to be determined and will likely require additional shared common resources and specimen banks [57▪].
Table 1.
Incidents of tubal precursors in HGSOC
| Author | % STIC in HGSOCa | # STIC | # HGSOC | SEE-FIM | Notes |
| Leeper et al. [12] | 60 | 3 | 5 | No | |
| Powell et al. [13] | 57 | 4 | 7 | No | |
| Carcangiu et al. [23] | 50 | 3 | 6 | No | |
| Finch et al. [14] | 86 | 6 | 7 | No | |
| Medeiros et al. [18] | 100 | 5 | 5 | Yes | |
| Callahan et al. [24] | 100 | 7 | 7 | No | |
| Kindelberger et al. [20] | 48 | 20 | 42 | Yes | |
| Carlson et al. [25] | 40 | 18 | 45 | Some | 47% with SEE-FIM, 35% without SEE-FIM |
| Hirst et al. [26] | 80 | 4 | 5 | Yes | |
| Jarboe et al. [27] | 23 | 5 | 22 | Yes | |
| Roh et al. [28] | 35 | 30 | 87 | Yes | |
| Maeda et al. [29] | 47 | 7 | 15 | Yes | |
| Przybycin et al. [30] | 59 | 24 | 41 | Yes | |
| Leonhardt et al. [31] | 33 | 3 | 9 | Yes | |
| Manchanda et al. [32] | 71 | 10 | 14 | No | |
| Diniz et al. [33] | 71 | 24 | 34 | Some | |
| Powell et al. [34] | 50 | 5 | 10 | No | |
| Seidman et al. [35] | 56 | 5 | 9 | Some | |
| Tang et al. [36] | 19 | 6 | 32 | Yes | |
| Gao et al. [37] | 92 | 107 | 116 | Yes | |
| Lee et al. [38] | 32 | 6 | 19 | No | |
| Reitsma et al. [39] | 75 | 3 | 4 | Some | Cases after 2006 are SEE-FIM |
| Conner et al. [40] | 74 | 14 | 19 | Yes | |
| Koc et al. [41] | 36 | 9 | 25 | Yes | |
| Mingels et al. [42] | 43 | 23 | 54 | Yes | |
| Sherman et al. [43] | 16 | 4 | 25 | No | |
| Gilks et al. [44] | 95 | 20 | 21 | Yes | |
| Munakata and Yamamoto [45] | 22 | 5 | 23 | Some | Only 10% SEE-FIM |
| Seidman [46] | 40 | 81 | 202 | Some | 1991–2007 no SEE-FIM, 2007–2011 half SEE-FIM |
| Malmberg et al. [47] | 61 | 8 | 13 | No | |
| Mittal et al. [48] | 22 | 7 | 32 | Yes | |
| Zakhour et al. [49▪] | 64 | 9 | 14 | Some |
HGSOC, high-grade serous ovarian carcinoma; SEE-FIM, Sectioning and Extensively Examining the FIMbriated end; STIC, serous tubal intraepithelial carcinoma.
aValues are in %.
The fallopian tube paradigm for HGSOC pathogenesis has motivated the development of new, robust, and tractable experimental model systems that focus on the fallopian tube as the site of origin. In particular, several mouse models were created by genetically manipulating murine oviductal cells [58–62]. Some of these models have recapitulated the development of tubal precursor lesions [58,60] and demonstrated that salpingectomy blocks tumor development [58,61]. More recently, Cho and colleagues developed a mouse in which the Ovgp1 promoter controls expression of a tamoxifen-regulated Cre recombinase in oviductal epithelium – the murine equivalent of human fallopian tube epithelium [59]. Deletion of Apc and Pten in this model was compared with a model in which an adenovirus expressing Cre was injected into the ovarian bursa to target the OSE. Tumors that emerged from the fallopian tube more closely resembled human endometrioid ovarian cancers than those from the OSE. The slow progression and late metastasis of oviductal tumors resemble the relatively indolent behavior characteristic of so-called Type I ovarian carcinomas in humans, for which endometrioid carcinoma is a prototype. This model emphasizes that importance of cellular context and the need to further understand the cell of origin for ovarian cancer [63].
Genomic landscape of high-grade serous ovarian carcinoma: the role of TP53
One of the hallmarks of HGSOC is the universal presence of mutations in the TP53 tumor suppressor gene [4,64,65▪▪,66▪▪]. The most common site of mutation of TP53 is the DNA-binding domain, but mutations in other regions have been identified [64]. Mutation of TP53 is the first known molecular event in the transformation of fallopian tube secretory cells, and can be identified in early tumor precursors [17]. Recent studies indicate that stabilizing TP53 missense mutations, but not loss of endogenous wildtype TP53, promote secretory cell survival and cell–cell aggregation under anchorage independent growth conditions. This mutant-mediated autocrine matrix deposition leads to the formation of cell clusters with mesothelial-intercalation capacity which is likely necessary for peritoneal dissemination [67▪]. Interestingly, it appears that the most common TP53 missense mutations, including R273H, R175H, and R248Q, express a large number and high amounts of shorter p53 protein isoforms that are translated from the mutated full-length p53 mRNA. These shorter isoforms, like Δ160p53, exhibit all the gain-of-function properties attributed to the mutant protein, including enhanced cell survival, proliferation, adhesion, and invasion [68▪]. These data suggest that early mutation of TP53 is necessary for HGSOC initiation. For these reasons, mutant p53 has re-emerged as an appealing therapeutic target in HGSOC. Small molecules that sculpt the mutant protein into a more wildtype confirmation are being evaluated in preclinical and clinical trials [69]. In addition, perturbation of pathways, like the mevalonate pathway, that lead to degradation of mutant p53 are being exploited for therapeutic gains [70].
Drugging BRCA in high-grade serous ovarian carcinoma
Despite the high frequency of TP53 mutations observed in the development of HGSOC, TCGA data suggest that recurrent mutations in other genes are relatively uncommon, with the exception of BRCA1 and BRCA2[4]. BRCA1 and BRCA2 are proteins that play a critical role in maintaining the integrity of the genome by orchestrating DNA repair through homologous recombination. Homologous recombination is a high-fidelity process and is considered to be an error-free mechanism of repairing double-stranded breaks (DSBs) because it uses the sister chromatid as a template for repairs. This mechanism is in contrast to the other major pathway, known as nonhomologous DNA end joining (NHEJ), which simply ligates DSB ends without a template and is more error-prone. Double-stranded DNA breaks occur most frequently during DNA replication, especially when the replication machinery encounters a single-stranded break (SSB), ultimately leading to genomic instability and cell death if unrepaired. Mutations in BRCA1 and BRCA2 cause homologous recombination deficiency (HRD), making cells rely much more heavily on the NHEJ pathway to repair DSB. Although germline and somatic mutations in the BRCA genes account for approximately 15–20% of all HGSOCs, dysfunction in the BRCA network and homologous recombination appears to be more widespread, with approximately 50% of HGSOC harboring alterations in genes involved in homologous recombination [4,65▪▪,71–73]. For instance, the promoter of BRCA1 can be highly methylated, resulting in loss of gene expression and mimicking the BRCA1 mutant phenotype [4]. In addition to the BRCA genes, there are several inherited DNA repair genes that likely contribute to HRD when mutated. These include genes in the Fanconi anemia complex, the RAD51 paralogs (RAD51B, RAD51C, and RAD51D), BRIP1, BARD1, PALB2, as well as RAD50, CHEK2, ATR, and ATM[74–76]. These alterations collectively display HRD and are often described as having a ‘BRCAness’ phenotype [77,78] because of the genomic instability associated with BRCA dysfunction [65▪▪,79–81].
Traditionally, ovarian cancers have been treated with cytotoxic agents, typically platinum-based chemotherapy, regardless of histological subtype. In fact, there are only two FDA approved targeted agents for use in ovarian cancer. The first is bevacizumab, a humanized monoclonal antibody against vascular endothelial growth factor (VEGF). This antiangiogenic therapy was approved for use in recurrent, platinum-resistant ovarian cancer [82–84]. The second is olaparib, a poly-ribose polymerase (PARP) inhibitor. Olaparib was approved in 2014 for use in patients with BRCA mutations and recurrent disease [85,86]. The success of PARP inhibition is grounded in the idea that loss of PARP1 function in the setting of HRD (i.e., BRCA1/2 mutation) causes an increase in DNA aberrations, not all of which could be repaired due to HRD, resulting in cell death via synthetic lethality [86–89]. Synthetic lethality occurs when there is an inactivation of two genes or pathways, neither of which produces lethal effects on its own, but when combined cause cell death. There are a number of mechanisms that may underlie PARP–BRCA synthetic lethality. First, PARP-1 is involved in the repair of single strand breaks (SSBs), which, in the presence of a PARP inhibitor, may persist and cause collapse of replication forks leading to DSBs. Because BRCA defective cancer cells lack homologous recombination, the resulting DSBs would be selectively toxic to the cancer cells. Another mechanism involves PARP trapping. PARP inhibitors trap PARP-1 onto SSBs that form spontaneously or during base excision repair. Trapped PARP-1 can pose an obstacle to replication that would require homologous recombination to resolve [90]. Interestingly, despite the selective activity of PARP inhibitors in BRCA mutant tumors, more patients responded to PARP inhibitor therapy than those individuals with confirmed BRCA mutations [91]. In fact, a recently published phase III clinical trial using the PARP inhibitor niraparib as maintenance therapy for patients with platinum-sensitive, recurrent ovarian cancer, demonstrated significantly prolonged progression free survival of patients regardless of their BRCA or HRD status [92▪▪]. These observations suggest that PARP inhibitors may have a broader role in ovarian cancer therapy. To date in the United States, only olaparib is FDA approved, although rucaparib was recently given breakthrough status by the FDA and others are expected to follow in the near future [93,94]. Currently, several clinical trials are progressing using different PARP inhibitors alone, or in combination with other drugs [95,96]. Studies like these show that PARP inhibitors have the potential to change the course of therapy for many individuals with ovarian cancer.
CCNE1: a unique opportunity in high-grade serous ovarian carcinoma
HGSOC is characterized by obligatory mutation of the TP53 gene, mutations in the homologous recombination DNA repair pathway, and widespread copy number alterations [4]. One of the most common copy number alterations in ovarian cancer is the amplification of the 19q12 locus. The Bowtell laboratory used a systematic knockdown of genes within the 19q12 amplicon to map CCNE1 as a key driver of the 19q12 amplicon [97]. CCNE1 encodes Cyclin E1, and it is amplified in a number of solid tumors (Fig. 2) and in approximately 20% of HGSOC cases (Fig. 1) [4,65▪▪]. Cyclin E1 protein levels vary during the cell cycle and play a major role in the G1-S phase transition by binding and activating the cyclin-dependent kinase 2 (CDK2) [98]. Aberrant Cyclin E1 expression is known to trigger unscheduled DNA replication, centrosome amplification, and chromosomal instability [99,100,101▪,102]. Importantly, CCNE1 amplification is associated with primary or refractory chemoresistant ovarian cancer [103] and poor overall survival [104,105]. Interestingly, amplification of CCNE1 and increased Cyclin E1 protein can be detected in STIC lesions, indicating that dysregulation of CCNE1 is an early event in the development of HGSOC [100,101▪,106].
FIGURE 2.
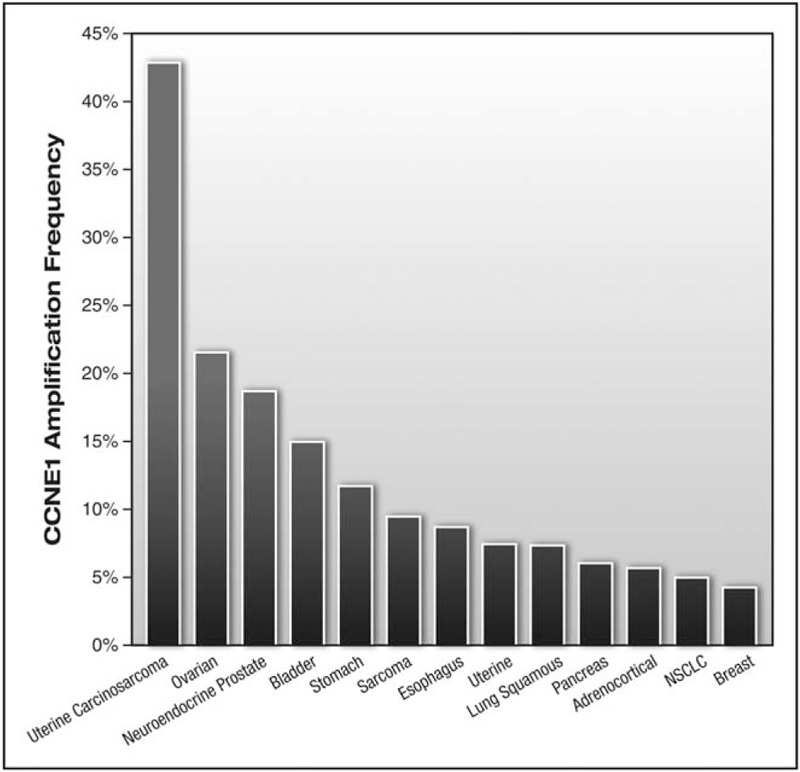
Amplification of CCNE1 across human cancers. The cbioportal (http://www.cbioportal.org) was queried for ‘CCNE1: AMP’ and the resulting bar graph was limited to tumors with at least 4% amplification.
Protein abundance of Cyclin E1 is controlled at several levels, including by ubiquitin-mediated proteolysis by E3 ligases FBXW7 and PARK2, both of which are frequently deleted in human tumors [107], and by PP2A-B55β, a phosphatase that also controls Cyclin E1 turnover [108]. Proteolytic cleavage of Cyclin E1 to low-molecular weight (LMW) isoforms by the elastase family of serine proteases enhances transformation [109,110] and increased expression of LMW isoforms is associated with poor outcome in breast cancer [111]. We recently showed that induced expression of CCNE1 in fallopian tube secretory epithelial cells harboring a TP53 missense mutation leads to increase proliferation, colony formation, loss of contact inhibition, centrosome amplification, and modest anchorage independent growth [100,101▪]. As expected, we detected increased DNA damage in these cells as measured by phosphorylation of histone H2AX and increased comet tails [100]. Expression analysis of these CCNE1-overexpressing cells revealed that they upregulate key factors involved in homologous recombination and replication fork protection. Most notable was the upregulation of BRCA1, FANCD2, CDC25C, BLM, and XRCC2 (a RAD51 paralog). Amazingly, a synthetic lethal screen identified many of the same proteins as essential in CCNE1-amplified HGSOC cell lines [112]. These findings strongly suggest that the chromosomal instability generated by defects in the homologous recombination pathway and amplification of CCNE1 cannot coexist within the same cell and at least one of these pathways must be functional for survival of the cell. It also suggests that inhibition of DNA repair and replication fork protection pathways may be a viable therapeutics strategy in CCNE1-amplified tumors.
Although CCNE1-amplified tumors represent a subset of HGSOC that deserve clinical attention, there are currently no targeted therapies for these tumors. The most obvious target is CDK2, the kinase partner of Cyclin E [113,114,115▪▪]. In fact, the development of small molecule inhibitors of CDKs has been an intense area of research [116] given their central role as regulators of cell division. Unfortunately, most compounds are not CDK2-specific and target multiple CDKs, eliciting dose-limiting toxicities that have slowed further clinical development [112,115▪▪,117]. However, the recent impressive findings with the CDK4/6 inhibitor palbociclib, targeted to Cyclin D1 and estrogen receptor-positive breast cancer [118], have renewed interest in the field [119–123]. In particular, a recent high-throughput compound screen in CCNE1-amplified ovarian cancer cell lines was performed to identify selective synergistic drug combinations with dinaciclib, a CDK1/2 inhibitor in clinical development. A synergistic therapeutic effect was elicited when dinaciclib was combined with an AKT2 inhibitor [115▪▪]. AKT2 and CCNE1 both reside on chromosome 19 and analysis of genomic data from TCGA demonstrated coamplification of CCNE1 and AKT2 in HGSOC. This finding suggests a specific dependency of CCNE1-amplified tumors for AKT activity, and points to a novel combination of dinaciclib and AKT inhibitors that may selectively target patients with CCNE1-amplified HGSOC, and possibly other solid tumors.
CONCLUSION
There is now significant clinical and experimental evidence pointing to the fallopian tube as the site of origin for a majority of HGSOCs. Next generation sequencing efforts have provided us with a panoramic view of HGSOCs and have revealed significant genomic heterogeneity. Alterations in the BRCA and CCNE1 pathways represent two distinct genotypes that exhibit unique vulnerabilities in DNA repair. The emergence of PARP inhibitors will change the clinical management of patients with BRCA mutant tumors as well as patients with tumors that are not HRD. Therapeutic approaches for CCNE1-amplified tumors are evolving and will likely exploit their dependency on homologous recombination and replication fork protection pathways.
Acknowledgements
We would like to thank Michael Cooper (www.cooper247.com) for assistance with graphic illustration.
Financial support and sponsorship
This work was supported by grants from the National Cancer Institute at the NIH P50-CA083636, NIH U01-CA152990, the CDMRP-OCRP (W81XWH-15-1-0160), the Dr Miriam and Sheldon G. Adelson Medical Research Foundation, and the Honorable Tina Brozman Foundation.
Conflicts of interest
R.D. serves on the Scientific Advisory Board of Siamab Therapeutics, Inc.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015; 136:E359–386. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin 2016; 66:7–30. [DOI] [PubMed] [Google Scholar]
- 3.Atlanta, American Cancer Society Cancer treatment & survivorship facts & figures 2016–2017. 2016. [Google Scholar]
- 4.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fathalla MF. Incessant ovulation – a factor in ovarian neoplasia? Lancet 1971; 2:163. [DOI] [PubMed] [Google Scholar]
- 6.Fathalla MF. Incessant ovulation and ovarian cancer – a hypothesis re-visited. Facts Views Vis Obgyn 2013; 5:292–297. [PMC free article] [PubMed] [Google Scholar]
- 7▪.Walker JL, Powell CB, Chen LM, et al. Society of Gynecologic Oncology recommendations for the prevention of ovarian cancer. Cancer 2015; 121:2108–2120. [DOI] [PubMed] [Google Scholar]; Update from Society of Gynecologic Oncology emphasizes the role of the fallopian tube in ovarian cancer etiology. Prevention strategies, including opportunitistic salpingectomy, are discussed.
- 8.Piek JM, Kenemans P, Verheijen RH. Intraperitoneal serous adenocarcinoma: a critical appraisal of three hypotheses on its cause. Am J Obstet Gynecol 2004; 191:718–732. [DOI] [PubMed] [Google Scholar]
- 9.Piek JM, van Diest PJ, Zweemer RP, et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J Pathol 2001; 195:451–456. [DOI] [PubMed] [Google Scholar]
- 10.Zweemer RP, van Diest PJ, Verheijen RH, et al. Molecular evidence linking primary cancer of the fallopian tube to BRCA1 germline mutations. Gynecol Oncol 2000; 76:45–50. [DOI] [PubMed] [Google Scholar]
- 11.Colgan TJ, Murphy J, Cole DE, et al. Occult carcinoma in prophylactic oophorectomy specimens: prevalence and association with BRCA germline mutation status. Am J Surg Pathol 2001; 25:1283–1289. [DOI] [PubMed] [Google Scholar]
- 12.Leeper K, Garcia R, Swisher E, et al. Pathologic findings in prophylactic oophorectomy specimens in high-risk women. Gynecol Oncol 2002; 87:52–56. [DOI] [PubMed] [Google Scholar]
- 13.Powell CB, Kenley E, Chen LM, et al. Risk-reducing salpingo-oophorectomy in BRCA mutation carriers: role of serial sectioning in the detection of occult malignancy. J Clin Oncol 2005; 23:127–132. [DOI] [PubMed] [Google Scholar]
- 14.Finch A, Shaw P, Rosen B, et al. Clinical and pathologic findings of prophylactic salpingo-oophorectomies in 159 BRCA1 and BRCA2 carriers. Gynecol Oncol 2006; 100:58–64. [DOI] [PubMed] [Google Scholar]
- 15.Carcangiu ML, Radice P, Manoukian S, et al. Atypical epithelial proliferation in fallopian tubes in prophylactic salpingo-oophorectomy specimens from BRCA1 and BRCA2 germline mutation carriers. Int J Gynecol Pathol 2004; 23:35–40. [DOI] [PubMed] [Google Scholar]
- 16.Shaw PA, Rouzbahman M, Pizer ES, et al. Candidate serous cancer precursors in fallopian tube epithelium of BRCA1/2 mutation carriers. Mod Pathol 2009; 22:1133–1138. [DOI] [PubMed] [Google Scholar]
- 17.Lee Y, Miron A, Drapkin R, et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J Pathol 2007; 211:26–35. [DOI] [PubMed] [Google Scholar]
- 18.Medeiros F, Muto MG, Lee Y, et al. The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am J Surg Pathol 2006; 30:230–236. [DOI] [PubMed] [Google Scholar]
- 19.Folkins AK, Jarboe EA, Saleemuddin A, et al. A candidate precursor to pelvic serous cancer (p53 signature) and its prevalence in ovaries and fallopian tubes from women with BRCA mutations. Gynecol Oncol 2008; 109:168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kindelberger DW, Lee Y, Miron A, et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: evidence for a causal relationship. Am J Surg Pathol 2007; 31:161–169. [DOI] [PubMed] [Google Scholar]
- 21.Kuhn E, Kurman RJ, Vang R, et al. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma – evidence supporting the clonal relationship of the two lesions. J Pathol 2012; 226:421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karst AM, Drapkin R. Ovarian cancer pathogenesis: a model in evolution. J Oncol 2010; 2010:932371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carcangiu ML, Peissel B, Pasini B, et al. Incidental carcinomas in prophylactic specimens in BRCA1 and BRCA2 germ-line mutation carriers, with emphasis on fallopian tube lesions: report of 6 cases and review of the literature. Am J Surg Pathol 2006; 30:1222–1230. [DOI] [PubMed] [Google Scholar]
- 24.Callahan MJ, Crum CP, Medeiros F, et al. Primary fallopian tube malignancies in BRCA-positive women undergoing surgery for ovarian cancer risk reduction. J Clin Oncol 2007; 25:3985–3990. [DOI] [PubMed] [Google Scholar]
- 25.Carlson JW, Miron A, Jarboe EA, et al. Serous tubal intraepithelial carcinoma: its potential role in primary peritoneal serous carcinoma and serous cancer prevention. J Clin Oncol 2008; 26:4160–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirst JE, Gard GB, McIllroy K, et al. High rates of occult fallopian tube cancer diagnosed at prophylactic bilateral salpingo-oophorectomy. Int J Gynecol Cancer 2009; 19:826–829. [DOI] [PubMed] [Google Scholar]
- 27.Jarboe EA, Miron A, Carlson JW, et al. Coexisting intraepithelial serous carcinomas of the endometrium and fallopian tube: frequency and potential significance. Int J Gynecol Pathol 2009; 28:308–315. [DOI] [PubMed] [Google Scholar]
- 28.Roh MH, Kindelberger D, Crum CP. Serous tubal intraepithelial carcinoma and the dominant ovarian mass: clues to serous tumor origin? Am J Surg Pathol 2009; 33:376–383. [DOI] [PubMed] [Google Scholar]
- 29.Maeda D, Ota S, Takazawa Y, et al. Mucosal carcinoma of the fallopian tube coexists with ovarian cancer of serous subtype only: a study of Japanese cases. Virchows Arch 2010; 457:597–608. [DOI] [PubMed] [Google Scholar]
- 30.Przybycin CG, Kurman RJ, Ronnett BM, et al. Are all pelvic (nonuterine) serous carcinomas of tubal origin? Am J Surg Pathol 2010; 34:1407–1416. [DOI] [PubMed] [Google Scholar]
- 31.Leonhardt K, Einenkel J, Sohr S, et al. p53 signature and serous tubal in-situ carcinoma in cases of primary tubal and peritoneal carcinomas and serous borderline tumors of the ovary. Int J Gynecol Pathol 2011; 30:417–424. [DOI] [PubMed] [Google Scholar]
- 32.Manchanda R, Abdelraheim A, Johnson M, et al. Outcome of risk-reducing salpingo-oophorectomy in BRCA carriers and women of unknown mutation status. BJOG 2011; 118:814–824. [DOI] [PubMed] [Google Scholar]
- 33.Diniz PM, Carvalho JP, Baracat EC, Carvalho FM. Fallopian tube origin of supposed ovarian high-grade serous carcinomas. Clinics (Sao Paulo) 2011; 66:73–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powell CB, Chen LM, McLennan J, et al. Risk-reducing salpingo-oophorectomy (RRSO) in BRCA mutation carriers: experience with a consecutive series of 111 patients using a standardized surgical-pathological protocol. Int J Gynecol Cancer 2011; 21:846–851. [DOI] [PubMed] [Google Scholar]
- 35.Seidman JD, Zhao P, Yemelyanova A. “Primary peritoneal” high-grade serous carcinoma is very likely metastatic from serous tubal intraepithelial carcinoma: assessing the new paradigm of ovarian and pelvic serous carcinogenesis and its implications for screening for ovarian cancer. Gynecol Oncol 2011; 120:470–473. [DOI] [PubMed] [Google Scholar]
- 36.Tang S, Onuma K, Deb P, et al. Frequency of serous tubal intraepithelial carcinoma in various gynecologic malignancies: a study of 300 consecutive cases. Int J Gynecol Pathol 2012; 31:103–110. [DOI] [PubMed] [Google Scholar]
- 37.Gao FF, Bhargava R, Yang H, et al. Clinicopathologic study of serous tubal intraepithelial carcinoma with invasive carcinoma: is serous tubal intraepithelial carcinoma a reliable feature for determining the organ of origin? Hum Pathol 2013; 44:1534–1543. [DOI] [PubMed] [Google Scholar]
- 38.Lee S, Nelson G, Duan Q, et al. Precursor lesions and prognostic factors in primary peritoneal serous carcinoma. Int J Gynecol Pathol 2013; 32:547–555. [DOI] [PubMed] [Google Scholar]
- 39.Reitsma W, de Bock GH, Oosterwijk JC, et al. Support of the ‘fallopian tube hypothesis’ in a prospective series of risk-reducing salpingo-oophorectomy specimens. Eur J Cancer 2013; 49:132–141. [DOI] [PubMed] [Google Scholar]
- 40.Conner JR, Meserve E, Pizer E, et al. Outcome of unexpected adnexal neoplasia discovered during risk reduction salpingo-oophorectomy in women with germ-line BRCA1 or BRCA2 mutations. Gynecol Oncol 2014; 132:280–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koc N, Ayas S, Uygur L. The association of serous tubal intraepithelial carcinoma with gynecologic pathologies and its role in pelvic serous cancer. Gynecol Oncol 2014; 134:486–491. [DOI] [PubMed] [Google Scholar]
- 42.Mingels MJ, van Ham MA, de Kievit IM, et al. Mullerian precursor lesions in serous ovarian cancer patients: using the SEE-Fim and SEE-End protocol. Mod Pathol 2014; 27:1002–1013. [DOI] [PubMed] [Google Scholar]
- 43.Sherman ME, Piedmonte M, Mai PL, et al. Pathologic findings at risk-reducing salpingo-oophorectomy: primary results from Gynecologic Oncology Group Trial GOG-0199. J Clin Oncol 2014; 32:3275–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilks CB, Irving J, Kobel M, et al. Incidental nonuterine high-grade serous carcinomas arise in the fallopian tube in most cases: further evidence for the tubal origin of high-grade serous carcinomas. Am J Surg Pathol 2015; 39:357–364. [DOI] [PubMed] [Google Scholar]
- 45.Munakata S, Yamamoto T. Incidence of serous tubal intraepithelial carcinoma (STIC) by algorithm classification in serous ovarian tumor associated with PAX8 expression in tubal epithelia: a study of single institution in Japan. Int J Gynecol Pathol 2015; 34:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seidman JD. Serous tubal intraepithelial carcinoma localizes to the tubal-peritoneal junction: a pivotal clue to the site of origin of extrauterine high-grade serous carcinoma (ovarian cancer). Int J Gynecol Pathol 2015; 34:112–120. [DOI] [PubMed] [Google Scholar]
- 47.Malmberg K, Klynning C, Floter-Radestad A, Carlson JW. Serous tubal intraepithelial carcinoma, chronic fallopian tube injury, and serous carcinoma development. Virchows Arch 2016; 468:707–713. [DOI] [PubMed] [Google Scholar]
- 48.Mittal N, Srinivasan R, Gupta N, et al. Secretory cell outgrowths, p53 signatures, and serous tubal intraepithelial carcinoma in the fallopian tubes of patients with sporadic pelvic serous carcinoma. Indian J Pathol Microbiol 2016; 59:481–488. [DOI] [PubMed] [Google Scholar]
- 49▪.Zakhour M, Danovitch Y, Lester J, et al. Occult and subsequent cancer incidence following risk-reducing surgery in BRCA mutation carriers. Gynecol Oncol 2016; 143:231–235. [DOI] [PubMed] [Google Scholar]; Documents the risk of developing pelvic serous carcinoma in women with benign adnexa versus STIC at time of risk-reducing surgery. Occult STIC lesions were associated with a higher incidence of pelvic serous carcinoma at follow-up.
- 50.Rabban JT, Garg K, Crawford B, et al. Early detection of high-grade tubal serous carcinoma in women at low risk for hereditary breast and ovarian cancer syndrome by systematic examination of fallopian tubes incidentally removed during benign surgery. Am J Surg Pathol 2014; 38:729–742. [DOI] [PubMed] [Google Scholar]
- 51.Mahe E, Tang S, Deb P, et al. Do deeper sections increase the frequency of detection of serous tubal intraepithelial carcinoma (STIC) in the “sectioning and extensively examining the FIMbriated end” (SEE-FIM) protocol? Int J Gynecol Pathol 2013; 32:353–357. [DOI] [PubMed] [Google Scholar]
- 52.Carlson JW, Jarboe EA, Kindelberger D, et al. Serous tubal intraepithelial carcinoma: diagnostic reproducibility and its implications. Int J Gynecol Pathol 2010; 29:310–314. [DOI] [PubMed] [Google Scholar]
- 53.Visvanathan K, Vang R, Shaw P, et al. Diagnosis of serous tubal intraepithelial carcinoma based on morphologic and immunohistochemical features: a reproducibility study. Am J Surg Pathol 2011; 35:1766–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vang R, Visvanathan K, Gross A, et al. Validation of an algorithm for the diagnosis of serous tubal intraepithelial carcinoma. Int J Gynecol Pathol 2012; 31:243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Novak M, Lester J, Karst AM, et al. Stathmin 1 and p16(INK4A) are sensitive adjunct biomarkers for serous tubal intraepithelial carcinoma. Gynecol Oncol 2015; 139:104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dubeau L, Drapkin R. Coming into focus: the nonovarian origins of ovarian cancer. Ann Oncol 2013; 24 Suppl 8:viii28–viii35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57▪.Sherman ME, Drapkin RI, Horowitz NS, et al. Rationale for developing a specimen bank to study the pathogenesis of high-grade serous carcinoma: a review of the evidence. Cancer Prev Res (Phila) 2016; 9:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]; Reviews our current understanding of ovarian cancer pathogenesis and makes an argument for developing an epidemiologically annotated national specimen resource to support studies on the fallopian tube.
- 58.Perets R, Wyant GA, Muto KW, et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 2013; 24:751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu R, Zhai Y, Kuick R, et al. Impact of oviductal versus ovarian epithelial cell of origin on ovarian endometrioid carcinoma phenotype in the mouse. J Pathol 2016; 240:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sherman-Baust CA, Kuhn E, Valle BL, et al. A genetically engineered ovarian cancer mouse model based on fallopian tube transformation mimics human high-grade serous carcinoma development. J Pathol 2014; 233:228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim J, Coffey DM, Creighton CJ, et al. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc Natl Acad Sci U S A 2012; 109:3921–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Y, Yen HY, Austria T, et al. A mouse model that reproduces the developmental pathways and site specificity of the cancers associated with the human BRCA1 mutation carrier state. EBioMedicine 2015; 2:1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.National Academies of Sciences EaM Ovarian cancers: evolving paradigms in research and care. Washington, DC: The National Academies Press; 2016. [PubMed] [Google Scholar]
- 64.Ahmed AA, Etemadmoghadam D, Temple J, et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol 2010; 221:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65▪▪.Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015; 521:489–494. [DOI] [PubMed] [Google Scholar]; This article presents whole-genome sequencing of tumor and germline DNA from 92 patients with refractory, resistant, and sensitive ovarian cancer. The study shows that gene breakage events are common in RB1, NF1, RAD51B, and PTEN, and that diverse molecular events are associated with acquired resistance.
- 66▪▪.Vang R, Levine DA, Soslow RA, et al. Molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinoma: a rereview of cases lacking TP53 mutations in The Cancer Genome Atlas Ovarian Study. Int J Gynecol Pathol 2016; 35:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]; The TCGA indicated that 96% of HGSOC harbor a TP53 mutation. A review of the 4% of cases without mutation showed that they were misclassified – hence, 100% of HGSOC have TP53 mutations.
- 67▪.Iwanicki MP, Chen HY, Iavarone C, et al. Mutant p53 regulates ovarian cancer transformed phenotypes through autocrine matrix deposition. JCI Insight 2016; 1:e86829. [DOI] [PMC free article] [PubMed] [Google Scholar]; Fallopian tube cells expressing a stabilizing p53 mutant acquire anchorage independence and mesothelial clearance ability that is not seen with loss of TP53.
- 68▪.Candeias MM, Hagiwara M, Matsuda M. Cancer-specific mutations in p53 induce the translation of Delta160p53 promoting tumorigenesis. EMBO Rep 2016; [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that the major mediators of mutant p53 function are the low-molecular weight isoforms encoded by the mutant allele.
- 69.Parrales A, Iwakuma T. Targeting oncogenic mutant p53 for cancer therapy. Front Oncol 2015; 5:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Freed-Pastor W, Prives C. Targeting mutant p53 through the mevalonate pathway. Nat Cell Biol 2016; 18:1122–1124. [DOI] [PubMed] [Google Scholar]
- 71.Mukhopadhyay A, Elattar A, Cerbinskaite A, et al. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin Cancer Res 2010; 16:2344–2351. [DOI] [PubMed] [Google Scholar]
- 72.Zhang S, Royer R, Li S, et al. Frequencies of BRCA1 and BRCA2 mutations among 1,342 unselected patients with invasive ovarian cancer. Gynecol Oncol 2011; 121:353–357. [DOI] [PubMed] [Google Scholar]
- 73.Alsop K, Fereday S, Meldrum C, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol 2012; 30:2654–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Walsh T, Casadei S, Lee MK, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A 2011; 108:18032–18037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Norquist BM, Harrell MI, Brady MF, et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol 2016; 2:482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Matulonis UA, Sood AK, Fallowfield L, et al. Ovarian cancer. Nat Rev Dis Primers 2016; 2:16061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer 2004; 4:814–819. [DOI] [PubMed] [Google Scholar]
- 78.Konstantinopoulos PA, Spentzos D, Karlan BY, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol 2010; 28:3555–3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Popova T, Manie E, Rieunier G, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res 2012; 72:5454–5462. [DOI] [PubMed] [Google Scholar]
- 80.Wang ZC, Birkbak NJ, Culhane AC, et al. Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clin Cancer Res 2012; 18:5806–5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abkevich V, Timms KM, Hennessy BT, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer 2012; 107:1776–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Burger RA, Brady MF, Bookman MA, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med 2011; 365:2473–2483. [DOI] [PubMed] [Google Scholar]
- 83.Perren TJ, Swart AM, Pfisterer J, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med 2011; 365:2484–2496. [DOI] [PubMed] [Google Scholar]
- 84.Aghajanian C, Blank SV, Goff BA, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol 2012; 30:2039–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015; 33:244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 2012; 366:1382–1392. [DOI] [PubMed] [Google Scholar]
- 87.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434:913–917. [DOI] [PubMed] [Google Scholar]
- 88.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917–921. [DOI] [PubMed] [Google Scholar]
- 89.Stover EH, Konstantinopoulos PA, Matulonis UA, Swisher EM. Biomarkers of response and resistance to DNA repair targeted therapies. Clin Cancer Res 2016; [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 90.Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 2011; 5:387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 2014; 15:852–861. [DOI] [PubMed] [Google Scholar]
- 92▪▪.Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med 2016; [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]; This is a landmark study of PARP1/2 inhibitor niraparib as maintenance therapy in platinum-sensitive, recurrent ovarian cancer. The study found that among those patients receiving niraparib, progression-free survival was significantly longer regardless of BRCA mutation or HRD status.
- 93.Miller RE, Ledermann JA. The status of poly(adenosine diphosphate-ribose) polymerase (PARP) inhibitors in ovarian cancer, part 1: olaparib. Clin Adv Hematol Oncol 2016; 14:619–627. [PubMed] [Google Scholar]
- 94.Ledermann JA. PARP inhibitors in ovarian cancer. Ann Oncol 2016; 27 Suppl 1:i40–i44. [DOI] [PubMed] [Google Scholar]
- 95.Konecny GE, Kristeleit RS. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: current practice and future directions. Br J Cancer 2016; 115:1157–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miller RE, Ledermann JA. The status of poly(adenosine diphosphate-ribose) polymerase (PARP) inhibitors in ovarian cancer, part 2: extending the scope beyond olaparib and BRCA1/2 mutations. Clin Adv Hematol Oncol 2016; 14:704–711. [PubMed] [Google Scholar]
- 97.Etemadmoghadam D, George J, Cowin PA, et al. Amplicon-dependent CCNE1 expression is critical for clonogenic survival after cisplatin treatment and is correlated with 20q11 gain in ovarian cancer. PLoS One 2010; 5:e15498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Koff A, Cross F, Fisher A, et al. Human cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell 1991; 66:1217–1228. [DOI] [PubMed] [Google Scholar]
- 99.Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature 1999; 401:297–300. [DOI] [PubMed] [Google Scholar]
- 100.Karst AM, Jones PM, Vena N, et al. Cyclin E1 deregulation occurs early in secretory cell transformation to promote formation of fallopian tube-derived high-grade serous ovarian cancers. Cancer Res 2014; 74:1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101▪.Kuhn E, Wang TL, Doberstein K, et al. CCNE1 amplification and centrosome number abnormality in serous tubal intraepithelial carcinoma: further evidence supporting its role as a precursor of ovarian high-grade serous carcinoma. Mod Pathol 2016; 29:1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study used FISH to show that CCNE1 gain/amplification occurs in early fallopian tube precursors (STICS) and precedes centrosome amplification.
- 102.Loeb KR, Kostner H, Firpo E, et al. A mouse model for cyclin E-dependent genetic instability and tumorigenesis. Cancer Cell 2005; 8:35–47. [DOI] [PubMed] [Google Scholar]
- 103.Etemadmoghadam D, deFazio A, Beroukhim R, et al. Integrated genome-wide DNA copy number and expression analysis identifies distinct mechanisms of primary chemoresistance in ovarian carcinomas. Clin Cancer Res 2009; 15:1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Farley J, Smith LM, Darcy KM, et al. Cyclin E expression is a significant predictor of survival in advanced, suboptimally debulked ovarian epithelial cancers: a Gynecologic Oncology Group study. Cancer Res 2003; 63:1235–1241. [PubMed] [Google Scholar]
- 105.Nakayama N, Nakayama K, Shamima Y, et al. Gene amplification CCNE1 is related to poor survival and potential therapeutic target in ovarian cancer. Cancer 2010; 116:2621–2634. [DOI] [PubMed] [Google Scholar]
- 106.Sehdev AS, Kurman RJ, Kuhn E, Shih Ie M. Serous tubal intraepithelial carcinoma upregulates markers associated with high-grade serous carcinomas including Rsf-1 (HBXAP), cyclin E and fatty acid synthase. Mod Pathol 2010; 23:844–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gong Y, Zack TI, Morris LG, et al. Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat Genet 2014; 46:588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tan Y, Sun D, Jiang W, et al. PP2A-B55β antagonizes cyclin E1 proteolysis and promotes its dysregulation in cancer. Cancer Res 2014; 74:2006–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bagheri-Yarmand R, Biernacka A, Hunt KK, Keyomarsi K. Low molecular weight cyclin E overexpression shortens mitosis, leading to chromosome missegregation and centrosome amplification. Cancer Res 2010; 70:5074–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Loeb KR, Chen X. Too much cleavage of cyclin E promotes breast tumorigenesis. PLoS Genet 2012; 8:e1002623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Duong MT, Akli S, Wei C, et al. LMW-E/CDK2 deregulates acinar morphogenesis, induces tumorigenesis, and associates with the activated b-Raf-ERK1/2-mTOR pathway in breast cancer patients. PLoS Genet 2012; 8:e1002538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Etemadmoghadam D, Weir BA, Au-Yeung G, et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc Natl Acad Sci U S A 2013; 110:19489–19494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Taylor-Harding B, Aspuria PJ, Agadjanian H, et al. Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget 2015; 6:696–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang L, Fang D, Chen H, et al. Cyclin-dependent kinase 2 is an ideal target for ovary tumors with elevated cyclin E1 expression. Oncotarget 2015; 6:20801–20812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115▪▪.Au-Yeung G, Lang F, Azar WJ, et al. Selective targeting of Cyclin E1 amplified high grade serous ovarian cancer by cyclin-dependent kinase 2 and AKT inhibition. Clin Cancer Res 2016; [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]; A high-throughput chemical screen was performed to identify selective synergistic drug combinations with dinaciclib, a CDK1/2 inhibitor. A synergistic therapeutic effect was elicited with an AKT inhibitor, suggesting that CCNE1 tumors depend on AKT activity and that the combination of CDK and AKT inhibitors may have selective activity in patients with CCNE1-amplified tumors.
- 116.Bruyere C, Meijer L. Targeting cyclin-dependent kinases in anti-neoplastic therapy. Curr Opin Cell Biol 2013; 25:772–779. [DOI] [PubMed] [Google Scholar]
- 117.Etemadmoghadam D, Au-Yeung G, Wall M, et al. Resistance to CDK2 inhibitors is associated with selection of polyploid cells in CCNE1-amplified ovarian cancer. Clin Cancer Res 2013; 19:5960–5971. [DOI] [PubMed] [Google Scholar]
- 118.Palbociclib ups PFS in HER2−/ER+ breast cancer. Cancer Discov 2014; 4:624–625. [DOI] [PubMed] [Google Scholar]
- 119.Konecny M, Milly M, Zavodna K, et al. Comprehensive genetic characterization of hereditary breast/ovarian cancer families from Slovakia. Breast Cancer Res Treat 2011; 126:119–130. [DOI] [PubMed] [Google Scholar]
- 120.Carlson BA, Dubay MM, Sausville EA, et al. Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res 1996; 56:2973–2978. [PubMed] [Google Scholar]
- 121.Bible KC, Peethambaram PP, Oberg AL, et al. A phase 2 trial of flavopiridol (Alvocidib) and cisplatin in platin-resistant ovarian and primary peritoneal carcinoma: MC0261. Gynecol Oncol 2012; 127:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fabre C, Gobbi M, Ezzili C, et al. Clinical study of the novel cyclin-dependent kinase inhibitor dinaciclib in combination with rituximab in relapsed/refractory chronic lymphocytic leukemia patients. Cancer Chemother Pharmacol 2014; 74:1057–1064. [DOI] [PubMed] [Google Scholar]
- 123.Tong WG, Chen R, Plunkett W, et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J Clin Oncol 2010; 28:3015–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]