

ABSTRACT
Coral tissue loss diseases, collectively known as white syndromes (WSs), induce significant mortality on reefs throughout the Indo-Pacific, yet definitive confirmation of WS etiologies remains elusive. In this study, we integrated ecological disease monitoring, bacterial community profiling, in situ visualization of microbe-host interactions, and cellular responses of the host coral through an 18-month repeated-sampling regime. We assert that the observed pathogenesis of WS lesions on acroporid corals at Lizard Island (Great Barrier Reef) is not the result of apoptosis or infection by Vibrio bacteria, ciliates, fungi, cyanobacteria, or helminths. Histological analyses detected helminths, ciliates, fungi, and cyanobacteria in fewer than 25% of WS samples, and helminths and fungi were also observed in 12% of visually healthy samples. The abundances of Vibrio-affiliated sequences (assessed using 16S rRNA amplicon sequencing) did not differ significantly between health states and never exceeded 3.3% of reads in any individual sample. In situ visualization detected Vibrio bacteria only in summer WS lesion samples and revealed no signs of these bacteria in winter disease samples (or any healthy tissue samples), despite continued disease progression year round. However, a 4-fold increase in Rhodobacteraceae-affiliated bacterial sequences at WS lesion fronts suggests that this group of bacteria could play a role in WS pathogenesis and/or serve as a diagnostic criterion for disease differentiation. While the causative agent(s) underlying WSs remains elusive, the microbial and cellular processes identified in this study will help to identify and differentiate visually similar but potentially distinct WS etiologies.
IMPORTANCE Over the past decade, a virulent group of coral diseases known as white syndromes have impacted coral reefs throughout the Indian and Pacific Oceans. This article provides a detailed case study of white syndromes to combine disease ecology, high-throughput microbial community profiling, and cellular-scale host-microbe visualization over seasonal time scales. We provide novel insights into the etiology of this devastating disease and reveal new diagnostic criteria that could be used to differentiate visually similar but etiologically distinct forms of white syndrome.
INTRODUCTION
Coral diseases have fundamentally altered Caribbean coral reef ecosystems and pose a growing threat to coral populations in the Indo-Pacific (1, 2). Coral tissue loss diseases, which are characterized by a spreading zone of tissue loss that exposes white coral skeleton directly adjacent to asymptomatic coral tissue, represent an important group of diseases worldwide because of their wide geographic distributions, diverse host ranges, and rapid and often irreversible damage (1, 3–6). The proliferation of names for macroscopically similar “white” diseases in the Caribbean has led to the adoption of the collective term white syndromes (WSs) in the Indo-Pacific until reliable diagnostic criteria are developed to differentiate potentially etiologically distinct diseases (2, 7). Given the relatively high susceptibility of often dominant and structurally important corals in the family Acroporidae, WSs have the potential to dramatically decrease coral cover, rugosity, and ecological complexity on impacted reefs (1, 2, 5, 6).
Despite Indo-Pacific-wide impacts (summarized in reference 8), definitive confirmation of WS etiologies remains elusive. WSs are likely comprised of multiple distinct diseases that share similar macroscopic disease signs (9–11), as has been shown for several “white” diseases in the Caribbean. Currently, many “white” diseases are distinguished solely by variable macroscopic characteristics of lesion shape and progression rate (12), but from a disease management perspective, it is critical to develop reliable criteria to differentiate distinct but visually similar tissue loss diseases (7–9). Standardized criteria for characterizing coral lesions at the gross, cellular, and microbial levels have proven to be instrumental in teasing apart disease causation in other animal systems, and standardized criteria for the description of coral diseases have been widely disseminated (13–15). However, the development of reliable and, importantly, comparable definitions of WS disease will not be possible unless these criteria are uniformly embraced (13, 16, 17). To identify and distinguish distinct WS disease etiologies, thorough case studies are needed that incorporate comprehensive characterization of both macroscopic and microscopic disease signs and offer feasible, testable hypotheses about how observed microscopic interactions give rise to macroscopic disease manifestations.
It is well established that infectious diseases are the outcome of interactions between susceptible hosts, disease agents, and the environments they live within (18). Despite this knowledge, few WS studies to date have investigated interactions between the coral host and potential bacterial and eukaryotic disease agents at the cellular level, and many focus on only a single time point, potentially missing important dynamics of disease etiology (10, 11, 19). In this study, we integrated bacterial community profiling, in situ visualization of microbe-host interactions, and cellular responses over seasonal time scales spanning 18 months. Coral colonies were monitored and repeatedly sampled to gain insights into temporal disease dynamics, with seasonal replication where possible, and ultimately provided a detailed case study of WS at Lizard Island in the northern sector of the Great Barrier Reef (GBR), Australia, that is easily comparable with other WS disease investigations worldwide.
RESULTS
Repeated sampling and disease progression on tagged coral colonies.
A total of 12 colonies exhibiting signs of WS were tagged and sampled throughout the study, although high rates of tissue loss often resulted in whole-colony mortality between sampling time points (Table 1). Five out of nine (55%) WS-affected colonies died after sampling at only a single time point, three colonies were sampled twice (33%), and one large diseased colony (colony D1 [surface area, 2.5 m2; maximum diameter, 2.45 m]) survived through 4 seasonal sampling time points (Table 1). Progression rates of WS lesions on tagged colonies ranged from 0.05 to 0.84 cm day−1, with an average (± standard error [SE]) of 0.34 ± 0.11 cm day−1 (Table 1). There were no clear seasonal patterns in lesion progression rates and no recorded instances of cessation of WS lesion progression, and all WS-affected colonies assessed at multiple time points eventually suffered whole-colony mortality.
TABLE 1.
Disease progression and fate of tagged coloniesa

Images show lesion progression on colony D1. White lines are shown just behind the lesion front, and the black scale bar denotes 50 cm. Abbreviations and symbols: H, colony visually healthy; WS, colony displaying active white syndrome disease signs; M, colony dead (mortality); *, sample not analyzed due to Drupella sp. infestation; ^ and #, bacterial community profiling data unavailable for healthy or HD samples, respectively, due to insufficient sequencing reads. Information bounded by parentheses indicates the average lesion progression rate since the previous sampling time point.
In addition, 11 healthy colonies were tagged and sampled at various time points throughout the 18-month study (Table 1). Four colonies (36%) died from unknown causes prior to the completion of the investigation (two died prior to the second sampling time point [summer 2011], and two died prior to the third [winter 2011]) (Table 1). These four colonies appeared visually healthy at the time of sampling. Only one colony (H1) remained visually healthy through all 5 seasonal sampling time points.
Bacterial community profiling of coral tissues and seawater.
Bacterial communities associated with coral tissues derived from healthy colonies (healthy), apparently healthy tissues on diseased colonies (HD), and WS lesion fronts (WS), as well as surface seawater samples, resulted in 179,283 classifiable, nonchimeric reads, with an average of 2,598 reads per sample. To account for unequal sequencing depth across samples, analyses were performed on a randomly selected subset of 400 sequences per sample. Operational taxonomic unit (OTU) richness (choa1) did not differ significantly among coral health states (PHSD > 0.05) (see Fig. S2 in the supplemental material). However, seawater samples (318 ± 25 [mean chao1 ± SE]) were significantly more OTU rich than healthy tissues on either healthy (181 ± 29) (PHSD = 0.003) or diseased (182 ± 25) (PHSD = 0.010) corals (df = 3, F = 4.4, P = 0.002) (Fig. S2). OTU richness did not differ significantly between seawater and WS samples (231 ± 23) (PHSD > 0.05). Microbial communities associated with seawater samples were significantly different from coral-associated communities regardless of coral health state (all Pperm and PMC < 0.001) (Fig. 1 and 2; Tables 2 and 3). The separation between coral- and seawater-associated bacterial communities can be visualized in the principal-coordinate analysis (PCoA) ordination as a partitioning of tightly clustered seawater samples away from more dispersed coral-derived samples along the second PCoA axis (Fig. 1).
FIG 1.
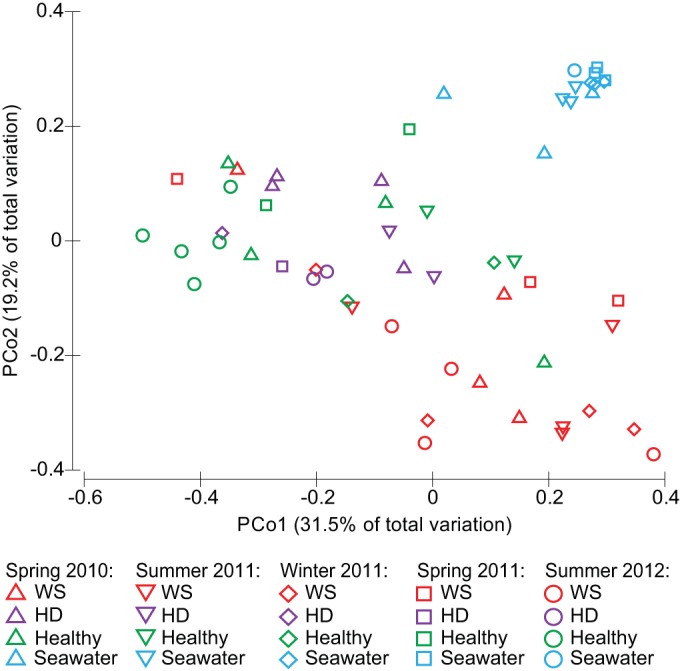
Principal-coordinate analysis (PCoA) plots based on weighted UniFrac distances of A. hyacinthus- and seawater associated bacterial communities across four sample types, i.e., WS-infected corals (WS) (red symbols), healthy tissues on WS-infected corals (HD) (purple symbols), healthy tissues on healthy corals (healthy) (green symbols), and surface seawater (seawater) (blue symbols), and five seasonal sampling time points, i.e., spring 2010 (upward-pointing triangles), summer 2011 (downward-pointing triangles), winter 2011 (diamonds), spring 2011 (squares), and summer 2012 (circles). Proximity of samples on the PCoA plot illustrates similarity of bacterial communities.
FIG 2.

Average abundances of the six most dominant bacterial classes identified through high-throughput sequencing of A. hyacinthus-associated and seawater-associated bacterial communities from four sample types: WS disease lesion fronts (WS), apparently healthy tissues on WS diseased corals (HD), healthy tissues on healthy corals (healthy), and surface seawater (seawater). Inset slices indicate the abundance of Rhodobacteraceae within a given sample type. Data from multiple seasonal sampling time points (spring 2010, summer 2011, winter 2011, spring 2011, and summer 2012) have been pooled for each sample type.
TABLE 2.
Summary statistics for permutational multivariate analysis of variance using weighted UniFrac distances of A. hyacinthus- and seawater-associated bacterial communitiese
| Difference compared and source | df | SS | MS | Pseudo-F | P value (permutation)a | Unique permutations | P value (Monte Carlo)a |
|---|---|---|---|---|---|---|---|
| Coral health stateb and seasonc | |||||||
| Health state | 2 | 1.1855 | 0.59276 | 3.5561 | 0.0006 | 9,935 | 0.0014 |
| Season | 4 | 1.2005 | 0.30013 | 1.8005 | 0.0103 | 9,885 | 0.0133 |
| Interaction (health state × season) | 8 | 1.2094 | 0.15117 | 0.9069 | 0.6678 | 9,858 | 0.656 |
| Residuals | 29 | 4.8340 | 0.16669 | ||||
| Total | 43 | 8.7951 | |||||
| Sampling time pointc for WS | |||||||
| Sampling time point | 4 | 0.6200 | 0.1550 | 0.8347 | 0.6821 | 9,898 | 0.6543 |
| Residuals | 14 | 2.5998 | 0.1857 | ||||
| Total | 18 | 3.2198 | |||||
| Seasonc for healthy and HD | |||||||
| Season | 4 | 1.1707 | 0.2927 | 1.8715 | 0.0036 | 9,857 | 0.0114 |
| Residuals | 20 | 3.1278 | 0.1564 | ||||
| Total | 24 | 4.2985 | |||||
| Sample typesd | |||||||
| Sample type (seawater vs coral) | 1 | 2.2334 | 2.2334 | 13.3010 | 0.0001 | 9,934 | 0.0001 |
| Residuals | 55 | 9.2351 | 0.1679 | ||||
| Total | 56 | 11.4690 |
Significant effects are shown in bold.
Healthy (healthy region on healthy colony), HD (healthy region on diseased colony), and WS (disease lesion on diseased colony).
Spring 2010, summer 2011, winter 2011, spring 2011, and summer 2012.
Seawater versus coral (healthy, HD, and WS).
SS, sum of squares; MS, mean square.
TABLE 3.
Pairwise comparisons for permutational multivariate analysis of variance using weighted UniFrac distances of A. hyacinthus- and seawater-associated bacterial communities
| Pairwise comparison and groupa | t statistic | P value (permutation)b | Unique permutations | P value (Monte Carlo)b |
|---|---|---|---|---|
| Differences between coral health states (healthy, HD, and WS) | ||||
| WS vs HD | 2.2247 | 0.0008 | 9,927 | 0.0016 |
| WS vs healthy | 2.3384 | 0.0004 | 9,930 | 0.0011 |
| Healthy vs HD | 0.94187 | 0.5054 | 9,910 | 0.4990 |
| Differences between sample types (seawater, healthy, HD, and WS) | ||||
| Seawater vs WS | 3.9129 | 0.0001 | 9,945 | 0.0001 |
| Seawater vs HD | 4.1780 | 0.0001 | 9,890 | 0.0001 |
| Seawater vs healthy | 3.8686 | 0.0001 | 9,933 | 0.0001 |
Healthy, healthy region on healthy colony; HD, healthy region on diseased colony; WS, disease lesion on diseased colony.
Significant effects are shown in bold.
Coral-associated bacterial communities fell into consistent phylogenetic clusters according to health state (df = 2, pseudo-F = 3.55, Pperm < 0.001, PMC = 0.001) and season (df = 4, pseudo-F = 1.80, Pperm = 0.010, PMC = 0.013). No significant differences were detected between healthy tissues sampled from healthy colonies and healthy tissues sampled from diseased corals away from the WS lesion (i.e., healthy and HD) (t statistic = 0.94, Pperm > 0.05, PMC > 0.05), although these bacterial communities did vary significantly among seasons (df = 4, pseudo-F = 1.87, Pperm = 0.004, PMC = 0.011) (Fig. 1 and 2; Tables 2 and 3). However, bacterial communities at WS lesion fronts were statistically distinct from those associated with healthy tissues of both diseased (t statistic = 2.25, Pperm < 0.001, PMC = 0.002) and healthy (t statistic = 2.34, Pperm < 0.001, PMC = 0.011) colonies throughout the study. These WS disease-associated bacterial communities did not vary significantly among seasonal sampling time points (df = 4, pseudo-F = 0.84, Pperm > 0.05, PMC > 0.05) (Fig. 1 and 2; Tables 2 and 3). Separation of bacterial communities among health states can be visualized as a loose clustering of lesion front samples toward the positive end of the first PCoA axis, transitioning toward a loose clustering of HD and healthy samples toward the center and negative end of this axis (Fig. 1).
Bacterial taxa belonging to the Rhodobacteraceae family (class Alphaproteobacteria) were significantly more abundant in WS lesion samples (36% ± 5.9% [mean ± SE]) than in healthy tissues derived from either diseased (4% ± 1.5%) (PHSD < 0.001) or healthy (10% ± 5.1%) (PHSD < 0.001) colonies (df = 2, F = 14.9, P < 0.001) (Fig. 2 and Table 4). These differences were not caused by a single OTU but were driven by combined changes among the 531 Rhodobacteraceae OTUs detected (Table 4; see Fig. S1 in the supplemental material). Among these OTUs, 15 contributed greater than 0.3% to the observed separation between bacterial communities associated with WS and healthy tissues (Fig. S1; Table 4). Twelve of these OTUs were more abundant in WS samples, while only three were more abundant in healthy samples (Table 4). The three OTUs that were more abundant in healthy samples all shared a lowest GreenGenes taxonomic assignment to the Rhodobacteraceae genus Paracoccus (Table 4), which was further supported by their phylogenetic position alongside previously isolated Paracoccus strains in the phylogenetic tree (Fig. S1a). Rhodobacteraceae OTUs that were more abundant in WS samples (i.e., those driving the 4-fold increase in Rhodobacteraceae abundance in WS samples) were fairly evenly distributed across the remainder of the Rhodobacteraceae phylogeny (Fig. S1).
TABLE 4.
SIMPER analysis identifying OTUs contributing >0.3% OTU-level dissimilarity between coral health statesa
| OTU | Phylum | Class | Order | Family | Genus | Mean abundance (%) |
Contribution to dissimilarity (%) |
||
|---|---|---|---|---|---|---|---|---|---|
| WS | Healthyb | Individual | Cumulative | ||||||
| 4455652 | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | 1.61 | 7.9 | 4.21 | 4.2 | |
| 3991527 | Firmicutes | Bacilli | Bacillales | Alicyclobacillaceae | Alicyclobacillus | 3.45 | 3.9 | 3.38 | 7.6 |
| NR_42 | Proteobacteria | Gammaproteobacteria | Oceanospirillales | Endozoicimonaceae | 2.29 | 2.2 | 2.18 | 9.8 | |
| NR_46 | Proteobacteria | Alphaproteobacteria | Rhizobiales | Hyphomicrobiaceae | 3.37 | 0.0 | 1.74 | 11.5 | |
| 875245 | Actinobacteria | Actinobacteria | Actinomycetales | Corynebacteriaceae | Corynebacterium | 0.09 | 3.1 | 1.58 | 13.1 |
| NR_23 | Proteobacteria | Gammaproteobacteria | Alteromonadales | 0 | 3.0 | 1.57 | 14.7 | ||
| 337783 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | 2.7 | 0.0 | 1.39 | 16.1 | |
| NR_38 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Paracoccus | 0 | 2.3 | 1.2 | 17.3 |
| 221108 | Proteobacteria | Gammaproteobacteria | Oceanospirillales | Endozoicimonaceae | 1.39 | 0.7 | 0.99 | 18.3 | |
| 355163 | Proteobacteria | Deltaproteobacteria | Spirobacillales | 1.79 | 0.0 | 0.92 | 19.2 | ||
| 4441357 | Proteobacteria | Gammaproteobacteria | Xanthomonadales | Xanthomonadaceae | 0.45 | 1.4 | 0.85 | 20.0 | |
| NCR_7 | Proteobacteria | Gammaproteobacteria | Oceanospirillales | Endozoicimonaceae | 0.67 | 1.0 | 0.82 | 20.8 | |
| 19540 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | 1.49 | 0.0 | 0.77 | 21.6 | |
| 364960 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | 1.33 | 0.0 | 0.68 | 22.3 | |
| NCR_1 | Proteobacteria | Gammaproteobacteria | Xanthomonadales | Xanthomonadaceae | 1.32 | 0.0 | 0.68 | 23.0 | |
| 219732 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | 1.25 | 0.0 | 0.64 | 23.6 | |
| 4381005 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | 1.11 | 0.1 | 0.58 | 24.2 | |
| NR_30 | Proteobacteria | Gammaproteobacteria | Oceanospirillales | Endozoicimonaceae | 0.36 | 0.9 | 0.57 | 24.8 | |
| 4301240 | Proteobacteria | Alphaproteobacteria | Sphingomonadales | Erythrobacteraceae | 0.29 | 0.9 | 0.57 | 25.3 | |
| 1106442 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | 0.96 | 0.0 | 0.49 | 25.8 | |
| 3016239 | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Serratia | 0.32 | 0.9 | 0.49 | 26.3 |
| 1000986 | Actinobacteria | Actinobacteria | Actinomycetales | Corynebacteriaceae | Corynebacterium | 0.03 | 0.9 | 0.48 | 26.8 |
| NR_37 | Proteobacteria | Gammaproteobacteria | Oceanospirillales | Endozoicimonaceae | 0.46 | 0.5 | 0.48 | 27.3 | |
| 937813 | Firmicutes | Clostridia | Clostridiales | [Tissierellaceae] | Anaerococcus | 0.04 | 0.9 | 0.47 | 27.7 |
| 904675 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | 0.86 | 0.0 | 0.44 | 28.2 | |
| 327709 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | 0.82 | 0.0 | 0.42 | 28.6 | |
| NR_96 | Proteobacteria | Gammaproteobacteria | Legionellales | Francisellaceae | Francisella | 0.82 | 0.0 | 0.42 | 29.0 |
| NCR_12 | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | 0 | 0.8 | 0.41 | 29.4 | |
| 4368477 | Cyanobacteria | Synechococcophycideae | Synechococcales | Synechococcaceae | Prochlorococcus | 0.11 | 0.7 | 0.39 | 29.8 |
| 4421429 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | 0.72 | 0.0 | 0.38 | 30.2 | |
| 4305517 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | 0.71 | 0.0 | 0.37 | 30.6 | |
| 2575651 | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | 0 | 0.7 | 0.36 | 30.9 | |
| 4473129 | Proteobacteria | Gammaproteobacteria | Pasteurellales | Pasteurellaceae | Haemophilus | 0.17 | 0.6 | 0.36 | 31.3 |
| 3244896 | Firmicutes | Bacilli | Lactobacillales | Streptococcaceae | Streptococcus | 0.51 | 0.2 | 0.36 | 31.6 |
| 4402516 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | 0.68 | 0.0 | 0.35 | 32.0 | |
| NR_57 | Cyanobacteria | Synechococcophycideae | Pseudanabaenales | Pseudanabaenaceae | 0.68 | 0.0 | 0.35 | 32.3 | |
| 1046541 | Actinobacteria | Actinobacteria | Actinomycetales | Corynebacteriaceae | Corynebacterium | 0.01 | 0.7 | 0.35 | 32.7 |
| 647864 | Proteobacteria | Alphaproteobacteria | Sphingomonadales | 0.05 | 0.6 | 0.35 | 33.0 | ||
| 166985 | Proteobacteria | Gammaproteobacteria | Oceanospirillales | Endozoicimonaceae | 0.5 | 0.2 | 0.34 | 33.4 | |
| 4352594 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | 0.64 | 0.0 | 0.33 | 33.7 | |
| 4395697 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Paracoccus | 0.01 | 0.6 | 0.31 | 34.0 |
| 826430 | Cyanobacteria | Synechococcophycideae | Synechococcales | Synechococcaceae | Synechococcus | 0.24 | 0.5 | 0.31 | 34.3 |
| NR_1 | Proteobacteria | Alphaproteobacteria | Rhodospirillales | Rhodospirillaceae | Skermanella | 0 | 0.6 | 0.31 | 34.7 |
| 1909414 | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Paracoccus | 0.14 | 0.5 | 0.31 | 35.0 |
Rhodobacteraceae OTUs are shown in bold. Abbreviations: SIMPER, similarity percentages; WS, disease lesion on diseased colony; NR, New.ReferenceOTU; NCR, New.CleanUp.ReferenceOTU.
Includes both HD (healthy region on diseased colony) and healthy (healthy region on healthy colony) states.
The Vibrionaceae abundance did not differ significantly among sample types (WS, 0.18% ± 0.17%; HD, 0.13% ± 0.13%; healthy, 0.0% ± 0.0%) (χ2Kruskal-Wallis = 1.63, df = 2, P > 0.05). When present, the relative abundance of Vibrionaceae-affiliated sequences was always low, never accounting for greater than 3.3% of reads in any one sample.
In situ visualization of host-microbe interactions and cellular responses to WS.
Histological analysis revealed no signs of seasonality in regard to cellular responses of host tissues or associated microscopic organisms in sections of either healthy or diseased coral tissues. Therefore, data from all time points per health category were pooled. Histological examination of healthy tissues (derived from both healthy and diseased colonies) did not show any evidence of necrosis, fragmentation, or swelling (the exception being minor tissue swelling in one healthy colony sample) throughout five seasonal sampling points (Table 5; Fig. 3). Helminths and fungi were observed in a small number of healthy tissue samples collected from healthy and WS-affected colonies (12% and 6%, respectively), while ciliates and cyanobacteria were absent from all healthy tissues (Table 5; Fig. 3).
TABLE 5.
Summary of FISH and histology results showing the percentage of samples of each tissue type harboring specific organisms and displaying host tissue responsesa
| Sample type | Tissue response, n ≥ 17 health state−1 (% of samples) |
Other organisms, n ≥ 17 health state−1 (% of samples) |
FISH, n = 3 health state−1 time point−1 (% of samples) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Necrosis | Fragmentation | Tissue swelling | Helminth | Ciliates | Fungi | Cyanobacteria | All bacteria |
Vibrio bacteria |
|||
| Summer 2011 | Winter 2011 | Summer 2011 | Winter 2011 | ||||||||
| Healthy | 0 | 0 | 6 | 12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| HDb | 0 | 0 | 0 | 6 | 0 | 6 | 0 | 0 | 0 | 0 | 0 |
| Diseasec | |||||||||||
| Healthy region | 0 | 0 | 0 | 6 | 0 | 11 | 0 | 0 | 0 | 0 | 0 |
| Lesion front | 100 | 78 | 0 | 6 | 22 | 22 | 17 | 100 | 100 | 66 | 0 |
| Disease region | 100 | 50 | 0 | 50 | 40 | 20 | 30 | 100 | 66 | 100 | 0 |
Seasonal (summer 2011 and winter 2011) patterns are shown for FISH, while histological data are compiled over 5 sampling time points (spring 2010, summer 2011, winter 2011, spring 2011, and summer 2012).
Healthy region on diseased colony greater than 5 cm from the WS disease lesion front.
Within 1 cm of the WS disease lesion front.
FIG 3.

Images of white syndrome (WS) lesions, comparing the appearances of healthy, lesion front, and diseased regions as viewed in the field (a), histologically when stained with hematoxylin and eosin (b and i), histologically when stained with Gomori trichrome (j and k), and with fluorescence in situ hybridization (FISH) staining for either total bacteria (c to e), or Vibrio sp. bacteria (f to h). FISH staining demonstrates bacterium-free healthy region (close to lesion front) tissues (c and f) and extensive bacterial infiltration into WS-affected tissues (d, e, g, and h) (red, bacteria; blue, coral tissue; green, endosymbiotic dinoflagellates). Histology shows normal tissue morphology in healthy regions (close to lesion fronts) (i) and eukaryotic microbe infiltration and extensive necrosis in lesion fronts (j) and diseased regions (k). Arrowheads and arrows indicate Symbiodinium (Sy), bacteria (Ba), Vibrio sp. bacteria (Vib), necrosis (Nec), ciliates (Cil), cyanobacteria (Cy), and helminths (He). Scale bars indicate 50 μm unless otherwise labeled.
Diseased tissue samples (collectively referred to as WS samples in other sections of this study) were divided into three subregions for histological and fluorescence in situ hybridization (FISH) analysis, based on the boundary between intact and compromised tissues demarcating the lesion border: “healthy region close to lesion front” (i.e., intact tissues less than 1 cm immediately ahead of the advancing border of the WS lesion), “lesion front” (i.e., the interface between intact and compromised tissues), and “disease region” (i.e., compromised tissues immediately behind the advancing border of the WS lesion) (see Fig. 3). Healthy region (close to lesion front) tissues were indistinguishable from other healthy tissue samples, displaying no signs of necrosis, fragmentation, tissue swelling, ciliates, or cyanobacteria, regardless of season (Table 5; Fig. 3). Similar to the case for other healthy tissues, helminths and fungi were observed in only a small number of samples (6% and 11%, respectively). High levels of necrosis (100% of samples) and tissue fragmentation (78%) characterized lesion front samples, while tissue swelling was completely absent (Table 5; Fig. 3). Ciliates (22%) and fungi (22%) were the most commonly observed organisms at WS lesion fronts, followed by cyanobacteria (17%) and helminths (6%). With the exception of fungi, associated organisms were more common within disease region tissues (less than 1 cm behind the border of WS lesions) (Table 5; Fig. 3). Tissue responses in the disease region were similar to those observed at the lesion front, with 100% of samples displaying necrosis, 50% showing fragmentation, and no signs of tissue swelling in any samples (Table 5, Fig. 3).
FISH revealed no accumulation of bacteria (Vibrio or otherwise) associated with healthy tissues from either healthy or WS-affected colonies, regardless of season. Similarly, no bacteria were observed in healthy region (close to lesion front) tissues (less than 1 cm ahead of WS lesion fronts), regardless of season (Table 5). With the exception of one winter 2011 disease region sample, bacteria were detected in all lesion front and disease region samples (i.e., both at and immediately behind the WS lesion front), regardless of season (Table 5; Fig. 3). However, while Vibrio bacteria were detected in 66% of lesion front and 100% of disease region samples in summer 2011, no vibrios were detected at either location in winter 2011 (Table 5; Fig. 3).
The ApopTag assays, employed to assess the abundance, distribution, and seasonality of apoptosis (i.e., programmed cell death) within coral tissues, revealed significantly different levels of apoptosis among seasons (df = 1, F = 6.1, P < 0.02) but not among health states (df = 2, F = 0.34, P > 0.05), with no interaction between the two factors (df = 2, F = 0.84, P > 0.05) (data not shown). Apoptosis levels were significantly higher in winter 2011 samples (63% ± 2% apoptotic nuclei) than in summer 2011 samples (52% ± 4%), regardless of health state (PHSD > 0.02).
DISCUSSION
The approach used in this study, of combining an 18-month, seasonally repeated, field-based sampling regime with bacterial community profiling, in situ visualization of host-microbe interactions, and physiological coral host responses, provides a detailed case study of WSs at reefs near Lizard Island in the northern GBR. Our results provide insights into the microbial dynamics of WS lesions affecting colonies of Acropora hyacinthus. WS-affected corals displayed nearly indistinguishable disease signs, both macroscopically and microscopically, regardless of season. Rapid tissue loss and whole-colony mortality were observed for all diseased corals monitored, highlighting the virulent nature of this disease and underscoring its potential to impact reef-building populations of corals that typically form the structural framework of Indo-Pacific reefs.
Tissue loss associated with Indo-Pacific WSs has been attributed to a number of etiological agents, including Vibrio bacteria (20–24), apoptosis (25), ciliates (11, 26), helminths (11), viruses (27), and intraspecific chimeric parasites (11, 28), suggesting that distinct and potentially distinguishable forms of WSs exist. Based on logical deduction, we contend that the WS lesions observed in this study are not the result of apoptosis or infection by Vibrio bacteria, ciliates, fungi, cyanobacteria, or helminths, because of the low levels and inconsistent presence of these organisms at WS lesion fronts. High-throughput bacterial profiling did, however, reveal distinct bacterial communities at WS disease lesion fronts relative to healthy tissues on both healthy and WS-affected corals.
The greater-than-4-fold increase in the proportion of Rhodobacteraceae-affiliated sequences in bacterial communities associated with WS lesions in this study suggests a potential role of this group in WS pathogenesis. Previous investigations have demonstrated elevated levels of Rhodobacteraceae in WS samples across species and ocean basins (29–31). Elevated levels of Rhodobacteraceae have also been associated with several other coral diseases, including black band and white band diseases, white plague, atramentous necrosis, and cyanobacterial patches (32). However, it is currently unclear if changes in Rhodobacteraceae emerge as a cause or consequence of the observed disease. For example, this group of bacteria has been identified as a core member of the coral microbiome (33, 34), and there is evidence to suggest that healthy corals have the ability to regulate Rhodobacteraceae bacteria through the production of antimicrobial compounds within the coral mucus (35). The presence of this taxon in healthy tissues, albeit at significantly lower levels, and the lack of bacterial infiltration into apparently healthy tissues immediately adjacent to (i.e., less than 1 cm ahead of) WS lesions suggest that Rhodobacteraceae bacteria rapidly colonize compromised tissues rather than drive the observed tissue loss. Members of the Rhodobacteraceae family are known to readily form biofilms in marine environments, which also suggests a role as opportunists rather than primary pathogens (36, 37). The hypothesis that overall disruptions to the “core coral microbiome” could drive elevated Rhodobacteraceae levels in diseased samples is further supported by the observation of reductions in specific bacterial taxa, most notably Actinobacteria, which are prominent antibiotic producers and proposed members of the “core” coral microbiome (38, 39). Nevertheless, increases in the abundance of a diverse array of Rhodobacteraceae OTUs in health-compromised coral samples alongside significant reductions in the abundances of several member of the Rhodobacteraceae genus Paracoccus suggests that greater attention should be devoted to determining the role of Rhodobacteraceae bacteria in coral health and disease. For example, targeted FISH approaches could be employed to determine the location and abundance of Rhodobacteraceae bacteria in healthy coral tissues and to determine how these dynamics are influenced by environmental stress and disease.
Members of the genus Vibrio have received a great deal of attention for their potential role in WS pathogenesis (20, 22, 23, 40–42). Vibrio bacteria have been associated with white band disease (40–42) and have been shown to cause WS-like disease signs in aquarium-based infection experiments (20, 22–24). In the current study, however, we found no significant difference in the relative abundance of sequences affiliated with Vibrio bacteria between health states. Moreover, when Vibrio-related sequences were detected, they were always retrieved in low relative abundance (i.e., <3.3% of reads). In situ visualization further highlighted the ephemeral nature of Vibrio associations, with FISH detecting Vibrio bacteria only in summer WS lesion samples and revealing no signs of these bacteria in winter disease samples (or any healthy tissue samples), despite continued disease progression year round (i.e., white, denuded skeleton at the lesion front indicative of active lesion progression). The seasonal presence of Vibrio bacteria at WS lesion fronts highlights the value of repeated seasonal sampling and supports the assertion that while Vibrio bacteria can proliferate opportunistically in WS lesions, they may not be an essential driver of WS disease progression in populations of A. hyacinthus at Lizard Island.
Ciliates (11, 26), helminths (11), and apoptosis (25) have also been suggested as drivers of WS diseases. While histological analyses revealed extensive necrosis and loss of tissue structure in nearly all lesion front samples, helminths, ciliates, fungi, or cyanobacteria were observed in fewer than 25% of WS samples, suggesting that these microbes are not likely to be the primary drivers of the lesions examined. This conclusion is further supported by the observation of helminths and fungi in healthy tissues samples. Similarly, assays measuring apoptotic cells showed no statistical difference in levels of programmed cell death between WS lesion and healthy tissue samples.
In contrast to previous investigations of white diseases in the Caribbean (43–45), WS-affected coral colonies monitored in this study showed no colony-wide microbial response to infection. Bacterial communities associated with healthy tissues on diseased colonies were statistically indistinguishable from communities associated with tissues collected from apparently healthy colonies (including one colony that was sampled 5 times over the course of the 18-month study). This inconsistency between the current study and studies of tissue loss diseases in the Caribbean could reflect differences in bacterial community profiling techniques (i.e., high-throughput sequencing versus clone libraries) or could provide evidence of distinct disease etiologies. Monitoring other indices of coral health (e.g., fatty acid and lipid reserves) could provide further insights into potential colony-wide responses to disease (46).
Conclusions.
Utilizing deductive logic, we posit that the WS disease signs observed in this study did not result from apoptosis or infection by Vibrio bacteria, ciliates, or helminths. While this conclusion may appear to contradict previous WS investigations, a more parsimonious explanation is that multiple forms of WSs with distinct and distinguishable etiologies exist (8, 11). The observation of a 4-fold increase in Rhodobacteraceae-affiliated bacterial sequences at WS lesion fronts suggests that this bacterium could play a role in WS pathogenesis and/or serve as a diagnostic criterion for disease differentiation. It is also important to note that viral dynamics were not assessed in the current study. There is evidence to suggest that the relative abundance of viruses differs between healthy and WS-affected coral tissues, with elevated numbers of small (<100-nm) virus-like particles observed on WS-affected tissues (27, 47). Characterizing the role of viruses in coral diseases is challenging but also represents a field that can provide important insights into the drivers of coral diseases. While the body of knowledge surrounding WS diseases is growing steadily, it is likely that published studies describe only a small sample of WS diseases throughout the Indo-Pacific. The development and dissemination of detailed and methodologically consistent case studies of WSs over wide geographic, temporal, and host ranges will help to identify and distinguish distinct WS etiologies and provide criteria to differentiate etiologically dissimilar diseases. While the current study does not definitely identify the underlying causes of WS etiologies afflicting colonies of A. hyacinthus near Lizard Island on the GBR, it provides a framework that characterizes the lesions at the cellular level over seasonal time scales and identifies areas where future studies may contribute to unlocking the mystery of this prevalent and virulent group of diseases.
MATERIALS AND METHODS
Study site and sample collection.
This study was conducted at reefs near Lizard Island (14°40′S, 145°27′E) in the northern sector of the Great Barrier Reef (GBR) between September 2010 and February 2012. Colonies of Acropora hyacinthus that were either healthy or showing signs of WSs (i.e., colonies displaying diffuse, acute to subacute areas of tissue loss revealing white, intact skeleton) were examined on SCUBA (1- to 12-m depth) at three reefs near Lizard Island (Bird Island, No Name, and Ribbon Reef no. 10). WS-affected and nearby conspecific healthy control colonies were tagged with plastic cattle tags inscribed with a unique colony identification number. Tagged colonies were photographed to quantify lesion progression rate (see below) and samples were collected at five seasonal time points: September 2010 (spring 2010), February 2011 (summer 2011), July 2011 (winter 2011), October 2011 (spring 2011), and February 2012 (summer 2012). Lesion progression rates were assessed using the image analysis software package ImageJ 1.45s (48). Linear progression rates of WS lesions were determined by overlaying photos of the same coral fragment at different time points (using topographical landmarks), haphazardly selecting five points along the lesion front, measuring the shortest distance between each point on the lesion fronts at time zero and the corresponding point on the lesion fronts at time x, and dividing the average of the five measurements by x. The final health state of tagged colonies was assessed in June 2012, when all remaining cattle tags were removed. Four healthy and four WS-affected colonies were tagged at the first sampling time point (spring 2010). When colonies suffered complete mortality between time points, new colonies were tagged in order to obtain samples from at least 3 healthy and 3 WS-affected colonies per time point (see Table 5 for details). Samples were collected from one location on each healthy colony and two locations on each WS-infected colony (i.e., one from the disease lesion interface and one from apparently healthy tissue approximately 10 cm away from the lesion). At each sampling location, one fragment (∼5 cm in length) was collected for bacterial community profiling, and a second, smaller fragment (∼3 cm) was collected for histological analyses using surgical bone cutters on SCUBA. Samples were placed in plastic bags underwater, and all tagged colonies were photographed before and after sample collection. Within 15 min of collection, fragments for bacterial community profiling were snap-frozen in liquid nitrogen and stored at −80°C until processing; fragments for histology were placed in sterile, freshly prepared 4% paraformaldehyde (Electron Microscopy Sciences, USA)–10 mM phosphate-buffered saline (PBS) solution at 4°C. In addition, two surface seawater samples (1 liter) were collected at each seasonal time point, immediately filtered through a 0.22-mm Millipore Sterivex filter unit (Sigma-Aldrich, St. Louis, MO, USA), and subsequently stored at −80°C until DNA extraction. Seawater samples were collected at the surface to avoid the potentially confounding influence of differing sampling depths among colonies and time points.
Histological processing and visualization.
After 8 to 10 h, the paraformaldehyde solution was exchanged for a 1:1 solution of PBS and ethanol and stored at 4°C. Samples were then embedded in 1.5% agarose to maintain tissues in their normal conformation during the decalcification process. Once set, excess agarose was trimmed and a small hole was punched through to the coral fragment to allow liquids to reach the sample. Agarose-embedded samples were placed in histological cassettes and decalcified in a 20% EDTA solution (pH 8.0); the solution was maintained at 4°C and exchanged approximately every 2 days until no skeleton remained (up to 2 weeks). Following decalcification, agarose-embedded samples were rinsed in PBS, dehydrated sequentially through a 70%, 80%, 90%, 95%, 100%, and 100% ethanol series (60 min each), and then processed through 3 30-min toluene rinses and embedded in Paraplast (paraffin wax). Samples were then washed three times in xylene and Paraplast paraffin wax (60 min each) prior to embedding in Paraplast wax. Paraffin-embedded samples were serially sectioned at 4 μm and collected on Superfrost Plus slides (Menzel, Germany).
Two serial sections from each sample were dewaxed in xylene (twice for 3 min each) and rehydrated through serial washes of three times for 5 min each in 100% ethanol, once for 5 min in 70% ethanol, and once for 2 min in water. Hydrated sections were counterstained in Mayer's hematoxylin for 8 min, rinsed in tap water (20 dips), differentiated in Scott's tap water substitute for 30 s for bluing, rinsed in water (2 min), and stained with hematoxylin-eosin-Gomori trichrome stain for 3 min. Stained sections were dehydrated through an ethanol series (once for 2 min in 70% ethanol and twice for 5 min each in 100% ethanol), washed in xylene (twice for 5 min each), and mounted in DPX mounting medium (Sigma-Aldrich, Australia). Observations at a magnification of ×20 were recorded using a DMI 6000B (Leica, Germany) or Eclipse (Nikon, Japan) light microscope, and photomicrograph images were processed using the LAS imaging software (Leica, Germany). Coral tissue responses (i.e., necrosis, fragmentation, and swelling) and associated organisms (i.e., helminths, ciliates, fungi, and cyanobacteria) were recorded at three haphazardly chosen locations within each sample. Additionally, diseased tissue samples were divided into three subregions for all microscopic analysis: “healthy region close to lesion front” (i.e., intact tissues immediately ahead of the advancing WS lesion), “lesion front” (i.e., the interface between intact and compromised tissues), and “disease region” (i.e., compromised tissues immediately behind the advancing WS lesion).
FISH and visualization.
Fluorescence in situ hybridization (FISH) was utilized to assess the density, distribution, and seasonal variability of total bacteria, and specifically of Vibrio bacteria, within coral samples collected in winter 2011 and summer 2011. Prior to staining, three serial tissue sections were dewaxed in xylene (twice for 3 min each), dehydrated through 100% ethanol washes (three times for 5 min each), and air dried. Dried sections were washed in a 0.2 M HCl solution for 12 min and a 20 mM Tris HCl solution (pH 8.0) for 10 min at room temperature. To digest bacterial cellular membranes and allow easier probe penetration into tissues, sections were mounted in a protease K (50 μg/ml)-20 mM Tris HCl solution (pH 8.0) for 5 min at 37°C and rinsed in 20 mM Tris HCl (pH 8.0) prior to probe hybridization. Oligonucleotide probes, including a broad-range total bacterial probe (EUB338 mix [5′-GCT GCC TCC CGT AGG AGT-3′, 5′-GCA GCC ACC CGT AGG TGT-3′, and 5′–GCT GCC ACC CGT AGG TGT-3′]), a Vibrio-specific probe (VibGV [5′–AGG CCA CAA CCT CCA AGT AG-3′]), and a nonsense, negative-control probe (NonEUB338 [5′-ACA TCC TAC GGG AGG C-3′]), were labeled with the Cy3 fluorochrome (Thermo Fisher Scientific, Germany) (49, 50). Tissue sections were covered with hybridization buffer (30% [vol/vol] formamide, 0.9 M NaCl, 20 mM Tris-HCl [pH 8.0], 0.01% SDS), oligonucleotide probes were added individually to a final concentration of 25 ng μl−1, and samples were incubated at 46°C for 1.5 h. Sections were washed in 50-ml Falcon tubes containing preheated wash buffer (0.112 M NaCl, 20 mM Tris-HCl [pH 8.0], 0.01% SDS, 5 mM EDTA) in a water bath at 48°C for 10 min. Following washing, sections were immediately soaked in cold, filtered water for 10 s to remove excess salts, air dried, and mounted in CitiFluor AF1 (ProScitech, Australia). As a negative control, one serial tissue section per sample was processed as described above, but no oligonucleotide probe was added. An LSM710 confocal laser scanning microscope (Carl Zeiss, Germany) combined with spectral emissions profiling was used to visualize tissue-associated, FISH-labeled bacterial communities, as described previously (51). Detection of the Cy3 fluorochrome label was in the emission range of 519 to 580 nm, and the target signal was recorded at 561 nm. Autofluorescence detection and spectral removal was targeted at emission ranges of 407 to 486 nm (to remove coral tissue autofluorescence) and 627 to 704 nm (to remove Symbiodinium autofluorescence). As described for histological processing, diseased tissue samples were divided into three regions: “healthy region close to lesion front,” “lesion front,” and “disease region.”
ApopTag in situ apoptosis detection.
The ApopTag peroxidase in situ apoptosis detection kit (S7100; Millipore, USA) was utilized to assess the abundance, distribution, and seasonality of apoptosis (i.e., programmed cell death) within coral samples collected in winter 2011 and summer 2011. The ApopTag technique, which was carried out according to the manufacturer's instructions (S7101; Chemicon International, Inc.), allows visual discrimination between apoptotic and necrotic cells by labeling apoptosis-associated DNA strand breaks through the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) method. Using the histology and light microscopy techniques outlined above, coral cells were recorded as apoptotic if the cell nucleus was positively labeled (i.e., stained red) or nonapoptotic if the nucleus was counterstained blue by hematoxylin. For each sample, three replicate counts of 100 cells per tissue layer were performed at randomly chosen regions within the epidermal and gastrodermal layers.
Genomic DNA extraction and PCR amplification of bacterial 16S rRNA gene.
Tissue was removed from thawed coral fragments by vigorous vortexing in 550 μl of PowerPlant bead solution (Mo Bio, Carlsbad, CA) for 5 min in individual sterile 50-ml Falcon tubes. Bacterial DNA was then extracted from the resulting coral slurry using the PowerPlant DNA isolation kit (MoBio, Carlsbad, CA) according to the manufacturer's instructions. Additionally, seawater sample DNA was extracted from Millipore Sterivex filter membranes using a method described previously (52). Purified DNA was stored at −80°C until PCR amplification. Bacterial tag-encoded FLX-titanium amplicon pyrosequencing based on the V1 to V3 region (Escherichia coli positions 27 to 519) of the small-subunit rRNA (16S rRNA) gene was performed on all samples with forward primer 27F (5′-GAG TTT GAT CNT GGC TCA G-3′) and reverse primer 519R (5′-GTN TTA CNG CGG CKG CTG-3′), as described previously (53, 54).
Sequence processing and OTU selection.
Sequence reads were processed using the Quantitative Insights Into Microbial Ecology (QIIME) pipeline (version 1.9.1), as described previously (55). Briefly, samples were separated by sample-specific barcodes, low quality reads and chimeric sequences were discarded (USEARCH [56]), and operational taxonomic units (OTUs) were identified (UCLUST method; threshold, 97% [56]). Representative sequences were chosen by consensus and assigned a taxonomic identity (UCLUST method; template, GreenGenes [57]), and a phylogenetic tree was constructed (FastTree method [58]). Prior to downstream analysis, sequences classified as “unknown” (i.e., sequences not classified at the kingdom level) or chloroplast and other potential contaminants were removed, and sequence data for all samples were rarefied to 400 reads to remove sequencing effort heterogeneity.
Statistical analyses.
Analysis of variance (ANOVA) was conducted on bacterial read data at the family level to determine if taxa were differentially abundant among health states and time points. ANOVA was also conducted on bacterial OTU richness (chao1) and ApopTag-derived abundance data to test for differences between health states and time points, as well as interactions between the two. Where main effects were identified by ANOVA, post hoc Tukey's honestly significant difference (HSD) tests were employed to assess pairwise differences. When assumptions of ANOVA could not be met, the nonparametric Kruskal-Wallis analysis of variance and Dunn's multiple-comparison post hoc test were employed. Unless otherwise stated, univariate analyses were performed using R v3.2.4 (59).
Beta diversity (i.e., between-sample diversity) was assessed using weighted UniFrac distance metrics (60). UniFrac distance is a phylogeny-based distance metric that avoids problems associated with using the so-called “star phylogeny,” the phylogeny that assumes all species are equally related, which all nonphylogenetic methods for comparing communities implicitly use. Weighted UniFrac distance is also desirable because it is quite sensitive to changes among abundant microbial lineages. UniFrac dissimilarity matrices based on bacterial sequences at the OTU level were used to construct unconstrained two-dimensional principal-coordinate analysis (PCoA) plots to visualize differences among bacterial community assemblages (61, 62). Permutational multivariate analysis of variance (PERMANOVA) (61, 62) was utilized to test for statistical differences in bacterial community assemblages among health states (i.e., healthy coral fragments [healthy], healthy fragments from diseased colonies [HD], fragments from disease lesion fronts [WS], and seawater samples) and time points (i.e., spring 2010, summer 2011, winter 2011, spring 2011, and summer 2012). This analysis was based on weighted UniFrac dissimilarity matrices, type III partial sums of squares, and 9,999 random permutations of the residuals under the reduced model. Additionally, P values were obtained using Monte Carlo random sampling from the asymptotic permutation distribution. Post hoc pairwise comparisons among locations and health states were conducted following significant main effects results. The similarity percentages (SIMPER) routine was utilized to investigate the contributions of individual bacterial taxa to the observed dissimilarity between sample groups. SIMPER analysis was based on a zero-adjusted Bray-Curtis similarity matrix (61) at the OTU level. All multivariate analyses were performed using Primer 6.0 statistical software (Primer-e Ltd., UK).
Phylogenetic trees showing the position of Rhodobacteraceae-affiliated OTUs and related type strains in the family Rhodobacteraceae were constructed based on the neighbor-joining method. Evolutionary relatedness was inferred by aligning gene sequences using ClustalW (63) and calculating similarity matrices and phylogenetic trees using MEGA5 (64). Evolutionary distance estimations were obtained using the Jukes-Cantor model (65).
Accession number(s).
The sequence data set was deposited in the NCBI Sequence Read Archive (SRA) database (accession number PRJNA308829).
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Sandra Binning, Alexandra Fennell, Sefano Katz, Hanna Kokko, Joleah Lamb, Raechel Littman, Aurelie Moya, Sam Noonan, Genevieve Phillips, Fred Pollock, Dylan Simonson, Melanie Trapon, and Jeroen van de Water for their field assistance at Lizard Island Research Station and Sefano Katz and Andy Muirhead for their assistance in the laboratory. We also acknowledge the staff of Lizard Island Research Station for their logistical support.
This work was funded by a Lizard Island Research Foundation Fellowship awarded to F.J.P. for study at Lizard Island Research Station, a facility of the Australian Museum.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02799-16.
REFERENCES
- 1.Aronson RB, Precht WF. 2001. White-band disease and the changing face of Caribbean coral reefs. Hydrobiologia 460:1–15. doi: 10.1023/A:1013103928980. [DOI] [Google Scholar]
- 2.Willis BL, Page CA, Dinsdale EA. 2004. Coral disease on the Great Barrier Reef, p 69–104. In Rosenberg E, Loya Y (ed), Coral health and disease. Springer, New York, NY. [Google Scholar]
- 3.Antonius A. 1985. Coral diseases in the Indo-Pacific: a first record. Mar Ecol 6:197–218. doi: 10.1111/j.1439-0485.1985.tb00322.x. [DOI] [Google Scholar]
- 4.Patterson KL, Porter JW, Ritchie KB, Polson SW, Mueller E, Peters EC, Santavy DL, Smith GW. 2002. The etiology of white pox, a lethal disease of the Caribbean elkhorn coral, Acropora palmata. Proc Natl Acad Sci U S A 99:8725–8730. doi: 10.1073/pnas.092260099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruno JF, Selig ER. 2007. Regional decline of coral cover in the Indo-Pacific: timing, extent, and subregional comparisons. PLoS One 2:e711. doi: 10.1371/journal.pone.0000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aeby GS, Ross M, Williams GJ, Lewis TD, Work TM. 2010. Disease dynamics of Montipora white syndrome within Kaneohe Bay, Oahu, Hawaii: distribution, seasonality, virulence, and transmissibility. Dis Aquat Organ 91:1–8. doi: 10.3354/dao02247. [DOI] [PubMed] [Google Scholar]
- 7.Pollock FJ, Morris PJ, Willis BL, Bourne DG. 2011. The urgent need for robust coral disease diagnostics. PLoS Pathog 7:e1002183. doi: 10.1371/journal.ppat.1002183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bourne DG, Ainsworth TD, Pollock FJ, Willis BL. 2015. Towards a better understanding of white syndromes and their causes on Indo-Pacific. Coral Reefs 34:233–242. doi: 10.1007/s00338-014-1239-x. [DOI] [Google Scholar]
- 9.Bourne DG, Garren M, Work TM, Rosenberg E, Smith GW, Harvell CD. 2009. Microbial disease and the coral holobiont. Trends Microbiol 17:554–562. doi: 10.1016/j.tim.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Work TM, Aeby GS. 2011. Pathology of tissue loss (white syndrome) in Acropora sp. corals from the Central Pacific. J Invertebr Pathol 107:127–131. doi: 10.1016/j.jip.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 11.Work TM, Russell R, Aeby GS. 2012. Tissue loss (white syndrome) in the coral Montipora capitata is a dynamic disease with multiple host responses and potential causes. Proc R Soc B Biol Sci 279:4334–4341. doi: 10.1098/rspb.2012.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bythell J, Pantos O, Richardson L. 2004. White plague, white band, and other “white” diseases, p 351–365. In Rosenberg E, Loya Y (ed), Coral health and disease. Springer, New York, NY. [Google Scholar]
- 13.Work TM, Aeby GS. 2006. Systematically describing gross lesions in corals. Dis Aquat Organ 70:155. doi: 10.3354/dao070155. [DOI] [PubMed] [Google Scholar]
- 14.Work TM, Richardson LL, Reynolds TL, Willis BL. 2008. Biomedical and veterinary science can increase our understanding of coral disease. J Exp Mar Biol Ecol 362:63–70. doi: 10.1016/j.jembe.2008.05.011. [DOI] [Google Scholar]
- 15.Work T, Meteyer C. 2014. To understand coral disease, look at coral cells. EcoHealth 11:610. doi: 10.1007/s10393-014-0931-1. [DOI] [PubMed] [Google Scholar]
- 16.Ainsworth TD, Fine M, Roff G, Hoegh-Guldberg O. 2008. Bacteria are not the primary cause of bleaching in the Mediterranean coral Oculina patagonica. ISME J 2:67–73. [DOI] [PubMed] [Google Scholar]
- 17.Downs CA, Kramarsky-Winter E, Woodley CM, Downs A, Winters G, Loya Y, Ostrander GK. 2009. Cellular pathology and histopathology of hypo-salinity exposure on the coral Stylophora pistillata. Sci Total Environ 407:4838–4851. doi: 10.1016/j.scitotenv.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 18.Bruckner AW. 2002. Priorities for effective management of coral diseases. NMFS-OPR-22. National Oceanic and Atmospheric Administration, Silver Spring, MD. [Google Scholar]
- 19.Roff G, Kvennefors ECE, Fine M, Ortiz J, Davy JE, Hoegh-Guldberg O. 2011. The ecology of “acroporid white syndrome,” a coral disease from the southern Great Barrier Reef. PLoS One 6:e26829. doi: 10.1371/journal.pone.0026829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sussman M, Willis BL, Victor S, Bourne DG. 2008. Coral pathogens identified for white syndrome (WS) epizootics in the Indo-Pacific. PLoS One 3:e2393. doi: 10.1371/journal.pone.0002393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arboleda M, Reichardt W. 2010. Vibrio sp. causing Porites ulcerative white spot disease. Dis Aquat Organ 90:93–104. doi: 10.3354/dao02222. [DOI] [PubMed] [Google Scholar]
- 22.Luna GM, Bongiorni L, Gili C, Biavasco F, Danovaro R. 2010. Vibrio harveyi as a causative agent of the White Syndrome in tropical stony corals. Environ Microbiol Rep 2:120–127. doi: 10.1111/j.1758-2229.2009.00114.x. [DOI] [PubMed] [Google Scholar]
- 23.Ushijima B, Smith A, Aeby GS, Callahan SM. 2012. Vibrio owensii induces the tissue loss disease Montipora white syndrome in the Hawaiian reef coral Montipora capitata. PLoS One 7:e46717. doi: 10.1371/journal.pone.0046717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ushijima B, Videau P, Burger AH, Shore-Maggio A, Runyon CM, Sudek M, Aeby GS, Callahan SM. 2014. Vibrio coralliilyticus strain OCN008 is an etiological agent of acute Montipora white syndrome. Appl Environ Microbiol 80:2102–2109. doi: 10.1128/AEM.03463-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ainsworth TD, Kramasky-Winter E, Loya Y, Hoegh-Guldberg O, Fine M. 2007. Coral disease diagnostics: what's between a plague and a band? Appl Environ Microbiol 73:981–992. doi: 10.1128/AEM.02172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sweet M, Bythell J. 2012. Ciliate and bacterial communities associated with white syndrome and brown band disease in reef-building corals: ciliate and bacterial pathogens in coral disease. Environ Microbiol 14:2184–2199. doi: 10.1111/j.1462-2920.2012.02746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollock F, Wood-Charlson E, van Oppen M, Bourne D, Willis B, Weynberg K. 2014. Abundance and morphology of virus-like particles associated with the coral Acropora hyacinthus differ between healthy and white syndrome-infected states. Mar Ecol Prog Ser 510:39–43. doi: 10.3354/meps10927. [DOI] [Google Scholar]
- 28.Work TM, Forsman ZH, Szabó Z, Lewis TD, Aeby GS, Toonen RJ. 2011. Inter-specific coral chimerism: genetically distinct multicellular structures associated with tissue loss in Montipora capitata. PLoS One 6:e22869. doi: 10.1371/journal.pone.0022869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sunagawa S, DeSantis TZ, Piceno YM, Brodie EL, DeSalvo MK, Voolstra CR, Weil E, Andersen GL, Medina M. 2009. Bacterial diversity and white plague disease-associated community changes in the Caribbean coral Montastraea faveolata. ISME J 3:512–521. doi: 10.1038/ismej.2008.131. [DOI] [PubMed] [Google Scholar]
- 30.Cárdenas A, Rodriguez-R LM, Pizarro V, Cadavid LF, Arévalo-Ferro C. 2012. Shifts in bacterial communities of two Caribbean reef-building coral species affected by white plague disease. ISME J 6:502–512. doi: 10.1038/ismej.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roder C, Arif C, Daniels C, Weil E, Voolstra CR. 2014. Bacterial profiling of white plague disease across corals and oceans indicates a conserved and distinct disease microbiome. Mol Ecol 23:965–974. doi: 10.1111/mec.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mouchka ME, Hewson I, Harvell CD. 2010. Coral-associated bacterial assemblages: current knowledge and the potential for climate-driven impacts. Integr Comp Biol 50:1–13. doi: 10.1093/icb/icq064. [DOI] [PubMed] [Google Scholar]
- 33.Morrow KM, Moss AG, Chadwick NE, Liles MR. 2012. Bacterial associates of two Caribbean coral species reveal species-specific distribution and geographic variability. Appl Environ Microbiol 78:6438–6449. doi: 10.1128/AEM.01162-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez-Lanetty M, Granados-Cifuentes C, Barberan A, Bellantuono AJ, Bastidas C. 2013. Ecological Inferences from a deep screening of the complex bacterial consortia associated with the coral, Porites astreoides. Mol Ecol 22:4349–4362. doi: 10.1111/mec.12392. [DOI] [PubMed] [Google Scholar]
- 35.Ritchie KB. 2006. Regulation of microbial populations by coral surface mucus and mucus-associated bacteria. Mar Ecol Prog Ser 322:1–14. doi: 10.3354/meps322001. [DOI] [Google Scholar]
- 36.Witt V, Wild C, Anthony KRN, Diaz-Pulido G, Uthicke S. 2011. Effects of ocean acidification on microbial community composition of, and oxygen fluxes through, biofilms from the Great Barrier Reef. Environ Microbiol 13:2976–2989. doi: 10.1111/j.1462-2920.2011.02571.x. [DOI] [PubMed] [Google Scholar]
- 37.Elifantz H, Horn G, Ayon M, Cohen Y, Minz D. 2013. Rhodobacteraceae are the key members of the microbial community of the initial biofilm formed in Eastern Mediterranean coastal seawater. FEMS Microbiol Ecol 85:348–357. doi: 10.1111/1574-6941.12122. [DOI] [PubMed] [Google Scholar]
- 38.Bull AT, Stach JEM. 2007. Marine actinobacteria: new opportunities for natural product search and discovery. Trends Microbiol 15:491–499. doi: 10.1016/j.tim.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Ainsworth TD, Krause L, Bridge T, Torda G, Raina J-B, Zakrzewski M, Gates RD, Padilla-Gamiño JL, Spalding HL, Smith C, Woolsey ES, Bourne DG, Bongaerts P, Hoegh-Guldberg O, Leggat W. 2015. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J 9:2261–2274. doi: 10.1038/ismej.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gladfelter WB. 1982. White-band disease in Acropora palmata: implications for the structure and growth of shallow reefs. Bull Mar Sci 32:639–643. [Google Scholar]
- 41.Ritchie KB, Smith GW. 1998. Type II white-band disease. Rev Biol Trop 46:199–203. [Google Scholar]
- 42.Gil-Agudelo DL, Smith GW, Weil E. 2006. The white band disease type II pathogen in Puerto Rico. Rev Biol Trop 54(Suppl 3):59–67.18457175 [Google Scholar]
- 43.Pantos O, Bythell JC. 2006. Bacterial community structure associated with white band disease in the elkhorn coral Acropora palmata determined using culture-independent 16S rRNA techniques. Dis Aquat Organ 69:79–88. doi: 10.3354/dao069079. [DOI] [PubMed] [Google Scholar]
- 44.Pantos O, Cooney RP, Le Tissier MD, Barer MR, O'Donnell AG, Bythell JC. 2003. The bacterial ecology of a plague-like disease affecting the Caribbean coral Montastrea annularis. Env Microbiol 5:370–382. doi: 10.1046/j.1462-2920.2003.00427.x. [DOI] [PubMed] [Google Scholar]
- 45.Wilson B, Aeby GS, Work TM, Bourne DG. 2012. Bacterial communities associated with healthy and Acropora white syndrome-affected corals from American Samoa. FEMS Microbiol Ecol 80:509–520. doi: 10.1111/j.1574-6941.2012.01319.x. [DOI] [PubMed] [Google Scholar]
- 46.Roff G, Hoegh-Guldberg O, Fine M. 2006. Intra-colonial response to acroporid “white syndrome” lesions in tabular Acropora spp. (Scleractinia). Coral Reefs 25:255–264. doi: 10.1007/s00338-006-0099-4. [DOI] [Google Scholar]
- 47.Soffer N, Brandt ME, Correa AM, Smith TB, Thurber RV. 2014. Potential role of viruses in white plague coral disease. ISME J 8:271–283. doi: 10.1038/ismej.2013.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abràmoff MD, Magalhães PJ, Ram SJ. 2004. Image processing with ImageJ. Biophotonics Int 11:36–42. [Google Scholar]
- 49.Wallner G, Amann R, Beisker W. 1993. Optimizing fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes for flow cytometric identification of microorganisms. Cytometry 14:136–143. doi: 10.1002/cyto.990140205. [DOI] [PubMed] [Google Scholar]
- 50.Daims H, Brühl A, Amann R, Schleifer K-H, Wagner M. 1999. The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst Appl Microbiol 22:434–444. doi: 10.1016/S0723-2020(99)80053-8. [DOI] [PubMed] [Google Scholar]
- 51.Ainsworth TD, Fine M, Blackall LL, Hoegh-Guldberg O. 2006. Fluorescence in situ hybridization and spectral imaging of coral-associated bacterial communities. Appl Environ Microbiol 72:3016–3020. doi: 10.1128/AEM.72.4.3016-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schauer M, Massana R, Pedros-Alio C. 2000. Spatial differences in bacterioplankton composition along the Catalan coast (NW Mediterranean) assessed by molecular fingerprinting. FEMS Microbiol Ecol 33:51–59. doi: 10.1111/j.1574-6941.2000.tb00726.x. [DOI] [PubMed] [Google Scholar]
- 53.Suchodolski JS, Dowd SE, Westermarck E, Steiner JM, Wolcott RD, Spillmann T, Harmoinen JA. 2009. The effect of the macrolide antibiotic tylosin on microbial diversity in the canine small intestine as demonstrated by massive parallel 16S rRNA gene sequencing. BMC Microbiol 9:210. doi: 10.1186/1471-2180-9-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Handl S, Dowd SE, Garcia-Mazcorro JF, Steiner JM, Suchodolski JS. 2011. Massive parallel 16S rRNA gene pyrosequencing reveals highly diverse fecal bacterial and fungal communities in healthy dogs and cats. FEMS Microbiol Ecol 76:301–310. doi: 10.1111/j.1574-6941.2011.01058.x. [DOI] [PubMed] [Google Scholar]
- 55.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 57.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Price MN, Dehal PS, Arkin AP. 2009. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.R Development Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 60.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clarke KR, Gorley RN. 2006. PRIMER v6: user manual/tutorial. PRIMER-E Ltd., Plymouth, UK. [Google Scholar]
- 62.Anderson MJ, Gorley RN, Clarke KR. 2008. PERMANOVA+ for PRIMER: guide to software and statistical methods. PRIMER-E Ltd., Plymouth, UK. [Google Scholar]
- 63.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 64.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jukes TH, Cantor CR. 1969. Evolution of protein molecules, p 21–132. In Munro HN. (ed), Mammalian protein metabolism. Academic Press, London, UK. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.