

ABSTRACT
Escherichia coli K-12 W3110 grows in the presence of membrane-permeant organic acids that can depress cytoplasmic pH and accumulate in the cytoplasm. We conducted experimental evolution by daily diluting cultures in increasing concentrations of benzoic acid (up to 20 mM) buffered at external pH 6.5, a pH at which permeant acids concentrate in the cytoplasm. By 2,000 generations, clones isolated from evolving populations showed increasing tolerance to benzoate but were sensitive to chloramphenicol and tetracycline. Sixteen clones grew to stationary phase in 20 mM benzoate, whereas the ancestral strain W3110 peaked and declined. Similar growth occurred in 10 mM salicylate. Benzoate-evolved strains grew like W3110 in the absence of benzoate, in media buffered at pH 4.8, pH 7.0, or pH 9.0, or in 20 mM acetate or sorbate at pH 6.5. Genomes of 16 strains revealed over 100 mutations, including single-nucleotide polymorphisms (SNPs), large deletions, and insertion knockouts. Most strains acquired deletions in the benzoate-induced multiple antibiotic resistance (Mar) regulon or in associated regulators such as rob and cpxA, as well as the multidrug resistance (MDR) efflux pumps emrA, emrY, and mdtA. Strains also lost or downregulated the Gad acid fitness regulon. In 5 mM benzoate or in 2 mM salicylate (2-hydroxybenzoate), most strains showed increased sensitivity to the antibiotics chloramphenicol and tetracycline; some strains were more sensitive than a marA knockout strain. Thus, our benzoate-evolved strains may reveal additional unknown drug resistance components. Benzoate or salicylate selection pressure may cause general loss of MDR genes and regulators.
IMPORTANCE Benzoate is a common food preservative, and salicylate is the primary active metabolite of aspirin. In the gut microbiome, genetic adaptation to salicylate may involve loss or downregulation of inducible multidrug resistance systems. This discovery implies that aspirin therapy may modulate the human gut microbiome to favor salicylate tolerance at the expense of drug resistance. Similar aspirin-associated loss of drug resistance might occur in bacterial pathogens found in arterial plaques.
KEYWORDS: acid, aspirin, benzoate, chloramphenicol, Escherichia coli, experimental evolution, salicylate, antibiotic resistance, low pH
INTRODUCTION
Pathogenic and commensal enteric bacteria maintain cytoplasmic pH homeostasis in the face of extreme external acid (pH 2 to 4 in the stomach) and the high concentrations of membrane-permeant organic acids in the colon (70 to 140 mM) (1–6). Many studies have focused on the response and recovery of Escherichia coli to external pH stress (1–5), but relatively few studies have focused on the genetic response to membrane-permeant organic acids (permeant acids) (7–9) despite the importance of permeant acids as food preservatives (10). Permeant acids depress cytoplasmic pH, while their anions accumulate in the cytoplasm (5, 7, 11). Some permeant acids also cross the membrane in the unprotonated form and decrease the proton motive force (PMF); an example is benzoic acid (benzoate), a food preservative found in soft drinks and acidic foods (12). The related molecule salicylic acid (salicylate) is a plant defense regulator (13, 14) as well as the primary active metabolite of acetylsalicylate (aspirin) (15–17). Salicylates enter the human diet from fruits and vegetables, leading to circulating plasma levels as high as 0.1 to 0.2 μM (18). Furthermore, aspirin therapy for cardioprotection and other metabolic conditions (19, 20) may generate plasma levels of 0.2 to 0.5 mM salicylate (16, 21, 22). Yet despite the important metabolic effects of aspirin, salicylate, and benzoate on plants and animals, there is surprisingly little research on the effects of benzoate and salicylate on the host microbiomes. In one study, aspirin inhibited the growth of Helicobacter pylori and enhanced the pathogen's sensitivity to antibiotics (23).
In E. coli, aromatic permeant acids such as salicylate and benzoate induce a large number of low-level multidrug efflux systems, governed by the Mar operon (marRAB) as well as additional, unidentified mechanisms (24). Benzoate and salicylate upregulate numerous genes of commensals and pathogens (25–28), including acrAB, tolC, and transport complexes that expel drugs across both the cytoplasmic and outer membranes. Mar family systems are widespread in bacterial genomes (29). Thus, in natural environments, aromatic acids may serve bacteria as early warning signals for the presence of antibiotic-producing competitors.
Mar family regulons commonly involve extensive upregulation of many genes by a small number of regulators. The E. coli regulator MarR represses expression of marRAB; repression is relieved when MarR binds salicylate (30) or one of several less potent inducers, such as benzoate or 2,4-dinitrophenol. The upregulated MarA is an AraC-type global regulator that differentially regulates approximately 60 genes (27, 31). Another AraC-type regulator, Rob, activates marRAB (26, 32). MarA downregulates the acid-inducible Gad acid fitness island (33). Gad includes glutamate decarboxylase (gadA) for extreme-acid survival (32–34), as well as periplasmic chaperone genes hdeA and hdeB (35) and multidrug resistance (MDR) loci mdtE and mdtF (36). Besides Mar, short-term benzoate exposure upregulates biofilm-associated genes (ymgABC and yhcN), the fimbrial phase variation regulator gene (fimB), and the cadmium stress protein gene yodA (37).
Thus, aromatic acid-inducible drug resistance incurs high energy costs associated with expression of so many genes, as well as the energy consumption by efflux pumps (38). Given the high energy cost, bacteria face a trade-off between inducible drug resistance and the toxicity of the drugs (39). One would expect a high selective pressure for regulator alleles that shift expression based on environmental factors. In fact, plate-based selection screens using lac fusions readily pick up mutations in marR and in MarR-regulated genes (8). Selective growth under antibiotic pressure leads to upregulation of marRAB (40).
A powerful tool for dissecting the long-term response to environmental stresses is experimental evolution (41, 42). Experimental evolution procedures with E. coli have included the adaptation to high temperatures (43), freeze-thaw cycles (44), high ethanol concentrations (45), and acid (46–48). We developed a microplate dilution cycle in order to generate evolving populations buffered at low pH (49). An advantage of our microplate dilution cycle is that we propagate a number of populations directly in the microplate, eliminating the intermediate stage of culture in flasks or tubes.
For the present study, we conducted experimental evolution of E. coli K-12 W3110 in microplate well populations containing media buffered at pH 6.5 and supplemented with increasing concentrations of benzoate (from 5 mM initially to 20 mM at 2,000 generations). We sequenced genomes of selected isolates and then identified genetic variants using the breseq pipeline (48–50). The breseq pipeline assembles a reference-based alignment to predict mutations compared to a previously sequenced genome (NCBI GenBank accession number NC_007779.1, E. coli K-12 W3110). Newer versions of breseq now predict structural variations, including large deletions, mobile element insertions, and gene duplications, all of which account for much of the genetic diversity in evolved clones (50–53).
Our analysis unexpectedly shows that genetic adaptation to benzoate is associated with loss or downregulation of benzoate- and salicylate-inducible genes, including those that encode multidrug resistance systems. The results have implications for evolution of the gut microbiome during aspirin therapy. More broadly, our results suggest a way to amplify the fitness costs of antibiotic resistance and possibly reverse antibiotic resistance in a microbiome (54).
RESULTS
Experimental evolution of benzoate-tolerant strains.
We conducted experimental evolution of Escherichia coli K-12 W3110 exposed to increasing concentrations of benzoic acid, as described in Materials and Methods (see Fig. S1 in the supplemental material). Over the course of the experiment, bacteria showed a progressive increase of benzoate tolerance, as measured by endpoint culture density. Clones were sampled from microplate populations frozen at intervals over the course of 0 to 2,900 generations (Fig. 1). The clones were cultured in microplate wells in medium containing 20 mM benzoate at pH 6.5, and the cell density was measured at 16 h. Over 1,400 to 2,900 generations, the growth endpoints of evolved clones increased significantly compared to that of the ancestor (Fig. 1A). A similar increase was observed for cell density during growth with 10 mM salicylate (Fig. 1B). Thus, overall, tolerance to benzoate and salicylate increased over generations of exposure.
FIG 1.

Growth measured after generations of repeated dilution and culture in 5 to 20 mM benzoate. Benzoate-evolved strains were isolated from frozen microplates, selecting 2 different clones from each of 4 populations. Clones from each plate generation were cultured at 37°C in a column of microplate wells; cell density values (OD600) were obtained at 16 h. (A to D) The colored dots represent lineages of evolved clones collected from distinct well populations (A1, C3, A5, and G5) after different numbers of generations as indicated on the x axis. Diamonds indicate median cell density for each generation tested. Asterisks indicate generations for which the 16-h cell density differed significantly from that of the ancestral strain W3110 in 2 out of 3 trials of the entire microplate experiment. For each microplate trial, the Friedman test was performed with post hoc Conover pairwise comparisons and Holm-Bonferroni-adjusted P values. LBK medium contained (A) 100 mM PIPES at pH 6.5 with 20 mM benzoate (diluted 1:200 from overnight cultures with 5 mM benzoate), (B) 100 mM PIPES at pH 6.5 with 10 mM salicylate (diluted 1:200 from overnight cultures in 2 mM salicylate), (C) 100 mM MOPS at pH 7.0 with 5 mM benzoate and 8 μg/ml chloramphenicol (diluted 1:200 from overnight cultures lacking chloramphenicol), or (D) 100 mM MOPS at pH 7.0 with 2 mM salicylate and 8 μg/ml chloramphenicol (diluted 1:200 from overnight cultures lacking chloramphenicol). (E) Plot of 16-h cell density values for 20 mM benzoate and for 5 mM benzoate–8 μg/ml chloramphenicol exposures. (F) Plot of 16-h cell density values for 10 mM salicylate and for 2 mM salicylate–8 μg/ml chloramphenicol exposures.
Since benzoate and salicylate induce multidrug resistance via the Mar regulon (24), it was of interest to test drug resistance of the evolved clones. We measured growth in chloramphenicol, an antibiotic that is effluxed by the MarA-dependent pump AcrA-AcrB-TolC (27), which confers low-level resistance. For our experiment, the same set of clones observed for growth in benzoate and salicylate were cultured in medium containing 8 μg/ml chloramphenicol (Fig. 1C and D). The medium was adjusted to pH 7.0 for maximal growth and contained a low concentration of benzoate or salicylate for induction of the Mar regulon. Later generations (generations 1,900 to 2,900) reached significantly lower cell density than the ancestor W3110.
Our results suggest that populations evolving with benzoate experienced a trade-off between benzoate-salicylate tolerance and inducible chloramphenicol resistance. This trade-off is confirmed by the plot of benzoate tolerance versus growth in chloramphenicol (Fig. 1E). Clones from the ancestral strain W3110 (red circles) and early generations (orange, generation 173; yellow, generation 399) showed little growth in 20 mM benzoate, but most grew in 8 μg/ml chloramphenicol (reached optical densities at 600 nm [OD600] of at least 0.05). Middle-generation clones (light green, 897; dark green, 1402) grew in benzoate to higher OD600 and showed variable growth in chloramphenicol. By 2,900 generations (purple), all clones reached OD600 values of at least 0.2 in 20 mM benzoate, but the clones barely grew at all in chloramphenicol. Growth of evolved clones in salicylate, versus growth in salicylate and chloramphenicol, showed a similar reciprocal relationship (Fig. 1F). Outliers appeared under all conditions, as expected under selection pressure (55).
Genome resequencing shows numerous SNPs, deletions, and IS5 insertion mutations.
After 2,000 generations, 8 clones showing benzoate tolerance were chosen for genome sequencing (as described in Materials and Methods). An additional 24 clones (one from each evolved population) were tested for chloramphenicol resistance; all were benzoate tolerant compared to strain W3110. Of these 24 clones, 8 were chosen for decreased resistance to chloramphenicol. Both sets of 8 clones (16 in all) were streaked for isolation and established as strains (Table 1). We used the breseq (version 0.27.1) computational pipeline to analyze the mutation predictions for our resequenced genomes compared to the ancestral E. coli W3110 strain (lab stock D13), assembled on the W3110 reference genome (56). More than 100 mutations were detected across the 16 sequenced genomes (see Table S1 in the supplemental material). Mutations found in our resequenced lab stock D13 compared to the reference W3110 (see Table S2 in the supplemental material) were filtered from the results. The types of mutations that accumulated across the 16 strains included single-nucleotide polymorphisms (SNPs), small indels, insertion sequences (IS), and large IS-mediated deletions. Both coding changes and intergenic mutations were frequent. A large number of insertion knockouts were mediated by mobile insertion sequence elements such as IS5 (57). An example, gadX::IS5 found in clone A5-1, is shown in Fig. 2. The inserted sequence, including insH plus IS5 flanking regions, was identical to those of 11 known IS5 inserts in the standard W3110 sequence; there was also a 4-bp duplication of the target site. Insertion sequence mobility is a major source of evolutionary change in E. coli (58).
TABLE 1.
Bacterial strains used in this study
| Strain | Population no. or relevant genotype | Source or reference |
|---|---|---|
| W3110 | Escherichia coli K-12 stock no. D13 | Fred Neidhardt |
| JLSK0001 | W3110 benzoate-evolved A1-1 | This study |
| JLSK0011 | W3110 benzoate-evolved A3-1 | This study |
| JLSK0022 | W3110 benzoate-evolved A5-1 | This study |
| JLSK0023 | W3110 benzoate-evolved A5-2 | This study |
| JLSK0002 | W3110 benzoate-evolved B1-1 | This study |
| JLSK0003 | W3110 benzoate-evolved C1-1 | This study |
| JLSK0014 | W3110 benzoate-evolved C3-1 | This study |
| JLSK0015 | W3110 benzoate-evolved C3-2 | This study |
| JLSK0026 | W3110 benzoate-evolved D5-1 | This study |
| JLSK0006 | W3110 benzoate-evolved E1-1 | This study |
| JLSK0007 | W3110 benzoate-evolved E1-2 | This study |
| JLSK0010 | W3110 benzoate-evolved H1-1 | This study |
| JLSK0020 | W3110 benzoate-evolved H3-1 | This study |
| JLSK0019 | W3110 benzoate-evolved G3-1 | This study |
| JLSK0030 | W3110 benzoate-evolved G5-1 | This study |
| JLSK0031 | W3110 benzoate-evolved G5-2 | This study |
| JW5249-1 | BW25113 marA752::kanR | 81 |
| JLS1506 | W3110 marA752::kanR | This study |
| MG1655 marR::kanR | MG1655 marR::kanR | Fred Blattner |
| JLS1610 | W3110 marR::kanR | This study |
| JW3484-1 | BW25113 gadX771::kanR | 81 |
| JLS1517 | W3110 gadX771::kanR | This study |
| JW5801-1 | BW25113 yjjX790::kanR | 81 |
| JLS1514 | W3110 yjjX790::kanR rob+ | This study |
| JW3487-1 | BW25113 treF774::kanR | 81 |
| JLS1612 | G5-2 treF774::kanR gadX+ | This study |
| JLS1613 | G5-2 yjjX790::frt rob+ treF774::kanR gadX+ | This study |
| JW3866-3 | BW25113 fdhD758::kanR | 81 |
| JLS1607 | A1-1 fdhD758::kanR cpxA+ | This study |
FIG 2.
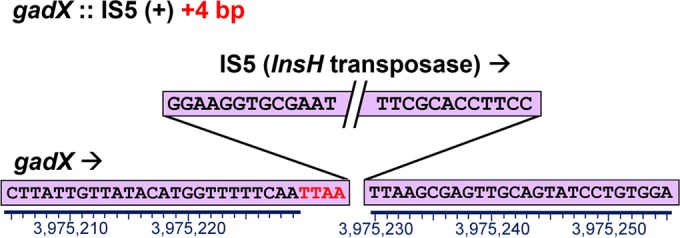
New insertion of IS5 within gadX, including a 4-bp duplication of the target site, in the A5-1 genome at position 3975230.
The 2,000-generation benzoate-adapted strains were grouped in six clades based on shared mutations (Table S1); a representative member of each clade is shown in Table 2. Most of the shared mutations originated within a population, as in the case of the two strains taken from each of the A5, E1, and C3 populations, and from inadvertent cross-transfer between microplate wells. For example, one population (G5) included a strain, G5-2, that shares mutations with the strains from the H1, H3, and G3 populations while sharing no mutations with strain G5-1, from the G5 population. Note also that isolated shared mutations can originate from the shared founder culture or can arise as independent genetic adaptations to a common stress condition (55).
TABLE 2.
Mutations in representative benzoate-evolved genomes compared to the genome of E. coli W3110a

E. coli W3110 was the NCBI reference strain (accession no. NC_007779.1). Δ, the mutation site is absent within a larger deleted region. Percent scores indicate breseq calls of less than 100%. Background colors designate strains. Mutation annotations: red, changed base pairs; green, synonymous mutation; blue, missense mutation; purple, IS-mediated deletion; *, nonsense mutation. Mutations present in our laboratory stock strain W3110-D13 compared to the NCBI reference strain (see Table S2 in the supplemental material) are omitted.
Mutations in Mar and other multidrug efflux systems.
Five of the six clades showed mutations affecting the Mar regulon, as well as other MDR genes (Table 2). Strain A5-1 had a 6,115-bp deletion including marRAB (ydeA, marRAB, eamA, and ydeEH). Regulators of Mar showed point mutations in strain G5-2 (mar paralog rob [26]) and in strain A1-1 (two-component activator cpxA [59]). Additional mutations appeared in other multidrug efflux systems: emrA and emrY (32, 60), mdtA and deletions covering mdtEF (60), and yeaS (leuE) leucine export (61).
The benzoate-evolved strains were tested by MIC assay for sensitivity to the antibiotics chloramphenicol and tetracycline, whose resistance is inducible by benzoate derivatives (Table 3). Sensitivity was assayed in the presence or absence of a Mar inducer, 2 mM salicylate. Salicylate increased the MIC for our ancestral strain W3110 for both chloramphenicol and tetracycline. The W3110 marA::kanR construct showed less of an increase in the MIC for chloramphenicol. For tetracycline, our marA knockout strain showed no loss of resistance; this may be due to induction of non-Mar salicylate-dependent resistance (24).
TABLE 3.
MICs of benzoate-evolved strains for the antibiotics chloramphenicol and tetracycline with or without salicylatea
| Strain | MIC (μg/ml)b |
|||
|---|---|---|---|---|
| Chloramphenicol |
Tetracycline |
|||
| No inducer | 2 mM salicylate | No inducer | 2 mM salicylate | |
| W3110 | 6 | 16 | 1 | 4 |
| W3110 marA::kanR | 6 | 8 | 1.5 | 4 |
| A1-1 | 6 | 8 | 1.5 | 2 |
| A5-1 | 6 | 8 | 2.5 | 1 |
| C3-1 | 6 | 8 | 1 | 3 |
| E1-1 | 6 | 16 | 1 | 4 |
| G5-1 | 7 | 12 | 1 | 2 |
| G5-2 | 4 | 8 | 1 | 2 |
Strains were cultured in LBK–100 mM MOPS (pH 7.0) with or without 2 mM salicylate for 22 h. Positive for growth was defined as an OD600 of ≥0.05 at 22 h. The results shown represent one of three trials overall.
Median values from 8 replicates are shown.
In the presence of salicylate, our evolved strains A1-1, A5-1, C3-1, and G5-2 showed MIC levels half that of ancestor W3110. These lowered MIC levels were comparable to that of a marA mutant, and only E1-1 showed chloramphenicol resistance equivalent to that of W3110. In the absence of salicylate, the ancestral and evolved strains all showed lower MIC values due to a lack of inducer. Strain G5-2 showed a slightly lower MIC than W3110 (4 μg/ml versus 6 μg/ml, respectively). Thus, it is possible that G5-2 has lost an additional component of chloramphenicol resistance that does not require salicylate induction.
For tetracycline, salicylate-inducible resistance was less than that of W3110 for all of our benzoate-evolved strains with the exception of strain E1-1. The tetracycline MIC values, however, showed some variability; for all strains, uninducible MIC levels varied among trials from 1 to 3 μg/ml. For further analysis, we focused on chloramphenicol.
Mutations in Gad acid resistance, RNA polymerase (RNAP), and fimbriae.
The Gad acid resistance genes are induced during cytoplasmic acidification (37) and show regulation intertwined with that of MDR systems (35). Strikingly, five of the six clades of our 16 sequenced strains showed a mutation in the Gad regulon (Table 2; Table S1). Strain E1-1 had a 14,146-bp deletion mediated by the insertion sequence insH flanking the gad acid fitness island (gadXW, mdtFE, gadE, hdeDAB, yhiDF, slp, insH, and yhiS). Similarly, strain A1-1 showed a 10,738-bp deletion covering most of the gad region. A1-1 also had an insertion in the ariR (ymgB) biofilm-dependent activator of Gad (37, 62). The mdtFE genes encode components of the efflux pump MdtF-MdeE-TolC, which confers resistance to chloramphenicol as well as fluoroquinolones and other drugs (63). Thus, gad deletion might explain the chloramphenicol sensitivity of strain A1-1, but not that of E1-1, which was chloramphenicol resistant despite the deletion. Other strains showed mutations in the gadX activator: A5-1 (IS5 insertion), G5-1 (missense L199F), and G5-2 (78-bp deletion). G5-2 also showed an hfq point substitution at a position known to affect the function of RpoS (64), which activates Gad (2).
The gad mutants showed different levels of gamma-aminobutyric acid (GABA) production by glutamate decarboxylase (GadA) during extreme-acid exposure (incubation at pH 2) (Fig. 3). The two strains with full Gad deletions (A1-1 and E1-1) produced no GABA, whereas strains with gadX mutations (A5-1, G5-1, and G5-2) produced significantly less GABA than did the ancestor W3110 (by the Friedman-Conover test). Only one representative strain, C3-1, showed no Gad-related mutation; the nearest mutation was an IS5 insertion adjacent to slp. This strain produced GABA in amounts comparable to those produced by W3110.
FIG 3.
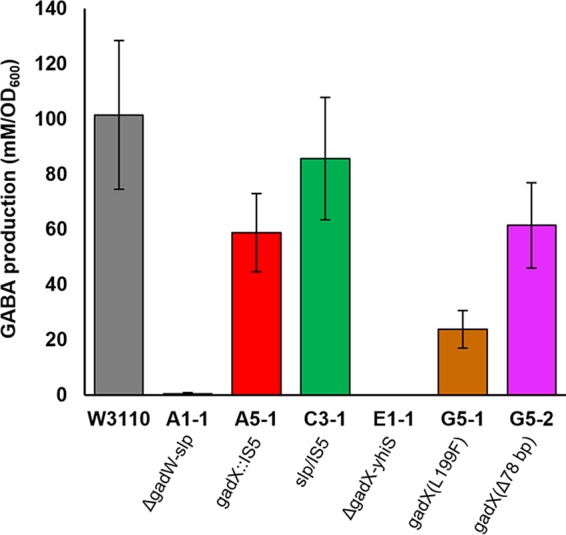
GABA produced by benzoate-evolved strains compared to W3110. Anaerobic overnight cultures in LB 10 mM glutamine–100 mM MES (pH 5.5) were adjusted with HCl to pH 2. After 2 h of incubation, bacteria were pelleted, and supernatant culture fluid was derivatized using EZ:faast (see Materials and Methods). GABA was quantified via GC-MS, and values were normalized to the cell density of the overnight culture. Error bars represent standard error of the mean (SEM) (n = 7 or 8). Genetic annotations are from Table 1.
Each benzoate-adapted strain also showed a mutation in an RNAP subunit gene (rpoB or rpoA), a sigma factor gene (rpoD or rpoS), or an RNAP-associated helicase gene (hepA). These mutations in the transcription apparatus are comparable to those we find under low-pH evolution (49). Four of the six clades had mutations in fimbria subunit gene fimA or in regulator gene fimB or fimE. Thus, benzoate exposure could select for loss of fimbria synthesis. Other interesting mutations affected cell division (ftsZ), cell wall biosynthesis (mrdA), and envelope functions (ecpD, lptD, ybbP, yejM, yfhM, yqiGH, and rfaY). The envelope mutations suggest responses to benzoate effects on the outer membrane and periplasm.
Benzoate-evolved strains show increased growth rates and stationary-phase cell densities.
In our evolution experiment, the microplate growth cycle (49) involves initially oxygenated cultures, which become semianaerobic and ultimately enter stationary phase for several hours. Thus, selection pressure occurs under changing conditions of oxygenation and cell density. Dynamic conditions are relevant to host environments such as the intestinal epithelium (65) and arterial plaque biofilms (66).
We observed the phases of growth for each benzoate-evolved strain in order to characterize the focus of selection pressure with respect to early growth rate, stationary-phase cell density, and death phase. Observing the entire growth curve provides more information than an endpoint MIC. Growth curves were determined in microplate wells for each of the six representative 2,000-generation benzoate-adapted strains. For each strain, eight replicate wells of the microplate were inoculated alongside eight replicate wells of strain W3110, as shown for strain G5-2 (Fig. 4A).
FIG 4.
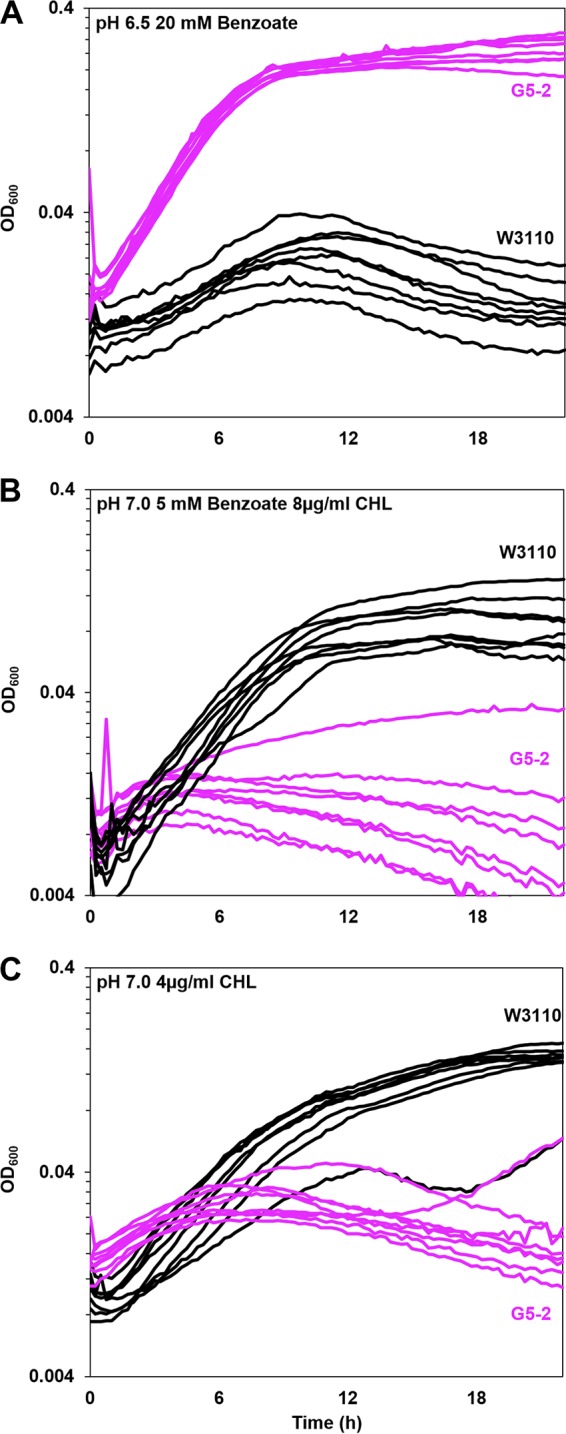
Benzoate-evolved strain G5-2 outgrows ancestor W3110 in the presence of benzoate but grows poorly in benzoate with chloramphenicol. The growth medium was LBK with (A) 100 mM PIPES and 20 mM benzoate (pH 6.5), (B) 100 mM MOPS, 5 mM benzoate (pH 7.0), and 8 μg/ml chloramphenicol (CHL), or (C) 100 mM MOPS (pH 7.0) and 4 μg/ml CHL. For each strain, 8 replicate curves from microplate wells are shown. For strains G5-2 and W3110 in each panel, cell density values post-log phase (OD600 at 16 h) were ranked and compared by using the Friedman test; post hoc Conover pairwise comparisons were conducted with Holm-Bonferroni-adjusted P values.
In the example shown, strain G5-2 maintained log-phase growth for approximately 4 h in the presence of 20 mM benzoate (0.42 ± 0.5 generation/h, measured over times 1 to 3 h). Strain G5-2 eventually reached a stationary-phase OD600 of approximately 1.0. In contrast, ancestral strain W3110 grew more slowly (0.18 ± 0.01 generation/h) and peaked at an OD600 of 0.5 to 0.7 by about 8 h. After 8 h, W3110 entered a death phase as the cell density declined. The presence of chloramphenicol, however, reversed the relative fitnesses of the two strains (Fig. 4B). The benzoate-evolved strain barely grew, and 7 of 8 replicates entered death phase by 3 to 4 h. In contrast, the ancestor grew steadily to an OD600 of 0.5 to 0.6, a level that was sustained for several hours.
At a lower concentration of chloramphenicol (4 μg/ml), the parental strain W3110 outgrew strain G5-2, even in the absence of benzoate inducer (Fig. 4C). This observation confirms the MIC result (Table 3) that G5-2 shows loss of an unidentified means of chloramphenicol resistance, independent of benzoate or salicylate. Other isolates showed only loss of benzoate-inducible resistance (presented below).
The effects of various permeant acids were tested in order to determine the specificity of acid tolerance (Fig. 5). For all growth curves, statistical comparison was performed using cell density values at 16 h. Each panel in Fig. 5 shows a curve with median cell density (at 16 h) for a benzoate-evolved strain as well as for strain W3110. Both benzoate and salicylate conditions showed a marked fitness advantage for all six benzoate-evolved strains (Fig. 5A and B). Five of the strains showed log-phase growth rates equivalent to each other, whereas G5-1 grew significantly more slowly during log phase (Friedman and Conover tests, P ≤ 0.05). All benzoate-evolved strains grew faster than strain W3110. All six benzoate-evolved strains reached equivalent plateau cell densities (OD600 values of approximately 1.0 with 20 mM benzoate and 0.9 with 10 mM salicylate). In contrast, in 20 mM benzoate, strain W3110 entered death phase by 10 h. This observation strongly suggests that death rates contribute to benzoate selection, besides the nominal 6.6 generations of growth per dilution cycle.
FIG 5.

Benzoate-evolved strains all outgrow the ancestor in benzoate or salicylate but not in acetate or sorbate. Growth curves for benzoate-evolved strains (colored curves) and the ancestor (black curve) in LBK–100 mM PIPES (pH 6.5) with (A) 20 mM benzoate, (B) 10 mM salicylate, (C) 20 mM acetate, or (D) 20 mM sorbate are shown. For each strain, a curve with median cell density at 16 h is shown. Panels A and B (but not C and D) showed significantly lower 16-h cell density for the ancestral W3110 strain than for benzoate-evolved strains (Friedman test and post hoc Conover pairwise comparisons with Holm-Bonferroni-adjusted P values).
In the presence of the aliphatic acid acetate or sorbate (Fig. 5C and D), no significant difference was seen between growth of the ancestral strain W3110 and that of the benzoate-evolved strains. Thus, the evolved fitness advantage is unlikely to result from cytoplasmic pH depression but appears to be specific to the presence of the aromatic acid benzoate or salicylate. Further testing with the uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP) at 40 μM showed no difference in growth rate or stationary-phase cell density among the strains (data not shown). Thus, while a decrease of proton motive force may be one factor, it cannot be the sole cause of the fitness advantage of our strains.
We also tested whether the benzoate-evolved strains showed any fitness advantage with respect to pH stress. The strains were cultured in media buffered at pH 7.0 (Fig. 6A) and at pH 4.8 or pH 9.0 (see Fig. S2 in the supplemental material). All of the benzoate-evolved strains grew similarly to the ancestor at external pH values across the full range permitting growth. Thus, the fitness advantage of the evolved strains was specific to the presence of benzoate or salicylate.
FIG 6.

Benzoate-evolved strains are sensitive to chloramphenicol. Growth curves for benzoate-evolved strains and the ancestor, compared to W3110 strains with marR and for marA deleted, are shown. Media contained LBK–100 mM MOPS (pH 7.0) with (A) no supplements, (B) 8 μg/ml chloramphenicol, (C) 5 mM benzoate and 8 μg/ml chloramphenicol, or (D) 2 mM salicylate and 8 μg/ml chloramphenicol. For each strain, a curve with median 16-h cell density is shown. Brackets indicate curves with cell densities at16 h that were lower than the cell density of ancestral strain W3110 (Friedman test and post hoc Conover pairwise comparisons with Holm-Bonferroni-adjusted P values). For panels C and D, Fig. S3 in the supplemental material shows all replicates of each strain plotted individually.
Chloramphenicol inhibits growth of benzoate-evolved clones, despite a benzoate fitness advantage.
Since each of the six clades showed a mutation in an MDR gene or regulator (discussed above), we characterized the growth profiles of all strains in the presence of chloramphenicol (Fig. 6B, C, and D). In the absence of a benzoate or salicylate inducer (Fig. 6B), all strains showed growth curves equivalent to that of W3110 or W3110 marA::kanR (which lacks the MarA activator of MDR efflux). Only the marR::kanR strains (constitutive for activator marA) showed resistance. In the presence of benzoate (Fig. 6C) or salicylate (Fig. 6D), the various benzoate-evolved strains reached different cell densities in the presence of chloramphenicol. Panels presenting results for all 8 replicates of each strain are in Fig. S3 and S4 in the supplemental material.
In the presence of chloramphenicol (with benzoate or salicylate), only strain E1-1 consistently grew at a rate comparable to that for W3110 (Fig. 6C and D). Strains A5-1 and G5-1 grew to a lower density, comparable to that for the marA::kanR strain. Strains C3-1 and G5-2 showed hypersensitivity to chloramphenicol, with cell densities significantly below that of the marA::kanR strain. The sensitivity of strain C3-1 is noteworthy given the absence of Mar or Gad mutations. Another strain, G5-2, shows chloramphenicol sensitivity greater than the level that would be predicted from loss of Rob activating MarA (25). Thus, the C3-1 and G5-2 genomes may reveal defects in other benzoate-inducible MDR genes, as yet unidentified.
Mutations in rob, gadX, and cpxA do not affect chloramphenicol sensitivity or benzoate tolerance.
Since five of the six benzoate-evolved strains showed defects affecting the Gad regulon, we tested the role of gadX in chloramphenicol resistance (Fig. 7). In the W3110 background, a gadX::kanR knockout (green) showed more sensitivity to chloramphenicol, as did the construct W3110 marA::kanR (gray). Thus, it is possible that GadX has some uncharacterized role in chloramphenicol efflux, either via activation of the Mar regulon or else via activation of mdtE and mdtF in the Gad fitness island (36).
FIG 7.
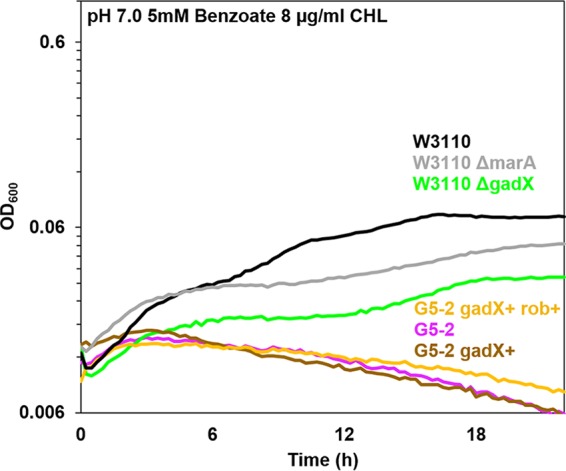
W3110 gadX::kanR shows sensitivity to chloramphenicol. Growth curves were determined in LBK–100 mM PIPES (pH 7.0) with 5 mM benzoate and 8 μg/ml chloramphenicol, as for Fig. 6. At 16 h, the gadX::kanR knockout mutant and the marA::kanR strain both grew significantly less than strain W3110. The cell density of strain G5-2 showed no difference from those of G5-2 gadX+ or G5-2 gadX+ rob+ (Friedman test and post hoc Conover pairwise comparisons with Holm-Bonferroni-adjusted P values).
We also tested the roles of gadX and rob in chloramphenicol sensitivity of the benzoate-evolved strain G5-2. The G5-2 mutant alleles of Mar activator rob (S34P) and Gad activator gadX (Δ78bp) were replaced by cotransduction. The ancestral alleles of rob and of gadX were each moved into G5-2 by cotransduction with linked markers yjjX790::kanR and treF774::kanR, respectively. Constructs of G5-2 with allele replacements gadX+ and rob+ gadX+ were cultured with chloramphenicol in the presence of 5 mM benzoate (Fig. 7). Both constructs were as sensitive to chloramphenicol as the parental G5-2 strain. Furthermore, strain G5-2 constructs with either rob+, gadX+, or rob+ gadX+ showed no significant loss of benzoate tolerance compared to that of the parental strain G5-2 (data not shown). Thus, both the benzoate fitness advantage and the chloramphenicol sensitivity of G5-2 must involve other, unidentified mutations in addition to rob or gadX.
We also tested the effect of cpxA+ on the benzoate fitness and chloramphenicol sensitivity of strain A1-1. The parental allele of MDR regulator cpxA was moved from W3110 into A1-1, replacing cpxA(N107S) with the marker fdhD::kanR linked to cpxA+ (see Fig. S5 in the supplemental material). Growth curves were determined as for Fig. 5 and 6, tested under the conditions of 20 mM benzoate (pH 6.5), benzoate with chloramphenicol, and salicylate with chloramphenicol. Under all conditions, the cell density of strain W3110 cpxA::kanR (Fig. S5, orange) showed no difference from that of W3110, and strain JLS1607 (A1-1 fdhD758::kanR cpxA+) (brown) showed no difference from JLSK0001 (A1-1) (blue). Thus, strain A1-1 must also contain benzoate-selected mutations in other, unknown MDR genes.
DISCUSSION
In order to identify candidate genes for the benzoate stress response, we sequenced the genomes of experimentally evolved strains (41, 42, 46, 49, 67). We observed over 100 distinct mutations in the sequenced isolates after 2,000 generations, including a surprising number of knockout alleles due to mobile elements (57, 58), in particular IS5 insertions and IS5-mediated deletions (52). The E. coli K-12 W3110 genome contains many IS elements, including eight copies of IS1, five copies of IS2, and copies of other less well-studied IS types, where the most prevalent are IS1 and IS5 (56, 68, 69). Transposition of IS5 may be induced by environmental factors such as motility conditions, which induce IS5 insertion upstream of the motility regulator gene flhD (70). Nonetheless, finding an additional 25 insertion sequences under benzoate selection is remarkable. Our detection of such IS inserts was enabled by use of the updated breseq pipeline (53).
Benzoate exposure decreases the cell's PMF while simultaneously upregulating several regulons, including many involved in drug resistance. Our data suggest that benzoate exposure selects for genetic changes in E. coli that result in, over time, the loss of energetically costly systems such as Mar and other MDR regulons, as well as the Gad acid-inducible extreme-acid regulon. MarA is a potent transcriptional factor in E. coli, upregulating numerous efflux pumps and virulence factors (8, 25, 27). The transcription and translation of so many gene products would result in a considerable energy strain on the individual cell.
Many MDR complexes are efflux pumps that spend PMF, which is diminished by partial uncouplers such as benzoate or salicylate. The decreased energy expense could explain the benzoate fitness advantage of strains that have broad-spectrum downregulation of Mar gene products, such as those we see in A5-1 and G5-2. At least one strain (G5-2) shows detectable loss of chloramphenicol resistance in the absence of Mar inducer, suggesting loss of constitutive MDR as well as the inducible system. Thus, it might be possible for benzoate and salicylate to select against a broad spectrum of drug resistance systems.
A similar energy load may occur under benzoate-depressed cytoplasmic pH, where the Gad regulon is induced. Gad includes expression of numerous gene products such as glutamate decarboxylase, whose activity enhances fitness only in extreme acid (pH 2) (35) and which breaks down valuable amino acids. Thus, the deletion of the gad region (seen in strains A1-1 and E1-1) could eliminate a fruitless energy drain. For comparison, we found similar Gad deletions in our pH 4.6 evolution experiment (A. He, S. R. Penix, P. J. Basting, J. M. Griffith, K. E. Creamer, D. W. Camperchioli, M. W. Clark, A. S. Gonzales, J. S. Chavez Erazo, N. S. George, A. A. Bhagwat, and J. L. Slonczewski, submitted), but not in our evolution experiment conducted at pH 9.2 (unpublished data). This implies that both pH 4.6 and benzoate/pH 6.5 induce Gad under conditions where glutamate decarboxylase fails to help the cell and thus the energy-expensive Gad expression is selected against.
The Gad region also includes genes encoding an MDR complex (mdtEF) (36), and a gadX knockout showed some loss of chloramphenicol resistance (Fig. 7). Note, however, that our E1-1 strain retains chloramphenicol resistance despite Gad deletion, whereas strain C3-1 (chloramphenicol sensitive) possesses the entire Gad region except for a possible defect in slp, encoding an acid resistance outer membrane protein. Thus, Gad mutations alone cannot explain the chloramphenicol sensitivities of our strains.
Another possible consequence of energy stress is the loss of fimbria synthesis (strains A5-1, C3-1, E1-1, and G5-1). Avoiding fimbria production could save energy for benzoate-stressed cells. Fim genes also show deletion under evolution at pH 9.2 (I. N. Hamdallah, unpublished data) but not at pH 4.6. This suggests a hypothesis that fimbria biosynthesis is dependent less on pH than on the proton motive force (PMF), which is depleted both by benzoate and at high external pH, a condition that decreases PMF (5).
The progressive loss of antibiotic resistance is a remarkable consequence of benzoate selection, evident at as early as 1,500 generations (Fig. 1). Several observations point to the existence of inducible MDR systems yet to be discovered. Strains C3-1 and G5-2 show hypersensitivity to chloramphenicol, beyond the level of sensitivity seen in a marA knockout (Fig. 6C and D; see Fig. S4 in the supplemental material). Furthermore, the reversion of mutant alleles of rob, gadX, and cpxA does not diminish the phenotypes of the 2,000-generation strains. It is likely that these alleles conferred a fitness advantage early on in our evolution experiment (55) but were then been superseded by further mutations in as-yet-unidentified players in drug resistance.
The fitness trade-off between drug resistance and benzoate/salicylate exposure has implications for the human microbiome. Mar and homologs such as Mex are reported in numerous bacteria, including proteobacteria and Bacteroides fragilis (71, 72). Salicylate is a plant defense molecule commonly obtained via human diets rich in fruits and vegetables (73, 74). Aspirin is deacetylated in the liver and stomach, forming salicylic acid, the primary therapeutic agent (75). As a membrane-permeant acid, salicylic acid permeates human tissues nonspecifically. Both food-related and aspirin-derived salicylates come in contact with enteric gut bacteria, where they would be expected to activate Mar-like antibiotic resistance systems. Commonly prescribed for cardiac health, aspirin releases salicylate at plasma levels of approximately 0.2 mM (16, 19, 20, 22).
Intestinal salicylate levels are poorly understood, but even lower concentrations of this antimicrobial agent could have fitness effects. For comparison, low concentrations of antibiotics, well below the MIC, can select for resistance (76, 77). Similarly, it may be that low concentrations of a resistance-reversing agent such as salicylate have a significant fitness cost for MDR bacteria. Furthermore, the effective concentration of permeant acids such as salicylate is amplified exponentially by the pH difference across the bacterial plasma membrane. Even mild acidity in the intestinal lumen (pH 6 to 6.5) could amplify the bacterial cytoplasmic concentration of a permeant acid by 10- to 30-fold.
Aspirin therapy is known to prevent clotting by inactivation of human cyclooxygenase, leading to suppression of prostaglandins. There is little attention, however, to the possible effects of aspirin on human-associated bacteria. Gram-negative pathogens such as Pseudomonas are found in arterial plaques and are associated with heart attacks (66). Aspirin-derived salicylate in plasma might provide a fitness cost for such bacteria. Aspirin also prevents colon cancer, by some unknown mechanism (16, 20). Colon cancer depends on colonic bacteria and the formation of biofilms (78, 79).
Long-term salicylate exposure via aspirin therapy may select a microbiome that is salicylate tolerant but drug sensitive. A salicylate-adapted microbiome may confer the benefit of excluding drug-resistant pathogens that lack salicylate tolerance. In blood plasma, salicylate levels might help exclude bacteria from arterial plaques. An adverse consideration, however, is that the salicylate-adapted microbiome of the colon may be more vulnerable to high-dose antibiotic therapy. For the future, we are testing these speculative possibilities in host microbial models.
MATERIALS AND METHODS
Bacterial strains and media.
Escherichia coli K-12 W3110 (80) was the ancestral strain of all benzoate-adapted populations. Additional strains derived from E. coli K-12 W3110 were isolated during the course of the evolution experiment (Table 1). Alleles with kanR insertions were obtained from the Keio collection (81) distributed by the E. coli Genetic Stock Center (CGSC). A marR::kanR strain was provided by Frederick R. Blattner, University of Wisconsin—Madison.
Bacteria were cultured in LBK (10 g/liter tryptone, 5 g/liter yeast extract, 7.45 g/liter KCl) (82). Culture media were buffered with either 100 mM piperazine-N,N′-bis(ethanesulfonic acid) (PIPES) (pKa = 6.8), 100 mM 2-(N-morpholino)ethanesulfonic acid (MES) (pKa = 5.96), 100 mM 3-morpholinopropane-1-sulfonic acid (MOPS) (pKa = 7.20), 100 mM homopiperazine-N,N′-bis-2-(ethanesulfonic acid) (HOMOPIPES) (pKa = 4.55, 8.12), or 150 mM N-tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid (TAPS) (pKa = 8.4). The pH of the medium was adjusted as necessary with either 5 M HCl or 5 M KOH. Potassium benzoate (referred to as benzoate), sodium salicylate (salicylate), potassium acetate, potassium sorbate, chloramphenicol, or tetracycline was added before filter sterilization for LBK media requiring various concentrations of acids or antibiotics. The temperature of incubation was 37°C unless noted otherwise.
Experimental evolution.
Experimental evolution was conducted according to the procedure for our low-pH laboratory evolution experiment (49) with modifications. Briefly, 24 cultures derived from the same ancestral strain (W3110, freezer stock D13) were cultured continuously in increasing concentrations of benzoate for 2,000 generations (see Fig. S1 in the supplemental material). An overnight culture of ancestral Escherichia coli K-12 W3110 was diluted 1:100 in LBK (pH 6.5)–100 mM PIPES–5 mM potassium benzoate. Growth was recorded over 22 h in a SpectraMax Plus384 MicroPlate reader (Molecular Devices). Every 15 min, the microplate was shaken for 3 s and the OD450 of each culture was recorded. The cultures were rediluted 1:100 into fresh benzoate growth medium at the end of the daily cycle. One hundred microliters of glycerol (50% glycerol, 100 mM MES, pH 6.5) was added to each well, after which the microplate was frozen at −80°F (49). The number of generations of the exposed cells was calculated based on the 1:100 daily dilution, resulting in a 100-fold daily growth to achieve approximately 6.6 generations of binary fission (83). In the course of the 22-hour cycle, all bacterial populations attained stationary-phase densities. Plating of representative cultures showed that during the dilution cycle, cell numbers increased from approximately 5 × 106 cells per ml to 5 × 108 cells per ml.
Over the course of the experiment, the benzoate concentration was increased in steps when all populations of the plate had attained approximately 100-fold stationary growth over multiple dilution cycles (Fig. S1). The benzoate was increased to 6 mM after 60 generations, to 10 mM after 90 generations, to 12 mM after 540 generations, to 15 mM after 1,020 generations, to 18 mM after 1,210 generations, and to 20 mM after 1,580 generations, to the conclusion of the experiment with a cumulative 3,000 generations of growth. If the strains had to be restarted from a frozen microplate, the frozen cultures were thawed and diluted 1:50 into fresh potassium benzoate growth medium.
After 2,000 generations, microplates were taken from the freezer and samples from specific wells were spread on LBK agar plates. Selected clones from each chosen well were streaked three times and stored as freezer stocks. Clones were cultured in medium at pH 6.5, with 5 mM benzoate; all clones showed increased growth compared to ancestral strain W3110 (data not shown). For genome sequencing, eight clones were chosen in pairs from each of four populations (clones A5-1, A5-2, C3-1, C3-2, E1-1, E1-2, G5-1, and G5-2). A total number of 24 clones (one from each population) were tested for sensitivity to chloramphenicol (8 μg/ml) in 5 mM benzoate medium, pH 7.0. From these, eight additional chloramphenicol-sensitive clones were selected for genome sequencing (A1-1, A3-1, B1-1, C1-1, D5-1, G3-1, H1-1, and H3-1). All 16 strains are identified in Table 1; and their genomic mutations (compared to ancestral strain W3110) are presented in Table S1 in the supplemental material. Mutations in six selected strains are presented in Table 2. These strains are color coded throughout our figures.
Growth assays.
Growth curves were measured in the microplate reader at 37°C for 22 h under various conditions of organic acids, pH values, and antibiotics. Strains were cultured overnight in LBK at pH 5.5 buffered with 100 mM MES, LBK at pH 6.5 buffered with 100 mM PIPES, LBK at pH 7.0 buffered with 100 mM MOPS, or LBK at pH 8.5 buffered with 150 mM TAPS. Supplements included benzoate, salicylate, acetate, or sorbate, as stated in the figure legends. Overnight cultures were diluted 1:100 (1:200 for the antibiotic growth assays) into the exposure media, which included LBK at pH 6.5 buffered with 100 mM PIPES; LBK at pH 4.8 with 100 mM HOMOPIPES, LBK at pH 7.0 with 100 mM MOPS, LBK at pH 9.0 with 150 mM TAPS, or LBK at pH 7.0 with 100 mM MOPS. Every 15 min, the plate was shaken for 3 s and an OD600 measurement was recorded. The growth rate k of each culture was calculated over the period of 1 to 3 h, approximately the log phase of growth (34). The cell density of each culture was measured at 16 h unless stated otherwise.
Genomic DNA extraction and sequencing.
Genomic DNA from benzoate-evolved clones and from the ancestral wild-type strain W3110 (freezer stock D13) was extracted using the DNeasy DNA extraction kit (Qiagen) and the MasterPure Complete DNA and RNA purification kit (Epicentre). The DNA purity was confirmed by measuring the 260 nm/280 nm and 260 nm/230 nm absorbance ratios using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific), and the concentration of the DNA was measured using both the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) and a Qubit 3.0 fluorometer (Thermo Fisher Scientific), according to the manufacturer's instructions.
The genomic DNA was sequenced by the Michigan State University Research Technology Support Facility Genomics Core. For Illumina MiSeq sequencing, libraries were prepared using the Illumina TruSeq Nano DNA library preparation kit. After library validation and quantitation, they were pooled and loaded on an Illumina MiSeq flow cell. Sequencing was done in a 2- by 250-bp paired-end format using an Illumina 500 cycle V2 reagent cartridge. Base calling was performed by Illumina Real Time Analysis (RTA) v1.18.54, and the output of RTA was demultiplexed and converted to FastQ format with Illumina Bcl2fastq v1.8.4.
Sequence assembly and analysis using the breseq computational pipeline.
The computational pipeline breseq version 0.27.1 was used to assemble and annotate the resulting reads of the evolved strains (50–52). The current breseq version detects IS element insertions and IS-mediated deletions, as well as SNPs and other mutations (53). The reads were mapped to the E. coli K-12 W3110 reference sequence (NCBI GenBank accession number NC_007779.1) (56). Mutations were predicted by breseq by comparing the sequences of the evolved isolates to that of the ancestral strain W3110, lab stock D13 (52). In order to visualize the assembly and annotations of our evolved isolate sequences mapped to the reference E. coli K-12 W3110 genome, we used the Integrative Genomics Viewer (IGV) from the Broad Institute at MIT (84). Sequence identity of clones was confirmed by PCR amplification of selected mutations.
P1 phage transduction and strain construction.
P1 phage transduction was conducted by standard procedures to replace a linked mutation with the ancestral nonmutated allele, as well to construct knockout strains (49). Strains with kanR insertions (81) were introduced into the evolved strain of choice or the ancestral strain W3110. Constructs were confirmed by PCR amplification and Sanger sequencing of key alleles of the donor and recipient.
MIC assays.
For assays of MICs of antibiotics (chloramphenicol or tetracycline), the strains were cultured in a microplate for 22 h in LBK–100 mM MOPS (pH 7.0)–2 mM salicylate. The medium contained a range of antibiotic concentrations (0, 1, 2, 4, 6, 8, 12, 16, or 24 μg/ml). A positive result for growth was defined as measurement of an OD600 of ≥0.05. Each MIC was reported as the median value from 8 replicates. For each antibiotic, three sets of 8 replicates were performed, that is, a total of 24 replicates per strain.
GABA assays.
The procedure for measuring GABA production via glutamate decarboxylase was modified from that described in reference 85. Strains were cultured overnight in LB medium (10 g/liter tryptone, 5 g/liter yeast extract, 100 mM NaCl) buffered with 100 mM MES, pH 5.5. Glutamine (10 mM), which the bacteria convert to glutamate, the substrate of glutamate decarboxylase (86), was included. For anaerobic culture, closed 9-ml screw-cap tubes nearly full of medium were incubated for 18 h at 37°C. The pH of each sample was lowered with HCl to 2.0 for extreme-acid stress (87), and samples were incubated for 2 h with rotation. Cell density (OD600) was measured in microplate wells in a SpectraMax plate reader. One milliliter of each culture was pelleted in a microcentrifuge. The supernatant was filtered and prepared for gas chromatography-mass spectrometry (GC-MS) by EZ:faast amino acid derivatization (88). The GABA concentration was calculated using a standard solution prepared at a concentration of 200 nm/ml. GABA and other compounds from the culture fluid were identified using NIST library analysis. GABA concentrations were normalized to OD600 values of the overnight cultures before assay.
Accession number(s).
Sequence data have been deposited in the NCBI Sequence Read Archive (SRA) under accession number SRP074501.
Supplementary Material
ACKNOWLEDGMENTS
We thank Zachary Blount, Jeff Barrick, Michael Harden, Rohan Maddamsetti, Peter Lund, Daniel Barich, and Bradley Hartlaub for valuable discussions. We thank Anna Tancredi for testing the gadX mutant.
This work was funded by grant MCB-1329815 from the National Science Foundation and by the Kenyon College Summer Science Scholars.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02736-16.
REFERENCES
- 1.Kanjee U, Houry WA. 2013. Mechanisms of acid resistance in Escherichia coli. Annu Rev Microbiol 67:65–81. doi: 10.1146/annurev-micro-092412-155708. [DOI] [PubMed] [Google Scholar]
- 2.Foster JW. 2004. Escherichia coli acid resistance: tales of an amateur acidophile. Nat Rev Microbiol 2:898–907. doi: 10.1038/nrmicro1021. [DOI] [PubMed] [Google Scholar]
- 3.Audia JP, Webb CC, Foster JW. 2001. Breaking through the acid barrier: an orchestrated response to proton stress by enteric bacteria. Int J Med Microbiol 291:97–106. doi: 10.1078/1438-4221-00106. [DOI] [PubMed] [Google Scholar]
- 4.Krulwich TA, Sachs G, Padan E. 2011. Molecular aspects of bacterial pH sensing and homeostasis. Nat Rev Microbiol 9:330–343. doi: 10.1038/nrmicro2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slonczewski JL, Fujisawa M, Dopson M, Krulwich TA. 2009. Cytoplasmic pH measurement and homeostasis in bacteria and archaea. Adv Microb Physiol 55:1–79, 317. doi: 10.1016/S0065-2911(09)05501-5. [DOI] [PubMed] [Google Scholar]
- 6.Qin J, Li R, Raes J, Arumugam M, Solvsten Burgdorf K, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J-M, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, et al. . 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–67. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slonczewski JL, Macnab RM, Alger JR, Castle AM. 1982. Effects of pH and repellent tactic stimuli on protein methylation levels in Escherichia coli. J Bacteriol 152:384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosner JL, Slonczewski JL. 1994. Dual regulation of inaA by the multiple antibiotic resistance (Mar) and superoxide (SoxRS) stress response systems of Escherichia coli. J Bacteriol 176:6262–6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee CH, Oon JSH, Lee KC, Ling MHT. 2012. Escherichia coli ATCC 8739 adapts to the presence of sodium chloride, monosodium glutamate, and benzoic acid after extended culture. ISRN Microbiol 2012:965356. doi: 10.5402/2012/965356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beales N. 2004. Adaptation of microorganisms to cold temperatures, weak acid preservatives, low pH, and osmotic stress: a review. Compr Rev Food Sci Food Saf 3:1–20. doi: 10.1111/j.1541-4337.2004.tb00057.x. [DOI] [PubMed] [Google Scholar]
- 11.Wilks JC, Slonczewski JL. 2007. pH of the cytoplasm and periplasm of Escherichia coli: rapid measurement by green fluorescent protein fluorimetry. J Bacteriol 189:5601–5607. doi: 10.1128/JB.00615-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lennerz B, Vafai SB, Delaney NF, Clish CB, Deik AA, Pierce KA, Ludwig DS, Mootha VK. 2015. Effects of sodium benzoate, a widely used food preservative, on glucose homeostatsis and metabolic profiles in humans. Mol Genet Metab 114:73–79. doi: 10.1016/j.ymgme.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.An C, Mou Z. 2011. Salicylic acid and its function in plant immunity. J Integr Plant Biol 53:412–428. doi: 10.1111/j.1744-7909.2011.01043.x. [DOI] [PubMed] [Google Scholar]
- 14.Du L, Ali GS, Simons KA, Hou J, Yang T, Reddy ASN, Poovaiah BW. 2009. Ca2+/calmodulin regulates salicylic-acid-mediated plant immunity. Nature 457:1154–1158. doi: 10.1038/nature07612. [DOI] [PubMed] [Google Scholar]
- 15.Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, Kemp BE, Sakamoto K, Steinberg GR, Hardie DG. 2012. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336:918–922. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paterson JR, Lawrence JR. 2001. Salicylic acid: a link between aspirin, diet and the prevention of colorectal cancer. QJM 94:445–448. doi: 10.1093/qjmed/94.8.445. [DOI] [PubMed] [Google Scholar]
- 17.Paterson JR, Baxter G, Dreyer JS, Halket JM, Flynn R, Lawrence JR. 2008. Salicylic acid sans aspirin in animals and man: persistence in fasting and biosynthesis from benzoic acid. J Agric Food Chem 56:11648–11652. doi: 10.1021/jf800974z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spadafranca A, Bertoli S, Fiorillo G, Testolin G, Battezzati A. 2007. Circulating salicylic acid is related to fruit and vegetable consumption in healthy subjects. Br J Nutr 98:802–806. doi: 10.1017/S0007114507744422. [DOI] [PubMed] [Google Scholar]
- 19.Patrono C, García Rodríguez LA, Landolfi R, Baigent C. 2005. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med 353:2373–2383. doi: 10.1056/NEJMra052717. [DOI] [PubMed] [Google Scholar]
- 20.Patrono C. 2013. Low-dose aspirin in primary prevention: cardioprotection, chemoprevention, both, or neither? Eur Heart J 34:3403–3411. doi: 10.1093/eurheartj/eht058. [DOI] [PubMed] [Google Scholar]
- 21.Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, Shulman GI. 2002. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest 109:1321–1326. doi: 10.1172/JCI0214955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmed N, Meek J, Davies GJ. 2010. Plasma salicylate level and aspirin resistance in survivors of myocardial infarction. J Thromb Thrombolysis 29:416–420. doi: 10.1007/s11239-009-0366-7. [DOI] [PubMed] [Google Scholar]
- 23.Wang WH, Wong WM, Dailidiene D, Berg DE, Gu Q, Lai KC, Lam SK, Wong BCY. 2003. Aspirin inhibits the growth of Helicobacter pylori and enhances its susceptibility of antimicrobial agents. Gut 52:490–495. doi: 10.1136/gut.52.4.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen SP, Levy SB, Foulds J, Rosner JL. 1993. Salicylate induction of antibiotic resistance in Escherichia coli: activation of the mar operon and a mar-independent pathway. J Bacteriol 175:7856–7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duval V, Lister IM. 2013. MarA, SoxS and Rob of Escherichia coli—global regulators of multidrug resistance, virulence and stress response. Int J Biotechnol Wellness Ind 2:101–124. doi: 10.6000/1927-3037.2013.02.03.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chubiz LM, Rao CV. 2010. Aromatic acid metabolites of Escherichia coli K-12 can induce the marRAB operon. J Bacteriol 192:4786–4789. doi: 10.1128/JB.00371-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruiz C, Levy SB. 2010. Many chromosomal genes modulate MarA-mediated multidrug resistance in Escherichia coli. Antimicrob Agents Chemother 54:2125–2134. doi: 10.1128/AAC.01420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Critzer FJ, Dsouza DH, Golden DA. 2008. Transcription analysis of stx1, marA, and eaeA genes in Escherichia coli O157:H7 treated with sodium benzoate. J Food Prot 71:1469–1474. [DOI] [PubMed] [Google Scholar]
- 29.Perera IC, Grove A. 2010. Molecular mechanisms of ligand-mediated attenuation of DNA binding by MarR family transcriptional regulators. J Mol Cell Biol 2:243–254. doi: 10.1093/jmcb/mjq021. [DOI] [PubMed] [Google Scholar]
- 30.Alekshun MN, Levy SB, Mealy TR, Seaton BA, Head JF. 2001. The crystal structure of MarR, a regulator of multiple antibiotic resistance, at 2.3 Å resolution. Nat Struct Mol Biol 8:710–714. doi: 10.1038/90429. [DOI] [PubMed] [Google Scholar]
- 31.Barbosa TM, Levy SB. 2000. Differential expression of over 60 chromosomal genes in Escherichia coli by constitutive expression of MarA. J Bacteriol 182:3467–3474. doi: 10.1128/JB.182.12.3467-3474.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grkovic S, Brown MH, Skurray RA. 2002. Regulation of bacterial drug export systems. Microbiol Mol Biol Rev 66:671–701. doi: 10.1128/MMBR.66.4.671-701.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruiz C, McMurry LM, Levy SB. 2008. Role of the multidrug resistance regulator MarA in global regulation of the hdeAB acid resistance operon in Escherichia coli. J Bacteriol 190:1290–1297. doi: 10.1128/JB.01729-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deininger KNW, Horikawa A, Kitko RD, Tatsumi R, Rosner JL, Wachi M, Slonczewski JL. 2011. A requirement of TolC and MDR efflux pumps for acid adaptation and GadAB induction in Escherichia coli. PLoS One 6:e18960. doi: 10.1371/journal.pone.0018960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mates AK, Sayed AK, Foster JW. 2007. Products of the Escherichia coli acid fitness island attenuate metabolite stress at extremely low pH and mediate a cell density-dependent acid resistance. J Bacteriol 189:2759–2768. doi: 10.1128/JB.01490-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishino K, Senda Y, Yamaguchi A. 2008. The AraC-family regulator GadX enhances multidrug resistance in Escherichia coli by activating expression of mdtEF multidrug efflux genes. J Infect Chemother 14:23–29. doi: 10.1007/s10156-007-0575-Y. [DOI] [PubMed] [Google Scholar]
- 37.Kannan G, Wilks JC, Fitzgerald DM, Jones BD, Bondurant SS, Slonczewski JL. 2008. Rapid acid treatment of Escherichia coli: transcriptomic response and recovery. BMC Microbiol 8:37. doi: 10.1186/1471-2180-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anes J, McCusker MP, Fanning S, Martins M. 2015. The ins and outs of RND efflux pumps in Escherichia coli. Front Microbiol 6:587. doi: 10.3389/fmicb.2015.00587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wood KB, Cluzel P. 2012. Trade-offs between drug toxicity and benefit in the multi-antibiotic resistance system underlie optimal growth of E. coli. BMC Syst Biol 6:48. doi: 10.1186/1752-0509-6-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiss SJ, Mansell TJ, Mortazavi P, Knight R, Gill RT. 2016. Parallel mapping of antibiotic resistance alleles in Escherichia coli. PLoS One 11:e0146916. doi: 10.1371/journal.pone.0146916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blount ZD, Borland CZ, Lenski RE. 2008. Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli. Proc Natl Acad Sci U S A 105:7899–7906. doi: 10.1073/pnas.0803151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blount ZD, Barrick JE, Davidson CJ, Lenski RE. 2012. Genomic analysis of a key innovation in an experimental Escherichia coli population. Nature 489:513–518. doi: 10.1038/nature11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riehle MM, Bennett AF, Lenski RE, Long AD. 2003. Evolutionary changes in heat-inducible gene expression in lines of Escherichia coli adapted to high temperature. Physiol Genomics 14:47–58. doi: 10.1152/physiolgenomics.00034.2002. [DOI] [PubMed] [Google Scholar]
- 44.Sleight SC, Lenski RE. 2007. Evolutionary adaptation to freeze-thaw-growth cycles in Escherichia coli. Physiol Biochem Zool 80:370–385. doi: 10.1086/518013. [DOI] [PubMed] [Google Scholar]
- 45.Goodarzi H, Bennett BD, Amini S, Reaves ML, Hottes AK, Rabinowitz JD, Tavazoie S. 2010. Regulatory and metabolic rewiring during laboratory evolution of ethanol tolerance in E. coli. Mol Syst Biol 6:378. doi: 10.1038/msb.2010.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dragosits M, Mattanovich D. 2013. Adaptive laboratory evolution—principles and applications for biotechnology. Microb Cell Fact 12:64. doi: 10.1186/1475-2859-12-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hughes BS, Cullum AJ, Bennett AF. 2007. Evolutionary adaptation to environmental pH in experimental lineages of Escherichia coli. Evolution (NY) 61:1725–1734. doi: 10.1111/j.1558-5646.2007.00139.x. [DOI] [PubMed] [Google Scholar]
- 48.Johnson MD, Bell J, Clarke K, Chandler R, Pathak P, Xia Y, Marshall RL, Weinstock GM, Loman NJ, Winn PJ, Lund PA. 2014. Characterization of mutations in the PAS domain of the EvgS sensor kinase selected by laboratory evolution for acid resistance in Escherichia coli. Mol Microbiol 93:911–927. doi: 10.1111/mmi.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harden MM, He A, Creamer K, Clark MW, Hamdallah I, Martinez KA, Kresslein RL, Bush SP, Slonczewski JL. 2015. Acid-adapted strains of Escherichia coli K-12 obtained by experimental evolution. Appl Environ Microbiol 81:1932–1941. doi: 10.1128/AEM.03494-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barrick JE, Colburn G, Deatherage DE, Traverse CC, Strand MD, Borges JJ, Knoester DB, Reba A, Meyer AG. 2014. Identifying structural variation in haploid microbial genomes from short-read resequencing data using breseq. BMC Genomics 15:1039. doi: 10.1186/1471-2164-15-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deatherage DE, Traverse CC, Wolf LN, Barrick JE. 2015. Detecting rare structural variation in evolving microbial populations from new sequence junctions using breseq. Front Genet 5:468. doi: 10.3389/fgene.2014.00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deatherage DE, Barrick JE. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol 1151:165–188. doi: 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tenaillon O, Barrick JE, Ribeck N, Deatherage DE, Blanchard JL, Dasgupta A, Wu GC, Schneider D, Lenski RE. 2016. Tempo and mode of genome evolution in a 50,000-generation experiment. bioRXiv Prepr doi: 10.1101/036806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andersson DI, Hughes D. 2010. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol 8:260–271. doi: 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- 55.Maddamsetti R, Lenski RE, Barrick JE. 2015. Adaptation, clonal interference, and frequency-dependent interactions in a long-term evolution experiment with Escherichia coli. Genetics 200:619–631. doi: 10.1534/genetics.115.176677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hayashi K, Morooka N, Yamamoto Y, Fujita K, Isono K, Choi S, Ohtsubo E, Baba T, Wanner BL, Mori H, Horiuchi T. 2006. Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol Syst Biol 2:2006.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schnetz K, Rak B. 1992. IS5: a mobile enhancer of transcription in Escherichia coli. Proc Natl Acad Sci U S A 89:1244–1248. doi: 10.1073/pnas.89.4.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneider D, Lenski RE. 2004. Dynamics of insertion sequence elements during experimental evolution of bacteria. Res Microbiol 155:319–327. doi: 10.1016/j.resmic.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 59.Weatherspoon-Griffin N, Yang D, Kong W, Hua Z, Shi Y. 2014. The CpxR/CpxA two-component regulatory system up-regulates the multidrug resistance cascade to facilitate Escherichia coli resistance to a model antimicrobial peptide. J Biol Chem 289:32571–32582. doi: 10.1074/jbc.M114.565762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li X-Z, Plésiat P, Nikaido H. 2015. The challenge of efflux-mediated antibiotic resistance in gram-negative bacteria. Clin Microbiol Rev 28:337–418. doi: 10.1128/CMR.00117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kutukova EA, Livshits VA, Altman IP, Ptitsyn LR, Zyiatdinov MH, Tokmakova IL, Zakataeva NP. 2005. The yeaS (leuE) gene of Escherichia coli encodes an exporter of leucine, and the Lrp protein regulates its expression. FEBS Lett 579:4629–4634. doi: 10.1016/j.febslet.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 62.Lee J, Page R, García-Contreras R, Palermino JM, Zhang XS, Doshi O, Wood TK, Peti W. 2007. Structure and function of the Escherichia coli protein YmgB: a protein critical for biofilm formation and acid-resistance. J Mol Biol 373:11–26. doi: 10.1016/j.jmb.2007.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bohnert A, Schuster S, Fahnrich E, Trittler R, Kern WV. 2007. Altered spectrum of multidrug resistance associated with a single point mutation in the Escherichia coli RND-type MDR efflux pump YhiV (MdtF). J Antimicrob Chemother 59:1216–1222. doi: 10.1093/jac/dkl426. [DOI] [PubMed] [Google Scholar]
- 64.Ziolkowska K, Derreumaux P, Folichon M, Pellegrini O, Boni IV, Hajnsdorf E. 2006. Hfq variant with altered RNA binding functions. Nucleic Acids Res 34:709–720. doi: 10.1093/nar/gkj464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng L, Kelly CJ, Colgan SP. 2015. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol 309:C350–C360. doi: 10.1152/ajpcell.00191.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lanter BB, Sauer K, Davies DG. 2014. Bacteria present in carotid arterial plaques are found as biofilm deposits which may contribute to enhanced risk of plaque rupture. mBio 5:e01206-14. doi: 10.1128/mBio.01206-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barrick JE, Lenski RE. 2013. Genome dynamics during experimental evolution. Nat Rev Genet 14:827–839. doi: 10.1038/nrg3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Herring CD, Raghunathan A, Honisch C, Patel T, Applebee MK, Joyce AR, Albert TJ, Blattner FR, van den Boom D, Cantor CR, Palsson BØ. 2006. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat Genet 38:1406–1412. doi: 10.1038/ng1906. [DOI] [PubMed] [Google Scholar]
- 69.Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 70.Wang X, Wood TK. 2011. IS5 inserts upstream of the master motility operon flhDC in a quasi-Lamarckian way. ISME J 5:1517–1525. doi: 10.1038/ismej.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pumbwe L, Skilbeck CA, Wexler HM. 2007. Induction of multiple antibiotic resistance in Bacteroides fragilis by benzene and benzene-derived active compounds of commonly used analgesics, antiseptics and cleaning agents. J Antimicrob Chemother 60:1288–1297. doi: 10.1093/jac/dkm363. [DOI] [PubMed] [Google Scholar]
- 72.Ueda O, Wexler HM, Hirai K, Shibata Y, Yoshimura F, Fujimura S. 2005. Sixteen homologs of the mex-type multidrug resistance efflux pump in Bacteroides fragilis. Antimicrob Agents Chemother 49:2807–2815. doi: 10.1128/AAC.49.7.2807-2815.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tomas-Barberan FA, Clifford MN. 2000. Dietary hydroxybenzoic acid derivatives—nature, occurrence and dietary burden. J Sci Food Agric 80:1024–1032. doi:. [DOI] [Google Scholar]
- 74.Blacklock CJ, Lawrence JR, Wiles D, Malcolm EA, Gibson IH, Kelly CJ, Paterson JR. 2001. Salicylic acid in the serum of subjects not taking aspirin. Comparison of salicylic acid concentrations in the serum of vegetarians, non-vegetarians, and patients taking low dose aspirin. J Clin Pathol 54:553–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Needs C, Brooks P. 1985. Clinical pharmacokinetics of the salicylates. Clin Pharmacokinet 10:164–177. doi: 10.2165/00003088-198510020-00004. [DOI] [PubMed] [Google Scholar]
- 76.Gullberg E, Cao S, Berg OG, Ilbäck C, Sandegren L, Hughes D, Andersson DI. 2011. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog 7:e1002158. doi: 10.1371/journal.ppat.1002158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Andersson DI, Hughes D. 2014. Microbiological effects of sublethal levels of antibiotics. Nat Rev Microbiol 12:465–478. doi: 10.1038/nrmicro3270. [DOI] [PubMed] [Google Scholar]
- 78.Sears CL, Garrett WS. 2014. Microbes, microbiota, and colon cancer. Cell Host Microbe 15:317–328. doi: 10.1016/j.chom.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnson CH, Dejea CM, Sears CL, Johnson CH, Dejea CM, Edler D, Hoang LT, Santidrian AF, Felding BH, Casero RA, Pardoll DM, White JR, Patti GJ, Sears CL, Siuzdak G. 2015. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab 21:891–897. doi: 10.1016/j.cmet.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith MW, Neidhardt FC. 1983. Proteins induced by aerobiosis in Escherichia coli. J Bacteriol 154:344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maurer LM, Yohannes E, Bondurant SS, Radmacher M, Slonczewski JL. 2005. pH regulates genes for flagellar motility, catabolism, and oxidative stress in Escherichia coli K-12. J Bacteriol 187:304–319. doi: 10.1128/JB.187.1.304-319.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lenski RE, Rose MR, Simpson SC, Tadler SC. 1991. Long-term experimental evolution in Escherichia coli. I Adaptation and divergence during 2,000 generations. Am Nat 138:1315–1341. [Google Scholar]
- 84.Thorvaldsdóttir H, Robinson JT, Mesirov JP, Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ma Z, Gong S, Richard H, Tucker DL, Conway T, Foster JW. 2003. GadE (YhiE) activates glutamate decarboxylase-dependent acid resistance in Escherichia coli K-12. Mol Microbiol 49:1309–1320. doi: 10.1046/j.1365-2958.2003.03633.x. [DOI] [PubMed] [Google Scholar]
- 86.Lu P, Ma D, Chen Y, Guo Y, Chen G-Q, Deng H, Shi Y. 2013. l-Glutamine provides acid resistance for Escherichia coli through enzymatic release of ammonia. Cell Res 23:635–644. doi: 10.1038/cr.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lin J, Smith MP, Chapin KC, Baik HS, Bennett GN, Foster JW. 1996. Mechanisms of acid resistance in enterohemorrhagic Escherichia coli. Appl Environ Microbiol 62:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Badawy AA-B. 2012. The EZ:Faast family of amino acid analysis kits: application of the GC-FID kit for rapid determination of plasma tryptophan and other amino acids. Methods Mol Biol 828:153–164. doi: 10.1007/978-1-61779-445-2_14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.