

ABSTRACT
In this study, we used multiple meta-omic approaches to characterize the microbial community and the active metabolic pathways of a stable industrial biogas reactor with food waste as the dominant feedstock, operating at thermophilic temperatures (60°C) and elevated levels of free ammonia (367 mg/liter NH3-N). The microbial community was strongly dominated (76% of all 16S rRNA amplicon sequences) by populations closely related to the proteolytic bacterium Coprothermobacter proteolyticus. Multiple Coprothermobacter-affiliated strains were detected, introducing an additional level of complexity seldom explored in biogas studies. Genome reconstructions provided metabolic insight into the microbes that performed biomass deconstruction and fermentation, including the deeply branching phyla Dictyoglomi and Planctomycetes and the candidate phylum “Atribacteria.” These biomass degraders were complemented by a synergistic network of microorganisms that convert key fermentation intermediates (fatty acids) via syntrophic interactions with hydrogenotrophic methanogens to ultimately produce methane. Interpretation of the proteomics data also suggested activity of a Methanosaeta phylotype acclimatized to high ammonia levels. In particular, we report multiple novel phylotypes proposed as syntrophic acetate oxidizers, which also exert expression of enzymes needed for both the Wood-Ljungdahl pathway and β-oxidation of fatty acids to acetyl coenzyme A. Such an arrangement differs from known syntrophic oxidizing bacteria and presents an interesting hypothesis for future studies. Collectively, these findings provide increased insight into active metabolic roles of uncultured phylotypes and presents new synergistic relationships, both of which may contribute to the stability of the biogas reactor.
IMPORTANCE Biogas production through anaerobic digestion of organic waste provides an attractive source of renewable energy and a sustainable waste management strategy. A comprehensive understanding of the microbial community that drives anaerobic digesters is essential to ensure stable and efficient energy production. Here, we characterize the intricate microbial networks and metabolic pathways in a thermophilic biogas reactor. We discuss the impact of frequently encountered microbial populations as well as the metabolism of newly discovered novel phylotypes that seem to play distinct roles within key microbial stages of anaerobic digestion in this stable high-temperature system. In particular, we draft a metabolic scenario whereby multiple uncultured syntrophic acetate-oxidizing bacteria are capable of syntrophically oxidizing acetate as well as longer-chain fatty acids (via the β-oxidation and Wood-Ljundahl pathways) to hydrogen and carbon dioxide, which methanogens subsequently convert to methane.
KEYWORDS: anaerobic digestion, metagenomics, metaproteomics, methane, microbial community
INTRODUCTION
Utilization of more sustainable approaches for waste disposal, rather than landfilling or incineration, has gathered global interest in recent years. Microbial anaerobic digestion (AD) of organic matter for the production of methane is a viable alternative, and many research initiatives address optimization of the efficiency of the AD process (for examples, see references 1–3). AD processes operating under thermophilic conditions have advantages over mesophilic processes with respect to digestion efficiency and substrate sanitation (4–6). However, the microbial community inherent to thermophilic AD conditions typically is less diverse and more vulnerable to environmental changes (7, 8).
Generally, AD proceeds via four major steps. A large consortium of bacteria performs the first three stages, called hydrolysis, acidogenesis, and acetogenesis, whereas a specialized group of archaea (methanogens) is responsible for the final step, methanogenesis (9, 10). In short, organic polymers such as polysaccharides, proteins, and lipids are hydrolyzed to oligomers and monomers in the initial step (hydrolysis). These compounds are in turn fermented to organic acids, such a volatile fatty acids (VFA) and amino acids (acidogenesis), which may be further degraded to acetate, hydrogen, carbon dioxide (CO2), and a few other one-carbon compounds (acetogenesis). Hydrogenotrophic methanogens subsequently convert CO2 and H2 into methane (CH4), whereas acetate can be converted to methane via two different pathways, namely, by direct conversion by acetoclastic methanogenesis (11) or by syntrophic acetate oxidation (SAO) (12), yielding CO2 and H2 to feed the hydrogenotrophic methanogens. Anaerobic oxidation of acetate and longer-chain fatty acids (FA) such as butyrate and propionate is thermodynamically unfavorable (ΔG > 0) under high partial pressure of H2 (13) and requires a syntrophic interplay with hydrogen-consuming microorganisms such as hydrogenotrophic methanogens. Syntrophic networks become particularly significant in thermophilic anaerobic digestion, where a combination of high levels of free ammonia and high temperature inhibits acetoclastic methanogens (14–16).
Despite the importance of these cooperative interactions in biogas processes, few cultivable syntrophic FA and acetate-oxidizing bacteria have been recovered and described (for examples, see references 17–19). Such bacteria typically grow poorly as a result of marginal energy acquisition shared between the oxidizing bacterium and hydrogen consumer, and their growth demands are difficult to simulate in laboratory cultures (20). Utilization of culture-independent technologies has recently enabled detailed characterizations of several putative syntrophic acetate-oxidizing bacteria (SAOBs) (21–23). In this study, we characterized the microbial community of a full-scale biogas plant (FrBGR) that has been operating efficiently and stably at an unusually high temperature of 60°C for a decade, with food waste as the dominant feedstock. Although food waste has a high potential for producing biogas through AD, it might inhibit certain microbial processes due to its high content of nitrogen-bearing material (24). We specifically sought to determine the metabolic roles of uncultured phylotypes and explored synergistic relationships that possibly play a role in stability of the AD process. For this cause, we used a combination of high-throughput 16S rRNA gene sequencing and total metagenome analyses, which allowed generation of population genome bins of both classified and novel phylotypes. These data were combined with extensive quantitative metaproteomics data, allowing us to assign abundance values to specific proteins for each bin. By combining these analyses, we identified key metabolic pathways and crucial microbes that drive the different steps of the AD in this thermophilic and stable reactor system.
RESULTS
Characteristics of the biogas reactor.
Table 1 shows the characteristics of the FrBGR biogas reactor. The reactor was operating at 60°C, and the average pH of the effluent was 8.0 ± 0.1. The alkalinity was relatively high (5,832 mg/liter CaCO3), indicating enough buffering capacity for neutralization of organic acids and ensuring pH stability. The concentrations of VFAs were generally low, and only acetate (91.0 ± 7.0 mg/liter) and propionate (32.0 ± 1.5 mg/liter) were detected in the effluent. Analysis of the organic content of the digester demonstrated that the major fraction of the chemical oxygen demand (COD) was in particulate form. The soluble COD fraction was only 7.5% of total COD (TCOD), implying an effective conversion of solubilized organics into biogas. The ammonium concentration in the reactor was 1,057 mg/liter NH4-N. Based on the temperature and pH of the digester, the free ammonia concentration was calculated as 367 mg/liter NH3-N (25). Overall, these analytical data are typical for a well-performing biogas reactor, apart from a slightly elevated ammonia level.
TABLE 1.
Characteristics of the FrBGR reactor
| Parameter (unit) | Value for FrBGR effluent |
|---|---|
| TS (%) | 4.4 ± 0.1 |
| VS (%) | 2.3 ± 0.1 |
| TCOD (mg/liter) | 38,615 ± 800 |
| TCOD/VS | 1.67 |
| SCOD (mg/liter) | 2,868 ± 345 |
| pH | 8.0 ± 0.1 |
| NH4-N (mg/liter) | 1,057 ± 12 |
| Propionate (mg/liter) | 32 ± 1.5 |
| Acetate (mg/liter) | 91 ± 7.0 |
| Alkalinity (mg/liter as CaCO3) | 5,832 ± 200 |
Data from combined meta-omics technologies.
More than 600,000 16S rRNA gene sequences were obtained using MiSeq sequencing of technical triplicates of the DNA sample. Total DNA metagenomic sequencing provided 54 million paired-end reads (MiSeq) and nearly 220,000 long reads (PacBio) (see Table S2 in the supplemental material). The hybrid assembly (MiSeq plus PacBio) resulted in 235,738 contigs (totaling 473 Mb), where 69 contigs were longer than 100 kb and 4,417 were longer than 10 kb. The longest contig was 541,616 bp long, with Dictyoglomus thermophilum as the closest phylogenomic relative. A total of 142,316 contigs were binned into 43 population bins. A total of 2,194 proteins were identified by metaproteomics and resulted in highly reproducible, label-free, quantitative data (LFQ; Pearson correlation [R] of 0.98) (Fig. S2). Of these 2,194, we selected 338 abundant proteins for further analysis, all being involved in key metabolic pathways for AD of organic matter. Detailed descriptions of the proteins are provided in the supplemental material (Table S3). The data assembled for 32 reconstructed population bins are summarized in Table 2.
TABLE 2.
Characteristics of population bins extracted from the FrBGR reactor
| ID | Population bin “species name”a | Estimated (%): |
Strain heterogeneity | Bin size (Mbp) | |
|---|---|---|---|---|---|
| Completeness | Contamination | ||||
| Dglo | Dictyoglomus thermophilum | 98 | 0 | 0 | 1.9 |
| Tyel | Thermodesulfovibrio yellowstonii | 98 | 9 | 95 | 2.2 |
| Swol | Syntrophomonas wolfei cluster 1 (Swol_c1) | 95 | 5 | 54 | 2.9 |
| Syntrophomonas wolfei cluster 2 (Swol_c2) | 20 | 9 | 67 | 2.1 | |
| Prot | Unclassified Proteobacteria | 94 | 3 | 33 | 4.1 |
| Tpha | Thermacetogenium phaeum cluster 1 (Tpha_c1) | 93 | 4 | 70 | 2.4 |
| Thermacetogenium phaeum cluster 2 (Tpha_c2) | 76 | 12 | 80 | 1.8 | |
| unFi | unFirm02_FrBGR cluster 2 (unFi_c2) | 86 | 5 | 100 | 2.1 |
| unFirm02_FrBGR cluster 1 (unFi_c1) | 82 | 2 | 0 | 2.2 | |
| Athe | Anaerolinea thermophila | 85 | 6 | 30 | 2.7 |
| Atri | Unclassified Atribacteria bacterium (OP9) cluster 1 (Atri_c1) | 84 | 43 | 98 | 2.7 |
| Unclassified Atribacteria bacterium (OP9) cluster 2 (Atri_c2) | 41 | 15 | 100 | 1.8 | |
| Cste | Clostridium stercorarium cluster 2 (Cste_c2) | 83 | 4 | 36 | 2.1 |
| Clostridium stercorarium cluster 1 (Cste_c1) | 72 | 11 | 6 | 1.8 | |
| Plan | Unclassified Planctomycetes | 83 | 8 | 82 | 4.1 |
| Lcri | Lactobacillus crispatus | 83 | 32 | 100 | 2.0 |
| Cthe | Clostridium thermocellum | 83 | 5 | 30 | 2.9 |
| Bado | Bifidobacterium adolescentis | 86 | 11 | 75 | 1.7 |
| Lraf | Lactococcus raffinolactis | 73 | 7 | 73 | 1.6 |
| Fpen | Fervidobacterium pennivorans cluster 1 (Fpen_c1) | 67 | 14 | 73 | 1.7 |
| Fervidobacterium pennivorans cluster 2 (Fpen_c2) | 58 | 16 | 12 | 1.2 | |
| Acti | Unclassified Actinobacteria | 55 | 18 | 12 | 2.4 |
| Ther | Unclassified Thermoanaerobacteraceae | 45 | 31 | 67 | 1.3 |
| Tace | Tepidanaerobacter acetatoxydans | 35 | 8 | 94 | 1.0 |
| Ccla | Clostridium clariflavum | 25 | 2 | 0 | 2.5 |
| Pthe | Pelotomaculum thermopropionicum | 20 | 3 | 0 | 1.0 |
| Fnod | Fervidobacterium nodosum | 16 | 2 | 0 | 0.5 |
| Mbt | Methanothermobacter thermautotrophicus | 15 | 1 | 100 | 0.3 |
| Mst | Methanosaeta thermophila | 8 | 0 | 0 | 0.1 |
| Cprob | Coprothermobacter spp. | 100 | 2,902 | 93 | 58 |
| Amobb | Anaerobaculum spp. | 100 | 1,100 | 84 | 24 |
| Slipb | Syntrophothermus spp. | 93 | 77 | 7 | 3.8 |
Taxonomic assignment of uncultured phylotypes was based on PhyloPythiaS+ analysis.
Higher-ranked population bin.
A microbiome strongly dominated by one proteolytic phylotype.
The 16S rRNA gene analysis revealed an uneven genus distribution within the FrBGR microbiome, whereby affiliates of the bacterial genus Coprothermobacter were predominant, representing approximately 76% of the total 16S rRNA gene inventory (Fig. 1). The second most abundant phylotype (∼7%) also was affiliated with the bacterial order Thermoanaerobacterales, but with the genus Thermacetogenium. An Anaerobaculum phylotype, affiliated with the bacterial Synergistales order, was ranked third, representing ∼6% of the total inventory. Moreover, the known cellulolytic bacterial genus Dictyoglomus represented ∼2% of the 16S rRNA gene sequences, whereas another 2% was assigned to Syntrophomonas.
FIG 1.

Phylogenetic distribution of the most dominant 16S rRNA gene sequences in FrBGR. Details for the Euryarchaeota (not visible in the major plot because of low abundance) are provided in a separate plot to the lower left. Scattered areas contain two or more phylotypes with low abundance. The phylotype assigned to the order Natranaerobiales in the 16S rRNA data set corresponds to the population genome bin named unFirm02_FrBGR in the metagenomic data set, as indicated. The plot was generated using Krona and then simplified (i.e., removal of low-abundance phylotypes) in order to reduce size.
Phylogenetic binning of the assembled contigs from the metagenome data set demonstrated that a population bin closely related to Coprothermobacter proteolyticus, here referred to as Coprothermobacter spp. (Cpro), accounted for the majority of assigned contigs. While the representative genome of Coprothermobacter proteolyticus DSM5256 is 2.42 Mbp (26), the size of the higher-ranked Cpro population bin was more than 10-fold larger (Table 2). The Cpro bin was examined for assembly and/or binning errors to explain this discrepancy. Closer inspection revealed gene sequences from more than one strain, which seemingly affected the assembly and binning of this phylogenetic group. In addition to Cpro, poor assembly and binning of a population related to Anaerobaculum mobile (Anaerobaculum spp. [Amob]) was observed. Visual assessment of PhyloPythiaS+-assigned contigs for both of these populations revealed no clustering based on coverage (Fig. S1a and b), whereas CheckM results revealed high levels of strain heterogeneity (Table 2). High-resolution investigation of the 16S rRNA gene inventory detected potential polymorphisms in the 16S rRNA data set, demonstrating that the operational taxonomic units (OTUs) assigned as Coprothermobacter comprised at least 11 oligotypes (Fig. S3a). Furthermore, an alignment between the Cpro population bin and the C. proteolyticus DSM5256 genome revealed multiple contigs that mapped against the same C. proteolyticus DSM5256 genome coordinates with various similarity (between 75 and 100%) (Fig. S3b).
A total of eight different proteases and peptidases were observed in the proteome of Cpro (Table S3). Of these proteases, an oligoendopeptidase was the most abundant, with a log10(LFQ) value of 8.4. Import/export of proteins and peptides were indicated by the detection of transporter systems related to oligopeptides, dipeptides, and branched amino acids [log10(LFQ) ranging from 6.6 to 9.2]. As expected, the Cpro proteome contained most enzymes of common amino acid-metabolizing pathways, dealing with glycine, serine, threonine, valine, leucine, and isoleucine. The proteomes of other FrBGR phylotypes also contained enzymes for amino acid metabolism, such as Anaerobaculum-affiliated phylotypes (Amob), that formed proteins inferred in glycine metabolism and arginine degradation via citrulline and ornithine (Table S3). Several Amob-affiliated proteins inferred in oligopeptide uptake and degradation were also detected in the proteome, indicating proteolytic activity (Table S3).
The FrBGR metagenome revealed a diverse group of microorganisms with cellulolytic potential, among them two uncultured phylotypes assigned to deeply branching and scarcely described bacterial clades, namely, phylum Planctomycetes and candidate phylum “Atribacteria” (lineage OP9) (Table S3). The proteome of the uncultured Atribacteria phylotype (Atri) included beta-glucoside-related glycosidase [log10(LFQ) = 7.3] in addition to galactose mutarotase, l-fucose isomerase, and xylose isomerase, indicating that this phylotype is participating in the hydrolysis of polysaccharides (e.g., xyloglucan oligomers) potentially derived from (hemi)cellulose degradation. Translocation of sugar compounds through the cell membrane was presumably mediated by ABC-type sugar transport systems [log10(LFQ) = 6.9], and almost all key enzymes needed for glycolysis (Embden-Meyerhof pathway [EMP]) were found in the proteome (Table S3). Moreover, peptide uptake [ABC-dipeptide transport system; log10(LFQ) = 8.8] and degradation [trypsin-like serine protease; log10(LFQ) = 7.2] systems were detected in the proteome of Atri, suggesting a broader hydrolytic role that also includes proteins and amino acids.
A deeply branched phylotype affiliated with the Planctomycetes (referred to as unclassified Planctomycetes [Plan]) was also detected, albeit at relatively low abundance (not shown in Fig. 1). The predicted metabolic role of Plan in FrBGR is linked to carbohydrate degradation, including hemicellulose. Proteome support for this prediction included proteins with carbohydrate binding domains [log10(LFQ) = 7.5], endoglucanases [log10(LFQ) = 6.7 and 6.5], beta-galactosidases [log10(LFQ) = 5.7], alpha-fucosidase [log10(LFQ) = 6.5], alpha-l-arabinofurosidase [log10(LFQ) = 6.5], as well as xylose- and l-arabinose isomerases [ranging from log10(LFQ) of 6.3 to 8.3]. Additionally, the proteome data showed that all genes needed for glycolysis (EMP pathway) were formed (Table S3). Several Plan-affiliated proteins were also assigned as proteases and peptidases with abundance [log10(LFQ)] ranging from 6.53 to 8.40 (Table S3).
A phylotype affiliated with Dictyoglomus thermophilum (Dglo) was predicted to degrade xylose via the isomerase pathway, whereby xylose is converted to xylulose by the action of xylose isomerase, followed by phosphorylation to xylulose-5-phosphate by xylulokinase. The Dglo-affiliated proteome comprised both a xylosidase/arabinosidase [log10(LFQ) = 7.3] and xylose isomerase [log10(LFQ) = 6.5], in addition to an α-amylase/α-mannosidase [log10(LFQ) = 8.6] and an endoglucanase [log10(LFQ) = 6.6]. A d-xylulose 5-phosphate/d-fructose 6-phosphate phosphoketolase (XFP) was also detected, potentially converting d-xylulose-5-phosphate to acetyl phosphate and glyceraldehyde-3-phosphate, which may enter the pentose phosphate pathway (PPP) and/or glycolysis (EMP) with acetate as an end product. Several other population bins that affiliated with known cellulolytic bacteria, e.g., Clostridium stercorarium and Clostridium thermocellum, were extracted from the FrBGR metagenome. However, only a few proteins from these reconstructed genomes were detected, indicating a reduced role in the biogas reactor.
Methanogenic population and the methanogenesis pathways.
Only 0.04% of the 16S rRNA sequences were affiliated with the Euryarchaeota, which were dominated by representatives of hydrogenotrophic methanogens (Methanothermobacter, 99% of Euryarchaeota sequences) and, to a much lesser extent, acetoclastic methanogens (Methanosaeta, 1%) (Fig. 1). Accordingly, the genomic bin of Methanosaeta thermophila was incomplete and therefore was supplemented by the corresponding reference genome (Methanosaeta thermophila PT; Table S1) for proteome analysis. Although the genomic bin of a Methanothermobacter thermoautotrophicus-affiliated phylotype (Mbt) also was incomplete, proteins related to hydrogenotrophic methanogenesis were observed at high abundance. These were mapped back to the Mbt bin, supplemented by the bins of Methanothermobacter, Methanobacteriales, and Archaea. Despite the discrepancy in relative abundance of hydrogenotrophic versus acetoclastic methanogens, proteins for both methanogenic pathways were detected (Fig. 2). Proteins related to hydrogenotrophic methanogenesis included the formylmethanofuran dehydrogenase cluster [log10(LFQ) = 9.7 to 10.4], methyl coenzyme M (CoM) reductase subunits [log10(LFQ) = 9.6 to 9.7], and several methyltransferase subunits [mtr cluster; log10(LFQ) = 7.8 to 9.3]. Proteins associated with acetoclastic methanogenesis (reference genome Methanosaeta thermophila PT) were generally detected at lower levels, ranging from log10(LFQ) values of 6.7 to 9.0 (acetyl coenzyme A [CoA] decarbonylase/synthase and acetyl-CoA synthetase, respectively) (Fig. 2; Table S3).
FIG 2.
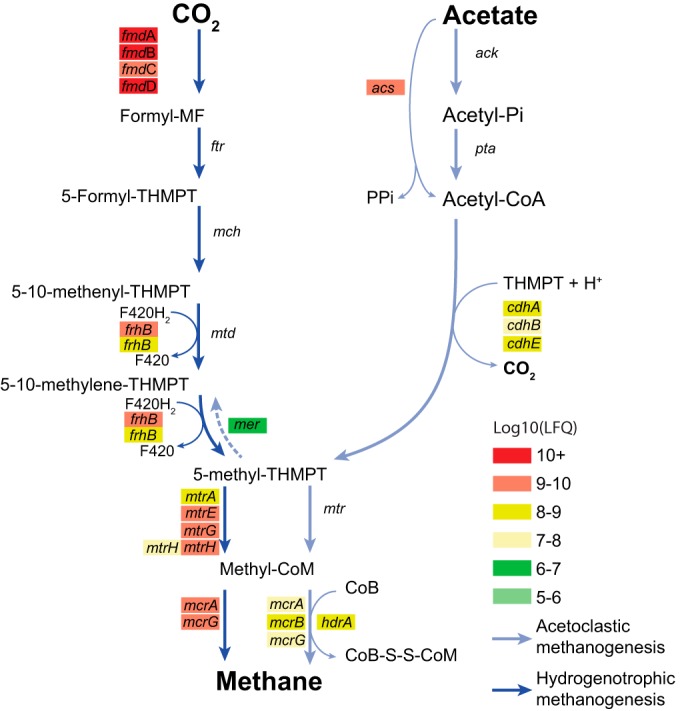
Methanogenesis pathways in FrBGR operating at high temperature and high ammonia concentration. Genes are shown as colored boxes, where the color indicates the protein abundance (MaxQuant LFQ values) ranging from high abundance (red) to low abundance (green). Acetoclastic methanogenesis is shown by light blue lines, where all proteins were mapped to the reference genome of Methanosaeta thermophila PT. Pathways illustrated by dark blue lines represent hydrogenotrophic methanogenesis, and the proteins were mapped to Methanothermobacter thermoautotrophicus supplemented by the population bins of Methanothermobacter, Methanobacteriales, and Archaea. Abbreviations used in this figure can be found in Table S3 in the supplemental material. Subunits of multimeric protein complexes are indicated, if detected (A, B, C, etc.).
Identification of proteins involved in syntrophic degradation of fermentation intermediates.
Syntrophic degradation of fermentation intermediates, especially the fatty acids butyrate, propionate, and acetate, is a key process in AD. Butyrate and longer-chain FA are oxidized through β-oxidation cycle(s), while oxidation of propionate proceeds through the methylmalonyl-CoA (MMC) pathway. Syntrophic oxidation of acetate, the major precursor in the process, is usually associated with the oxidative direction of the Wood-Ljungdahl (WL) pathway. Phylotypes affiliated with syntrophic bacteria from the genus Syntrophomonas were among the most prominent in the FrBGR 16S rRNA data set, and two population bins were reconstructed for phylotypes closely affiliated with Syntrophomonas wolfei. For the S. wolfei-affiliated phylotype (Swol_c1), all four enzyme classes required for β-oxidation of butyrate (acyl-CoA dehydrogenase, enoyl-CoA hydratase, 3-hydroxyacyl-CoA dehydrogenase, 3-ketoacyl-CoA thiolase) were formed, demonstrating the phylotype's activity within FrBGR. A higher-ranked population bin was also recovered for several closely related phylotypes affiliated with Syntrophothermus lipocalidus (Syntrophothermus spp.; Slip), although Syntrophothermus was not present among the abundant genera in the FrBGR 16S rRNA data set. A long-chain acyl-CoA synthase was identified [log10(LFQ) = 6.8] in the proteome of Slip, indicating uptake and degradation of long-chain FA (LCFA), which could emerge from lipid hydrolysis.
A phylotype affiliated with the thermophilic, syntrophic propionate-degrading bacterium Pelotomaculum thermopropionicum (27) was detected in FrBGR, but metabolic reconstruction of this phylotype was not possible due to low genome coverage. The incorporation of the reference genome of P. thermopropionicum (Table S1) improved proteome mapping and suggested metabolism of propionate, as both the MMC cluster and propionate CoA transferase (PCT) cluster were detected with relatively high abundance [up to log10(LFQ) of 8.1 and 7.5, respectively; Table S3].
The most numerically abundant phylotype inferred in acetate oxidation was affiliated with two phylotypes closely related to Thermacetogenium phaeum (Tpha-c1 and Tpha_c2) that collectively represented approximately 7% of the 16S rRNA gene inventory. Genome reconstruction and proteome analysis of both Tpha_c1 and Tpha_c2 revealed a relatively high abundance of proteins that constitute the WL pathway, which is the mainstay in most syntrophic acetate oxidizers (i.e., formyltetrahydrofolate synthase, 5,10-methylenetetrahydrofolate dehydrogenase, methylenetetrahydrofolate reductase, trimethylamine-corrinoid methyltransferase, carbon monoxide dehydrogenase–acetyl-CoA, phosphotransacetylase, and acetate kinase). In addition, proteomic analyses of two uncultured phylotypes affiliated with the Firmicutes phylum (referred to as unFirm02_FrBGR clusters 1 and 2; unFi_c1 and unFi_c2) also gave strong indications toward its role as an SAOB. This was based on the proteome detection of nearly all enzymes required for the WL pathway (Fig. 3; Table S3 and Fig. S4). In general, unFi_c2 showed higher protein abundance than unFi_c1. Moreover, the four β-oxidation enzyme classes needed for the degradation of FA to acetyl-CoA were also detected (28), where an acyl-CoA synthetase protein had the highest abundance [log10(LFQ) = 9.0]. Notably, the protein abundance of β-oxidation-related enzymes was generally higher for unFirm02_FrBGR than for both Slip and Swol (Table S3). However, the broad functioning of many enzymes in these classes made further predictions into the specific FA being oxidized overly speculative and therefore was not attempted. The proteome also revealed that proteins only related to the lower part of the unidirectional EMP were observed (Fig. 3; Table S3). A protein cluster encoding Fe-S oxidoreductase and electron transfer flavoprotein (ETF) α- and β-subunits was identified in the unFi_c2 proteome, which, based on previous reports (28), suggests a mechanism for electron transfer that is important in anaerobic FA oxidation. Thus, we hypothesize that the unFi populations import and oxidize FA to acetyl-CoA, which is further oxidized to CO2 and H2 through an oxidative WL pathway, as illustrated in Fig. 3. Searching the 16S rRNA data set against the metagenome data demonstrated that this population was among the 10 most abundant microbes in FrBGR, comprising 0.5% of the total 16S rRNA gene sequences. Its representative OTU could not be assigned further than to the order Natranaerobiales ML1228J-1 in our 16S rRNA gene sequencing analysis (using RDP classifier). A phylogenetic comparison of a 16S rRNA gene sequence encoded within the unFi_c1 population genome revealed a closer relationship to the thermotolerant SAOB Tepidanaerobacter acetoxydans than T. phaeum (Fig. S5). The closest relative to the representative sequence for unFi was “Candidatus Contubernalis alkalaceticum” clone Z-7904, with only 91% 16S rRNA gene sequence similarity (using BLAST).
FIG 3.

Selected metabolic pathways of the putative novel SAOB unFirm02_FrBGR cluster 2. The pathways are proposed based on genome and proteome comparison, and protein abundances are indicated by color, ranging from high abundance (red) to low abundance (green). All enzymes needed for β-oxidation of fatty acids, in addition to most enzymes associated with the Wood-Ljungdahl pathway, were detected for unFirm02_FrBGR cluster 2 (unFi_c2). Parts of both pathways were also detected for unFirm02_FrBGR cluster 1 (unFi_c1), albeit at a lower detection level (see Fig. S4 in the supplemental material). Only proteins affiliated to the lower part of EMP were represented in the proteome. Acetate kinase (ack) was detected in the genome but not in the proteome. More details on the proteins detected can be found in Table S3, including abbreviations.
DISCUSSION
Biomass degradation in an ammonia-rich biogas reactor operating at 60°C.
Genome reconstruction and functional interpretation of uncultured phylotypes identified participants within key central metabolic pathways in FrBGR, including hydrolysis of macromolecules, downstream fermentation, syntrophic degradation of intermediates, and methanogenesis via two pathways (syntrophic acetate oxidation-hydrogenotrophic methanogenesis and acetoclastic methanogenesis) (summarized in Fig. 4; see also Table S3 in the supplemental material). The high content of protein-rich material in the substrate was reflected by a strong dominance of Cpro and Amob populations, both proven to form high levels of enzymes essential for protein and amino acid degradation. C. proteolyticus is frequently reported at various abundances in thermophilic digesters treating protein-rich biomass, and its proteolytic activity is clearly supported by the literature (29, 30). Our data introduce additional complexities for C. proteolyticus-like populations by indicating the presence of multiple strains; however, the specific diversity and functional interplay that exists remains to be elucidated.
FIG 4.
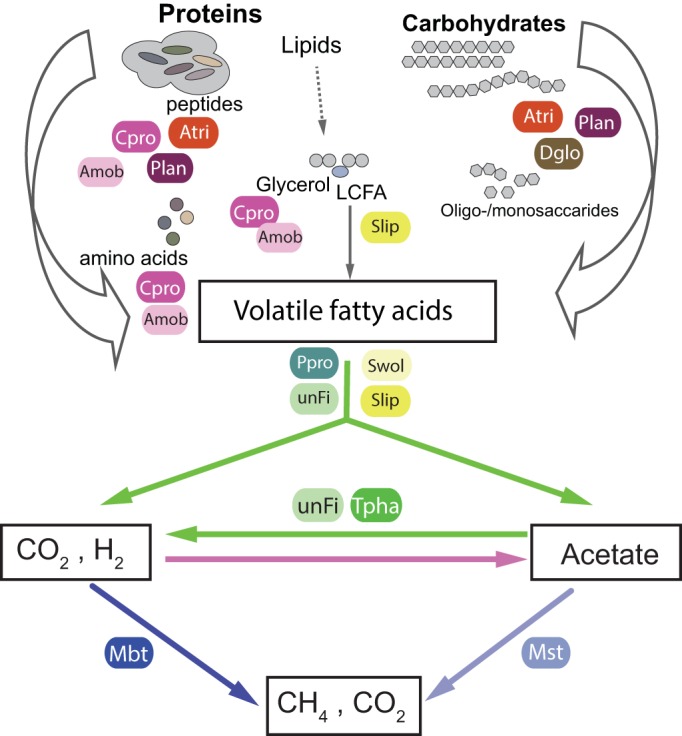
Hypothetical model of the carbon flux in FrBGR, with functional roles of dominant phylotypes inferred from comparison of metagenome and metaproteome data sets. Metabolic pathways showing the key stages (arrows) of acetogenesis (pink) and methanogenesis (blue), in addition to syntrophic metabolic processes (green). Only the most prominent (with regard to relative abundance in the 16S rRNA gene sequence inventory and protein abundance) phylotypes in the FrBGR microbial community were evaluated, and it should be noted that a rare portion of the population might account for underlying key metabolic pathways not shown here. Organism abbreviations used in this figure correspond to the population bin IDs listed in Table 2.
Several phylotypes were identified to actively produce enzymes needed for degradation of polysaccharide, another major compartment of food waste (31). A deeply branching bacterium, D. thermophilum, was observed at relatively high abundance (2% of all 16S rRNA gene sequences; Fig. 1) and with high abundance levels of proteins related to xylan degradation, in accordance with literature (32). In addition, functional insight was generated for two phylotypes affiliated with deeply branching phyla, Planctomycetes and the candidate phylum Atribacteria. Representatives from Planctomycetes are only sporadically observed in anaerobic digesters (33, 34), while candidate phylum Atribacteria (formerly termed OP9) are widespread within these systems (7, 15, 35, 36). Recent studies have demonstrated carbohydrate-degrading characteristics of uncultivated OP9 members (Atribacteria) (37, 38), and the metabolic insight gained in this study also suggests proteolytic activity. Moreover, the results imply that the Plan group is metabolically active in the hydrolysis of both proteins and carbohydrates.
A syntrophic network enables complete degradation of fatty acids.
Propionate and longer-chain FA are important intermediates in anaerobic digestion, and a rapid degradation of these compounds to acetate, formate, or hydrogen is essential to enable a complete degradation of organic matter to methane. Anaerobic degradation of FA in concert with hydrogen production is highly endergonic under standard conditions (propionate ΔG°′ of +76.1 kJ mol−1; butyrate ΔG°′ of +48.6 kJ mol−1) (39) and is possible only when hydrogen is kept at low partial pressure by a secondary organism, e.g., a hydrogenotrophic methanogen. Propionate, originating from, e.g., amino acid degradation, polysaccharide fermentation, or as a by-product from β-oxidation of LCFA, is often one of the major VFAs in AD processes, and insufficient removal can lead to instability (40). Most of the characterized syntrophic propionate-oxidizing bacteria degrade propionate through the unidirectional methylmalonyl-CoA (MMC) pathway. Within FrBGR, Pelotomaculum thermopropionicum (27) was seemingly the only organism forming most of the MMC-related enzymes. Involvement of the aforementioned Atribacteria in syntrophic propionate degradation also has been suggested previously (41); however, no evidence for this was found in our data.
Degradation of butyrate and longer-chain FA to acetyl-CoA and acetate proceeds through β-oxidation cycles, of which Swol_c1, unFi_c1, unFi_c2, and the Slip higher-ranked populations formed enzymes from the required classifications (acyl-CoA dehydrogenase, enoyl-CoA hydratase, 3-hydroxyacyl-CoA dehydrogenase, and 3-ketoacyl-CoA thiolase). Both S. wolfei and S. lipocalidus are well-known FA degraders, whereby the former can utilize FA of four to eight carbons (C4 to C8) (42, 43) and the latter can degrade C4 to C10 FA (44). Interestingly, unFi_c2 exerted higher acyl-CoA synthetase and acyl-CoA dehydrogenase protein abundance than either Swol or Slip, suggesting this novel phylotype is an important FA degrader in FrBGR.
Protein levels indicate activity of a low-abundance acetoclastic methanogen.
Generally, acetate serves as one of the most important intermediates in AD processes (10). Acetate-utilizing populations such as acetoclastic methanogens or SAOB are therefore essential to ensure stable carbon flow via acetate with methane as a final product. The operating temperature (60°C) of FrBGR has been previously proposed as a critical balance temperature for an efficient anaerobic digestion (45). Moreover, similar ammonia levels (367 mg/liter NH3-N) and operating temperatures for methanogenic bioreactors are associated with a shift in the methanogenic population, from acetoclastic methanogens to hydrogenotrophic methanogens. Low detection of acetoclastic methanogenesis was therefore expected, as well as promoted importance of SAOB and hydrogenotrophic methanogens (46, 47).
The overall population structure of methanogens in FrBGR was predominated by the obligate hydrogenotrophic methanogen, M. thermoautotrophicus. As expected, low population levels were associated with the obligate acetoclastic methanogenic genus Methanosaeta in FrBGR; however, surprisingly several Methanosaeta thermophila enzymes associated with acetoclastic methanogenesis were detected in the FrBGR metaproteome. This illustrated that despite its low 16S rRNA abundance, M. thermophila is metabolically active. According to the wide range of NH3 inhibition coefficients (KI,NH3) reported in the literature (14, 15, 48), the degree of process inhibition is highly dependent on the operational history of a digester and whether the microbial community within the digester has acclimatized to high ammonia exposure. Previous studies have shown that communities can adapt to stress and remain tolerant over several generation times (49). Since FrBGR has been running steadily for several years, the populations of acetoclastic methanogens in FrBGR are seemingly acclimatized to the relatively high free ammonia levels calculated for the reactor. The genomic reconstruction of phylotypes affiliated with the well-known SOAB T. phaeum (50) was accompanied by high proteomic detection of the WL pathway, which collectively suggests that acetate turnover is occurring predominately via syntrophic acetate oxidation in coexistence with hydrogen-consuming Methanothermobacter thermoautotrophicus-like phylotypes. This scenario well suites the hypothesis, suggested by Ho et al. (45), that some levels of acetoclastic methanogenesis are necessary for removal of residual acetate, while SAOB remove the bulk in order to enable efficient degradation.
Indications for a novel thermophilic acetate-oxidizing bacteria, taking longer-chain fatty acids all the way to CO2.
Very few SAOB have been successfully isolated and characterized to date. This limited collection is represented by only two thermophiles (T. phaeum and Thermotoga lettingae [51]), one that is thermotolerant (Tepidanaerobacter acetoxydans [19]), and two mesophiles (Clostridium ultunense [17] and Syntrophaceticus schinkii [52]). Recent studies have emphasized that the appearance of SAOB in anaerobic systems is more widespread than those characterized by culture-dependent methods, and several genomes representing uncultured putative SAOB have been deduced (21, 22, 41). In this study, we identified genomic data affiliated with T. phaeum and T. acetoxydans; however, only a few proteins were mapped to the latter, indicating low metabolic activity. Phylotypes affiliated with the genus Thermacetogenium were the second most abundant in the 16S rRNA gene inventory, and two population genomes were additionally reconstructed. In addition, Tpha-affiliated proteins related to the WL pathway were detected at high levels, indicating a prominence of SAOB in FrBGR. Aside from relatively well-known SAOB, our analysis suggested a metabolic role for two uncultured and novel thermophilic SAOB, unFi_c1 and unFi_c2. Interestingly, these two phylotypes encode the necessary genes and pathways to syntrophically oxidize acetate as well as longer-chain FA. This combination of pathways is documented for nonsyntrophic sulfate reducers (53, 54) but, to the best of our knowledge, not for the characterized SAOB. Genomic annotation indicates that unFi is not a sulfate reducer, as no genes for sulfate reduction were found. Proteomics showed that nearly all proteins essential for the WL pathway were detected with particularly high levels of formyltetrahydrofolate synthetase (FTHFS) and CO dehydrogenase (AcsB). The expression of a gene cluster encoding Fe-S oxidoreductase and electron transfer flavoprotein (ETF) α and β subunits has been previously shown to serve as an electron transfer system in syntrophic fatty acid-oxidizing bacteria (28). Finally, phylogenetic analysis confirmed that unFi was most closely related to “Candidatus Contubernalis alkalaceticum,” an uncultivated obligate syntrophic acetate oxidizer (55). Although cultivation-based efforts are required to completely validate the metabolic traits and ecological role of unFi, we believe that the discussed FrBGR observations infer a syntrophic lifestyle. Overall, this study demonstrates a thermophilic bacterium that seemingly utilizes β-oxidation to degrade longer-chain FA to acetate, followed by a further oxidation of acetate to CO2 through the Wood-Ljungdahl pathway.
Conclusion.
Collectively, we present an increased understanding of uncultured phylotypes that engage in key microbial processes within a stable thermophilic biogas reactor. Multiple strains were detected within reconstructed genomes for several key phylotypes, introducing an additional level of complexity seldom explored in biogas studies. Particular focus on uncultivated and scarcely described microbial groups, such as the candidate phylum Atribacteria (Atri), Planctomycetes (Plan), and a novel putative SAOB (unFi), complements ongoing efforts to characterize the microbial processes that control biogas production. Key observations enabled metabolic predictions that novel populations (unFi) are capable of both β-oxidation of longer-chain fatty acids as well as acetate oxidation via WL pathway. The putative connection of these two pathways expands our knowledge into the intricate syntrophic networks that are required to convert important fermentation intermediates into methane. Culture-based confirmation is still required and presents an ongoing challenge.
MATERIALS AND METHODS
Source and basic analysis of the sample.
The sample was collected in June 2014 from a 2,200-m3 thermophilic biogas plant (FrBGR) in Fredrikstad, Norway. This plant has been operating stably for a decade at 60°C, mainly using food waste as the substrate. The composition of this food waste has been published previously (15), but briefly it has a TCOD and ammonia content of around 270 g/liter and 500 mg/liter, respectively. Total solids (TS), volatile solids (VS), total chemical oxygen demand (TCOD), and soluble COD (SCOD) were analyzed according to standard methods (56). For measurement of SCOD and VFAs, an aliquot of the sample was first centrifuged at 18,800 × g for 1 min and then filtered using 0.45-μm filters. Ammonium was determined in the centrifuged filtered samples using an ammonium-sensitive electrode (Orion 93 electrode; Thermo Scientific, USA). VFAs were analyzed by reversed-phase high-performance liquid chromatography (HPLC) (Dionex, Sunnyvale CA, USA) using a Zorbax Eclipse Plus C18 column (150- by 2.1-mm column; 3.5-μm particles; Agilent, USA) equipped with a guard column (12.5 by 2.1 mm; 5-μm particles). The pH was adjusted to 2.5 using 2.5 mM H2SO4 prior to VFA analysis of the sample.
DNA extraction.
An aliquot of 1 ml slurry from the biogas reactor was frozen immediately after sampling and kept frozen (at −20°C) until extraction of protein and DNA. DNA was extracted and processed according to Rosewarne et al. (57), with minor modifications. DNA concentrations were quantified using a Qubit fluorometer and the Quant-iT dsDNA BR assay kit (Invitrogen, Carlsbad, CA, USA).
16S rRNA gene sequencing.
16S rRNA gene amplicons for the Illumina MiSeq system (Illumina Inc.) were prepared as described in Zamanzadeh et al. (15), and sequencing was conducted using paired-end, 2× 300-bp cycle runs with the MiSeq reagent kit v3 on an Illumina MiSeq sequencing system. All 16S rRNA gene sequences were processed using the QIIME version 1.8.0 software package (58). Paired ends were joined (using join_paired_ends.py) and quality filtered as follows. Only three sequential low-quality (Phred quality score of <20) bases were allowed per sequence before truncating, and reads with <75% (of total length) consecutive high-quality base calls were discarded. No N characters or barcodes were allowed in the sequence. Chimeric sequences were removed from the data set using UCHIME incorporated in USEARH (59), and a threshold of 3% dissimilarity between 16S rRNA gene sequences was used to cluster sequences into de novo operational taxonomic units (OTUs) (60). Taxonomy (up to rank genus) was assigned to each OTU using the Usearch-based consensus taxonomy assigner implemented in QIIME with default parameters. The Krona visualization tool (61) (available from http://krona.sourceforge.net) was used to visualize the microbial diversity in the biogas reactor sample. Nucleotide variation among 16S rRNA gene sequences assigned to selected taxonomic phylotypes (e.g., Coprothermobacter) was detected with oligotyping in order to explore the occurrence of multiple strains not detected by the 3% clustering method or taxonomic classification (62).
Total DNA metagenomics.
Shotgun metagenomic sequencing of genomic DNA was performed using two different sequencing technology platforms, Illumina MiSeq and the Pacific Biosciences (PacBio) RS II single-molecule, real-time (SMRT) DNA sequencing system (63). TruSeq DNA PCR-free sample preparation and paired-ended (2× 300-bp) MiSeq sequencing were performed at the Norwegian Sequencing Center (NSC; Oslo, Norway). PacBio circular consensus sequencing (CCS) of a total of 8 SMRT cells (minimum accuracy, 0.99) using P4-C2 chemistry was performed at the same facility. The PowerClean DNA clean-up kit (MoBio Laboratories, Carlsbad, CA, USA) was used to purify extracted microbial DNA prior to PacBio sequencing in order to avoid enzymatic inhibition during downstream sample library preparation. MiRA assembler (version 4) was applied for hybrid assembly of reads from MiSeq and PacBio. Only contigs above 1 kb were considered for the downstream analysis. Open reading frames (ORFs) identified with MetaGeneMark v.1 (64) were screened for protein-encoding marker genes, and these were subsequently converted into a multiple FASTA file (aa_from_gff.pl, included in MetaGeneMark v.1). Comparison of the protein sequences against a set of 31 AMPHORA marker genes using HMMSCAN (HMMER 3.0) was then performed.
GC content and sequencing coverage were calculated for each contig, and contigs containing 16S rRNA gene fragments were identified. This information was used to generate high-quality training data for phylogenetic annotation (binning) using PhyloPythiaS+ (65), resulting in 43 population bins at phylum, species, and phylotype levels. All population bins were manually inspected and refined based on visual %GC versus coverage plots (see Fig. S1 in the supplemental material). CheckM was used to estimate the quality of the bins (66). All hybrid MiSeq/PacBio contigs (>1 kb) were uploaded to Integrated Microbial Genomes (IMG) Expert Review for functional annotation. Scaffold sets where resubmitted to specify genomic bins using the IMG Workspace tool. Nucleotide MUMmer (NUCmer) was used for alignment of FrBGR genomic bins and closely related reference genomes.
Metaproteomics.
Proteins were extracted from the FrBGR sample by two approaches, one tailored for extracting proteins from within the bacterial cells and one for proteins residing in the extracellular liquid. The proteins identified with these two methods were combined into one proteome.
In the first method, cells and substrate were pelleted at 16,600 × g for 2 min and liquid was removed. In order to separate cells from substrate, the pellet was dissolved in 1% (vol/vol) MeOH, 1% (vol/vol) tert-butanol, 0.1% (vol/vol) Tween 80, pH 2.0, and substrate pelleted by gentle centrifugation at 100 × g for 20 s. The cell-containing supernatant was transferred to a new tube and the pellet washed again. This was repeated three times to increase the cell count. Cells, now dissociated from the substrate, were finally pelleted and washed in 10 mM Tris-HCl, 1 M NaCl, pH 8.0, prior to cell lysis. Lysis was performed using a bead-beating approach where glass beads (size, ≤106 μm) were added together with lysis buffer (50 mM Tris-HCl, 0.1% [vol/vol] Triton X-100, 200 mM NaCl, 1 mM dithiothreitol [DTT]), and cells were disrupted in 3 60-s cycles using a FastPrep24 (MP Biomedicals, Santa Ana, CA, USA). Debris were removed by centrifugation at 16,600 × g for 20 min, and proteins were precipitated overnight in 16% ice-cold trichloroacetic acid (TCA). The next day, proteins were dissolved in SDS sample buffer, separated by SDS-PAGE using an Any-kD Mini-Protean gel (Bio-Rad Laboratories, Hercules, CA, USA), and stained using Coomassie brilliant blue R250. The gel was cut in 16 slices and reduced, alkylated, and digested as described previously (67). Prior to mass spectrometry, peptides were desalted using C18 ZipTips (Merck Millipore, Darmstadt, Germany) according to the manufacturer's instructions.
A second method was also used, but it was optimized to extract proteins from the culture liquid. An aliquot of the FrBGR sample (15-ml biomass) was centrifuged at 4,500 × g for 10 min, and the supernatants were transferred to a 10-kDa-molecular-mass cutoff filter and concentrated to 500 μl. Proteins were then processed for mass spectrometric analysis while residing in the cutoff filter according to the FASP procedure (68). In brief, denaturing, alkylation, and digestion were accomplished by subsequently passing through 8 M urea, 50 mM iodoacetamide, and 2 μg trypsin in Tris-HCl, pH 7.8. Trypsination was performed overnight on a filter, and peptides were collected the next day by centrifugation, as these would now pass through the cutoff filter. Peptides were desalted by C18 ZipTips as described above.
The extracted proteins were denatured, reduced, carbamidomethylated, and further processed into peptides using trypsin. The peptides were analyzed by nano-liquid chromatography-tandem mass spectrometry as described previously using a Q-Exactive hybrid quadrupole Orbitrap mass spectrometer (Thermo Scientific, Bremen, Germany) (67), and acquired raw data were analyzed using MaxQuant (69), version 1.4.1.2. Proteins were quantified using the MaxLFQ algorithm (70). The data were searched against a sample-specific database (607.516 protein sequences), generated from the FrBGR metagenomic contigs that had been organized into phylogenomic bins using PhyloPythiaS+ (described above), and supplemented with reference genomes (www.ncbi.nlm.nih.gov) included in Table S1. In addition, common contaminants such as human keratins, trypsin, and bovine serum albumin were concatenated to the database, as were reversed sequences of all protein entries for estimation of false discovery rates. Protein N-terminal acetylation, oxidation of methionine, conversion of glutamine to pyroglutamic acid, and deamination of asparagine and glutamine were used as variable modifications, while carbamidomethylation of cysteine residues was used as a fixed modification. Trypsin was used as a digestion enzyme, and two missed cleavages were allowed. All identifications were filtered in order to achieve a protein false discovery rate of 1% using the target-decoy strategy. For a protein to be considered valid, we required the protein to be both identified and quantified in both replicates. In addition, we required at least one unique peptide per protein and at least two peptides in total for every protein. For every protein group reported by MaxQuant, we applied Occam's Razor in order to reduce the redundancy of homologous protein identifications. In order to apply this reduction across different taxonomic assignments, we utilized metabolic pathways as guiding lines. If a protein could be mapped to two different species and the protein belonged to a pathway, it was assigned to the species that had the most other protein identification supporting the same pathway.
Accession number(s).
Data sets are available at the NCBI Sequence Read Archive under accession number SRP076292. The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository (71) with the data set identifier PXD004538.
Supplementary Material
ACKNOWLEDGMENTS
This work was financially supported by the Norwegian Research Council (project no. 228747). J.A.F., M.Ø.A., and P.B.P. were supported by a grant from the European Research Council (336355-MicroDE).
Reactor material from an industrial anaerobic biogas reactor and operating parameter information were provided by the municipal renovation enterprise Fredrikstad Vann, Avløp og Renovasjonsforetak FREVAR FK, Fredrikstad, Norway. The sequencing service was provided by the Norwegian Sequencing Centre (NSC) (www.sequencing.uio.no). A special thanks to NSC staff member Ave Tooming-Klunderud for expertise regarding sequencing. We thank Abigail A. Salyers from the University of Illinois for her helpful advice and correspondence.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01955-16.
REFERENCES
- 1.Ward AJ, Hobbs PJ, Holliman PJ, Jones DL. 2008. Optimisation of the anaerobic digestion of agricultural resources. Bioresour Technol 99:1–15. doi: 10.1016/j.biortech.2008.02.044. [DOI] [PubMed] [Google Scholar]
- 2.Regueiro L, Carballa M, Lema JM. 2016. Microbiome response to controlled shifts in ammonium and LCFA levels in co-digestion systems. J Biotechnol 220:35–44. doi: 10.1016/j.jbiotec.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Vivekanand V, Olsen EF, Eijsink VG, Horn SJ. 2013. Effect of different steam explosion conditions on methane potential and enzymatic saccharification of birch. Bioresour Technol 127:343–349. doi: 10.1016/j.biortech.2012.09.118. [DOI] [PubMed] [Google Scholar]
- 4.Zabranska J, Stepova J, Wachtl R, Jenicek P, Dohanyos M. 2000. The activity of anaerobic biomass in thermophilic and mesophilic digesters at different loading rates. Water Sci Technol 42:49–56. [Google Scholar]
- 5.Bolzonella D, Cavinato C, Fatone F, Pavan P, Cecchi F. 2012. High rate mesophilic, thermophilic, and temperature phased anaerobic digestion of waste activated sludge: a pilot scale study. Waste Manag 32:1196–1201. doi: 10.1016/j.wasman.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 6.Sanchez E, Borja R, Weiland P, Travieso L, Martin A. 2000. Effect of temperature and pH on the kinetics of methane production, organic nitrogen and phosphorus removal in the batch anaerobic digestion process of cattle manure. Bioprocess Biosyst Eng 22:247–252. doi: 10.1007/s004490050727. [DOI] [Google Scholar]
- 7.Levén L, Eriksson AR, Schnürer A. 2007. Effect of process temperature on bacterial and archaeal communities in two methanogenic bioreactors treating organic household waste. FEMS Microbiol Ecol 59:683–693. doi: 10.1111/j.1574-6941.2006.00263.x. [DOI] [PubMed] [Google Scholar]
- 8.Karakashev D, Batstone DJ, Angelidaki I. 2005. Influence of environmental conditions on methanogenic compositions in anaerobic biogas reactors. Appl Environ Microbiol 71:331–338. doi: 10.1128/AEM.71.1.331-338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferry JG. 1993. Methanogenesis: ecology, physiology, biochemistry & genetics. Chapman & Hall, New York, NY. [Google Scholar]
- 10.Gujer W, Zehnder AJB. 1983. Conversion processes in anaerobic digestion. Water Sci Technol 15:127–167. [Google Scholar]
- 11.Thauer RK. 1998. Biochemistry of methanogenesis: a tribute to Marjory Stephenson. Microbiology 144:2377–2406. doi: 10.1099/00221287-144-9-2377. [DOI] [PubMed] [Google Scholar]
- 12.Schnürer A, Zellner G, Svensson BH. 1999. Mesophilic syntrophic acetate oxidation during methane formation in biogas reactors. FEMS Microbiol Ecol 29:249–261. doi: 10.1016/S0168-6496(99)00016-1. [DOI] [Google Scholar]
- 13.McInerney MJ, Struchtemeyer CG, Sieber J, Mouttaki H, Stams AJM, Schink B, Rohlin L, Gunsalus RP. 2008. Physiology, ecology, phylogeny, and genomics of microorganisms capable of syntrophic metabolism, p 58–72. In Wiegel J, Maier RJ, Adams MWW (ed), Incredible anaerobes: from physiology to genomics to fuels, vol 1125 Blackwell Publishing, Oxford, United Kingdom. [DOI] [PubMed] [Google Scholar]
- 14.Angelidaki I, Ahring BK. 1993. Thermophilic anaerobic digestion of livestock waste: the effect of ammonia. Appl Microbiol Biotechnol 38:560–564. [DOI] [PubMed] [Google Scholar]
- 15.Zamanzadeh M, Hagen LH, Svensson K, Linjordet R, Horn SJ. 2016. Anaerobic digestion of food waste—effect of recirculation and temperature on performance and microbiology. Water Res 96:246–254. doi: 10.1016/j.watres.2016.03.058. [DOI] [PubMed] [Google Scholar]
- 16.Sasaki K, Morita M, Sasaki D, Nagaoka J, Matsumoto N, Ohmura N, Shinozaki H. 2011. Syntrophic degradation of proteinaceous materials by the thermophilic strains Coprothermobacter proteolyticus and Methanothermobacter thermautotrophicus. J Biosci Bioeng 112:469–472. doi: 10.1016/j.jbiosc.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Schnürer A, Schink B, Svensson BH. 1996. Clostridium ultunense sp. nov., a mesophilic bacterium oxidizing acetate in syntrophic association with a hydrogenotrophic methanogenic bacterium. Int J Syst Bacteriol 46:1145–1152. doi: 10.1099/00207713-46-4-1145. [DOI] [PubMed] [Google Scholar]
- 18.Hattori S, Galushko AS, Kamagata Y, Schink B. 2005. Operation of the CO dehydrogenase/acetyl coenzyme A pathway in both acetate oxidation and acetate formation by the syntrophically acetate-oxidizing bacterium Thermacetogenium phaeum. J Bacteriol 187:3471–3476. doi: 10.1128/JB.187.10.3471-3476.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westerholm M, Roos S, Schnürer A. 2011. Tepidanaerobacter acetatoxydans sp. nov., an anaerobic, syntrophic acetate-oxidizing bacterium isolated from two ammonium-enriched mesophilic methanogenic processes. Syst Appl Microbiol 34:260–266. doi: 10.1016/j.syapm.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 20.Schink B. 1997. Energetics of syntrophic cooperation in methanogenic degradation. Microbiol Mol Biol Rev 61:262–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mosbæk F, Kjeldal H, Mulat DG, Albertsen M, Ward AJ, Feilberg A, Nielsen JL. 2016. Identification of syntrophic acetate-oxidizing bacteria in anaerobic digesters by combined protein-based stable isotope probing and metagenomics. ISME J 10:2405–2418. doi: 10.1038/ismej.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Müller B, Sun L, Westerholm M, Schnürer A. 2016. Bacterial community composition and fhs profiles of low-and high-ammonia biogas digesters reveal novel syntrophic acetate-oxidising bacteria. Biotechnol Biofuels 9:1. doi: 10.1186/s13068-015-0423-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frank JA, Arntzen MØ, Sun L, Hagen LH, McHardy AC, Horn SJ, Eijsink VGH, Schnürer A, Pope PB. 2016. Novel syntrophic populations dominate an ammonia-tolerant methanogenic microbiome. mSystems 1:e00092-16. doi: 10.1128/mSystems.00092-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartmann H, Ahring BK. 2005. Anaerobic digestion of the organic fraction of municipal solid waste: influence of co-digestion with manure. Water Res 39:1543–1552. doi: 10.1016/j.watres.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Anthonisen AC, Loehr RC, Prakasam TBS, Srinath EG. 1976. Inhibition of nitrification by ammonia and nitrous acid. J Water Pollut Control Fed 48:835–852. [PubMed] [Google Scholar]
- 26.Alexiev A, Coil DA, Badger JH, Enticknap J, Ward N, Robb FT, Eisen JA. 2014. Complete genome sequence of Coprothermobacter proteolyticus DSM 5265. Genome Announc 2:e00470-14. doi: 10.1128/genomeA.00470-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kosaka T, Uchiyama T, Ishii S-I, Enoki M, Imachi H, Kamagata Y, Ohashi A, Harada H, Ikenaga H, Watanabe K. 2006. Reconstruction and regulation of the central catabolic pathway in the thermophilic propionate-oxidizing syntroph Pelotomaculum thermopropionicum. J Bacteriol 188:202–210. doi: 10.1128/JB.188.1.202-210.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sieber JR, Sims DR, Han C, Kim E, Lykidis A, Lapidus AL, McDonnald E, Rohlin L, Culley DE, Gunsalus R. 2010. The genome of Syntrophomonas wolfei: new insights into syntrophic metabolism and biohydrogen production. Environ Microbiol 12:2289–2301. doi: 10.1111/j.1462-2920.2010.02237.x. [DOI] [PubMed] [Google Scholar]
- 29.Gagliano MC, Braguglia CM, Petruccioli M, Rossetti S. 2015. Ecology and biotechnological potential of the thermophilic fermentative Coprothermobacter spp. FEMS Microbiol Ecol 91:fiv018. doi: 10.1093/femsec/fiv018. [DOI] [PubMed] [Google Scholar]
- 30.Tandishabo K, Nakamura K, Umetsu K, Takamizawa K. 2012. Distribution and role of Coprothermobacter spp. in anaerobic digesters. J Biosci Bioeng 114:518–520. doi: 10.1016/j.jbiosc.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 31.Zhang R, El-Mashad HM, Hartman K, Wang F, Liu G, Choate C, Gamble P. 2007. Characterization of food waste as feedstock for anaerobic digestion. Bioresource Technol 98:929–935. doi: 10.1016/j.biortech.2006.02.039. [DOI] [PubMed] [Google Scholar]
- 32.Saiki T, Kobayashi Y, Kawagoe K, Beppu T. 1985. Dictyoglomus thermophilum gen. nov., sp. nov., a chemoorganotrophic, anaerobic, thermophilic bacterium. Int J Syst Bacteriol 35:253–259. [Google Scholar]
- 33.Nelson MC, Morrison M, Yu ZT. 2011. A meta-analysis of the microbial diversity observed in anaerobic digesters. Bioresour Technol 102:3730–3739. doi: 10.1016/j.biortech.2010.11.119. [DOI] [PubMed] [Google Scholar]
- 34.Riviere D, Desvignes V, Pelletier E, Chaussonnerie S, Guermazi S, Weissenbach J, Li T, Camacho P, Sghir A. 2009. Towards the definition of a core of microorganisms involved in anaerobic digestion of sludge. ISME J 3:700–714. doi: 10.1038/ismej.2009.2. [DOI] [PubMed] [Google Scholar]
- 35.Hagen LH, Vivekanand V, Pope PB, Eijsink VGH, Horn SJ. 2015. The effect of storage conditions on microbial community composition and biomethane potential in a biogas starter culture. Appl Microbiol Biotechnol 99:5749–5761. doi: 10.1007/s00253-015-6623-0. [DOI] [PubMed] [Google Scholar]
- 36.Tang Y-Q, Ji P, Hayashi J, Koike Y, Wu X-L, Kida K. 2011. Characteristic microbial community of a dry thermophilic methanogenic digester: its long-term stability and change with feeding. Appl Microbiol Biotechnol 91:1447–1461. doi: 10.1007/s00253-011-3479-9. [DOI] [PubMed] [Google Scholar]
- 37.Dodsworth JA, Blainey PC, Murugapiran SK, Swingley WD, Ross CA, Tringe SG, Chain PSG, Scholz MB, Lo C-C, Raymond J. 2013. Single-cell and metagenomic analyses indicate a fermentative and saccharolytic lifestyle for members of the OP9 lineage. Nat Commun 4:1854. doi: 10.1038/ncomms2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nobu MK, Dodsworth JA, Murugapiran SK, Rinke C, Gies EA, Webster G, Schwientek P, Kille P, Parkes RJ, Sass H. 2016. Phylogeny and physiology of candidate phylum “Atribacteria” (OP9/JS1) inferred from cultivation-independent genomics. ISME J 10:273–286. doi: 10.1038/ismej.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thauer RK, Jungermann K, Decker K. 1977. Energy-conservation in chemotrophic anaerobic bacteria. Bacteriol Rev 41:100–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagen LH, Vivekanand V, Linjordet R, Pope PB, Eijsink VG, Horn SJ. 2014. Microbial community structure and dynamics during co-digestion of whey permeate and cow manure in continuous stirred tank reactor systems. Biores Technol 171:350–359. doi: 10.1016/j.biortech.2014.08.095. [DOI] [PubMed] [Google Scholar]
- 41.Nobu MK, Narihiro T, Rinke C, Kamagata Y, Tringe SG, Woyke T, Liu W-T. 2015. Microbial dark matter ecogenomics reveals complex synergistic networks in a methanogenic bioreactor. ISME J 9:1710–1722. doi: 10.1038/ismej.2014.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McInerney MJ, Bryant MP, Pfennig N. 1979. Anaerobic bacterium that degrades fatty acids in syntrophic association with methanogens. Arch Microbiol 122:129–135. doi: 10.1007/BF00411351. [DOI] [PubMed] [Google Scholar]
- 43.McInerney MJ, Bryant MP, Hespell RB, Costerton JW. 1981. Syntrophomonas wolfei gen. nov., sp. nov., an anaerobic, syntrophic, fatty acid-oxidizing bacterium. Appl Environ Microbiol 41:1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sekiguchi Y, Kamagata Y, Nakamura K, Ohashi A, Harada H. 2000. Syntrophothermus lipocalidus gen. nov., sp. nov., a novel thermophilic, syntrophic, fatty-acid-oxidizing anaerobe which utilizes isobutyrate. Int J Syst Evol Microbiol 50:771–779. doi: 10.1099/00207713-50-2-771. [DOI] [PubMed] [Google Scholar]
- 45.Ho D, Jensen P, Batstone D. 2014. Effects of temperature and hydraulic retention time on acetotrophic pathways and performance in high-rate sludge digestion. Environ Sci Technol 48:6468–6476. doi: 10.1021/es500074j. [DOI] [PubMed] [Google Scholar]
- 46.Müller B, Sun L, Schnürer A. 2013. First insights into the syntrophic acetate-oxidizing bacteria—a genetic study. Microbiologyopen 2:35–53. doi: 10.1002/mbo3.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hattori S. 2008. Syntrophic acetate-oxidizing microbes in methanogenic environments. Microbes Environ 23:118–127. doi: 10.1264/jsme2.23.118. [DOI] [PubMed] [Google Scholar]
- 48.Yenigün O, Demirel B. 2013. Ammonia inhibition in anaerobic digestion: a review. Process Biochem 48:901–911. doi: 10.1016/j.procbio.2013.04.012. [DOI] [Google Scholar]
- 49.Fotidis IA, Karakashev D, Kotsopoulos TA, Martzopoulos GG, Angelidaki I. 2013. Effect of ammonium and acetate on methanogenic pathway and methanogenic community composition. FEMS Microbiol Ecol 83:38–48. doi: 10.1111/j.1574-6941.2012.01456.x. [DOI] [PubMed] [Google Scholar]
- 50.Hattori S, Kamagata Y, Hanada S, Shoun H. 2000. Thermacetogenium phaeum gen. nov., sp. nov., a strictly anaerobic, thermophilic, syntrophic acetate-oxidizing bacterium. Int J Syst Evol Microbiol 50:1601–1609. doi: 10.1099/00207713-50-4-1601. [DOI] [PubMed] [Google Scholar]
- 51.Balk M, Weijma J, Stams AJ. 2002. Thermotoga lettingae sp. nov., a novel thermophilic, methanol-degrading bacterium isolated from a thermophilic anaerobic reactor. Int J Syst Evol Microbiol 52:1361–1368. doi: 10.1099/00207713-52-4-1361. [DOI] [PubMed] [Google Scholar]
- 52.Westerholm M, Roos S, Schnürer A. 2010. Syntrophaceticus schinkii gen. nov., sp. nov., an anaerobic, syntrophic acetate-oxidizing bacterium isolated from a mesophilic anaerobic filter. FEMS Microbiol Lett 309:100–104. [DOI] [PubMed] [Google Scholar]
- 53.Strittmatter AW, Liesegang H, Rabus R, Decker I, Amann J, Andres S, Henne A, Fricke WF, Martinez-Arias R, Bartels D. 2009. Genome sequence of Desulfobacterium autotrophicum HRM2, a marine sulfate reducer oxidizing organic carbon completely to carbon dioxide. Environ Microbiol 11:1038–1055. doi: 10.1111/j.1462-2920.2008.01825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Callaghan A, Morris B, Pereira I, McInerney M, Austin RN, Groves JT, Kukor J, Suflita J, Young L, Zylstra G. 2012. The genome sequence of Desulfatibacillum alkenivorans AK-01: a blueprint for anaerobic alkane oxidation. Environ Microbiol 14:101–113. doi: 10.1111/j.1462-2920.2011.02516.x. [DOI] [PubMed] [Google Scholar]
- 55.Zhilina TN, Zavarzina DG, Kolganova TV, Tourova TP, Zavarzin GA. 2005. “Candidatus Contubernalis alkalaceticum,” an obligately syntrophic alkaliphilic bacterium capable of anaerobic acetate oxidation in a coculture with Desulfonatronum cooperativum. Microbiology 74:695–703. doi: 10.1007/s11021-005-0126-4. [DOI] [PubMed] [Google Scholar]
- 56.Clescerl LS, Greenberg AE, Eaton AD (ed). 1999. Standard methods for the examination of water and wastewater, 20th ed American Public Health Association, Washington, DC. [Google Scholar]
- 57.Rosewarne CP, Pope PB, Denman SE, McSweeney CS, O'Cuiv P, Morrison M. 2010. High-yield and phylogenetically robust methods of DNA recovery for analysis of microbial biofilms adherent to plant biomass in the herbivore gut. Microb Ecol 61:448–454. doi: 10.1007/s00248-010-9745-z. [DOI] [PubMed] [Google Scholar]
- 58.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 61.Ondov B, Bergman N, Phillippy A. 2011. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. doi: 10.1186/1471-2105-12-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eren AM, Zozaya M, Taylor CM, Dowd SE, Martin DH, Ferris MJ. 2011. Exploring the diversity of Gardnerella vaginalis in the genitourinary tract microbiota of monogamous couples through subtle nucleotide variation. PLoS One 6:e26732. doi: 10.1371/journal.pone.0026732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frank JA, Pan Y, Tooming-Klunderud A, Eijsink VG, McHardy AC, Nederbragt AJ, Pope PB. 2016. Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence data. Sci Rep 6:25373. doi: 10.1038/srep25373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu W, Lomsadze A, Borodovsky M. 2010. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res 38:e132. doi: 10.1093/nar/gkq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gregor I, Dröge J, Schirmer M, Quince C, McHardy AC. 2014. PhyloPythiaS+: a self-training method for the rapid reconstruction of low-ranking taxonomic bins from metagenomes. arXiv arXiv:14067123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arntzen MØ, Karlskås IL, Skaugen M, Eijsink VGH, Mathiesen G. 2015. Proteomic investigation of the response of Enterococcus faecalis V583 when cultivated in urine. PLoS One 10:e0126694. doi: 10.1371/journal.pone.0126694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wisniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat Methods 6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 69.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 70.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. 2014. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vizcaíno JA, Côté RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J. 2013. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res 41:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.