

ABSTRACT
Filarial parasites cause functional impairment of host dendritic cells (DCs). However, the effects of early infection on individual DC subsets are not known. In this study, we infected BALB/c mice with infective stage 3 larvae of the lymphatic filarial parasite Brugia malayi (Bm-L3) and studied the effect on fluorescence-activated cell sorter (FACS)-sorted DC subsets. While myeloid DCs (mDCs) accumulated by day 3 postinfection (p.i.), lymphoid DCs (LDCs) and CD8+ plasmacytoid DCs (pDCs) peaked at day 7 p.i. in the spleens and mesenteric lymph nodes (mLNs) of infected mice. Increased tumor necrosis factor alpha (TNF-α) but reduced interleukin 12 (IL-12) and Toll-like receptor 4 (TLR4), -6, and -9 and reciprocal secretion of IL-4 and IL-10 were also observed across all DC subsets. Interestingly, Bm-L3 increased the expression of CD80 and CD86 across all DC subsets but decreased that of major histocompatibility complex class II (MHC-II) on mDCs and pDCs, resulting in their impaired antigen uptake and presentation capacities, but maximally attenuated the T-cell proliferation capacity of only mDCs. Furthermore, Bm-L3 increased phosphorylated p38 (p-p38), but not p-ERK, in mDCs and LDCs but downregulated them in pDCs, along with differential modulation of protein tyrosine phosphatases SHP-1, TCPTP, PTEN, and PTP1B across all DC subsets. Taken together, we report hitherto undocumented effects of early Bm-L3 infection on purified host DC subsets that lead to their functional impairment and attenuated host T-cell response.
KEYWORDS: filariasis, myeloid dendritic cells, lymphoid dendritic cells, plasmacytoid dendritic cells, flow cytometry, MAP kinases, Toll-like receptors
INTRODUCTION
Parasites have evolved many diverse and novel strategies to evade the host immune response (1). These include dampening of the host's proinflammatory cytokine response, attenuating the functions of innate immune cells; generating regulatory T cells; and impairing the activation, maturation, and functions of host dendritic cells (DCs), leading to their functional impairment (2–9). Impairment of host Langerhans cells has also been reported during lymphatic filarial infections, which suggests that parasites not only interfere with the functions of Langerhans cells, but have also developed means to expertly evade antigen (Ag)-presenting cell (APC) detection in the host skin (1, 10). We recently documented the role of lung eosinophils and functional impairment of lung macrophages during filarial manifestation of tropical pulmonary eosinophilia (TPE) (11). This is important, as macrophages, along with DCs, not only are a heterogeneous group of APCs, but immature DCs circulate in the peripheral blood, where they capture, process, and present antigens (12). Also, encounter of DCs with the pathogen results in their maturation, characterized by increased expression of major histocompatibility complex class II (MHC-II) and costimulatory molecules, CD40, CD80, and CD86, and their subsequent migration to the secondary lymphoid organs, where they help in the initiation of specific immune responses, including adaptive immunity (12, 13).
Though a few reports have looked into the role of primary host DCs during the very early stages of filarial infection (7, 8), to our knowledge, not only are DC subset-specific studies lacking, it is also not clear to what extent these subsets are affected during the early stages of filarial infection. In the present study, we infected BALB/c mice with infective larval stage 3 (L3) of the filarial nematode Brugia malayi (Bm-L3) and studied the effect of this inoculation on the recruitment patterns of different DC subsets, viz., myeloid DCs (mDCs), lymphoid DCs (LDCs), CD8+ plasmacytoid DCs (pDCs), and CD8− pDCs, in the spleens and mesenteric lymph nodes (mLNs) of mice at 12 h, 3 days, 5 days, 7 days, and 10 days postinfection (p.i.). We found that Bm-L3 infection led to differential accumulation of host DC subsets in the spleens and mLNs of infected mice and caused variable secretion of both Th1 and Th2 cytokines, resulting in the generation of a mixed cytokine milieu. Furthermore, we also observed differential maturation and activation patterns of different DC subsets that correlated with their impaired antigen uptake and presentation capacities and led to selective attenuation of their T-cell proliferation capacities. Infection with Bm-L3 also caused moderate to severe downmodulation of various TLRs; members of host MAP kinases (MAPKs) p38 and ERK; and protein tyrosine phosphatases, viz., SHP-1, TCPTP, PTEN, and PTP1B, depending on the DC subset in question.
Collectively, our study demonstrates that early modulation of the immune response by the filarial parasite is not restricted to merely skewing the host immune response to a type 2 response but that other underlying complex mechanisms also occur, which differentially impair the functional capacities of individual host DC subsets, rendering them ineffective in initiating a strong adaptive immune response. Interventions that can boost the early host immune response of host DCs during the very early stages of filarial infection can therefore provide relief to patients suffering from filariasis.
RESULTS
Dendritic cell subsets accumulate in the secondary lymphoid organs of mice following Bm-L3 infection.
Using specific combinations of fluorochrome-tagged monoclonal antibodies, we successfully identified four major subsets of DCs (mDCs, LDCs, CD8+ pDCs, and CD8− pDCs) present in the spleens and mLNs of mice and sorted them to high purity (≥98%) using a FACS Aria cell sorter, as described in Materials and Methods and outlined in Fig. 1. After sorting analysis and cytospins, the preparations confirmed the purity of the sorted DC subsets (Fig. 1, far right). Detailed analysis of various DC subsets showed increased accumulation of mDCs (CD11c+, CD11b+, and CD8a−) in the spleens of infected mice starting from 12 h postinfection (46% ± 5%) until day 10 postinfection (53% ± 5%) (Fig. 2A), while there was no major change in the percentages of splenic LDCs (CD11c+, CD11b−, and CD8a+) during the first 10 days of Bm-L3 infection (Fig. 2A). Interestingly, the recruitment patterns of mDCs and LDCs in the mLNs was quite different from that observed in the spleens; while mDCs increased soon after infection and attained their peak by day 3 postinfection (32% ± 2%), they declined rapidly thereafter and were present at their lowest levels by day 10 postinfection (9.0% ± 5%). However, LDCs in the mLNs first declined by day 3 postinfection (26% ± 4%) but rapidly increased thereafter and reached their peak levels by day 10 postinfection (51% ± 6%) (Fig. 2B).
FIG 1.

Immunophenotyping and sorting of host dendritic cell subsets. A gating strategy for identification and sorting of different host dendritic cell subsets present in the CD11c-positive cell fraction from mouse spleens is shown. A similar strategy was adopted for mLNs, except that the cells were not sorted. Post-sort dot plots and May-Grunwald-Giemsa-stained cytospins (right) illustrate very high purity (≥98%) of the sorted cells.
FIG 2.

Kinetics of host dendritic cell subset accumulation in the secondary lymphoid organs of mice. (A and B) Percentages of various host DC subsets as enumerated by flow cytometry in spleens (A) and mLNs (B) of mice at different time points post-Bm-L3 infection. (C and D) Absolute quantification of each DC subset, using TruCount beads, in spleens (C) and mLNs (D) of mice at day 3 and day 7 post-Bm-L3 infection. The data represent means ± SD of the results of three independent experiments with at least 5 or 6 animals/group. P values of ≤0.05 (*) and ≤0.01 (**) were considered significant and highly significant, respectively. Cont, control.
Similarly, the recruitment patterns of CD8a+ and CD8− pDCs in the spleens and mLNs of mice also showed an interesting trend. While CD8a+ pDCs declined marginally in the spleens by day 5 postinfection, they reached their peak by day 7 postinfection (10% ± 2%), only to decline abruptly by day 10 postinfection (3% ± 1%) to a point approximately 3-fold lower than in uninfected mice (Fig. 2A). However, CD8a− splenic pDCs moderately increased after infection and attained their peak between day 7 and day 10 postinfection (8% ± 3%) (Fig. 2A). Similarly, CD8a+ pDCs in the mLNs initially declined by day 3 postinfection (7% ± 2%) but then increased rapidly to reach their peak levels by day 7 postinfection (23% ± 2%), only to decline suddenly by day 10 postinfection (9% ± 2%) (Fig. 2B). However, quite surprisingly, just like splenic LDCs, CD8a− pDCs in the mLNs did not show any major change during the entire course of infection (Fig. 2B). Quite interestingly, we observed a largely similar trend in the recruitment kinetics of various DC subsets at day 3 and day 7 post-Bm-L3 infection when absolute quantification of these subsets was done using TruCount beads (BD Biosciences, San Jose, CA) (Fig. 2C and D).
Taken together, these results show that infection with Bm-L3 leads to differential recruitment kinetics of various DC subsets in the secondary lymphoid organs of mice, which might either be correlated with the establishment of L3 infection within the host or due to the molting of Bm-L3 to the L4 stage, with a bearing on the consequences of initiation of adaptive immunity in the host during the early days of filarial infection.
Bm-L3 differentially modulates the cytokine secretion patterns of different DC subsets.
Secretion of Th1 (tumor necrosis factor alpha [TNF-α] and interleukin 12 [IL-12]) and Th2 (IL-4 and IL-10) cytokines was analyzed in mDCs, LDCs, and pDCs post-Bm-L3 infection, as outlined in Materials and Methods. Our results, shown as percentages of cytokine-secreting cells in Fig. 3, show increased TNF-α secretion by all DC subsets post-Bm-L3 infection, with a more prominent increase at day 7 postinfection than in uninfected mice (P ≤ 0.05 for mDCs and LDCs and P ≤ 0.001 for pDCs at day 7). However, quite contrary to the heightened pattern of TNF-α secretion, secretion of IL-12 mostly decreased across all DC subsets at day 3 but increased at day 7 postinfection compared to uninfected mice (P ≤ 0.01 for mDCs at day 3 and day 7, P ≤ 0.001 for LDCs at day 3 and day 7, and P ≤ 0.05 for pDCs at day 3). Similarly, while reduced secretion of IL-10 was seen in mDCs and LDCs at day 3 p.i. compared to uninfected controls (P ≤ 0.05 for mDCs and LDCs at day 3), it increased by day 7 compared to day 3 postinfection (P ≤ 0.001 for mDCs and P ≤ 0.01 for LDCs at day 7). Interestingly, quite contrary to observations in mDCs and LDCs, secretion of IL-10 was higher in pDCs at both day 3 and day 7 p.i. than in uninfected controls (P ≤ 0.01 at day 3 and P ≤ 0.001 at day 7). Also interesting was the observation of highly elevated levels of IL-4 across all DC subsets at day 3 postinfection compared to uninfected controls (P ≤ 0.001 at day 3 for all DC subsets). Although levels of IL-4 decreased significantly by day 7 p.i. compared to day 3 p.i. (P ≤ 0.001 at day 7 for all DC subsets), they remained at higher levels than in uninfected controls (P ≤ 0.01 at day 7 for all DC subsets). These results suggest that Bm-L3 differentially affects the cytokine-secreting potentials of different DC subsets during the initial stages of infection, which might have a bearing on the recruitment patterns of DCs and other leukocytes in the secondary lymphoid organs of infected mice.
FIG 3.
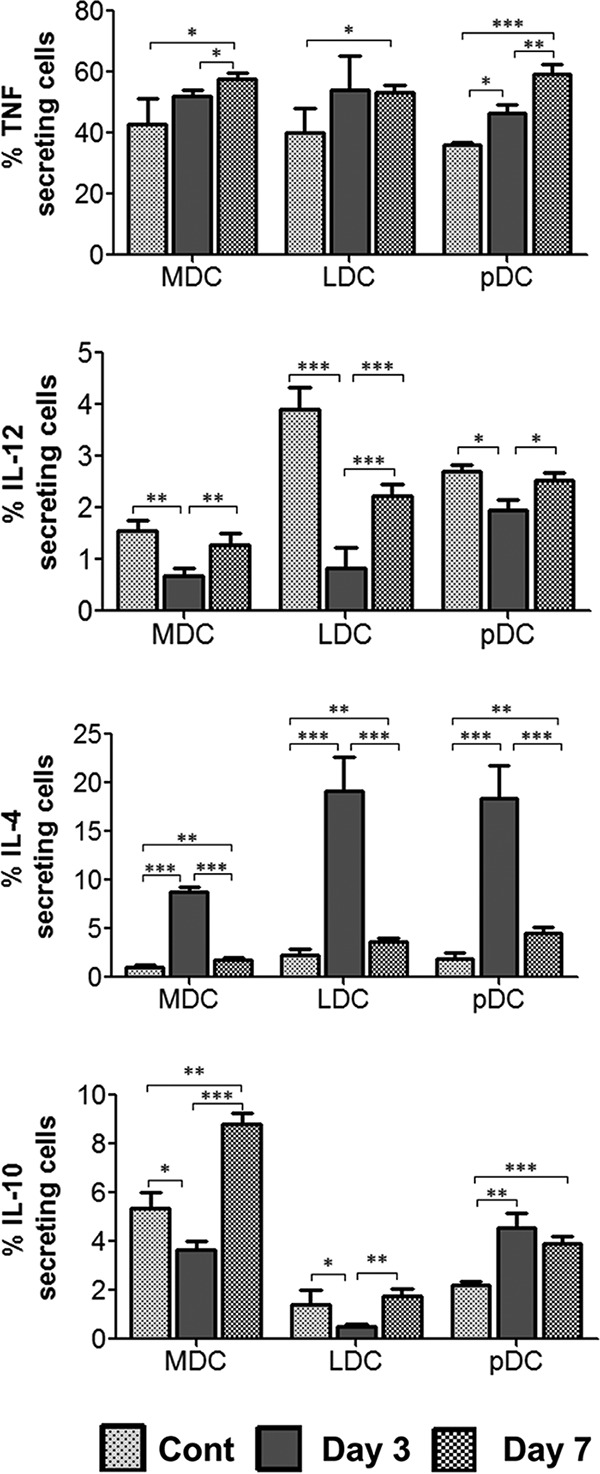
Estimation of cytokine secretion by host dendritic cell subsets. A CD11c-positive cell fraction from mouse spleens was subjected to intracellular staining using monoclonal antibodies against TNF-α, IL-4, IL-10, and IL-12, and the cells were subsequently acquired on a BD FACS Aria flow cytometer, as described in Materials and Methods. The data represent means and SD of the DC subsets secreting the respective cytokines at day 3 and day 7 post-Bm-L3 infection from the results of three independent experiments with 5 or 6 mice/group. P values of ≤0.05 (*), ≤0.01 (**), and ≤0.001 (***) were considered significant, highly significant, and very highly significant, respectively.
Toll-like receptors are downregulated following infection with Bm-L3.
Transcript levels of different Toll-like receptors (TLRs), viz., TLR2, -4, -6, and -9, were analyzed in flow-sorted mDCs, LDCs, and pDCs at day 3 and day 7 post-Bm-L3 infection using real-time reverse transcription (RT)-PCR, as described in Materials and Methods. The results show that, compared to uninfected mice, infection with Bm-L3 leads to significant downregulation of nearly all TLRs across all DC subsets, with the exception of TLR2, which was moderately elevated (approximately 2.3-fold and 4.7-fold in LDCs and pDCs, respectively) at day 3 postinfection (Fig. 4A) (P ≤ 0.001 in LDCs and P ≤ 0.01 in pDCs). Interestingly, mDCs showed maximal 10-fold downregulation of TLR2 and TLR4 by day 7 postinfection and 21-fold and 29-fold downregulation of TLR6 and TLR9 at day 3 postinfection (P ≤ 0.01 for both TLRs). Similarly, LDCs showed 9-fold downregulation of TLR4 (P ≤ 0.05) and TLR9 (P ≤ 0.01) at day 3 postinfection and 8-fold downregulation of TLR6 by day 7 postinfection. Likewise, pDCs reported approximately 9-fold downregulation of TLR9 on day 3 postinfection (P ≤ 0.05) and about 12-fold downregulation of TLR6 by day 7 postinfection. To lend weight to our findings, we also studied the expression patterns of these TLRs at the protein level using flow cytometry, as described in Materials and Methods. Our results, plotted as histograms in Fig. 4B, largely matched the results obtained at the mRNA level with a few exceptions. The mean fluorescence intensity (MFI) of each TLR present on the host DC subset is given in Table 1. These results show that infection with Bm-L3 rapidly downmodulated the expression patterns of different TLRs present on different host DC subsets, which might have a bearing on the development of the overall inflammatory response of the host during the early stages of filarial infection.
FIG 4.

Estimation of TLRs in host dendritic cell subsets. (A) Real-time RT-PCR was used to measure transcript levels of different TLRs in flow-sorted mDCs, LDCs, and pDCs at day 3 and day 7 post-Bm-L3 infection. The values represent fold changes (means ± SD) compared to an uninfected control. (B) Surface expression of TLR2, -4, and -6 and intracellular expression of TLR9 on each DC subset at day 3 and day 7 post-Bm-L3 infection, as assessed by flow cytometry. The data are representative of the results of three independent experiments with 5 or 6 mice/group. P values of ≤0.05 (*), ≤0.01 (**), and ≤0.001 (***) were considered significant, highly significant, and very highly significant, respectively.
TABLE 1.
MFIs of different TLRs on DC subsets in sham-infected and Bm-L3-infected mice at day 3 and day 7 postinfection
| Cell type | Infectiona | MFI |
|||
|---|---|---|---|---|---|
| TLR2 | TLR4 | TLR6 | TLR9 | ||
| mDC | Cont | 3,306 ± 706 | 1,319 ± 77 | 20,503 ± 238 | 2,741 ± 233 |
| Day 3 | 2,644 ± 392 | 1,184 ± 146 | 9,858 ± 1,295 | 2,622 ± 307 | |
| Day 7 | 3,649 ± 244 | 1,131 ± 145 | 8,628 ± 21 | 2,270 ± 368 | |
| LDC | Cont | 1,670 ± 170 | 760 ± 21 | 11,567 ± 786 | 1,444 ± 36 |
| Day 3 | 1,965 ± 475 | 624 ± 78 | 8,422 ± 2,947 | 1,495 ± 95 | |
| Day 7 | 1,776 ± 46 | 742 ± 17 | 4,236 ± 796 | 1,514 ± 212 | |
| pDC | Cont | 2,298 ± 135 | 949 ± 52 | 14,569 ± 1,249 | 4,325 ± 258 |
| Day 3 | 2,915 ± 656 | 907 ± 109 | 13,687 ± 87 | 3,162 ± 44 | |
| Day 7 | 2,776 ± 712 | 1,190 ± 274 | 11,896 ± 557 | 4,125 ± 217 | |
Cont, sham-infected (control) mice; Day 3 and Day 7, Bm-L3-infected mice at day 3 and day 7 postinfection.
Bm-L3 modulates the expression of costimulatory and maturation markers on host DC subsets.
To evaluate the effects of Bm-L3 infection on the costimulatory and maturation markers CD80, CD86, and MHC-II present on DCs, flow-sorted DC subsets were processed as described in Materials and Methods. The results shown in Fig. 5A reveal that expression of CD80 increased significantly at day 3 postinfection in mDCs (P ≤ 0.01) and LDCs (P ≤ 0.05) and increased even further by day 7 postinfection in mDCs (P ≤ 0.001), LDCs (P ≤ 0.01), and pDCs (P ≤ 0.001). Interestingly, expression of CD86 also increased significantly in mDCs and pDCs by day 7 postinfection (P ≤ 0.05 for both), but surprisingly, there was no change in the expression of CD86 on LDCs during the course of infection. Of note, the expression of MHC-II decreased in mDCs and pDCs by day 7 postinfection (P ≤ 0.01 for mDCs and P ≤ 0.05 for pDCs), but once again, no change was reported in LDCs during the course of infection (Fig. 5A). These results show that Bm-L3 differentially modulates the expression patterns of costimulatory and maturation markers on host DCs, depending on the dendritic cell subset in question.
FIG 5.

Assessment of maturation markers and T-cell proliferation capacities of host dendritic cell subsets. (A) Expression of maturation and costimulatory markers was assessed on flow-sorted mDCs, LDCs, and pDCs at day 3 and day 7 post-Bm-L3 infection using flow cytometry. Representative histograms show MFI values of DC subsets at the given time points. (B and C) Antigen uptake (phagocytosis) and antigen presentation capacities of flow-sorted mDCs, LDCs, and pDCs were assessed at day 3 and day 7 post-Bm-L3 infection by incubating the cells with either FITC-dextran or DQ-ovalbumin, respectively, followed by acquisition on a BD FACS Aria flow cytometer, as described in Materials and Methods. Shown are percentages of phagocytic cells (B) and MFIs (C) of DC subsets at the given time points. (D) Mitochondrial activity, as a measure of T-cell proliferation, was measured by XTT assay by coculturing flow-sorted mDCs, LDCs, and pDCs with CD4+ T cells, as described in Materials and Methods. Shown are optical densities at 450 nm (OD450) of DC subsets at the given time points. The data are representative of three independent experiments with at least 5 or 6 animals/group. P values of ≤0.05 (*), ≤0.01 (**), and ≤0.001 (***) were considered significant, highly significant, and very highly significant, respectively.
Bm-L3 infection impairs the antigen uptake, antigen presentation, and T-cell proliferation capacities of dendritic cell subsets.
The effects of Bm-L3 on the antigen uptake (phagocytosis), antigen presentation, and T-cell proliferation capacities of different flow-sorted DC subsets were studied, as described in Materials and Methods. Our results show that Bm-L3 selectively decreased the antigen uptake capacity of pDCs at both day 3 and day 7 postinfection (Fig. 5B) (P ≤ 0.05), but in contrast, it marginally increased that of mDCs at day 7 postinfection (P ≤ 0.05); however, no major change was observed in the antigen uptake capacity of LDCs compared to uninfected controls (Fig. 5B). Interestingly, Bm-L3 selectively decreased the antigen presentation capacity of mDCs at day 7 postinfection (Fig. 5C) (P ≤ 0.05) and that of pDCs at day 3 and day 7 postinfection (P ≤ 0.05), but once again, the antigen presentation capacity of LDCs remained unaltered during the course of infection (Fig. 5C). These results show that Bm-L3 selectively modulates the antigen uptake and presentation capacities of mDCs and pDCs but does not affect that of LDCs.
To further elucidate the effects of impaired antigen uptake and presentation capacities of host DC subsets on the priming of the T-cell response, flow-sorted DC subsets were cocultured with naive CD4+ T cells as described in Materials and Methods, and our results showed that infection with Bm-L3 leads to maximal impairment of the T-cell proliferation capacity of mDCs at day 7 postinfection (Fig. 5D) (P ≤ 0.01). Although we did observe some reduction in the T-cell proliferation capacities of LDCs and pDCs at day 7 postinfection compared to day 3 postinfection (P ≤ 0.01 for LDCs and P ≤ 0.001 for pDCs) (Fig. 5D), no major effect was seen in LDCs and pDCs compared to controls. These results again imply that Bm-L3 causes selective attenuation of the T-cell proliferation capacity of host DC subsets, which might affect the overall induction of an adaptive host immune response in infected animals.
Bm-L3 impairs activation of MAP kinases in host DC subsets.
Filarial antigens modulate the expression of several kinases, including MAP kinases, which affects the overall proinflammatory signaling cascade of the host cellular machinery. We elucidated the effect of Bm-L3 on the expression patterns of two key members of the MAP kinase pathway, phosphorylated p38 (p-p38) and p-ERK, in flow-sorted mDCs, LDCs, and pDCs, as described in Materials and Methods. Our results show that compared to uninfected controls, infection with Bm-L3 led to significant upregulation of p-p38 in mDCs and LDCs at day 3 postinfection (Fig. 6A) (P ≤ 0.05 for mDCs and P ≤ 0.01 for LDCs); even though this decreased slightly in mDCs by day 7 postinfection, it was still at higher levels than in controls (P ≤ 0.05). However, unlike mDCs and LDCs, significant downregulation of p-p38 was observed in pDCs at day 7 postinfection (P ≤ 0.01). Interestingly, in contrast to p-p38, significant downregulation of p-ERK was noted in mDCs and pDCs at day 7 postinfection (Fig. 6A) (P ≤ 0.01 for mDCs and pDCs), but no significant change was reported in LDCs during this period. Furthermore, to gain deeper insights into the role of negative regulators of the MAP kinase signal transduction pathway, we measured the transcript levels of protein tyrosine phosphatases SHP-1, TCPTP, PTEN, and PTP1B in flow-sorted DC subsets and found moderately upregulated levels of SHP-1, TCPTP, PTEN, and PTP1B at day 3 postinfection only in mDCs (Fig. 6B) (P ≤ 0.05 for SHP-1 and PTEN); however, quite surprisingly, all protein tyrosine phosphatases were downregulated at day 3 postinfection in LDCs and pDCs (P ≤ 0.01 for TCPTP and PTP1B in LDCs and P ≤ 0.05 for SHP-1 and PTEN in pDCs), although some moderately upregulated levels of TCPTP and PTP1B were seen in LDCs at day 7 postinfection (P ≤ 0.01) and of SHP-1 and PTEN in pDCs at day 7 postinfection (P ≤ 0.05). Taken together, these results show that Bm-L3 differentially modulates not only the expression of key members of the MAP kinase signaling pathway, but also that of protein tyrosine phosphatases during the early phase of filarial infection.
FIG 6.

Activation of MAP kinases and protein tyrosine phosphatases. (A) Phosphorylation of p38 and ERK was assessed in flow-sorted mDCs, LDCs, and pDCs at day 3 and day 7 post-Bm-L3 infection using flow cytometry, as described in Materials and Methods. Shown are MFIs of cells at the given time points. (B) Real-time RT-PCR was used to measure transcript levels of different protein tyrosine phosphatases (SHP-1, TCPTP, PTEN, and PTP1B) in flow-sorted mDCs, LDCs, and pDCs at day 3 and day 7 post-Bm-L3 infection. The values represent fold changes (means ± SD) compared to an uninfected control. The data are representative of three independent experiments with 5 or 6 mice/group. P values of ≤0.05 (*) and ≤0.01 (**) were considered significant and highly significant, respectively.
DISCUSSION
Filarial parasites cause chronic infections that are usually clinically asymptomatic and without significant pathology (10). However, the effects of filarial worms on various subsets of host DCs during the very early stages of infection remain obscure. Recently, using a mouse model of TPE, we showed that the pathogenesis of TPE is characterized by functional impairment of alveolar macrophages (AMϕs), alternative activation of lung macrophages, and upregulation of antiapoptotic genes by eosinophils, which led to severe lung inflammation and compromised host lung immunity (11). Extrapolating our findings even further, in the present study, we infected BALB/c mice with infective Bm-L3 and studied the effect of this infection on flow-sorted DC subsets present in the secondary lymphoid organs of mice. We observed that, compared to uninfected controls, Bm-L3 infection disturbed the homeostasis of DC subpopulations present in the secondary lymphoid organs of infected mice, as contrasting trends of mDC and LDC accumulation were noted in the secondary lymphoid organs of infected mice. While mDCs slightly increased in spleens during the course of infection, LDCs, after an initial drop at day 3 p.i. in mLNs, showed preferential accumulation between day 5 and day 10 p.i. However, it is important to note that the slight increase in the mDC pool after Bm-L3 infection might be due to the infiltration and subsequent differentiation of circulating inflammatory monocytes into the spleen. However, concrete evidence for this is lacking, and further investigations into the phenomenon using chimeric mice are needed to substantiate our hypothesis. Also, the initial loss of LDCs might be due to the preferential apoptosis of the cells, as LDCs are responsible for activating CD8+ T cells and subsequent initiation of a cytotoxic immune response against the parasite; therefore, we believe that this might be an evasion strategy honed by the parasite during the initial stages of infection. Furthermore, preferential accumulation of CD8− pDCs was also observed only in the spleens of infected mice at day 10 p.i., but not in the mLNs, which shows that Bm-L3 differentially modulates the recruitment pattern of host DC subsets during the early stages of filarial infection. This observation is in line with an earlier report documenting an expanded pool of circulating mDCs during patent human filarial infection (14) and selective migration of DC subsets during other parasitic infections (7).
Interestingly, the percentages of CD8+ pDCs peaked at day 7 p.i. in both the spleens and mLNs of mice, which correlates with a previous report in which it was shown that following antigenic challenge, activated pDCs express high levels of CD8, regardless of their previous expression of CD4 (15). However, thereafter, CD8+ pDCs showed an abrupt fall in their percentages by day 10 postinfection in both the secondary lymphoid organs, which might be due to selective apoptosis of host APCs, as reported previously (6), or could be correlated with the molting of infective L3 larvae into stage L4, resulting in altered kinetics of DC subset recruitment, as reported previously (16, 17).
Quite importantly, our absolute quantification data for various DC subsets obtained using TruCount beads largely matched the trend of preferential accumulation of these subsets observed in the spleens and mLNs of infected mice. Though there were some minor exceptions, occasional disparities observed between the percentage of a particular cell obtained through hierarchical gating and the absolute number of the same cell obtained using TruCount beads have been documented previously by several other researchers (18). Furthermore, our results showing increased TNF-α secretion by all DC subsets correlates with an earlier report of increased TNF-α production during parasite infections (7). Also, we believe that increased TNF production across all DC subsets can also be due to involvement of TNF-inducible nitric oxide synthase (iNOS)-secreting (TiP) DCs; however, to substantiate this hypothesis, further investigation is required. Also, the contrasting patterns of IL-4 and IL-10 secretion are in line with a previous report showing that during murine filariasis, IL-10 counterregulates IL-4-dependent effector mechanisms, thereby providing causal evidence of existing antagonistic activity between IL-4 and IL-10 in vivo (19). It is also worth noting that while mature DCs synthesize high levels of IL-12 (20), which resist the suppressive effects of IL-10 required for parasite establishment (21), release of several excretory-secretory (ES) products by the infective larvae also impairs both the Th1 and Th2 pathways (22). Boonstra and colleagues have also suggested that distinct murine DC subsets are preprogrammed to direct either Th1 or Th2 development (23, 24), which suggests that helminths modulate host immunity by modifying DC responsiveness in a manner that favors a Th2 outcome (7). Collectively, we believe that a very complex scenario emerges soon after the onset of Bm-L3 infection in which different DC subsets and a rapidly evolving cytokine milieu act together to shape the overall immune response of the host (15, 25).
Considering the importance of TLRs during filarial and other parasitic infections (26–30), we investigated the mRNA and protein expression patterns of different TLRs in flow-sorted host DC subsets and found heavy downregulation of TLR6 and TLR9 in mDCs, LDCs, and pDCs compared to TLR2 and TLR4, which is in agreement with a previous report in which it was shown that TLR6 is required for full activation of DCs in order to induce a robust proinflammatory cytokine response, which means that Bm-L3 might have subverted the early host proinflammatory response by downregulating TLR6 expression (31). Daehnel and colleagues have also reported that activation of DCs is dependent on Toll-like receptor 2 (32), while others have shown TLR2-dependent functional anarchy of Mϕs during secondary challenge with B. malayi (33). Also, downregulation of TLR4 has been correlated with alternative activation of Mϕs (34, 35). Altogether, we believe that differentially modulated expression patterns of TLRs on host DCs might have been one of the factors behind differential secretion of Th1 and Th2 cytokines, as TLR2 ligands have been shown to stimulate a Th2 response (36), while TLR6 has been shown to induce TNF-α and IL-12 secretion (31), and both TLR2 and TLR4 have been shown to differentially activate human dendritic cells (37).
Parasitic infections are also known to induce distinct DC phenotypes in terms of costimulatory molecule expression and cytokine production (7). In our case, we observed increased expression of CD80 and CD86, along with decreased expression of MHC-II, on mDCs and pDCs, which meant that Bm-L3 differentially altered the expression of these markers on DC subsets. Increased expression of costimulatory molecules supports the idea that inhibitory signals are also delivered by CD80 and CD86 to T cells to prevent their differentiation and also for induction of regulatory T cells (38). While increased TNF-α secretion has been linked to DC maturation (35), the fact that not all DC subsets showed similar expression patterns of maturation and costimulatory markers or had similarly modulated antigen uptake and presentation capacities suggests that some other factors, possibly excretory-secretory products released by the infective larvae, might have been responsible for the phenomenon (5, 39, 40). Importantly, support for this notion is evident from the work carried out by Everts et al., who showed that human mDCs infected with Schistosoma haematobium had lower levels of HLA-DR expression than those of uninfected individuals (8).
Furthermore, since DC maturation is associated with reduction in phagocytic ability but enhanced antigen presentation capacity, we checked the two functions using flow cytometry. While pDCs and mDCs showed both reduced antigen uptake and reduced antigen presentation, they were not significantly altered in LDCs, which was not surprising, as LDCs, despite having the most efficient endocytosing ability of all splenic DC subsets (41), use a different mechanism of antigen uptake, e.g., mannose receptor (MR), which is exclusively expressed by them (42, 43), as documented during some bacterial infections (44).
Also, studies have shown that some pathogens induce normal maturation of cells while effectively containing their antigen presentation capacities, which might provide an answer to the differential maturation, antigen uptake, and presentation capacities of individual DC subsets observed in our case (39, 45). Taking the data at hand and previously published studies together, it is not clear whether the impaired Ag-presenting capacity of some DC subsets observed in our case can be explained by the decreased expression of MHC class II and other costimulatory molecules alone or whether additional mechanisms are involved, as it has been documented that signals controlling the expression of costimulatory molecules on DCs are distinct and depend on the pathogen stimulus in question (46, 47).
Nevertheless, the differential immunomodulation of DC subsets prompted us to ask if it would translate into impairment of the T-cell-activating capacities of host DC subsets. Our results showed significantly attenuated T-cell proliferation capacity of mDCs at day 7 p.i. compared to other DC subsets, which was in line with our observation of the reduced MHC-II expression and reduced antigen-presenting capacity of these cells. While additional studies are needed to specifically identify the role of enhanced IL-10 secretion by these cells at day 7 p.i. as a reason behind the attenuated T-cell proliferation capacity, previous reports have shown that IL-10-modulated DCs do induce antigen-specific anergy in CD4+ and CD8+ T cells (48). Collectively, we believe that the parasite's interference with the antigen-presenting machinery of DCs and their ability to prime naive T cells are crucial for the decision as to whether tolerance or immunity will be induced (8, 49, 50).
To further understand the molecular basis behind the mixed cytokine response and attenuated T-cell proliferation capacity of host DCs, we investigated the expression patterns of key members of the MAP kinase signaling pathway, viz., ERK and p38, as they have been implicated in the maturation and activation of DCs (30, 51–54). In fact, the roles of TLR2- and TLR9-mediated proinflammatory cytokine induction and activation of the MAPK and NF-kappa B pathways have been documented in the development of pathology in human lymphatic filariasis (55). Also, B. malayi is known to produce a p38 MAP kinase ortholog (Bm-MPK1) that modulates the activity of host MAP kinases, leading to dampened cytokine production (56, 57).
We observed elevated levels of p-p38 expression in mDCs and LDCs at day 3 postinfection. While p-p38 expression was still higher in mDCs at day 7 p.i. than in uninfected controls, it dropped in pDCs at day 7 p.i. Similarly, levels of p-ERK dropped in mDCs and pDCs at day 7, while they remained largely unchanged in LDCs. These data are intriguing if correlated with the expression of IL-10 and IL-12, as it has been shown that not only is ERK activation linked to upregulation of IL-10 in mouse DCs (58), but the ERK pathway is also involved in the negative regulation of IL-12 production by immature DCs (iDCs), but not mature DCs, while the p38 MAPK pathway appears to positively regulate IL-12 production in iDCs but not in mature DCs, suggesting that the roles of ERK and p38 MAPK in IL-12 production are developmentally changed in murine DCs (59). In our case, too, different DC subsets exhibited different maturation statuses at different time points, which might be correlated with their differentially regulated patterns of MAPK activation.
Moreover, since MAPK signaling is regulated by phosphotyrosine phosphatases (60–63), we asked ourselves whether infection with Bm-L3 would result in activation of phosphotyrosine phosphatases. To address this question, we elucidated the mRNA expression levels of tyrosine phosphatases in flow-sorted DC subsets and observed upregulated levels of protein tyrosine phosphatases, particularly SHP-1, TCPTP, PTEN, and PTP1B, in mDCs at day 3 postinfection. This observation fits well with data reported by Martin-Granados and colleagues, who have shown that kinases Erk1/Erk2 and SAPK/JNK are regulated by SHP-1, whose absence results in increased phosphorylation, hinting at a regulatory strategy, and specifically a novel immune evasion strategy, that the parasites have evolved to evade the host immune response (64). It is also worth noting that while PTP1B regulates the maturation of dendritic cells (64), SHP-1 has been implicated in regulating CD40 signaling reciprocity (65). Furthermore, studies have underlined the importance of SHP-1-mediated negative regulation in maintaining NO homeostasis, (66), which lends weight to the notion that parasites have developed means to modulate the signaling pathways in the host in order to establish themselves inside the host (51).
In summary, working with purified host DC subsets, we showed that complex interactions occur between infective larvae of B. malayi and host DC subsets during the very early stages of filarial infection. These interactions are not only distinct and diverse in nature, but intriguingly complex, as they involve paradoxical functions that affect cytokine release, maturation of cells, expression of TLRs, phagocytosis, and the antigen-presenting capability of cells that either stimulate or halt T-cell responses, leading to the control of infection or progression of disease. We also observed that the magnitude of activation or the functional impairment of various DC subsets investigated in this study showed considerable variation, which we believe depended on the molting process of the infective larvae within the host, the developmental stage of the DC subsets in question, or the exposure of the DCs to the various excretory-secretory products released by the infective larvae, which might have provided variable stimuli, resulting in sometimes weak or transient activation of one subset but not the other and differential activation and selective modulation of various DC functions that led to impaired T-cell proliferation. All this shows that the infective larvae have cleverly mastered the art of evading the initial host immune response to further enhance their establishment inside the host. Though the exact effect exerted on each subset by the infective larvae cannot be clearly ascertained without the use of loss/gain of function, what is clear from the data is that the molecular basis of parasite-derived and DC-derived differences remains enigmatic, as the parasite employs selective and differential modulation of host DC subsets, including alterations in DC signaling pathways, as an evasion strategy for its establishment. Hence, immunoregulatory or tolerogenic strategies that can modulate the numbers and/or functions of individual host DC subsets might help in optimizing the host immune response during the earliest host-parasite interface, which would help to advance our understanding of the host-parasite interactions.
MATERIALS AND METHODS
Animals and parasite.
Six- to eight-week-old BALB/c mice were used for all experiments, in accordance with our Institutional Animal Ethical Committee (IAEC) guidelines. B. malayi was maintained in Mastomys coucha, and the third infective larval stage of the parasite (Bm-L3) (n = 50) recovered from infected Aedes aegypti mosquitos was used to infect mice via the intraperitoneal (i.p.) route. Uninfected control animals were administered (i.p.) only sterile phosphate-buffered saline (PBS).
Reagents.
The cDNA synthesis kit; SYBR green master mix; TRIzol reagent; DQ ovalbumin; anti-mouse monoclonal antibodies directed against F4/80, TLR2, TLR4, TLR6, and TLR9; and fluorescein isothiocyanate (FITC)-labeled secondary IgG antibody were purchased from Thermo Fisher Scientific (Waltham, MA). May-Grunwald-Giemsa stain was purchased from Merck & Co. (Darmstadt, Germany). CD11c and CD4 magnetic cell separation (MACS) kits were purchased from Miltenyi Biotec (Bergisch-Gladbach, Germany). PDCA-1 anti-mouse monoclonal antibody was purchased from eBioscience (San Diego, CA). The fixation and permeabilization kit; brefeldin A; cell strainers; red blood cell (RBC) lysis buffer; TruCount tubes; and other anti-mouse monoclonal antibodies, viz., CD11c, CD11b, CD8a, CD45R, CD80, CD86, MHC-II, TNF-α, IL-4, IL-12, and IL-10, were purchased from BD Biosciences (San Jose, CA). p-p38 and p-ERK antibodies were purchased from Cell Signaling Technology (Danvers, MA), while FITC-dextran was purchased from Sigma (St. Louis, MO).
Preparation of single-cell suspension.
Mice were euthanized at different time points, i.e., at 12 h, day 3, day 5, day 7, and day 10 post-Bm-L3 infection, along with uninfected controls, and their spleens and mLNs were carefully excised and used for preparation of single-cell suspensions. Briefly, spleens and mLNs were gently minced and passed over a 40-μm cell strainer to remove clumps. Thereafter, the cell suspension was centrifuged at 300 × g for 10 min at 4°C, and contaminating erythrocytes in the splenocyte fraction were lysed using RBC lysis buffer. Next, the cells were centrifuged and suspended in ice-cold MACS buffer (1× PBS, pH 7.2, 0.5% bovine serum albumin [BSA], 2 mM EDTA), and CD11c-positive cells were enriched using CD11c magnetic beads, as described below.
Purification of CD11c-positive cells.
CD11c-positive cells present in the spleens and mLNs were purified from single-cell suspensions from control and infected mice at different time points using a CD11c magnetic cell separation kit following the instructions of the manufacturer. Briefly, single-cell suspensions were incubated with Fc block on ice for 10 min to avoid Fc receptor-mediated antibody binding. Thereafter, the cells were washed, centrifuged, and incubated with CD11c beads (1 μl beads/106 cells) for 20 min at 4°C. After subsequent washes, the cells were passed over a MACS MS column that was finally flushed with MACS buffer to obtain CD11c-positive cells, as described recently (67). Aliquots of CD11c-positive cells were assessed for purity using a BD FACS Aria flow cytometer and were consistently found to contain ∼90% CD11c-positive cells.
Immunophenotyping of CD11c-positive cells.
CD11c-positive cell fractions from spleens and mLNs (containing mostly dendritic cells and Mϕs) were separately incubated with an antibody cocktail containing the following monoclonal antibodies: CD45R-allophycocyanin (APC), CD11c- peridinin chlorophyll protein (PerCP)-Cy5.5, CD11b-phycoerythrin (PE)-Cy7, CD8a-FITC, F4/80-Pacific Blue, and PDCA1-PE for 20 min at 4°C. After incubation, flow cytometric data were acquired on 5-decade log-scale dot plots displaying the forward scatter (FSC) area versus the side scatter (SSC) area. First, hierarchy gates were set in FSC-A–versus–SSC-A dot plots to exclude contaminating dead cells and cell debris; a second hierarchy gate was set in an FSC-A–versus–FSC-H dot plot to exclude cell doublets; and thereafter, cell populations within the FSC-A–versus–FSC-H dot plot were segregated according to their differential expression profiles of the above-mentioned markers.
Briefly, as outlined in Fig. 1, a fluorescence-activated cell sorter (FACS) dot plot of F4/80+ against CD11b helped us in gating out splenic Mϕs (CD11cpos, CD11bpos, and F4/80pos) from other CD11c-positive cells (nonsplenic-Mϕ pool). Furthermore, since the latter fraction contained both conventional dendritic cells (cDCs) and non-cDCs, the two populations were separated from each other by plotting CD45R against CD11c, which helped us in differentiating between cDCs (CD11chi and CD45Rneg) and non-cDCs (CD11cmid and CD45Rpos). The CDCs were further subgated into either mDCs or LDCs based on the expression of CD11b and CD8a surface markers: while mDCs were CD11cpos, CD11bpos, and CD8aneg, LDCs were CD11cpos, CD11bneg, and CD8apos. Similarly, from the non-cDC pool, a dot plot of PDCA-1 against CD8a yielded two different subsets of pDCs, viz., PDCA-1pos CD8apos pDCs and PDCA-1pos CD8aneg pDCs. However, since the numbers of individual pDC subsets were low, for sorting purposes, we sorted pDCs as a single population by plotting PDCA-1 against CD45R, which yielded a very distinct PDCA-1pos CD45Rpos pDC pool (Fig. 1). Taken together, this gating strategy helped us in discriminating splenic Mϕs from splenic DCs and also helped us in identifying four prominent splenic DC subsets, i.e., mDCs, LDCs, CD8apos pDCs, and CD8aneg pDCs, in the spleens of mice. A similar gating strategy was used for mLNs (data not shown). Data acquisition and compensation were done on a FACS Aria flow cytometer, and analysis was done using FACS DIVA software (BD Biosciences).
Absolute quantification of dendritic cell subsets in the spleens and mLNs.
To elucidate to what extent infection with Bm-L3 influenced the numbers of each dendritic cell subset present in the secondary lymphoid organs of mice, we used commercially available TruCount beads (BD Biosciences) and quantified the absolute numbers of each DC subset present in the spleens and mLNs of control mice and Bm-L3-infected mice at day 3 and day 7 postinfection, following the instructions of the manufacturer. Briefly, DCs were enriched from both the spleens and mLNs, using CD11c beads as described above, and all the CD11c+ cells were suspended in 50 μl of FACS buffer. Thereafter, the cell suspension was added to TruCount tubes containing a calculated number of beads (in this case, 47,000), followed by addition of 20 μl of antibody cocktail containing CD11c-FITC, CD45R-APC, CD11b-PE-Cy7, PDCA-1-PE, CD8a-PerCP-Cy5.5, and F4/80-Pacific Blue. The tubes were then incubated for 15 min in the dark at room temperature, and the total volume was adjusted to 500 μl using FACS buffer. The cells were subsequently acquired on a BD FACS Aria flow cytometer, and absolute quantification was done, following the instructions of the manufacturer.
Flow cytometry-assisted cell sorting of different dendritic cell subsets.
While we studied the kinetics of recruitment for all four prominent DC subsets, i.e., mDCs, LDCs, CD8apos pDCs, and CD8aneg pDCs, in the spleens and mLNs of infected mice at 12 h, day 3, day 5, day 7, and day 10 post-Bm-L3 infection, sorting and other detailed functional investigations were carried out only on splenic DCs at day 3 and day 7 post-Bm-L3 infection because of the insufficient numbers of DC subsets present in the mLNs. Also, since the numbers of individual pDC subsets (CD8apos and CD8aneg pDCs) were low, the subsets were sorted as a single population (referred to below as sorted pDCs) using the PDCA-1–versus–CD45R dot plot, as shown in Fig. 1. Flow cytometry-assisted cell sorting was carried out with uninfected and Bm-L3-infected mice at two different time points, i.e., day 3 and day 7 post-Bm-L3 infection, because Bm-L3 organisms undergo their first molting (to L4) inside the host in approximately a week, so we wanted to evaluate the effect of Bm-L3 infection on host DCs at early (day 3) and late (day 7) time points that coincided with their molting process. Briefly, single-cell suspensions of spleens from at least 6 to 8 identically treated mice were pooled, and CD11c-positive cells were enriched using CD11c beads, as described above. Sorting was carried out using a high-speed FACS Aria flow cytometer (BD Biosciences) fitted with a 70-μm nozzle, as described recently (11, 67). After sorting, the sorted cells were subjected to analysis to ascertain their purity, and a small fraction was used to prepare cytospins (Fig. 1, extreme right), while the remaining cells were used to carry out other immunological assays, as detailed below.
Estimation of intracellular cytokines.
Intracellular cytokines secreted by splenic mDCs, LDCs, and pDCs were estimated by ex vivo intracellular staining of the respective cells, as described recently (68). Briefly, ∼4 × 106 CD11cpos cells enriched from mouse spleens at day 3 and day 7 post-Bm-L3 infection, along with uninfected controls, were incubated with 10 μg/ml of brefeldin A in the dark for 6 h in a CO2 incubator set at 37°C. After incubation, the cells were washed and stained with the following monoclonal antibodies: CD45R-APC, CD11c-PerCP-Cy5.5, CD11b-PE-Cy7, CD8a-FITC, and F4/80-Pacific Blue. Next, they were fixed, permeabilized, and distributed equally into four separate tubes, followed by addition of PE-labeled anti-mouse monoclonal antibodies directed against TNF-α, IL-12, IL-4, and IL-10. Data acquisition and compensation were done on a FACS Aria flow cytometer, and analysis was done using FACS DIVA software, as described recently (11).
Analysis of costimulatory molecules and the phagocytosis and antigen presentation capacities of dendritic cell subsets.
Expression of maturation and costimulatory molecules was assessed on flow-sorted mDCs, LDCs, and pDCs by incubating the flow-sorted cells (n = 50,000) with anti-mouse monoclonal antibodies directed against CD80, CD86, and MHC-II for 20 min in the dark at 4°C. After incubation, the cells were washed and acquired on a FACS Aria flow cytometer. Similarly, the phagocytosis and antigen presentation capacities of flow-sorted DC subsets were measured by incubating each DC subset with either FITC-dextran (1 mg/ml) or DQ-ovalbumin (0.5 mg/ml) in separate tubes for 1 h in a CO2 incubator at 37°C, and the increase in FITC fluorescence was subsequently measured by flow cytometry (11).
T-cell proliferation capacities of dendritic cell subsets.
The T-cell proliferation capacities of FACS-sorted mDCs, LDCs, and pDCs were estimated by coculturing each DC subset (n = 5,000) with 2 × 105 CD4+ T cells that were purified from the spleens of naive mice using CD4 magnetic beads (Miltenyi Biotec). The cells were cocultured in 96-well flat-bottom plates (Corning) for 48 h in a CO2 incubator at 37°C, and mitochondrial activity, as a measure of T-cell proliferation, was subsequently measured by XTT assay as described recently (11).
Real-time RT-PCR.
Total cellular RNA was isolated, quantified, and reverse transcribed from flow-sorted mDCs, LDCs, and pDCs as described previously (69). Reactions were run on a Step One Plus thermal cycler (Applied Biosystems) using SYBR green master mix. The β-actin gene was used as the reference gene, and mean fold changes were calculated according to the 2−ΔΔCT method (70). After the reactions were over, melting-curve analysis was performed to confirm the specificity of amplicon formation. The primer sequences used for RT-PCR are listed in Table 2.
TABLE 2.
Primer sequences used for real-time RT-PCR
| Gene | Primer (5′-3′) |
|
|---|---|---|
| Forward | Reverse | |
| β-Actin | TGGAATCCTGTGGCATCCATGAA | TAAAACGCAGCTCAGTAACAGTC |
| SHP-1 | AGTCTCACGCTACCCTGCTA | AGTCTATCGGGTGGAAGCCT |
| TCPTP | TTAACACAGGGCCCACTTCC | CTGTCATCCGTTGGCCAGTA |
| PTEN | TCCTGCAGAAAGACTTGAAGGT | AGGATACTGTGCAACTCTGC |
| PTP1B | TTACCAGGACATTCGACATGAA | CTCCTCTGGGCTTCTTCCAT |
| TLR2 | CACCACTGCCCGTAGATGAAG | AGGGTACAGTCGTCGAACTCT |
| TLR4 | ATGGCATGGCTTACACCACC | GAGGCCAATTTTGTCTCCACA |
| TLR6 | TGAGCCAAGACAGAAAACCCA | GGGACATGAGTAAGGTTCCTGTT |
| TLR9 | ATGGTTCTCCGTCGAAGGACT | GAGGCTTCAGCTCACAGGG |
Expression of TLRs on DC subsets by flow cytometry.
CD11c+ purified cells were collected from spleens of uninfected and Bm-L3-infected mice at day 3 and day 7 postinfection and stained with the following anti-mouse monoclonal antibodies: CD45R-APC, CD11c-PE, CD11b-PE-Cy7, CD8a-PerCP-Cy5.5, and F4/80-Pacific Blue. The labeled cells were then distributed equally into four different tubes, and each tube was further incubated with FITC-labeled anti-mouse TLR2, TLR4, TLR6, or TLR9. Appropriate gating strategies, as shown in Fig. 1, were used to identify each DC, and TLR expression was analyzed using FACS DIVA software.
MAP kinase assay.
Estimation of MAP kinase activity was carried out as described previously (8). Briefly, freshly sorted DC subsets (n = 200,000 cells) were fixed, permeabilized, and stained with primary antibodies against p-p38 and p-ERK for 20 min at 4°C, followed by addition of FITC-labeled secondary IgG antibody. After a brief incubation, the cells were washed again with ice-cold PBS and acquired on a FACS Aria flow cytometer. Data analysis was done using FACS DIVA software.
Statistics.
The data are presented as means and standard deviations (SD) derived from the results of at least three independent experiments with at least 6 to 8 animals/group. Statistical analysis was carried out using Student's t test, and P values of ≤0.05, ≤0.01, and ≤0.001 between different groups or time points were considered significant, highly significant, and very highly significant, respectively.
ACKNOWLEDGMENTS
We acknowledge the excellent technical support provided by Shikha Mishra and O. P. Yadav in maintaining B. malayi infection in the laboratory.
P.S. and A.S. acknowledge fellowship support from the University Grants Commission (UGC) and the Council of Scientific and Industrial Research (CSIR), New Delhi, India, respectively. This work was supported by grants provided under the CSIR-Network projects New Approaches toward Understanding of Disease Dynamics and To Accelerate Drug Discovery (UNDO) and Emerging and Reemerging Challenges in Infectious Diseases: Systems Based Drug Design for Infectious Diseases (SPLenDID) to M.S. The funders had no role in study design, data collection, analysis, decision to publish, or preparation of the manuscript.
We declare no conflict of interest.
This is communication number 9375 from CSIR-CDRI.
REFERENCES
- 1.Boyd A, Bennuru S, Wang Y, Sanprasert V, Law M, Chaussabel D, Nutman TB, Semnani RT. 2013. Quiescent innate response to infective filariae by human Langerhans cells suggests a strategy of immune evasion. Infect Immun 81:1420–1429. doi: 10.1128/IAI.01301-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henri S, Vremec D, Kamath A, Waithman J, Williams S, Benoist C, Burnham K, Saeland S, Handman E, Shortman K. 2001. The dendritic cell populations of mouse lymph nodes. J Immunol 167:741–748. doi: 10.4049/jimmunol.167.2.741. [DOI] [PubMed] [Google Scholar]
- 3.Maizels RM, Yazdanbakhsh M. 2003. Immune regulation by helminth parasites: cellular and molecular mechanisms. Nat Rev Immunol 3:733–744. doi: 10.1038/nri1183. [DOI] [PubMed] [Google Scholar]
- 4.Schuster P, Thomann S, Werner M, Vollmer J, Schmidt B. 2015. A subset of human plasmacytoid dendritic cells expresses CD8alpha upon exposure to herpes simplex virus type 1. Front Microbiol 6:557. doi: 10.3389/fmicb.2015.00557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Segura M, Su Z, Piccirillo C, Stevenson MM. 2007. Impairment of dendritic cell function by excretory-secretory products: a potential mechanism for nematode-induced immunosuppression. Eur J Immunol 37:1887–1904. doi: 10.1002/eji.200636553. [DOI] [PubMed] [Google Scholar]
- 6.Semnani RT, Liu AY, Sabzevari H, Kubofcik J, Zhou J, Gilden JK, Nutman TB. 2003. Brugia malayi microfilariae induce cell death in human dendritic cells, inhibit their ability to make IL-12 and IL-10, and reduce their capacity to activate CD4+ T cells. J Immunol 171:1950–1960. doi: 10.4049/jimmunol.171.4.1950. [DOI] [PubMed] [Google Scholar]
- 7.Balic A, Smith KA, Harcus Y, Maizels RM. 2009. Dynamics of CD11c(+) dendritic cell subsets in lymph nodes draining the site of intestinal nematode infection. Immunol Lett 127:68–75. doi: 10.1016/j.imlet.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Everts B, Adegnika AA, Kruize YC, Smits HH, Kremsner PG, Yazdanbakhsh M. 2010. Functional impairment of human myeloid dendritic cells during Schistosoma haematobium infection. PLoS Negl Trop Dis 4:e667. doi: 10.1371/journal.pntd.0000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Overtvelt L, Vanderheyde N, Verhasselt V, Ismaili J, De Vos L, Goldman M, Willems F, Vray B. 1999. Trypanosoma cruzi infects human dendritic cells and prevents their maturation: inhibition of cytokines, HLA-DR, and costimulatory molecules. Infect Immun 67:4033–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cotton RN, McDonald-Fleming R, Boyd A, Spates K, Nutman TB, Tolouei Semnani R. 2015. Brugia malayi infective larvae fail to activate Langerhans cells and dermal dendritic cells in human skin. Parasite Immunol 37:79–91. doi: 10.1111/pim.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma P, Sharma A, Vishwakarma AL, Agnihotri PK, Sharma S, Srivastava M. 2016. Host lung immunity is severely compromised during tropical pulmonary eosinophilia: role of lung eosinophils and macrophages. J Leukoc Biol 99:619–628. doi: 10.1189/jlb.4A0715-309RR. [DOI] [PubMed] [Google Scholar]
- 12.Banchereau J, Steinman RM. 1998. Dendritic cells and the control of immunity. Nature 392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 13.Larsen CP, Ritchie SC, Pearson TC, Linsley PS, Lowry RP. 1992. Functional expression of the costimulatory molecule, B7/BB1, on murine dendritic cell populations. J Exp Med 176:1215–1220. doi: 10.1084/jem.176.4.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Semnani RT, Mahapatra L, Dembele B, Konate S, Metenou S, Dolo H, Coulibaly ME, Soumaoro L, Coulibaly SY, Sanogo D, Seriba Doumbia S, Diallo AA, Traore SF, Klion A, Nutman TB, Mahanty S. 2010. Expanded numbers of circulating myeloid dendritic cells in patent human filarial infection reflect lower CCR1 expression. J Immunol 185:6364–6372. doi: 10.4049/jimmunol.1001605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naik SH, Corcoran LM, Wu L. 2005. Development of murine plasmacytoid dendritic cell subsets. Immunol Cell Biol 83:563–570. doi: 10.1111/j.1440-1711.2005.01390.x. [DOI] [PubMed] [Google Scholar]
- 16.Kim PK, Kwon YG, Chung HT, Kim YM. 2002. Regulation of caspases by nitric oxide. Ann N Y Acad Sci 962:42–52. doi: 10.1111/j.1749-6632.2002.tb04054.x. [DOI] [PubMed] [Google Scholar]
- 17.Semnani RT, Nutman TB. 2004. Toward an understanding of the interaction between filarial parasites and host antigen-presenting cells. Immunol Rev 201:127–138. doi: 10.1111/j.0105-2896.2004.00196.x. [DOI] [PubMed] [Google Scholar]
- 18.Ma L, Scheers W, Vandenberghe P. 2004. A flow cytometric method for determination of absolute counts of myeloid precursor dendritic cells in peripheral blood. J Immunol Methods 285:215–221. doi: 10.1016/j.jim.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 19.Specht S, Volkmann L, Wynn T, Hoerauf A. 2004. Interleukin-10 (IL-10) counterregulates IL-4-dependent effector mechanisms in murine filariasis. Infect Immun 72:6287–6293. doi: 10.1128/IAI.72.11.6287-6293.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koch F, Stanzl U, Jennewein P, Janke K, Heufler C, Kampgen E, Romani N, Schuler G. 1996. High level IL-12 production by murine dendritic cells: upregulation via MHC class II and CD40 molecules and downregulation by IL-4 and IL-10. J Exp Med 184:741–746. doi: 10.1084/jem.184.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urban JF Jr, Madden KB, Svetic A, Cheever A, Trotta PP, Gause WC, Katona IM, Finkelman FD. 1992. The importance of Th2 cytokines in protective immunity to nematodes. Immunol Rev 127:205–220. doi: 10.1111/j.1600-065X.1992.tb01415.x. [DOI] [PubMed] [Google Scholar]
- 22.Babu S, Blauvelt CP, Kumaraswami V, Nutman TB. 2006. Regulatory networks induced by live parasites impair both Th1 and Th2 pathways in patent lymphatic filariasis: implications for parasite persistence. J Immunol 176:3248–3256. doi: 10.4049/jimmunol.176.5.3248. [DOI] [PubMed] [Google Scholar]
- 23.Boonstra A, Asselin-Paturel C, Gilliet M, Crain C, Trinchieri G, Liu YJ, O'Garra A. 2003. Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential Toll-like receptor ligation. J Exp Med 197:101–109. doi: 10.1084/jem.20021908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carvalho LP, Pearce EJ, Scott P. 2008. Functional dichotomy of dendritic cells following interaction with Leishmania braziliensis: infected cells produce high levels of TNF-alpha, whereas bystander dendritic cells are activated to promote T cell responses. J Immunol 181:6473–6480. doi: 10.4049/jimmunol.181.9.6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Keeffe M, Hochrein H, Vremec D, Caminschi I, Miller JL, Anders EM, Wu L, Lahoud MH, Henri S, Scott B, Hertzog P, Tatarczuch L, Shortman K. 2002. Mouse plasmacytoid cells: long-lived cells, heterogeneous in surface phenotype and function, that differentiate into CD8(+) dendritic cells only after microbial stimulus. J Exp Med 196:1307–1319. doi: 10.1084/jem.20021031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hise AG, Daehnel K, Gillette-Ferguson I, Cho E, McGarry HF, Taylor MJ, Golenbock DT, Fitzgerald KA, Kazura JW, Pearlman E. 2007. Innate immune responses to endosymbiotic Wolbachia bacteria in Brugia malayi and Onchocerca volvulus are dependent on TLR2, TLR6, MyD88, and Mal, but not TLR4, TRIF, or TRAM. J Immunol 178:1068–1076. doi: 10.4049/jimmunol.178.2.1068. [DOI] [PubMed] [Google Scholar]
- 27.Venugopal PG, Nutman TB, Semnani RT. 2009. Activation and regulation of Toll-like receptors (TLRs) by helminth parasites. Immunol Res 43:252–263. doi: 10.1007/s12026-008-8079-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Babu S, Blauvelt CP, Kumaraswami V, Nutman TB. 2005. Diminished expression and function of TLR in lymphatic filariasis: a novel mechanism of immune dysregulation. J Immunol 175:1170–1176. doi: 10.4049/jimmunol.175.2.1170. [DOI] [PubMed] [Google Scholar]
- 29.Krug A, Towarowski A, Britsch S, Rothenfusser S, Hornung V, Bals R, Giese T, Engelmann H, Endres S, Krieg AM, Hartmann G. 2001. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur J Immunol 31:3026–3037. doi: 10.1002/1521-4141(2001010)31:10<3026::AID-IMMU3026>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 30.Kane CM, Cervi L, Sun J, McKee AS, Masek KS, Shapira S, Hunter CA, Pearce EJ. 2004. Helminth antigens modulate TLR-initiated dendritic cell activation. J Immunol 173:7454–7461. doi: 10.4049/jimmunol.173.12.7454. [DOI] [PubMed] [Google Scholar]
- 31.de Almeida LA, Macedo GC, Marinho FA, Gomes MT, Corsetti PP, Silva AM, Cassataro J, Giambartolomei GH, Oliveira SC. 2013. Toll-like receptor 6 plays an important role in host innate resistance to Brucella abortus infection in mice. Infect Immun 81:1654–1662. doi: 10.1128/IAI.01356-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daehnel K, Gillette-Ferguson I, Hise AG, Diaconu E, Harling MJ, Heinzel FP, Pearlman E. 2007. Filaria/Wolbachia activation of dendritic cells and development of Th1-associated responses is dependent on Toll-like receptor 2 in a mouse model of ocular onchocerciasis (river blindness). Parasite Immunol 29:455–465. doi: 10.1111/j.1365-3024.2007.00962.x. [DOI] [PubMed] [Google Scholar]
- 33.Turner JD, Langley RS, Johnston KL, Egerton G, Wanji S, Taylor MJ. 2006. Wolbachia endosymbiotic bacteria of Brugia malayi mediate macrophage tolerance to TLR- and CD40-specific stimuli in a MyD88/TLR2-dependent manner. J Immunol 177:1240–1249. doi: 10.4049/jimmunol.177.2.1240. [DOI] [PubMed] [Google Scholar]
- 34.Orr JS, Puglisi MJ, Ellacott KL, Lumeng CN, Wasserman DH, Hasty AH. 2012. Toll-like receptor 4 deficiency promotes the alternative activation of adipose tissue macrophages. Diabetes 61:2718–2727. doi: 10.2337/db11-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goodridge HS, Marshall FA, Else KJ, Houston KM, Egan C, Al-Riyami L, Liew FY, Harnett W, Harnett MM. 2005. Immunomodulation via novel use of TLR4 by the filarial nematode phosphorylcholine-containing secreted product, ES-62. J Immunol 174:284–293. doi: 10.4049/jimmunol.174.1.284. [DOI] [PubMed] [Google Scholar]
- 36.Dillon S, Agrawal A, Van Dyke T, Landreth G, McCauley L, Koh A, Maliszewski C, Akira S, Pulendran B. 2004. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J Immunol 172:4733–4743. doi: 10.4049/jimmunol.172.8.4733. [DOI] [PubMed] [Google Scholar]
- 37.Re F, Strominger JL. 2001. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J Biol Chem 276:37692–37699. doi: 10.1074/jbc.M105927200. [DOI] [PubMed] [Google Scholar]
- 38.Thauland TJ, Koguchi Y, Dustin ML, Parker DC. 2014. CD28-CD80 interactions control regulatory T cell motility and immunological synapse formation. J Immunol 193:5894–5903. doi: 10.4049/jimmunol.1401752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manoury B, Gregory WF, Maizels RM, Watts C. 2001. Bm-CPI-2, a cystatin homolog secreted by the filarial parasite Brugia malayi, inhibits class II MHC-restricted antigen processing. Curr Biol 11:447–451. doi: 10.1016/S0960-9822(01)00118-X. [DOI] [PubMed] [Google Scholar]
- 40.Sun Y, Liu G, Li Z, Chen Y, Liu Y, Liu B, Su Z. 2013. Modulation of dendritic cell function and immune response by cysteine protease inhibitor from murine nematode parasite Heligmosomoides polygyrus. Immunology 138:370–381. doi: 10.1111/imm.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pooley JL, Heath WR, Shortman K. 2001. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8− dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J Immunol 166:5327–5330. doi: 10.4049/jimmunol.166.9.5327. [DOI] [PubMed] [Google Scholar]
- 42.Burgdorf S, Lukacs-Kornek V, Kurts C. 2006. The mannose receptor mediates uptake of soluble but not of cell-associated antigen for cross-presentation. J Immunol 176:6770–6776. doi: 10.4049/jimmunol.176.11.6770. [DOI] [PubMed] [Google Scholar]
- 43.Burgdorf S, Kautz A, Bohnert V, Knolle PA, Kurts C. 2007. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 316:612–616. doi: 10.1126/science.1137971. [DOI] [PubMed] [Google Scholar]
- 44.Autenrieth SE, Soldanova I, Rosemann R, Gunst D, Zahir N, Kracht M, Ruckdeschel K, Wagner H, Borgmann S, Autenrieth IB. 2007. Yersinia enterocolitica YopP inhibits MAP kinase-mediated antigen uptake in dendritic cells. Cell Microbiol 9:425–437. doi: 10.1111/j.1462-5822.2006.00800.x. [DOI] [PubMed] [Google Scholar]
- 45.Cheminay C, Mohlenbrink A, Hensel M. 2005. Intracellular Salmonella inhibit antigen presentation by dendritic cells. J Immunol 174:2892–2899. doi: 10.4049/jimmunol.174.5.2892. [DOI] [PubMed] [Google Scholar]
- 46.Pulendran B, Smith JL, Caspary G, Brasel K, Pettit D, Maraskovsky E, Maliszewski CR. 1999. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc Natl Acad Sci U S A 96:1036–1041. doi: 10.1073/pnas.96.3.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pejawar SS, Parks GD, Alexander-Miller MA. 2005. Abortive versus productive viral infection of dendritic cells with a paramyxovirus results in differential upregulation of select costimulatory molecules. J Virol 79:7544–7557. doi: 10.1128/JVI.79.12.7544-7557.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mahnke K, Johnson TS, Ring S, Enk AH. 2007. Tolerogenic dendritic cells and regulatory T cells: a two-way relationship. J Dermatol Sci 46:159–167. doi: 10.1016/j.jdermsci.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Jenkins SJ, Mountford AP. 2005. Dendritic cells activated with products released by schistosome larvae drive Th2-type immune responses, which can be inhibited by manipulation of CD40 costimulation. Infect Immun 73:395–402. doi: 10.1128/IAI.73.1.395-402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alba Soto CD, Mirkin GA, Solana ME, Gonzalez Cappa SM. 2003. Trypanosoma cruzi infection modulates in vivo expression of major histocompatibility complex class II molecules on antigen-presenting cells and T-cell stimulatory activity of dendritic cells in a strain-dependent manner. Infect Immun 71:1194–1199. doi: 10.1128/IAI.71.3.1194-1199.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Puig-Kroger A, Relloso M, Fernandez-Capetillo O, Zubiaga A, Silva A, Bernabeu C, Corbi AL. 2001. Extracellular signal-regulated protein kinase signaling pathway negatively regulates the phenotypic and functional maturation of monocyte-derived human dendritic cells. Blood 98:2175–2182. doi: 10.1182/blood.V98.7.2175. [DOI] [PubMed] [Google Scholar]
- 52.Lim MX, Png CW, Tay CY, Teo JD, Jiao H, Lehming N, Tan KS, Zhang Y. 2014. Differential regulation of proinflammatory cytokine expression by mitogen-activated protein kinases in macrophages in response to intestinal parasite infection. Infect Immun 82:4789–4801. doi: 10.1128/IAI.02279-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klotz C, Ziegler T, Figueiredo AS, Rausch S, Hepworth MR, Obsivac N, Sers C, Lang R, Hammerstein P, Lucius R, Hartmann S. 2011. A helminth immunomodulator exploits host signaling events to regulate cytokine production in macrophages. PLoS Pathog 7:e1001248. doi: 10.1371/journal.ppat.1001248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karakhanova S, Meisel S, Ring S, Mahnke K, Enk AH. 2010. ERK/p38 MAP-kinases and PI3K are involved in the differential regulation of B7-H1 expression in DC subsets. Eur J Immunol 40:254–266. doi: 10.1002/eji.200939289. [DOI] [PubMed] [Google Scholar]
- 55.Babu S, Anuradha R, Kumar NP, George PJ, Kumaraswami V, Nutman TB. 2011. Filarial lymphatic pathology reflects augmented Toll-like receptor-mediated, mitogen-activated protein kinase-mediated proinflammatory cytokine production. Infect Immun 79:4600–4608. doi: 10.1128/IAI.05419-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patel A, Chojnowski AN, Gaskill K, De Martini W, Goldberg RL, Siekierka JJ. 2011. The role of a Brugia malayi p38 MAP kinase ortholog (Bm-MPK1) in parasite anti-oxidative stress responses. Mol Biochem Parasitol 176:90–97. doi: 10.1016/j.molbiopara.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 57.Nakahara T, Moroi Y, Uchi H, Furue M. 2006. Differential role of MAPK signaling in human dendritic cell maturation and Th1/Th2 engagement. J Dermatol Sci 42:1–11. doi: 10.1016/j.jdermsci.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 58.Jin Y, Wi HJ, Choi MH, Hong ST, Bae YM. 2014. Regulation of anti-inflammatory cytokines IL-10 and TGF-beta in mouse dendritic cells through treatment with Clonorchis sinensis crude antigen. Exp Mol Med 46:e74. doi: 10.1038/emm.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kikuchi K, Yanagawa Y, Iwabuchi K, Onoe K. 2003. Differential role of mitogen-activated protein kinases in CD40-mediated IL-12 production by immature and mature dendritic cells. Immunol Lett 89:149–154. doi: 10.1016/S0165-2478(03)00134-2. [DOI] [PubMed] [Google Scholar]
- 60.Nandan D, Lo R, Reiner NE. 1999. Activation of phosphotyrosine phosphatase activity attenuates mitogen-activated protein kinase signaling and inhibits c-FOS and nitric oxide synthase expression in macrophages infected with Leishmania donovani. Infect Immun 67:4055–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Contreras I, Estrada JA, Guak H, Martel C, Borjian A, Ralph B, Shio MT, Fournier S, Krawczyk CM, Olivier M. 2014. Impact of Leishmania mexicana infection on dendritic cell signaling and functions. PLoS Negl Trop Dis 8:e3202. doi: 10.1371/journal.pntd.0003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gomez MA, Contreras I, Halle M, Tremblay ML, McMaster RW, Olivier M. 2009. Leishmania GP63 alters host signaling through cleavage-activated protein tyrosine phosphatases. Sci Signal 2:ra58. doi: 10.1126/scisignal.2000213. [DOI] [PubMed] [Google Scholar]
- 63.Niedzielska M, Raffi FA, Tel J, Muench S, Jozefowski K, Alati N, Lahl K, Mages J, Billmeier U, Schiemann M, Appelt UK, Wirtz S, Sparwasser T, Hochrein H, Figdor CG, Keyse SM, Lang R. 2015. Selective expression of the MAPK phosphatase Dusp9/MKP-4 in mouse plasmacytoid dendritic cells and regulation of IFN-beta production. J Immunol 195:1753–1762. doi: 10.4049/jimmunol.1400658. [DOI] [PubMed] [Google Scholar]
- 64.Martin-Granados C, Prescott AR, Le Sommer S, Klaska IP, Yu T, Muckersie E, Giuraniuc CV, Grant L, Delibegovic M, Forrester JV. 2015. A key role for PTP1B in dendritic cell maturation, migration, and T cell activation. J Mol Cell Biol 7:517–528. doi: 10.1093/jmcb/mjv032. [DOI] [PubMed] [Google Scholar]
- 65.Khan TH, Srivastava N, Srivastava A, Sareen A, Mathur RK, Chande AG, Musti KV, Roy S, Mukhopadhyaya R, Saha B. 2014. SHP-1 plays a crucial role in CD40 signaling reciprocity. J Immunol 193:3644–3653. doi: 10.4049/jimmunol.1400620. [DOI] [PubMed] [Google Scholar]
- 66.Blanchette J, Abu-Dayyeh I, Hassani K, Whitcombe L, Olivier M. 2009. Regulation of macrophage nitric oxide production by the protein tyrosine phosphatase Src homology 2 domain phosphotyrosine phosphatase 1 (SHP-1). Immunology 127:123–133. doi: 10.1111/j.1365-2567.2008.02929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Srivastava M, Meinders A, Steinwede K, Maus R, Lucke N, Buhling F, Ehlers S, Welte T, Maus UA. 2007. Mediator responses of alveolar macrophages and kinetics of mononuclear phagocyte subset recruitment during acute primary and secondary mycobacterial infections in the lungs of mice. Cell Microbiol 9:738–752. doi: 10.1111/j.1462-5822.2006.00824.x. [DOI] [PubMed] [Google Scholar]
- 68.Pathak M, Verma M, Srivastava M, Misra-Bhattacharya S. 2015. Wolbachia endosymbiont of Brugia malayi elicits a T helper type 17-mediated pro-inflammatory immune response through Wolbachia surface protein. Immunology 144:231–244. doi: 10.1111/imm.12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Srivastava M, Jung S, Wilhelm J, Fink L, Buhling F, Welte T, Bohle RM, Seeger W, Lohmeyer J, Maus UA. 2005. The inflammatory versus constitutive trafficking of mononuclear phagocytes into the alveolar space of mice is associated with drastic changes in their gene expression profiles. J Immunol 175:1884–1893. doi: 10.4049/jimmunol.175.3.1884. [DOI] [PubMed] [Google Scholar]
- 70.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]