

ABSTRACT
An outer membrane vesicle (OMV)-based cholera vaccine is highly efficacious in preventing intestinal colonization in the suckling mouse model. Immunity from OMVs comes from immunoglobulin (Ig), particularly IgG, in the milk of mucosally immunized dams. Anti-OMV IgG renders Vibrio cholerae organisms immotile, thus they pass through the small intestine without colonizing. However, the importance of motility inhibition for protection and the mechanism by which motility is inhibited remain unclear. By using both in vitro and in vivo experiments, we found that IgG inhibits motility by specifically binding to the O-antigen of V. cholerae. We demonstrate that the bivalent structure of IgG, although not required for binding to the O-antigen, is required for motility inhibition. Finally, we show using competition assays in suckling mice that inhibition of motility appears to be responsible for most, if not all, of the protection engendered by OMV vaccination, thus providing insight into the mechanism of immune protection.
KEYWORDS: Vibrio cholerae, cholera, flagellar motility, immunity, lipopolysaccharide, mucosal vaccines, outer membrane vesicles
INTRODUCTION
Cholera is an infectious, intestinal disease caused by the Gram-negative bacterium Vibrio cholerae. It is a prevalent problem in developing countries and can be lethal if not treated quickly. Every year, it infects millions and causes over 100,000 deaths, particularly in young children (1). Cholera is contracted by ingesting V. cholerae-contaminated food or water in regions where sanitation is poor. Patients with cholera present with massive secretory diarrhea, the so-called rice water stool, and shed up to one liter per hour (2). This can lead to extensive dehydration, hypotensive shock, and death within 12 h of the onset of symptoms. Currently, rehydration via oral or intravenous fluids is the mainstream treatment. Although the standard care can reduce the mortality rate to less than 1%, this is often not possible in many countries with poor health care infrastructure (2). Thus, many resources have been devoted to the prevention of cholera by vaccination. The existing vaccines for cholera (reviewed in reference 3) include whole-cell-killed (WCK) and live attenuated vaccines, both delivered orally. It has long been known that the O-antigen component of the lipopolysaccharide (LPS) is a critical component of these vaccines (4), and that natural immunity following cholera correlates with the titer of anti-O-antigen antibodies (5).
Recent work has been done in our laboratory to develop a compositionally simpler cholera vaccine composed of outer membrane vesicles (OMVs) (6–9). OMVs are naturally released exocytic vesicles of Gram-negative bacteria that serve a number of functions, including in biofilm formation, virulence, protein secretion, and cell-to-cell communication (10). However, because of their immunogenicity (they contain cell surface epitopes, including lipopolysaccharide [LPS]) and ability to be taken up by mammalian cells, they are increasingly being explored as vaccines (11).
Infant mice and infant rabbits are susceptible to V. cholerae colonization of the small intestine, whereas adult animals (with the exception of humans) are resistant. Thus, infant mice and rabbits are used as animal models for challenge studies. Previous studies have shown that infant mice born to OMV-immunized dams are protected from infection, and that the protection seems to be associated primarily with IgG in the milk (6, 7). By measuring anti-OMV titers for various classes of antibody, it was shown that IgG levels increased significantly, while IgM and IgA showed no significant increase (7). Thus, it is hypothesized that OMV-conferred immunity in this animal model is associated with an increase in IgG.
Of the two possible means of transferring protective antibodies to their pups, placentally or in milk, it was shown that the latter is the main source of IgG and the source of protection (7). Furthermore, it was shown that milk from immunized dams inhibited motility of V. cholerae in vitro (8). Because V. cholerae requires motility to colonize the intestinal villi (12, 13), we recently hypothesized that IgG ingested in milk blocks motility, causing the bacteria to pass out of the small intestine (9).
How IgG is blocking motility, and what specific epitopes are sufficient, remain unknown. Furthermore, because antibodies can have multiple effects on bacteria, it is unclear if protection is mediated solely through blocking of motility or is instead multifactorial. Because motility is inhibited at antibody concentrations too low to cause agglutination, we hypothesized that IgG binds to the O-antigen of the outer membrane sheath surrounding the V. cholerae polar flagellum and imposes a crimping force, or alternatively it cross-links the sheath and flagellum to the cell body, as recently demonstrated for IgA (14), so that the flagellum no longer functions properly. This suggests that the bivalent structure of IgG is required for motility inhibition, much like its property for agglutination. In this study, we show that motility inhibition is the primary mechanism of protection in suckling mice. We also use OMVs purified from rough (no O-antigen) and deep rough (no O-antigen or core oligosaccharide) mutants to show that the O-antigen is the critical protective epitope. Finally, we show that the bivalent nature of IgG is required for motility inhibition.
RESULTS
Inhibition of V. cholerae motility is the primary mechanism of protection.
Intranasal immunization of adult female mice with 25 μg of OMVs has been proven effective in protection of their suckling mice from challenge, whether using laboratory-grown or hyperinfectious V. cholerae shed in cholera patient stools for challenge (6–9). Furthermore, it was suggested that protection is due to inhibition of motility through binding of antibodies in the milk of the immunized dam (9). To further test this hypothesis, competition assays of the wild type and two different nonmotile strains were performed. Our rationale was that if protection is primarily mediated by inhibition of motility, then the competitive advantage the wild type has over nonmotile mutants should be negated in immune mice. Adult female BALB/c mice were immunized intranasally with 25 μg OMVs purified from wild-type strain E7946 as described previously (6), and approximately 5 weeks later their 5-day-old pups were challenged with a 1:1 mixture of wild-type and nonmotile ΔmotY (flagellated and sheathed) or ΔflaACEDB (aflagellar and lacking a sheath) strains. Both mutant strains exhibit the same doubling time and grow to the same final density as the wild-type strain in shaking LB broth cultures in vitro (data not shown). The competitive index (CI) was calculated as the ratio of nonmotile to wild-type cells after 24 h of infection of the small intestine. As expected (13), we observed an ∼16-fold attenuation (CI of ∼0.06) for both nonmotile strains when competed in mock-immunized, naive mice (Fig. 1A and B). In contrast, there was no competitive defect detected for either nonmotile strain (CI of ∼1) when competed in immune mice (Fig. 1A and B). A 100-fold higher inoculum dose was used for infecting immune infant mice than for naive infant mice to allow recovery of sufficient numbers of bacteria for calculating a competitive index. These data confirm the importance of bacterial motility for colonization in this animal model and support our hypothesis that the mechanism of protection afforded by OMV immunization is primarily, if not entirely, antibody-mediated inhibition of motility.
FIG 1.

Inhibition of V. cholerae motility is the primary mechanism of immune protection. Two types of nonmotile mutant, flagellated but nonmotile (ΔmotY) (A) and aflagellate (ΔflaACEDB) (B), were competed against the motile wild-type strain (WT) in either naive or immune pups. Five-day-old suckling mice born to mice mock immunized or immunized with OMVs produced by strains indicated on the x axes were challenged with equal mixtures of the competing V. cholerae strains. The competitive index was calculated as the ratio of nonmotile to wild type after correcting for the input ratio. Each symbol represents sample from one mouse. Bars are medians. Statistical analysis was done using Mann-Whitney U test with Kruskal-Wallis test for multiple-variable groups (*, P < 0.0001).
Characteristics of OMVs and LPS from mutant strains of V. cholerae.
It was recently shown that OMVs purified from a lipid A-detoxified mutant strain of V. cholerae maintain vaccine efficacy; therefore, this strain represents a safer vaccine (15). We utilized this strain for the studies described below. To determine what LPS components are required for vaccine efficacy, we purified OMVs from rough and deep rough mutants (Table 1). Prior to testing them as vaccines, we assessed the protein content and morphology of the OMVs produced. Five micrograms of OMV was purified from each mutant strain and, for comparison, from strain AC4280, which makes a lipid A-detoxified but otherwise normal LPS, and were visualized on a silver-stained SDS-PAGE gel. Pretreatment of OMVs with SDS and proteinase K was done to remove protein content, thus allowing for immunization with just the outer membrane lipids. The protein content of OMVs was highly similar between the different strains (Fig. 2A). As expected, the rough and deep rough mutants lack the major LPS band, owing to their lacking O-antigen. In addition, the deep rough mutant strain lacks the lipid A plus core oligosaccharide band, as expected. When purified LPS from the rough mutant was spotted on a nitrocellulose membrane and probed with O-antigen-specific antibody, we confirmed that the O-antigen was lacking (Fig. 2B).
TABLE 1.
Strains used in this study
| V. cholerae strain | Genotype and antibiotic resistance | Relevant description | Reference or source |
|---|---|---|---|
| Wild type | |||
| AC53 | El Tor O1 Ogawa | Spontaneous Smr derivative of V. cholerae strain E7946 | (6) |
| AC2995 | O139 | V. cholerae strain MO10 | Karl Klose |
| Mutant | |||
| ΔompA ΔctxAB ΔmsbB | Derivative of AC53 | (16, 17) | |
| motY::neo | Derivative of AC53 | This study | |
| ΔflaACEDB | Derivative of AC53 | This study | |
| ΔlacZ | Derivative of AC53 | (18) | |
| ΔwbeL | Rough mutant derivative of AC53 | (15) | |
| SP27459waaF::pGP | Deep rough mutant derivative of AC53 | J. Reidl |
FIG 2.
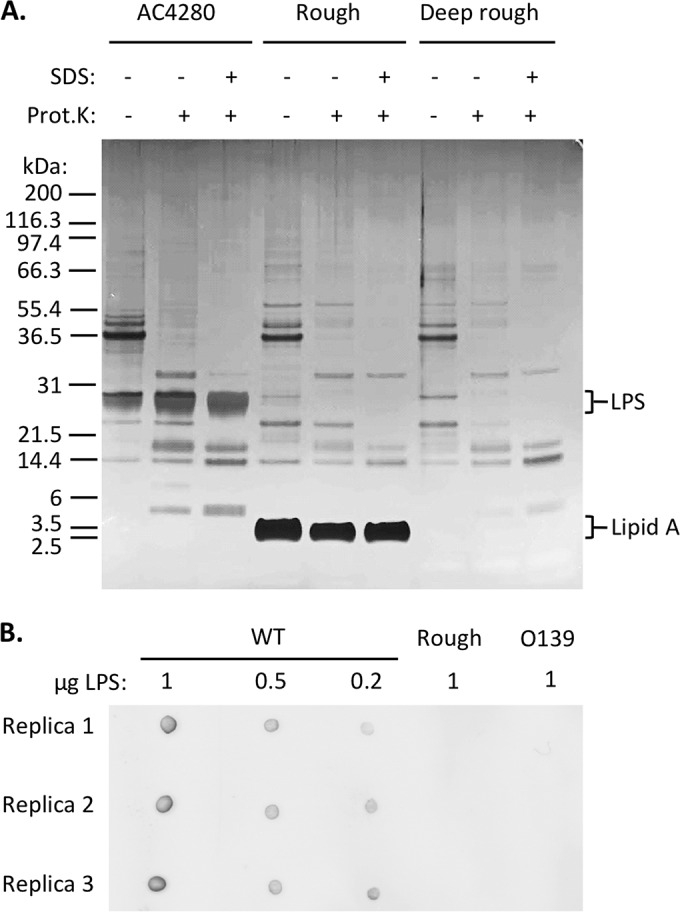
Both OMV and LPS from mutant strains of V. cholerae lack O antigen. (A) Protein and LPS content of outer membrane vesicles produced by mutant strains of V. cholerae. Five micrograms of OMVs with or without SDS and proteinase K (Prot. K) were incubated at 55°C overnight. An aliquot of 2.5 μg was loaded on a 4 to 12% gradient SDS-PAGE gel. After electrophoretic separation, the gel was stained with silver. (B) Ten microliters of serial dilutions of purified LPS from wild-type O1 El Tor E7946, a rough mutant derivative (ΔwbeL), and O139 serogroup strain MO10 were spotted in triplicate onto a nitrocellulose membrane and air dried. The membrane was probed using primary anti-O1-antigen antibody and Cy5-conjugated, anti-IgG secondary antibody.
Because LPS forms the bulk of the outer leaflet of the outer membrane and the rough and deep rough strains have major truncations in their LPS, we wanted to examine whether these mutant strains make OMVs with normal morphology. We used transmission electron microscopy (TEM) images of negatively stained OMVs to assess this. OMVs of all strains were comparable in size and shape (Fig. 3). This was also true for OMVs depleted of proteins. The only observable phenotype we noted by TEM was that the deep rough OMVs readily aggregate, as shown in Fig. 3.
FIG 3.

Transmission electron microscopy examination of outer membrane vesicle morphology. OMVs purified from the strains indicated were diluted to a concentration of 1 μg/μl in PBS. Grids were floated in OMV solution for 1 min, washed with 2% acidic uranyl acetate, and blotted dry before visualization under TEM. Scale bars are 100 nm.
Anti-O-antigen antibody is the source of protection.
We know that anti-OMV antibody inhibits motility and that the O-antigen is a critical epitope for immune protection. To further assess the importance of the O-antigen in protection and to distinguish its role from other LPS components, namely, the core oligosaccharide and lipid A, OMVs from the rough and deep rough strains were tested in parallel as vaccines in the mouse model described above, in which immunity is assessed by determining the CI of the ΔflaACEDB strain. In contrast to the CI of ∼1 observed in immune mice, the CIs for the rough and deep rough OMV-immunized groups were low, comparable to that observed in naive mice (Fig. 1B). This suggests that the O1 antigen, but not the core oligosaccharide or lipid A, is the important LPS antigen for protection.
It was previously reported that the protein content of OMVs is not important for immunity (8). We confirmed and extended this finding by showing that protein depletion of OMVs purified from the detoxified lipid A strain AC4280 does not affect vaccine efficacy (Fig. 1B).
Multivalent structure of antibody is required for inhibition of motility.
Previous work has shown that the main type of antibody present in the milk of OMV-immunized mice is IgG1 (6), which contains two antigen binding sites. We hypothesized that the divalent nature of this antibody is important for inhibition of V. cholerae motility and, thus, protection. To test this hypothesis, we examined the motility of both anti-O-antigen antibody and Fab fragments generated from it. When whole anti-O-antigen IgG was added at a subagglutinating concentration, it completely inhibited motility by 4 min of incubation (Fig. 4A), whereas an isotype control anti-CD22 antibody maintained 100% motility. The cellular morphology of V. cholerae as assessed by TEM did not change after binding of anti-O-antigen IgG (data not shown). In contrast to intact anti-O-antigen antibody, an equivalent 2-fold number of Fab, or even 10-fold, failed to inhibit motility at 4 min (Fig. 4A) or up to 1 h (data not shown).
FIG 4.
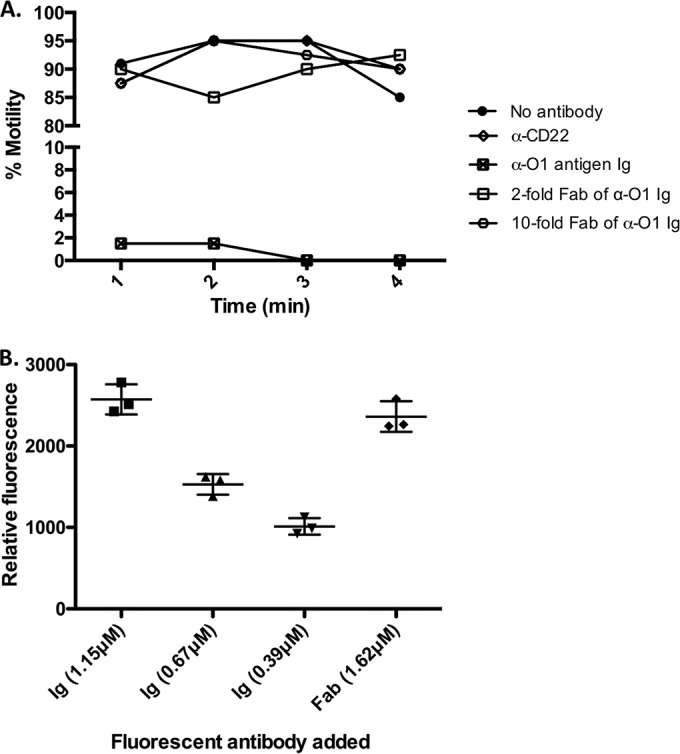
Inhibition of V. cholerae motility requires intact IgG antibody binding. Wild-type V. cholerae was incubated with commercial IgG that is specific against O-antigen at subagglutinating concentrations. Percentages of motile bacteria after antibody addition were recorded (A), and the relative amounts of IgG or Fab bound to bacteria were calculated using fluorescently labeled antibody (B). Each symbol represents samples from one experiment. Bars are means with standard deviations.
To ensure that the lack of inhibition of motility is due to the inherent monovalent structure of Fab fragments (according to our hypothesis) and not simply a failure to bind, IgG was fluorescently labeled on free amines and Fabs were purified from it. A dose-response binding of known concentrations of labeled IgG was done to identify concentrations that are within the linear range of the assay (Fig. 4B, first three columns). The labeled IgG at 1.15 µM inhibited V. cholerae motility completely, while 0.67 and 0.39 µM resulted in partial inhibition (data not shown). A higher concentration of labeled Fab (1.62 µM) bound efficiently to V. cholerae (Fig. 4B) but failed to inhibit motility (data not shown). These results show that Fab fragments are bound to V. cholerae but do not block motility.
DISCUSSION
There is a need for improved vaccines against V. cholerae and other mucosal pathogens. Although currently licensed WCK oral vaccines are effective against cholera, for various reasons, including incomplete immunity and short duration of protection, they have not been widely implemented. A V. cholerae OMV vaccine delivered intranasally represents an alternative vaccine approach; however, much remains to be learned in terms of its efficacy, safety, and mechanism of protection. Here, we address primarily the question of mechanism of protection.
It was previously shown that purified OMVs provide long-term protection in the infant mouse model of V. cholerae intestinal colonization, primarily through IgG in the milk. Furthermore, it was hypothesized that inhibition of motility, a trait of V. cholerae that is required for efficient colonization of the small intestine, contributes to this protection. Here, we show, using competition assays in naive versus immune mice, that inhibition of motility is the primary, if not the sole, mechanism of protection. Although it remains possible that additional mechanisms of protection via antibodies occur during human immunity to cholera, it is tempting to speculate that inhibition of motility plays an important role, not only in breast-fed infants but also in older humans with elevated mucosal anti-O-antigen antibody titers.
Since the concentrations of antibody we used here were subagglutinating, we hypothesize that antibody inhibits motility by binding to O-antigen in the flagellar sheath and causing deformation of the sheathed flagellum, or alternatively but not exclusively through cross-linking the sheathed flagellum to the cell body. Recently, support for the latter mechanism was reported using a monoclonal anti-LPS antibody (14). However, in that study, Fab fragments generated from IgA maintained their ability to inhibit motility, raising questions as to what the mechanism of motility inhibition is. Here we focused on IgG because of its physiological relevance for protection in the infant mouse model and its potential role in contributing to immunity to V. cholerae in humans. In contrast to the report using monoclonal IgA (14), we found that multivalent antibody binding is required for motility inhibition, since monovalent Fab fragments readily bound to the surface of V. cholerae but did not inhibit motility. Thus, our work supports either of the two mechanisms described above for how antibodies inhibit V. cholerae motility, i.e., deformation of the flagellum and sheath or cross-linking of the flagellum and sheath to the cell body.
Overall, our study has identified the O-antigen component of the LPS as the necessary antigen and subsequent target for IgG binding in motility inhibition and subsequent immune protection. This and future work should allow us to gain a better understanding of the immune response against OMVs and how the resulting passive protection works in suckling mice. This in turn will provide a foundation for future testing of OMV-based cholera vaccines in humans for immunity in adults and passive protection of breast-fed infants.
MATERIALS AND METHODS
Strains and culture.
V. cholerae O1 El Tor Ogawa strain E7946 and its isogenic derivatives were cultured on Luria-Bertani (LB) agar plates with 100 μg/ml streptomycin overnight at 30°C. V. cholerae was subsequently suspended in 1 ml LB broth and the concentration adjusted to an optical density at 600 nm (OD600) of 0.1. V. cholerae strains used are listed in Table 1. The motY::neo mutant strain was constructed by allelic exchange via natural transformation of competent E7946 transformed with genomic DNA of strain AJB32 (19). The ΔflaACEDB mutant strain was constructed by consecutive deletions of the two loci harboring the five V. cholerae flagellin genes (VC2142-VC2143-VC2144 and VC2187-VC2188) using the FLP recombination target (FRT)/FLP system as described previously (20). The final strain harbors deletions from the start codon of VC2142 to the stop codon of VC2144 and from the start codon of VC2187 to the stop codon of VC2188, each replaced with an FRT scar.
Preparation of OMVs.
OMVs were isolated from late-exponential-phase cultures as described previously (6). Briefly, 10-ml cultures of V. cholerae were used to inoculate 1 liter of LB and grown to late exponential phase for 8 h. Bacteria were removed by centrifugation (4,500 × g, 15 min, 4°C), and the supernatant was filtered through a 0.22-μm filter. One milliliter of the filtrate was plated on an LB agar plate to ensure complete absence of viable bacterial cells. OMVs were then purified by ultracentrifugation (140,000 × g, 4 h, 4°C) using a Beckmann SW32Ti rotor. The resulting pellet was washed once with phosphate-buffered saline (PBS), repelleted, and finally resuspended in 625 μl of PBS. The protein concentration of each OMV preparation was determined by using a modified Lowry protein assay kit (Pierce) and adjusted to 2.5 μg/μl using PBS. Purified OMVs were stored at −80°C.
Competition assay.
All animal experiments were performed in accordance with NIH guidelines, the Animal Welfare Act, and U.S. federal law. The experimental protocol used for this study was approved by the Tufts University School of Medicine's Institutional Animal Care and Use Committee. Mice used for these experiments were housed in an AAALAC-accredited research animal facility. Immunization and challenge with V. cholerae were done according to a previously established protocol (9). In brief, adult female BALB/c mice 16 to 22 weeks of age were anesthetized with 2% isoflurane gas and intranasally immunized with 25 μg of OMV in 10 μl of PBS and boosted by the same route 2 weeks later. Two weeks after the boost, these mice were mated. Infant mice born to mock- or OMV-vaccinated mice were challenged at day 5 of age. On day 5, pups were separated from their dams for 1 h, anesthetized by 2% isoflurane gas, and then intragastrically inoculated with 50 μl of V. cholerae suspension in PBS. Pups were then returned to their dams for 24 h and sacrificed by humane measures. The small bowel was then removed by dissection and mechanically homogenized. Competition assays in naive infant mice were done using an inoculum of approximately 105 CFU of a 1:1 mixture of motile and nonmotile strains, whereas those done in immune infant mice used an inoculum of approximately 107 CFU. The higher inoculum dose was necessary to allow recovery of enough CFU at the end of the infection to calculate a competitive index. The nonmotile strains express the endogenous lacZ gene, whereas the motile competing strain has the gene deleted, thus allowing the input and output ratios to be determined by plating on agar plates supplemented with the chromogenic substrate 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal). Because of variations in litter size and day of delivery, the data from multiple litters infected on different days were pooled for analysis.
Antibody and Fab purification.
The source of antibody used for purification of IgG was rabbit polyclonal Vibrio cholerae antiserum O1 Ogawa (BD Biosciences). For IgG purification and removal of bovine serum albumin (BSA) and other interfering components, NAB protein A columns were used (Pierce) according to the manufacturer's instructions. Fab fragment purification was done on purified IgG using the Fab preparation kit (Pierce) according to the manufacturer's instructions. Briefly, purified samples of anti-O1 IgG were first desalted and then digested with immobilized papain bound on agarose resin for 5 h at 37°C. To isolate Fab fragments, the antibody mixture after digestion was incubated with protein A beads at room temperature for 10 min and then the supernatant was collected after centrifugation. Fc fragments and undigested IgG were eluted with 40 μl of 1 M Tris-HCl, pH 8. The other antibody used was rabbit polyclonal anti-CD22 (Santa Cruz Biotechnologies).
SDS-PAGE and dot blotting.
The protein concentration of OMVs was measured by NanoDrop (Thermo Scientific), and known amounts were loaded onto 4 to 12% gradient SDS-PAGE gels. Reducing conditions were achieved by using 5× loading dye with β-mercaptoethanol and boiling for 10 min. Silver staining of LPS and protein was also performed by running SDS-PAGE gel for 35 min and then staining with the SilverQuest staining kit (Invitrogen) according to the manufacturer's instructions. Gel images were acquired using a Fujifilm FLA-9000 image reader and Fujifilm Multi-Gauge software.
LPS O1-antigen was qualitatively measured by dot blot. Aliquots of 10 μl of serial dilutions made in PBS of purified LPS from wild-type, rough mutant, and negative-control strain MO10 (O139 serogroup) were spotted in triplicate onto a nitrocellulose membrane and air dried. The membrane was blocked by incubation in 5% skim milk in PBS buffer with 0.1% Tween (PBS-T) at 4°C overnight. The primary Ig (purified anti-O1 IgG) was diluted 1:10,000 in 5% BSA in PBS-T, incubated at 4°C overnight, and washed three times for 5 min with PBS-T. The secondary Ig (goat anti-mouse IgG1-Cy5; SouthernBiotech) was diluted 1:1,000 in PBS-T, applied to the membrane, and incubated at room temperature for 2 h with gentle rocking. The membrane was washed three times for 5 min with PBS-T. Binding of Cy5-conjugated secondary antibody was detected using a Fujifilm FLA-9000 image reader (Life Sciences).
Motility inhibition assay.
V. cholerae cells were streaked on agar plates with 100 μg/ml of streptomycin and incubated overnight at 30°C. Bacterial colonies were transferred to 1 ml LB broth, and the OD600 was adjusted to 0.1. Small aliquots of V. cholerae were then mixed with an equal volume of antibody or Fab solution. Aliquots of 10 μl of the mixtures were taken at different time points for phase-contrast microscopy to assess motility.
TEM.
Transmission electron microscopy (TEM) images were obtained of V. cholerae or purified OMVs visualized using negative stain. In brief, grids were floated on V. cholerae at a concentration of 105 CFU/μl or OMVs at a concentration of 1 μg/μl on Parafilm for 1 min and then washed with 2% acidic uranyl acetate. Grids were blotted dry and examined under an FEI Tecnai Spirit transmission electron microscope.
Antibody labeling.
Purified IgG antibodies were buffer exchanged to remove all free amines through Zeba desalting spin columns (Pierce). Whole antibody was labeled with a CF633 SE protein labeling kit (Biotium) according to the manufacturer's instructions. Extra dye molecules were removed by centrifuging using ultrafiltration columns with a molecular mass cutoff of 10 kDa (provided by the kit). The degree of labeling was then calculated by measuring absorbance at 280 nm and 630 nm on a NanoDrop and calculated using equations provided in the CF633 SE protein labeling kit (Biotium) protocol. Labeled Fab and Fc fragments were then generated from this labeled antibody mixture.
Quantification of bound antibody.
V. cholerae was grown on LB agar plates and resuspended in LB solution, and density was adjusted to an OD600 of 0.1. Aliquots of 10 μl of cells were pelleted at 6,000 × g for 10 min and washed once with 1 ml of PBS. The cell pellet was then resuspended in 10 μl of solution containing labeled, known amounts of antibody or Fab fragments. The mixture was incubated at room temperature for 10 min to allow for antibody or Fab binding to cells and then centrifuged at 6,000 × g to pellet cells. The supernatant containing unbound antibody or Fab was removed and saved. The cells were washed once with 1 ml of PBS, pelleted as described above, and finally resuspended in 10 μl of PBS. The resuspended cells and initial supernatants containing unbound antibody or Fab were spotted as 10-μl droplets onto a polystyrene petri dish. The whole dish was scanned using a Fujifilm Starion imager by excitation with a 633-nm red laser and filtering with the corresponding long-pass filter.
Statistical analysis.
Data from competition assays in mice were not normally distributed due to biological variation, therefore we used nonparametric tests for these experiments. Comparison between two categorical values was done using Mann-Whitney U test with Kruskal-Wallis test for multiple-variable groups. Prism 6 was used for all statistical analyses. A P value of <0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Nicki Watson from the Whitehead Institute for her help with electron microscopy preparation and imaging and Heather Kamp for constructing the ΔflaACEDB strain.
Support for this work came from the National Institutes of Health (AI055058) and the Howard Hughes Medical Institute.
REFERENCES
- 1.Ali M, Lopez AL, You YA, Kim YE, Sah B, Maskery B, Clemens J. 2012. The global burden of cholera. Bull World Health Organ 90:209–218. doi: 10.2471/BLT.11.093427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris JB, LaRocque RC, Qadri F, Ryan ET, Calderwood SB. 2012. Cholera. Lancet 379:2466–2476. doi: 10.1016/S0140-6736(12)60436-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bishop AL, Camilli A. 2011. Vibrio cholerae: lessons for mucosal vaccine design. Expert Rev Vaccines 10:79–94. doi: 10.1586/erv.10.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finkelstein RA. 1962. Vibriocidal antibody inhibition (VAI) analysis: a technique for the identification of the predominant vibriocidal antibodies in serum and for the detection and identification of Vibrio cholerae antigens. J Immunol 89:264–271. [Google Scholar]
- 5.Mosley WH, Ahmad S, Benenson AS, Ahmed A. 1968. The relationship of vibriocidal antibody titre to susceptibility to cholera in family contacts of cholera patients. Bull World Health Organ 38:777–785. [PMC free article] [PubMed] [Google Scholar]
- 6.Schild S, Nelson EJ, Camilli A. 2008. Immunization with Vibrio cholerae outer membrane vesicles induces protective immunity in mice. Infect Immun 76:4554–4563. doi: 10.1128/IAI.00532-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schild S, Nelson EJ, Bishop AL, Camilli A. 2009. Characterization of Vibrio cholerae outer membrane vesicles as a candidate vaccine for cholera. Infect Immun 77:472–484. doi: 10.1128/IAI.01139-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bishop AL, Schild S, Patimalla B, Klein B, Camilli A. 2010. Mucosal immunization with Vibrio cholerae outer membrane vesicles provides maternal protection mediated by antilipopolysaccharide antibodies that inhibit bacterial motility. Infect Immun 78:4402–4420. doi: 10.1128/IAI.00398-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bishop AL, Tarique AA, Patimalla B, Calderwood SB, Qadri F, Camilli A. 2012. Immunization of mice with Vibrio cholerae outer-membrane vesicles protects against hyperinfectious challenge and blocks transmission. J Infect Dis 205:412–421. doi: 10.1093/infdis/jir756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwechheimer C, Kuehn MJ. 2015. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat Rev Microbiol 13:605–619. doi: 10.1038/nrmicro3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins BS. 2011. Gram-negative outer membrane vesicles in vaccine development. Discov Med 12:7–15. [PubMed] [Google Scholar]
- 12.Freter R, O'Brien PC. 1981. Role of chemotaxis in the association of motile bacteria with intestinal mucosa: fitness and virulence of nonchemotactic Vibrio cholerae mutants in infant mice. Infect Immun 34:222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butler SM, Camilli A. 2004. Both chemotaxis and net motility greatly influence the infectivity of Vibrio cholerae. Proc Natl Acad Sci U S A 101:5018–5023. doi: 10.1073/pnas.0308052101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levinson KJ, De Jesus M, Mantis NJ. 2015. Rapid effects of a protective O-polysaccharide-specific monoclonal IgA on Vibrio cholerae agglutination, motility, and surface morphology. Infect Immun 83:1674–1683. doi: 10.1128/IAI.02856-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leitner DR, Feichter S, Schild-Prüfert K, Rechberger GN, Reidl J, Schild S. 2013. Lipopolysaccharide modifications of a cholera vaccine candidate based on outer membrane vesicles reduce endotoxicity and reveal the major protective antigen. Infect Immun 81:2379–2393. doi: 10.1128/IAI.01382-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song T, Mika F, Lindmark B, Liu Z, Schild S, Bishop A, Zhu J, Camilli A, Johansson J, Vogel J, Wai SN. 2008. A new Vibrio cholerae sRNA modulates colonization and affects release of outer membrane vesicles. Mol Microbiol 70:100–111. doi: 10.1111/j.1365-2958.2008.06392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leitner DR, Lichtenegger S, Temel P, Zingl FG, Ratzberger D, Roier S, Schild-Prüfert K, Feichter S, Reidl J, Schild S. 2015. A combined vaccine approach against Vibrio cholerae and ETEC based on outer membrane vesicles. Front Microbiol 6:823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradley ES, Bodi K, Ismail AM, Camilli A. 2011. A genome-wide approach to discovery of small RNAs involved in regulation of virulence in Vibrio cholerae. PLoS Pathog 7:e1002126. doi: 10.1371/journal.ppat.1002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silva AJ, Leitch GJ, Camilli A, Benitez JA. 2006. Contribution of hemagglutinin/protease and motility to the pathogenesis of El Tor biotype cholera. Infect Immun 74:2072–2079. doi: 10.1128/IAI.74.4.2072-2079.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Souza Silva O, Blokesch M. 2010. Genetic manipulation of Vibrio cholerae by combining natural transformation with FLP recombination. Plasmid 64:186–195. doi: 10.1016/j.plasmid.2010.08.001. [DOI] [PubMed] [Google Scholar]