

Abstract
CD8+ cells from simian immunodeficiency virus (SIV)-infected long-term non-progressors and some uninfected macaques can suppress viral replication in vitro without killing the infected cells. The aim of this study was to identify factors responsible for non-cytolytic viral suppression by transcriptional profiling and to investigate their potential impact on SIV replication. Results of microarray experiments and further validation with cells from infected and uninfected macaques revealed that FAM26F RNA levels distinguished CD8+ cells of controllers and non-controllers (P=0.001). However, FAM26F was also expressed in CD4+ T-cells and B-cells. FAM26F expression increased in lymphocytes after in vitro IFN-γ treatment on average 40-fold, and ex vivo FAM26F RNA levels in peripheral blood mononuclear cells correlated with plasma IFN-γ but not with IFN-α. Baseline FAM26F expression appeared to be stable for months, albeit the individual expression levels varied up to tenfold. Investigating its role in SIV-infection revealed that FAM26F was upregulated after infection (P<0.0008), but did not directly correlate with viral load in contrast to MX1 and CXCL10. However, pre-infection levels of FAM26F correlated inversely with overall plasma viral load (AUC) during the acute and post-acute phases of infection (e.g. AUC weeks post infection 0–8; no AIDS vaccine: P<0.0001, Spearman rank correlation coefficient (rs)=−0.89, n=16; immunized with an AIDS vaccine: P=0.033, rs=−0.43; n=25). FAM26F transcript levels prior to infection can provide information about the pace and strength of the antiviral immune response during the early stage of infection. FAM26F expression represented, in our experiments, one of the earliest prognostic markers, and could supplement major histocompatibility complex (MHC)-typing to predict disease progression before SIV-infection.
Keywords: SIV, HIV, FAM26F, innate immune response, IFN-gamma, acute infection
Introduction
Infection with the human immunodeficiency virus (HIV-1) is one of the world's most significant public health challenges. In contrast to most untreated HIV-infected patients, a small subset of patients, called long-term non-progressors (LTNPs) can maintain low or undetectable viraemia and physiological CD4+ T-cell levels and remain clinically asymptomatic for years (Deeks & Walker, 2007; Poropatich & Sullivan, 2011; Sheppard et al., 1993). CD8+ lymphocytes are well documented to have a pivotal role in direct control of viral replication, e.g. by classical major histocompatibility complex (MHC) class I restricted antigen-specific cytotoxic T lymphocyte (CTL) effector mechanism and non-classical CD8 T-cell mediated non-cytolytic antiviral response (CNAR) (Mackewicz et al., 2003; Walker et al., 1986, 1991). CNAR refers to the property of CD8+ T-lymphocytes to suppress HIV-1 replication in acutely infected CD4+ cells without killing them (Levy, 2003; Stranford et al., 1999; Vella & Daniels, 2003; Walker et al., 1991; Wiviott et al., 1990). High CNAR has been reported to correlate with an asymptomatic healthy clinical state and long-term survival of HIV-infected individuals (Blackbourn et al., 1996; Castelli et al., 2002; Greco et al., 1998; Mackewicz et al., 1991; Yuan et al., 2009).
Whole transcriptome analyses have been performed to identify the gene(s) mediating CNAR. The first study involved a pair of HIV-1 discordant identical twins (Diaz et al., 2003). Another study included four CNAR-positive and four CNAR-negative HIV-infected individuals (Martinez-Mariño et al., 2007). There was no overlap between the genes identified in these two reports. Comparison of gene expression profiles from CD8+ cells of three children with differential CNAR identified more than 7000 differentially expressed genes belonging to diverse cellular processes (Katz et al., 2011). While there was a significant overlap in differential gene expression between this and the two former studies, unfortunately no gene represented a strong candidate gene for CNAR.
Simian immunodeficiency virus (SIV)-infected macaques represent the most accepted model for HIV-infection. Our previous studies showed that CNAR can be detected over years in SIV-infected LTNPs and is inversely correlated with plasma viral load (Javed et al., 2015). The present study focused on the identification of factors associated with non-cytolytic viral suppression using CD8+ cells displaying high CNAR activity and CD8+ cells that lack CNAR. Microarray experiments were performed, notably including CNAR(+) cells from infected and uninfected macaques. The most significantly differentially expressed gene, FAM26F, appeared to be related to immune activation, which provided the rationale for further studies. Investigations into a potential role in SIV-infection revealed that the transcript levels of FAM26F were predictive of overall plasma viral load during acute and post-acute phase of SIV-infection.
Results
Gene expression profile comparison of CNAR(+) and CNAR(−) CD8 cells
Global gene expression profiles were determined from CD8+ cells of CNAR(+) and CNAR(−) SIV-infected LTNPs and uninfected macaques after performing a viral inhibition test. Uninfected macaques were included to minimize the effects that might potentially be related to long-term SIV-infection and distinguishes this work from previously published experiments in HIV-infected humans (Diaz et al., 2003; Katz et al., 2011; Killian et al., 2013; Martinez-Mariño et al., 2007). Comparison between CNAR(−) versus CNAR(+) animals revealed 143 differentially regulated genes (differential expression over twofold, P<0.1), which belonged to diverse biological processes (Table S1, Fig. S1, available in the online Supplementary Material). Only 14 genes were upregulated in CNAR(+) animals.
Furthermore, except for glutamate-ammonia ligase (GLUL) none of the genes was previously identified as differentially regulated in CNAR(+) individuals (Diaz et al., 2003; Katz et al., 2011; Martinez-Mariño et al., 2007). Notably, no soluble factors such as chemokines or cytokines known to be associated with HIV or SIV replication were differentially expressed when combined data from SIV-infected and non-infected monkeys were analysed (Table S1).
Some of the downregulated genes in CNAR(+) CD8+ cells have already been investigated in HIV-infection. Lower expression of protein disulfide isomerase family A member 4 (PDIA4), dipeptidylpeptidase 4 (DPP4/CD26) or tumour necrosis factor (ligand) superfamily member 13b (TNFSF13B/BAFF) may contribute to inhibition of HIV in CD4+ T-cells but does not explain how this should affect anti-HIV/SIV CD8 T-cell function (Bernier et al., 2013; Fan et al., 2012; Gallina et al., 2002; Jager et al., 2012; Reiser et al., 2012; Rotger et al., 2011; Songok et al., 2010; Stantchev et al., 2012). Notably, many (26 % of downregulated, 46 % of upregulated) differentially expressed genes encode membrane proteins belonging to activation/signalling pathways and/or are involved in cell adhesion.
Lower in vitro expression of FAM26F in CD8+ cells from CNAR(+) macaques
Differential expression of 15 genes from microarray data was further investigated by quantitative reverse transcription PCR (RT-PCR) in CD8+ cells from six CNAR(+) and four CNAR(−) LTNPS, as well as from four CNAR(+) and five CNAR(−) uninfected monkeys (Fig. 1a). Since CNAR has been ascribed to proteinase activity (Mackewicz et al., 2003) we also included CMA1, which was found to be upregulated fourfold in microarray analysis of uninfected CNAR(+) animals (data not shown). For validation purposes, the same protocol as for generation of the samples for the microarray analysis was employed.
Fig. 1.

Differential expression of 15 genes in CD8+ cells from CNAR(+) animals compared with CNAR(−) animals. (a) RNA was extracted from re-isolated CD8+ cells of six CNAR(+) and four CNAR(−) SIV-infected LTNPS, as well as from four CNAR(+) and five CNAR(−) uninfected monkeys after the seventh day of co-culture with SIV-infected MHC-mismatched CD4+ cells. Fold difference in gene expression was calculated using the 2−ΔΔCt formula. Mean with standard deviation is shown. (b) Relative FAM26F RNA levels compared with the housekeeping gene GAPDH in CD8+ cells after overnight concanavalin A (ConA) activation and after day 7 of co-culture. (c) Relative FAM26F RNA levels in CD8+ cells from combined infected and uninfected CNAR(+) (n=10) and CNAR(−) (n=9) macaques in day 7 co-cultures. Mean, standard error of the mean and significant P values are depicted (Mann Whitney U test).
Four genes were found to be more than twofold downregulated in SIV-infected CNAR(+) animals, and six genes in CD8+ cells from uninfected CNAR(+) macaques as compared with the respective CNAR(−) animals (Fig. 1a). Finally, only FAM26F RNA levels were significantly lower in CD8+ CNAR(+) compared with CNAR(−) cells from uninfected macaques (P=0.0159, Mann Whitney test; Fig. 1b). Combined data from uninfected and infected macaques confirmed that FAM26F RNA levels were significantly lower in CD8+ cells from CNAR(+) macaques compared with those from CNAR (−) macaques (P=0.001, Mann Whitney test, Fig. 1c).
Differential expression of FAM26F emerged after co-cultivation with SIV-infected CD4+ cells
Next, we investigated whether the differential transcript levels of FAM26F in CD8+ cells emerged as a consequence of co-cultivation with SIV-infected CD4+ cells during the viral inhibition test or whether these differences were present before the start of the experiment. FAM26F RNA levels did not differ significantly either in CD8+ cells after concanavalin A (ConA) stimulation before initiation of the co-cultures (P>0.4, Mann Whitney test; Fig. 1b) or in ex vivo-isolated CD8+ cells between CNAR(+) and CNAR(−) monkeys (P>0.2, Mann Whitney test; data not shown). However, upon co-cultivation of CD8+ T-cells with SIV-infected CD4+ T-cells, FAM26F RNA levels increased up to tenfold (Fig. 1b). The increase after co-cultivation was on average three to four times stronger in cells from CNAR(−) monkeys as compared with cells from CNAR(+) monkeys and resulted in a lower expression of FAM26F in CNAR(+) animals. These findings indicated that FAM26F might not necessarily be related to CNAR but to immune activation and provided the rationale for further investigations into FAM26F expression and its potential role as a prognostic marker in SIV-infection.
Expression of FAM26F in lymphocytes
Expression of FAM26F was analysed by RT-PCR in three major lymphocyte populations, namely CD8+, CD4+ and CD20+ (B-cells), of four naive animals. RNA levels of the innate immune genes MX1 and CXCL10 were also measured. Maximum expression of FAM26F was observed in CD4+ cells. MX1 and CXCL10 were barely expressed, with highest levels in CD20+ cells (Fig. 2). The results demonstrate that FAM26F represents a gene that is well expressed in many lymphocyte populations. Because of this broad expression profile and the function of FAM26F via homophilic interactions (Ebihara et al., 2010), we used peripheral blood mononuclear cells (PBMCs) in the further studies.
Fig. 2.
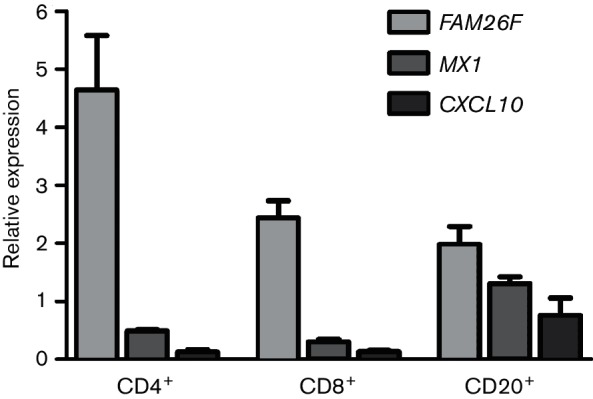
FAM26F, MX1 and CXCL10 expression in CD8+, CD4+ and CD20+ cells of four naive animals. Cell populations were isolated by magnetic bead purification, RNA was isolated and relative expression was quantified by RT-PCR. Mean and standard error of the mean are shown.
FAM26F expression is linked to IFN-γ expression
To gain insight into FAM26F regulation, its expression was quantified after stimulation of PBMCs from eight LTNPs, and two and three uninfected monkeys with IFN-γ and IFN-α2, respectively.
MX1 and CXCL10/IP-10 as interferon type I and type II regulated genes were also investigated. At 6 to 12 h after IFN-γ stimulation, FAM26F RNA increased on average 40-fold following kinetics similar to those of CXCL10 (Fig. 3a). MX1 RNA levels increased only fourfold upon IFN-γ stimulation (Fig. 3a). Six hours after IFN-α2 stimulation of PBMCs from LTNPs, MX1 RNA levels increased on average 30-fold, whereas FAM26F and CXCL10 RNA levels increased only 7- and 12-fold, respectively (Fig. 3b). Maximum increase of FAM26F transcripts occurred 6 h after IFN-γ stimulation in cells from uninfected monkeys pre-treated with low concentrations of ConA (10 ng ml−1) in contrast to IFN-α2 stimulation (Fig. 3c, d).
Fig. 3.
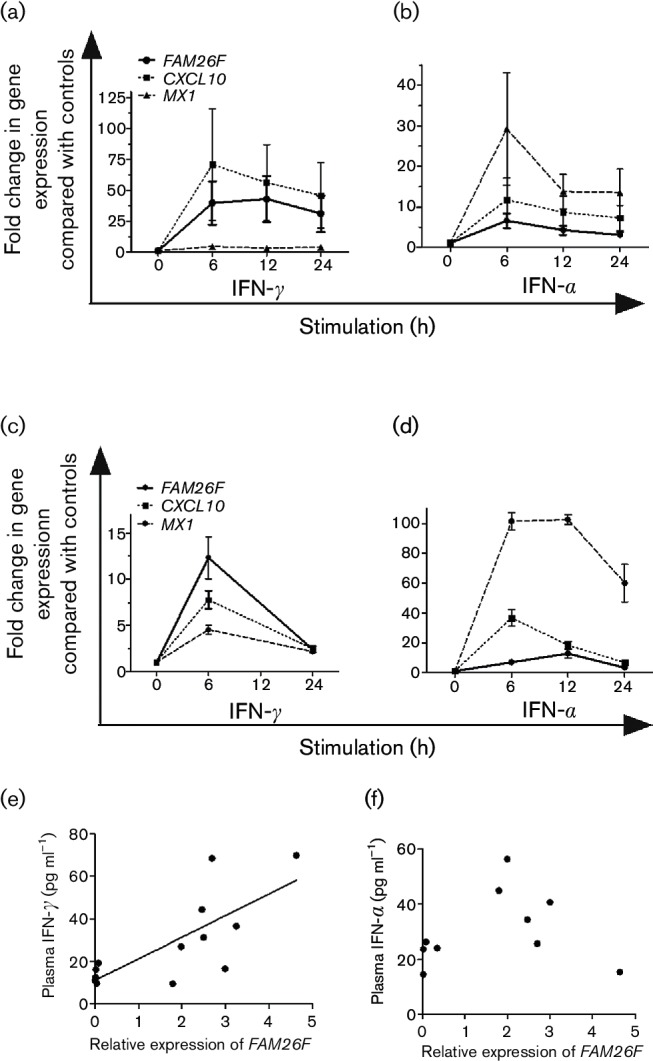
FAM26F expression is linked to IFN-γ pathway. IFN-γ was added to PBMCs from (a) eight LTNPs without and (c) two naive macaques with prior low-dose ConA activation (10 ng ml−1 overnight). IFN-α2 was added to PBMCs from (b) eight LTNPs without and (d) three naive macaques with prior low-dose ConA activation. At the time points indicated, RNA was extracted and fold differences in gene expression of FAM26F, CXCL10 and MX1 compared with controls not treated with interferons were determined. Mean and standard error of the mean are shown. (e) Plasma IFN-γ and (f) plasma IFN-α were quantified by ELISA while relative expression of FAM26F and CXCL10 was quantified by RT-PCR in PBMCs. FAM26F expression correlated with plasma IFN-γ (P=0.0031, Pearson r=0.7508; n=13) but not with plasma IFN-α (n=10).
In addition, relative RNA levels of FAM26F were quantified in blood samples of macaques immunized with viral vectors. As with other genes of innate immunity, expression of FAM26F increased significantly within 24 h compared with pre-immunization levels (data not shown). In parallel, plasma IFN-γ and IFN-α were quantified by ELISA. FAM26F RNA correlated significantly with plasma IFN-γ (P=0.0031, Pearson r=0.7508; n=13) (Fig. 3e) but not with IFN-α (P=0.66, Pearson r=0.16, n=10) (Fig. 3f). In summary, our results suggest that FAM26F RNA levels are linked to the IFN-γ pathway.
FAM26F expression increases after SIV-infection
The expression pattern of FAM26F before and after infection was investigated in two independent AIDS vaccine experiments termed E1 and E2 and one infection study (E3) comprising a total of 51 animals (see Methods and Table S2). FAM26F RNA in PBMCs was quantified at defined time points along with MX1 and CXCL10. In all experiments, FAM26F transcripts were significantly elevated after SIV-infection as compared with pre-infection values until the end of the study period (P<0.0008 from week 2 up to weeks 32–48 post infection, Mann Whitney test, data for E1; Fig. 4).
Fig. 4.
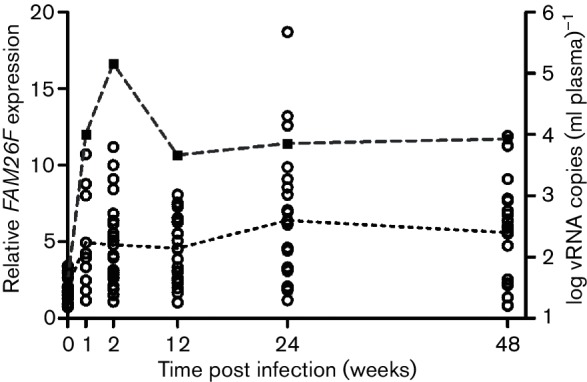
FAM26F gene expression after SIV-infection in relation to viral load. (a) Relative FAM26F transcript levels were determined at specified time points, before and after SIV-infection of rhesus macaques. Each time point shows data from 9 to 24 macaques. Expression of FAM26F (open circles) was significantly upregulated after SIV-infection as compared with its pre-infection values until the end of the study period (P<0.0008, Mann Whitney test). Log10 of the geometric mean of plasma viral RNA (vRNA) copies is shown as a dashed line and filled squares. Mean of relative FAM26F gene expression is depicted as a dotted line.
We also investigated whether FAM26F RNA levels could be related to plasma viral load. However, FAM26F RNA did not correlate with viral load (e.g. weeks 2, 12, 24, 48 post infection; Fig. S2a). In contrast, but in line with previous publications, MX1 did so (P≤0.0016, Spearman rank correlation coefficient (rs)>0.66 at week 24 post infection as example; Fig. S2b) (Mussil et al., 2011). Relative expression of CXCL10 correlated with viral load in two of the three experiments at week 24 post infection (Fig. S2c).
Pre-infection RNA levels of FAM26F correlate robustly with overall plasma viral load in acute and post-acute infection in unimmunized macaques
To study if the differential expression of FAM26F can be an early predictor of viral load of the chronic phase of infection, potential correlations between FAM26F RNA levels and viral load were investigated. Investigating the predictive potential of RNA levels at 2 weeks post infection (wpi) revealed that FAM26F, MX1 and CXCL10 were predictive of chronic phase set-point viral load only when peak viraemia correlated with the further course of infection (Fig. S3).
Next, we investigated whether pre-infection transcript levels could have a prognostic value. Since immunization can influence early viral replication kinetics, we first analysed unvaccinated macaques. Pre-infection samples were available from E2 (Fig. 5a; n=6) and E3 (Fig. 5b; n=10). In each experiment, pre-infection RNA levels of FAM26F correlated inversely with overall (AUC) plasma viral load during the acute and post-acute phases of infection [e.g. area under curve (AUC) plasma viral load wpi 0–8; E2: rs=−0.88, P=0.019; E3: rs=−0.74, P=0.013; Fig. 5a, b]. Combining both data sets confirmed the highly significant inverse correlation of FAM26F expression with the overall plasma viral load during the early phase of infection (Fig. 5c; e.g. AUC viral load wpi 0–8: rs=−0.89, P<0.0001). Note that the overall viral load is mainly (albeit not exclusively) influenced by the peak and post-peak (e.g. 4 wpi) viral load so that RNA copy numbers at later time points add relatively little to the overall plasma viral load. Consequently, the pre-infection RNA levels can also correlate with AUC values earlier or later than 8 wpi depending on the experiment (Fig. 5a, b). For E2, pre-infection RNA levels of FAM26F correlated with overall plasma viral load from AUC 0–8 to AUC 0–32 wpi, which was the latest available time point for all six animals (Fig. 5a). In E3, AUC values from 0–4 to 0–16 wpi correlated with pre-infection FAM26F RNA levels (Fig. 5b).
Fig. 5.

FAM26F gene expression at day of first SIV exposure correlates with the extent of early viral replication. (a) Log plasma viral RNA copies of unvaccinated macaques (n=6) from E2 are shown. Asterisks (*) mark significant correlation of FAM26F RNA levels at day of first SIV exposure with overall plasma viral load from infection to this time point. Significant correlations with overall viral load: AUC 0–8 to AUC 0–20: P=0.033, rs=−0.89; AUC 0–24 and AUC 0–32: P=0.017, rs=−0.94. (b) Log plasma viral RNA copies of unimmunized macaques (n=10) from E3 are shown. Asterisks (*) mark significant correlation of FAM26F RNA levels at day of first SIV exposure with overall plasma viral load from infection to this time point. Significant correlations with overall viral load: AUC 0–4: P=0.02, rs=−0.71; AUC 0–6: P=0.016, rs=−0.73; AUC 0–8: P=0.013, rs=−0.74; AUC 0–12 and AUC 0–16: P=0.029, rs=−0.68. (c) Relative expression of FAM26F in PBMCs at day of first SIV exposure correlated with overall plasma viral load 0–8 wpi in non-immunized macaques from two independent experiments (AUC wpi 0–8: E2: rs=−0.88, P=0.033, n=6; E3: rs=−0.74, P=0.013, n=10); P value and Spearman's correlation coefficient rs for the combined data set are shown in the figure. (d) Correlation of individual relative FAM26F RNA levels in PBMCs before immunization with those of day of first SIV exposure 10 months later in immunized (n=12) and unvaccinated control macaques (n=6). Significance and Pearson r are indicated in the figure. (e) Log plasma viral RNA copies of unvaccinated macaques (from E1 and E3, n=25) are shown as median (solid line) and upper and lower range (dashed lines). Asterisks (*) mark significant correlation of FAM26F RNA levels at day of first SIV exposure with overall plasma viral load from infection to this time point. Significant correlations with overall viral load: AUC 0–6: P=0.036, rs=−0.42; AUC 0–8: P=0.032, rs=−0.43; AUC 0–12: P=0.017, rs=−0.46; AUC 0–16: 0.017, rs=−0.48; AUC 0–24: P=0.014, rs=−0.45; AUC 0–32: P=0.029, rs=−0.43). (f) Relative expression of FAM26F in PBMCs at day of first SIV exposure correlated with overall plasma viral load 0–8 wpi (AUC wpi 0–8) in macaques from two experiments (E1, E2) immunized with an experimental AIDS vaccine (n=25). P value and Spearman rank correlation coefficient (rs) are shown.
Next we investigated whether the presence of MHC class I alleles known to be associated with slower disease progression had influenced the results. In each experiment (E2, E3) 50 % of the macaques carried Mamu-A1*001, which is in our experiments significantly associated with slow disease progression (Muhl et al., 2002; Sauermann et al., 2008). Combining the results from E2 and E3 showed that FAM26F RNA levels were associated with overall viral load (AUC 0–8) in both Mamu-A1*001-positive and -negative monkeys (Mamu-A1*001-positive: P=0.021, rs=0.81, n=8; Mamu-A1*001-negative: P=0.0004, rs=−0.9762, n=8; Fig. S4a). Furthermore, neither the relative gene expression of FAM26F (P=0.38; Fig. S4b) nor the total viral load (AUC 0–8, P=0.44, Mann Whitney test; Fig. S4c) differed significantly between the groups, excluding a systematic influence of the MHC genotype on our data set.
CXCL10 RNA levels before infection correlated with overall plasma viral load only after combining both data sets and with a lower significance level (e.g. AUC wpi 0–8: rs=−0.72, P=0.0017; Fig. S4d). Pre-infection MX1 RNA levels did not correlate with overall plasma viral load (Fig. S4e).
Pre-infection RNA levels of FAM26F did not correlate with plasma viral load later on. However, overall early plasma viral load (AUC wpi 0–8) correlated with chronic phase plasma viral RNA copies at wpi 12, 16, 24 and 32 (r>0.52, P<0.043, n=16; Pearson correlation for a linear relationship; Fig. S4f), which represented the latest available data for all non-immunized animals. Taken together, pre-infection RNA levels of FAM26F were robustly associated with overall plasma viral load in the early phase of infection, which was in turn associated with chronic phase viral load.
Pre-infection FAM26F RNA levels correlate with extent of early viral replication in vaccinated macaques
A potential association of pre-infection gene expression of FAM26F, CXCL10 and MX1 with viral replication in macaques immunized with experimental AIDS vaccines from E1 and E2 was also investigated.
Immunization had a strong impact on early viral replication (E2: reduction of overall plasma vial load wpi 0–8 in vaccinees compared with controls >1 log, P=0.0057; E1: varying effects depending on the vaccine formulation; Tenbusch et al., 2012). Strong associations were therefore not expected. In addition, FAM26F RNA levels could be analysed for E2 before the start of the immunization and 10 months later, on the day of first SIV exposure. Relative expression of FAM26F prior to immunization and infection correlated significantly (Pearson r=0.95, P<0.0001; Fig. 5d) indicating that expression was stable for months and that FAM26F expression levels on the day of first exposure were comparable to baseline.
In neither immunized macaques from E1 (available pre-infection samples: n=14) nor those from E2 (n=11) did relative expression of FAM26F, MX1 and CXCL10 before infection correlate with the extent of early viral replication. However, combining both data sets revealed that only RNA levels of FAM26F correlated with the extent of early viral replication (e.g. AUC 0–8, FAM26F: P=0.033, rs=−0.43; n=25; Fig. 5c, d). The association between pre-infection FAM26F RNA levels and overall plasma viral load was observed in the time intervals ranging from 0–4 to 0–32 wpi (Fig. 5e). Furthermore, combined data sets also revealed a correlation between pre-infection levels of FAM26F RNA and plasma viral RNA copies at weeks 12 and 24–26 post infection (Fig S5a). These results confirm an association of FAM26F RNA levels with chronic phase viral load (P<0.046, rs>−0.4) in immunized macaques. Neither pre-infection CXCL10 (Fig. S5b) nor MX1 (Fig. S5c) transcript levels correlated with overall early plasma viral load.
No significant association of early viral replication (AUC wpi 0–8) with chronic phase viral load was evident for E1 (Fig. S5d). However, in E2 it correlated with plasma viral load at weeks 24 and 32 (Pearson r>0.97, P<0.0001, n=11; Fig. S5e).
Discussion
This study initially aimed at the identification of factors responsible for non-cytolytic viral suppression in SIV-infected rhesus macaques by transcriptional profiling. Since in vitro viral inhibition tests for identification of CNAR(+) and CNAR(−) animals showed that CD8+ T-cells of non-infected animals can display CNAR, CD8+ cells from both SIV-infected and uninfected macaques were investigated (Javed et al., 2015). This two-pronged strategy increased the probability of identifying the differentially expressed genes that were not influenced by previous SIV-infection. After replicating microarray results of selected genes in 19 macaques, FAM26F RNA levels could be distinguished in CD8+ cells from CNAR(+) macaques compared with those from CNAR(−) animals. FAM26F RNA levels increased in CD8+ cells after co-cultivation with SIV-infected CD4+ cells, but no differences were evident in ex vivo isolated CD8+ cells or in CD8+ cells after ConA activation. Investigating FAM26F transcript levels in SIV-infection showed that they increased also after SIV-infection. Higher FAM26F RNA levels in CNAR(−) compared with CNAR(+) cells after a 7-day culture with SIV-infected cells could therefore be explained by the higher SIV replication and probably stronger immune activation in CNAR(−) compared with CNAR(+) cell cultures. If the interpretation that FAM26F might not be necessarily related to CNAR but to immune activation is correct, our results are in line with other attempts that failed to identify by microarray analysis a strong candidate gene contributing to CNAR (Diaz et al., 2003; Katz et al., 2011; Martinez-Mariño et al., 2007). Perhaps conspicuous gene expression differences are not the hallmark that distinguishes CNAR(+) and CNAR(–) CD8+ T-cells but rather the equipment with distinct polymorphic genes. In this respect, a recent report has uncovered mechanisms underlying cell-contact-mediated CNAR (Lu et al., 2016) by showing that particular regulatory CD8+ T-cells in carriers of KIR3DL1 and HLA-B : Bw4-80Ile can suppress viral replication in CD4+ T-cells. However, in the interaction of the specialized subset of CD8+ cells with CD4+ cells, FAM26F could play a role, and a low expression may contribute to a reduced activation of CD4+ T-cells.
Our aim was then to investigate whether this is of potential prognostic value. We found that FAM26F gene expression in PBMCs before infection correlated inversely with overall plasma viral load during acute and post-acute phases of infection. This was most robust in unvaccinated monkeys but an effect was still evident in vaccinated macaques and may indicate that a certain pre-activation status could contribute to limit viral replication during early infection. FAM26F is a tetraspanin-like membrane glycoprotein (Ebihara et al., 2010) and shows sequence similarity to an ion transporter (NCBI gene information; www.ncbi.nlm.nih.gov/gene).
FAM26F can make homophilic interactions by which synapses between cells are potentially established (Ebihara et al., 2010). To date, no other interacting partner is known (NCBI gene information; www.ncbi.nlm.nih.gov/gene). FAM26F expression was required on both natural killer (NK) cells and myeloid dendritic cells to activate NK cells, and the cytoplasmatic tail was required for directed signal transmission from bone marrow derived dendritic cells to NK cells (Ebihara et al., 2010).
Detailed knowledge about its function is largely lacking, but differential FAM26F expression has been detected by several whole-transcriptome analyses. FAM26F expression was one of the top classifiers for prognosis or diagnosis in a wide range of clinical settings, most of them related to inflammation (Defamie et al., 2008; Kim et al., 2009; Pankla et al., 2009; Shahzad et al., 2010). Increased FAM26F expression was also associated with clearance of hepatitis C virus (Grimes et al., 2013) and clinical benefit through immune-based MAGE-A3 therapy in melanoma patients (Ulloa-Montoya et al., 2013).
Insight into the transcriptional regulation of FAM26F is beginning to emerge. Its expression is upregulated by numerous signalling pathways that are likely to act synergistically (Chmielewski et al., 2014). FAM26F expression on dendritic cells increases after TLR3 via polyI : C or TLR4 stimulation (Chmielewski et al., 2014; Ebihara et al., 2010; Lee et al., 2014), upon IFN-β exposure (Lee et al., 2014), by stimulating the dectin-1 pathway (Chiba et al., 2014) and after murine cytomegalovirus (MCMV) infection (Manh et al., 2013). Furthermore, deletion of IFNRA1 (IFN-α and IFN-β receptor) in mice resulted in abrogation of FAM26F induction by polyI : C (Kasamatsu et al., 2014). Depending on the initial signal, IRF-3 and TICAM-1/TRIF (Ebihara et al., 2010) or IRF-5 (Chiba et al., 2014) are required, since their deletion results in loss of or markedly reduced FAM26F expression in dendritic cells/macrophages and impaired activation and cytolytic function of NK cells. These pathways can merge in STAT1 activation, which probably switches on FAM26F transcription (Chmielewski et al., 2014; Lee et al., 2014).
FAM26F expression is also linked to the IFN-γ pathway. We found that FAM26F gene expression in macaques correlated with plasma IFN-γ and increased in lymphocyte cultures after addition of IFN-γ. IFN-α could also stimulate FAM26F transcription. However, its effect was moderate and is likely to depend on the pre-activation status of the lymphocytes. In other species, IFN-γ alone (Brown et al., 2010; Chmielewski et al., 2014) or IFN-γ in combination with lipopolysaccharide or IFN-β (Chmielewski et al., 2014; Zhang et al., 2010) also leads to an increase of FAM26F RNA in different immune cells. Depletion of IFN-γ with a monoclonal antibody in human patients resulted in downregulation of FAM26F in blood cells (Welcher et al., 2015). In addition, direct knock-out of FAM26F in mice resulted in significantly reduced initial production of IFN-γ after polyI : C treatment and reduced elimination of tumour cells (Kasamatsu et al., 2014). These and our results indicate that FAM26F transcription is stimulated by IFN-γ but that FAM26F itself can also augment IFN-γ responses.
Furthermore, FAM26F is rapidly induced after stimulation as shown in the in vitro assays, in vivo studies in human PBMCs after vaccination (Matsumiya et al., 2014) and in mice 5 h after staphylococcal enterotoxin B exposure (Ferreyra et al., 2014) and represents therefore an early/immediate IFN-induced gene. Current knowledge on regulation of FAM26F transcription and action as well as its potential effects on T-cells is shown in Fig. 6.
Fig. 6.
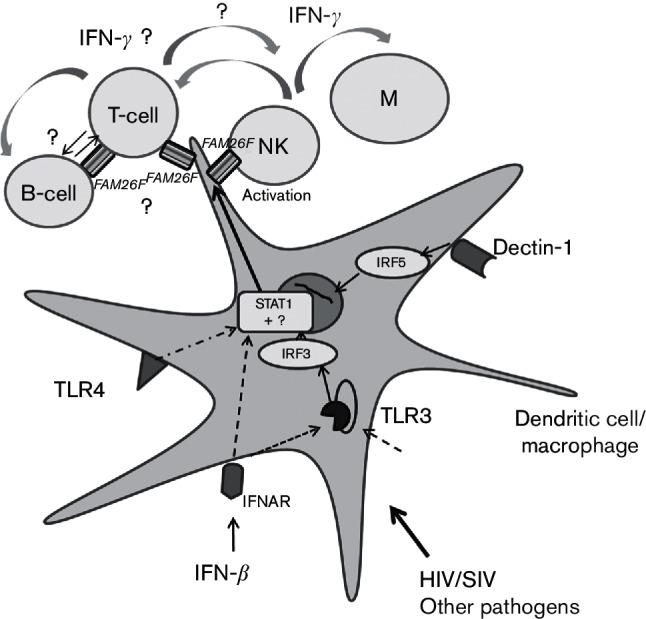
Schematic view illustrating current knowledge and hypothetical role of FAM26F in immune response induction. Activation of dendritic cells/macrophages via IFN-α/β receptor (IFNAR), TLR3, TLR4 and/or dectin-1 and pathogens results in increased FAM26F expression probably through activation of STAT1 (Chiba et al., 2014; Chmielewski et al., 2014; Ebihara et al., 2010; Kasamatsu et al., 2014; Lee et al., 2014; Zhang et al., 2010). Deletion of IRF-3 or IRF-5 reduces FAM26F expression in mice (Chiba et al., 2014; Ebihara et al., 2010). Expression of FAM26F contributes to early IFN-γ secretion and tumour reduction by NK cells (Kasamatsu et al., 2014). The effect of FAM26F expression in T-cells, B-cells and other immune cells in terms of further augmentation of an IFN-γ response through FAM26F-mediated cell–cell contact is hypothetical and requires further investigation.
So far, detailed investigations have focused on the interaction of FAM26F in dendritic cells with NK cells for cancer therapy (Chiba et al., 2014; Ebihara et al., 2010; Kasamatsu et al., 2014). Investigations concerning its role in activation of T-cells and adaptive immune responses are still lacking. The present study showed that pre-infection RNA levels of FAM26F in blood lymphocytes represent a strong prognostic marker for viral replication during the acute and post-acute phase of SIV-infection. Notably, FAM26F gene expression did not correlate with peak viraemia but with the overall viral load during the early phase of infection. This phase includes reduction of viraemia by innate immune system and cytolytic responses mediated by adaptive immunity. Assuming that FAM26F plays an important role in the initiation of IFN-γ-governed adaptive and innate immune responses, high FAM26F expression before infection could contribute to or facilitate a fast activation of a collection of antiviral activities that limit early viral replication and determine further disease progression. Given that early innate responses are already inhibited during the first days of SIV-infection, a certain pre-activation status may be crucial to compensate for some of these effects (Barouch et al., 2016). IFN-γ plays a key role in macrophage, NK cell and CD8+ T-cell activation, and is recognized as a hallmark of the subset 1 of helper T-cells (Th1). Moreover, SIV-specific CTLs are detectable at 2 weeks post infection and peak 1 week later (Allen et al., 2000; O'Connor et al., 2012). Plasma viral loads in SIV-infected macaques that control viral replication and those that do not start to diverge as early as 4 weeks post infection and differ significantly by 8 weeks post infection (Mudd & Watkins, 2011). During this early phase of infection the immune system can become irreversibly damaged. Among the pronounced effects is the massive depletion of gut-associated lymphoid tissue (GALT) CD4+ T-cells (Barouch et al., 2016; Lim et al., 1993; Mattapallil et al., 2005; Schneider et al., 1995). The acute phase thus represents a critical stage in HIV/SIV-infection, and the extent of early viral replication shapes the further course of infection. Albeit formally not proven, our results highlight that IFN-γ responses are of prominent importance for early control of viral replication.
Our results also have practical implications. FAM26F transcript levels represented in our experiments one of the earliest prognostic markers for disease progression after SIV-infection. Furthermore, the individual transcript levels differed significantly, and in the experimental setting, FAM26F expression at baseline appeared to be stable for months. In humans, polymorphisms close to FAM26F in the promotor region have been identified and are presumably linked to basal expression differences (Julià et al., 2014; Lee et al., 2014). The stable differences in FAM26F expression at baseline suggest that differential FAM26F expression may also be genetically determined in macaques. Since FAM26F expression is independent of the MHC genotype, it has a strong potential to supplement MHC-typing as a means to predict disease progression after SIV-infection and to evenly allocate macaques with similar immunobiological profile among the different experimental groups.
Assuming that FAM26F gene expression level is at least a hallmark for the differential speed and spread of early IFN-γ-guided immune responses, future investigations into its role on the initiation of innate and adaptive immune responses are pivotal. In addition, its potential role in local amplification of an immune response by cell–cell contact shall be a subject for further investigation and may also be important in the context of other viral infections. Importantly, appropriate tools to investigate FAM26F protein expression in humans and macaques need to be established.
Methods
Animal experiments.
Blood samples of 24 macaques of Indian origin were derived from an AIDS vaccine study described previously (experiment 1, E1) (Tenbusch et al., 2012). An additional 18 macaques were from an AIDS vaccine study (experiment 2, E2) that included three groups, namely one control group and two vaccine groups consisting of six animals each (unpublished results). Following the immunization period, macaques were subjected weekly to low-dose intrarectal (i.r.) challenges with 120 TCID50 of SIVmac251 until >100 viral RNA copies (ml plasma)−1 were detected. Further details regarding the virus and the challenge procedure have been described previously (Tenbusch et al., 2012). The third group consisted of 10 naive macaques (experiment 3, E3) that had been pre-exposed to escalating low doses of a SIVmac variant but had remained uninfected. After an 18-week waiting period, they became infected with SIVmac251 using the same virus stock and the same repeated low-dose i.r. challenge protocol as described above for E2. One to six inoculations were required to infect the monkeys not pretreated with an AIDS vaccine. The number of inoculations did not differ between E2 and E3 (log rank test, P=0.75). The LTNPs used for this study have been described before (Javed et al., 2015) The LTNP status was defined as survival post SIV-infection for more than 1 year and plasma viral RNA copies below the detection limit of our assay or as a survival of the SIV-infection more than 3 years post infection (Javed et al., 2015). All macaques were MHC-typed using standard techniques (Muhl et al., 2002; Sauermann et al., 2008).
Ethical statement.
Rhesus macaques of Indian origin were housed at the German Primate Centre under standardized conditions according to the national and European Union guidelines on the use of non-human primates in biomedical research. The animal experiments were approved by the Lower Saxony State Office for Consumer Protection and Food Safety and performed with the project licences AZ 33.14-4502-04-017/07, AZ 33.14-42502-04-072/08, 33.14-42502-04-11/0626 and AZ 33.9-42502-04-12/0820.
The macaques had constant access to water and were fed twice a day with monkey dry food supplemented with fresh fruits and foraging or task-oriented feeding methods (e.g. treats, food puzzle). Physical examinations, bleedings and immunizations were carried out under ketamine anaesthesia [10 mg (kg body weight)−1]). In cases of suffering predefined by a scoring system of termination criteria that was approved by the external ethics committee and corresponds to the IACUC endpoint guidelines, monkeys were humanely euthanized in deep anaesthesia by an overdose of pentobarbitone sodium (Narcoren; Merial).
Microarray experiment.
PBMCs were isolated through density gradient centrifugation (lymphocyte separation medium; PAA Laboratories) from 18 ml citrated blood of two CNAR(+) SIV-infected LTNPs, two CNAR(−) SIV-infected slow progressors (total survival of SIV-infection: 115 and 342 weeks post infection), and two CNAR(+) and two CNAR(−) non-infected monkeys, respectively. CD8+ lymphocytes were purified using the CD8+ isolation kit (Miltenyi). CD4+ cells from uninfected macaques were isolated from 9 ml blood using the CD4+ isolation kit (Miltenyi). CD8+ and CD4+ cells were re-suspended in complete RPMI medium [RPMI 1640 (PAN Biotech,), supplemented with 20 % FBS (PAN Biotech), 100 U penicillin ml−1 (PAN Biotech) and 100 µg streptomycin ml−1 (PAN Biotech)] and stimulated overnight with ConA (10 µg ml−1; SERVA Electrophoresis). CD4+ cells were infected with SIVmac239 as described previously (Ochieng et al., 2009) and re-suspended in 1.5 ml complete RPMI medium. ConA-activated CD8+ cells were washed twice and re-suspended in 2 ml complete RPMI medium supplemented with 100 U recombinant human IL-2 ml−1 (PeproTech) and co-cultured with SIV-infected CD4+ cells. Fifty percent of the medium was replaced on days 3 and 6. On day 7, CD4+ cells were re-isolated with the CD4+ cell isolation kit (Miltenyi). Flow-through containing CD8+ cells was collected, centrifuged and re-suspended in 300 µl RNAprotect Cell Reagent (Qiagen) and frozen at −80 °C until further use.
RNA was isolated using the RNeasy Mini kit (Qiagen), digested with DNase (Qiagen) and purified again using the RNAeasy Mini kit. Quantity, purity and integrity were checked using a NanoDrop (Agilent Technologies) and an Agilent 2100 Bioanalyzer. cRNA synthesis, amplification and labelling were performed with 1 µg RNA using ‘Low RNA Input Linear Amplification kit PLUS, One Color’ (Agilent Technologies) and the Agilent ‘RNA One Color Spike-In kit’ following the manufacturer's instructions. Quality, quantity and efficiency of the labelling were checked with the Agilent 2100 Bioanalyzer and NanoDrop NS-1000. The fragmented cRNA of each sample was hybridized on 4×44 K monkey arrays (Agilent Technologies) in an ozone free environment (Hybridization Oven, Agilent). Each RNA sample was tested in duplicate. Washing and staining of the arrays were performed according to the manufacturer's recommendations. Cy3 intensities were detected by one-colour scanning using an Agilent microarray scanner at 5 µm resolution. Scanned image files revealed no artefacts upon visual inspection.
Viral inhibition assay and re-isolation of CD8+ T-cells.
Samples from CNAR(+) and CNAR(−) macaques as well as the samples from which we re-isolated the CD8+ cells were from viral inhibition assays described in a previous study on CNAR (Javed et al., 2015). Briefly, for the viral inhibition assays, CD4+ T-cells from SIV-uninfected, MHC-mismatched donors and CD8+ T-cells from SIV-infected and uninfected experimental animals were used. CD4+ T-cells and CD8+ T-cells were isolated from PBMCs by the magnetic bead separation technique for nonhuman primates (MACS) (Miltenyi Biotech) and activated with ConA in complete RPMI 1640 medium for 24 h. CD4+ cells were infected with SIVmac239 at an m.o.i. of 0.001 TCID50. In vitro infected CD4+ T-cells were either cultured alone (controls) or co-cultured with ConA-activated CD8+ cells from experimental animals in duplicate or triplicate at a 2 : 1 cell input ratio. Supernatants were collected on days 5 and 7 for viral RNA isolation. CD8+ cells were re-isolated at day 7 post infection by MACS techniques and frozen in RNAprotect Cell Reagent (Qiagen) until further use. Macaques were regarded as CNAR(+) when CD8+ cells inhibited viral replication >50 fold (Javed et al., 2015).
Cellular RNA extraction and cDNA synthesis.
For RT-PCR quantification of selected genes, cellular RNA from CD8+ T-cells was isolated by using the RNeasy Mini kit (Qiagen). Quality of RNA was checked by using an Agilent 2100 Bioanalyzer and average RIN (RNA integrity number) values for most samples were greater than 8.5. One hundred nanograms RNA was reverse transcribed using the Quantitect Reverse Transcription kit (Qiagen) and random primers. A no-reverse-transcriptase (NRT) reaction was included to ensure that there was no genomic DNA contamination. Total cellular RNA was isolated from whole blood stored in PAXgene Blood RNA tubes (BD Diagnostics) using the PAXgene Blood RNA kit (Qiagen). RNA was reverse transcribed by using the QuantiTect Reverse Transcription kit (Qiagen).
Real-time PCR quantification of cellular genes.
Expression of cellular genes was quantified by RT-PCR in an ABI Prism 7500 cycler. Reactions were performed in 96 well MicroAmp Optical Reaction Plates (Applied Biosystems). Each 25 µl reaction mixture contained 12.5 µl Immomix (Bioline), 6.6 µl 10× ROX (Bioline), 0.5 µl SYBR Green (Lonza), 2 µM each of forward and reverse primers, and 1 µl of 1 : 5 diluted cDNA products. Cycling conditions were as follows: initial denaturation at 95 °C (7 min) followed by 40 cycles at 95 °C (15 s) and 57 °C (33 s). An additional melting/dissociation step was included to monitor amplification specificity. NRT controls were performed. Non-template controls and a cDNA as positive control were included in each PCR run. Primer sequences (Table 1) for validation of results of microarray experiments were designed using NCBI primer3online software (http://www.ncbi.nlm.nih.gov). Discrepancies in gene annotation between NCBI and Ensembl are listed in Table 1 where applicable. Primers for TNFSF13B amplify the longer transcript variant 1 that has been found in humans and other macaque species, but is formally not present in the rhesus macaque genome databases. Amplification efficiency was checked using serially diluted cDNA. Efficiencies between 94 and 98 % were regarded as sufficient for further analysis. Primer sequences for quantification of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH), MX1 and CXCL10/IP-10 were taken from the report of Mussil et al. (2011). RNA levels of GAPDH were used as reference to calculate the relative expression levels of the target genes. All samples were run at least in duplicates. The results were analysed by using Sequence Detection Software (ABI). The relative expression levels of target genes were calculated as ΔCt = mean Ct (target gene) − mean Ct (GAPDH). ΔΔCt was calculated as: ΔCt of respective gene in CNAR(+) animals – ΔCt of respective gene in CNAR(−) animals (Fig. 1) or control cultures (Fig. 4). Fold difference in gene expression was calculated as 2−ΔΔCt. For other comparisons, relative gene expression was determined by using the formula rE=100×2−ΔCt. GraphPad Prism version 5 was used to plot the graphs.
Table 1. Rhesus macaque gene symbols and names, reference IDs and primer sequences.
| Gene symbol | Gene name | NCBI reference sequence/Ensembl ID | Primer sequence (5′ to 3′) |
|---|---|---|---|
| CISH | Cytokine inducible SH2-containing protein | NM_001258075.1 | F: GGATGTGGTCAGCCTTGT R: CAGGCAGTGCTGGATCATTA |
| CMA1 | Chymase 1, mast cell | NM_001194633.1 | F: CACAGAATGCAAGCCACACT R: TGTCAGCACAAAGTTCCGTC |
| CST6 | Cystatin E/M | XM_001111664.2 | F: TACAACATGGGCAGCAACAG R: CCATCTCCATCGTCAGGAA |
| DPP4 | Dipeptidyl-peptidase 4, CD26 | NM_001039190.2 | F: CAAATTGAAGCAGCCAGACA R: TCCCAGGACCATTGAGGTTA |
| EPHB6 | EPH receptor B6 | NM_001265762.1 | F: ATGACCAGGCAGAAGACG R: CTCGCACCTGGAAACCATAG |
| FAM26F | Family with sequence similarity 26, member F | XM_001111520.2 | F: TGTTGGGCTGGATCTTGATAG R: GCTCCTGTTCCAAATAGATTTTCC |
| LOC721572/GAL-201 | Uncharacterized LOC721572/(galanin) | XR_091573.1/ENSMMUT00000033055 | F: CCATGCCTGAGAACAATATCA R: GACCGCTCCATGTCTTCT |
| GLUL | Glutamate-ammonia ligase | NM_001266013.2 | F: CCGGATTAGAAACCAAGCAT R: GCAGAAACCCAGAAGTGGTC |
| GSTO1 | Glutathione S-transferase omega 1 | XM_001099994.2 | F: GGCTGGAAGCAATGAGGTTA R: GGCTGAGACTGTGGGATCTT |
| MMP25 | Matrix metallopeptidase 25 | XM_001091146.2/ENSMMUG00000009483 | F: CTCACCCTGAATCCTCACTCA R: AACACCACGAGACAGTCAGCT |
| PON3 | Paraoxonase 3 | XM_001095770.2 | F: TGCAGCCATTAGAACCTGAA R: AAGCCAGCCCACTAGGAAGT |
| PSAT1 | Phosphoserine aminotransferase 1 | XM_001101767.2 | F: CCTGCTTATTTTGCCTTTGC R: TGTGTTCCCATGACTCCAGA |
| RPL13 | 60S ribosomal protein L13-like (LOC700603) | XM_002802597.1 | F: CAAAGCCTTCGCTAGTCTCC R: TTTCAACATCCTGTTCTGCG |
| SLAMF8-201 | SLAM family member 8 | XM_001117299.1/ENSMMUT00000015880 | F: GCAGATCCACACTGCTCAAA F: ATTGGTCTCACGGAAGCACT |
| TNSF13B | Tumour necrosis factor (ligand) superfamily, member 13b, transcript variant 1 | XM_005586225.1 | F: GCGATAAGTGGAGTCAGAGTTTC R: GCAAAAGGCAATGAAGGTTT |
IFN stimulation.
PBMCs from four SIV-infected LTNPs (plasma viral RNA copies <40 ml−1) were purified by density centrifugation using lymphocyte separation medium (PAA Laboratories), washed with PBS and incubated overnight at a concentration of 2.5×106 cells ml−1 in RPMI complete medium at 37 °C. Cells were then plated in duplicate at a density of 2×106 cells m in RPMI complete medium supplemented with IL-2 and rhesus IFN-α2 (5–10 ng ml−1; PBL Assay Science) or recombinant rhesus macaque IFN-γ (100 ng ml−1; R&D Systems) was added. In parallel, unstimulated PBMCs were used as controls. Cells were harvested by centrifugation at different time points and frozen in RNAprotect Cell Reagent (Qiagen). In a second set of experiments, PBMCs were activated by ConA (10 ng ml−1; SERVA Electrophoresis) for 8 h. After washing twice, cells were treated as described above. ΔΔCt was calculated by subtracting ΔCt of the stimulated samples from ΔCt of unstimulated controls. Fold difference in gene expression was calculated as 2−ΔΔCt. Values at the start of experiments were set as 1.
Viral RNA quantification.
Viral RNA was extracted from cell culture supernatant or plasma with the QIAamp Viral RNA Mini kit following the instructions of the manufacturer (Qiagen). Purified SIV RNA was quantified using RT-PCR (QuantiTect Probe RT-PCR kit; Qiagen) and the 7500 Real-Time PCR systems (Applied Biosystems) as described (Negri et al., 2004).
Quantification of IFN in plasma samples.
Plasma IFN-α and IFN-γ were quantified by ELISA using pan-specific antibodies for IFN-α (Mabtech) and rhesus macaque-specific antibodies for IFN-γ (Mabtech). Briefly, plasma was added to high binding microtitre plates (Greiner Bio-One) coated with the respective antibodies. Plasma IFNs were detected through the addition of biotinylated anti-IFN-γ monoclonal antibodies, streptavidin–horseradish peroxidase conjugate and tetramethylbenzidine substrate (Sigma-Aldrich). CXCL10/IP-10 was quantified using a cross-reactive human CXCL10/IP-10 ELISA kit (R&D Systems). Absorbance was measured at 405 nm with a 550 microplate reader (Bio-Rad).
Statistical analysis.
Statistical analysis of differential gene expression of microarray data included ANOVA modelling, non-linear local regression normalization and component-wise t-tests. P values were adjusted for high-dimensional microarray data to control family-wise error rate or false discovery rate. Principal component analysis was applied to search for major differences (R-Bioconductor).
For comparison of two groups, the two-tailed nonparametric Mann Whitney U test was applied. For identification of potential correlation, Spearman's rank calculations were carried out.
Linear correlations were identified using Pearson correlation. The log-rank Mantel Cox test was used to calculate potential differences in the number of exposures required to infect all macaques.
Acknowledgements
We thank the animal caretakers and the veterinarians of the German Primate Center, especially Dr Monika Franz, Dr Katharina Raue, Dr Antonina Klippert and Maria Daskalaki for providing blood samples of monkeys and Dr Nicole Stolte-Leeb for co-supervision of the animal experiments. We also thank Heidi Meyer and Jessica Schlüter for expert technical support.
Footnotes
Two supplementary tables and five supplementary figures are available with the online Supplementary Material.
Supplementary Data
References
- Allen T. M., O'Connor D. H., Jing P., Dzuris J. L., Mothé B. R., Vogel T. U., Dunphy E., Liebl M. E., Emerson C., et al. (2000). Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407386–390. 10.1038/35030124 [DOI] [PubMed] [Google Scholar]
- Barouch D. H., Ghneim K., Bosche W. J., Li Y., Berkemeier B., Hull M., Bhattacharyya S., Cameron M., Liu J., et al. (2016). Rapid inflammasome activation following mucosal SIV infection of rhesus monkeys. Cell 165656–667. 10.1016/j.cell.2016.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier A., Cleret-Buhot A., Zhang Y., Goulet J. P., Monteiro P., Gosselin A., DaFonseca S., Wacleche V. S., Jenabian M. A., et al. (2013). Transcriptional profiling reveals molecular signatures associated with HIV permissiveness in Th1Th17 cells and identifies peroxisome proliferator-activated receptor gamma as an intrinsic negative regulator of viral replication. Retrovirology 10160. 10.1186/1742-4690-10-160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackbourn D. J., Mackewicz C. E., Barker E., Hunt T. K., Herndier B., Haase A. T., Levy J. A.(1996). Suppression of HIV replication by lymphoid tissue CD8+ cells correlates with the clinical state of HIV-infected individuals. Proc Natl Acad Sci U S A 9313125–13130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J., Wallet M. A., Krastins B., Sarracino D., Goodenow M. M.(2010). Proteome bioprofiles distinguish between M1 priming and activation states in human macrophages. J Leukoc Biol 87655–662. 10.1189/jlb.0809570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli J. C., Deeks S. G., Shiboski S., Levy J. A.(2002). Relationship of CD8+ T cell noncytotoxic anti-HIV response to CD4+ T cell number in untreated asymptomatic HIV-infected individuals. Blood 994225–4227. 10.1182/blood-2001-11-0078 [DOI] [PubMed] [Google Scholar]
- Chiba S., Ikushima H., Ueki H., Yanai H., Kimura Y., Hangai S., Nishio J., Negishi H., Tamura T., et al. (2014). Recognition of tumor cells by dectin-1 orchestrates innate immune cells for anti-tumor responses. Elife 3e04177. 10.7554/eLife.04177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielewski S., Olejnik A., Sikorski K., Pelisek J., Błaszczyk K., Aoqui C., Nowicka H., Zernecke A., Heemann U., et al. (2014). STAT1-dependent signal integration between IFNγ and TLR4 in vascular cells reflect pro-atherogenic responses in human atherosclerosis. PLoS One 9e113318. 10.1371/journal.pone.0113318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks S. G., Walker B. D.(2007). Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27406–416. 10.1016/j.immuni.2007.08.010 [DOI] [PubMed] [Google Scholar]
- Defamie V., Cursio R., Le Brigand K., Moreilhon C., Saint-Paul M. C., Laurens M., Crenesse D., Cardinaud B., Auberger P., et al. (2008). Gene expression profiling of human liver transplants identifies an early transcriptional signature associated with initial poor graft function. Am J Transplant 81221–1236. 10.1111/j.1600-6143.2008.02249.x [DOI] [PubMed] [Google Scholar]
- Diaz L. S., Stone M. R., Mackewicz C. E., Levy J. A.(2003). Differential gene expression in CD8+ cells exhibiting noncytotoxic anti-HIV activity. Virology 311400–409. [DOI] [PubMed] [Google Scholar]
- Ebihara T., Azuma M., Oshiumi H., Kasamatsu J., Iwabuchi K., Matsumoto K., Saito H., Taniguchi T., Matsumoto M., Seya T.(2010). Identification of a polyI : C-inducible membrane protein that participates in dendritic cell-mediated natural killer cell activation. J Exp Med 2072675–2687. 10.1084/jem.20091573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H., Tansi F. L., Weihofen W. A., Böttcher C., Hu J., Martinez J., Saenger W., Reutter W.(2012). Molecular mechanism and structural basis of interactions of dipeptidyl peptidase IV with adenosine deaminase and human immunodeficiency virus type-1 transcription transactivator. Eur J Cell Biol 91265–273. 10.1016/j.ejcb.2011.06.001 [DOI] [PubMed] [Google Scholar]
- Ferreyra G. A., Elinoff J. M., Demirkale C. Y., Starost M. F., Buckley M., Munson P. J., Krakauer T., Danner R. L.(2014). Late multiple organ surge in interferon-regulated target genes characterizes staphylococcal enterotoxin B lethality. PLoS One 9e88756. 10.1371/journal.pone.0088756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina A., Hanley T. M., Mandel R., Trahey M., Broder C. C., Viglianti G. A., Ryser H. J.(2002). Inhibitors of protein-disulfide isomerase prevent cleavage of disulfide bonds in receptor-bound glycoprotein 120 and prevent HIV-1 entry. J Biol Chem 27750579–50588. 10.1074/jbc.M204547200 [DOI] [PubMed] [Google Scholar]
- Greco G., Barker E., Levy J. A.(1998). Differences in HIV replication in CD4+ lymphocytes are not related to β-chemokine production. AIDS Res Hum Retroviruses 141407–1411. 10.1089/aid.1998.14.1407 [DOI] [PubMed] [Google Scholar]
- Grimes C. Z., Hwang L. Y., Wei P., Shah D. P., Volcik K. A., Brown E. L.(2013). Differentially regulated gene expression associated with hepatitis C virus clearance. J Gen Virol 94534–542. 10.1099/vir.0.047738-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S., Cimermancic P., Gulbahce N., Johnson J. R., McGovern K. E., Clarke S. C., Shales M., Mercenne G., Pache L., et al. (2012). Global landscape of HIV-human protein complexes. Nature 481365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javed A., Leuchte N., Neumann B., Sopper S., Sauermann U.(2015). Noncytolytic CD8+ cell mediated antiviral response represents a strong element in the immune response of simian immunodeficiency virus-infected long-term non-progressing rhesus macaques. PLoS One 10e0142086. 10.1371/journal.pone.0142086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julià A., Domènech E., Chaparro M., García-Sánchez V., Gomollón F., Panés J., Mañosa M., Barreiro-De Acosta M., Gutiérrez A., et al. (2014). A genome-wide association study identifies a novel locus at 6q22.1 associated with ulcerative colitis. Hum Mol Genet 236927–6934. 10.1093/hmg/ddu398 [DOI] [PubMed] [Google Scholar]
- Kasamatsu J., Azuma M., Oshiumi H., Morioka Y., Okabe M., Ebihara T., Matsumoto M., Seya T.(2014). INAM plays a critical role in IFN-γ production by NK cells interacting with polyinosinic-polycytidylic acid-stimulated accessory cells. J Immunol 1935199–5207. 10.4049/jimmunol.1400924 [DOI] [PubMed] [Google Scholar]
- Katz B. Z., Salimi B., Gadd S. L., Huang C. C., Kabat W. J., Kersey D., McCabe C., Heald-Sargent T., Katz E. D., Yogev R.(2011). Differential gene expression of soluble CD8+ T-cell mediated suppression of HIV replication in three older children. J Med Virol 8324–32. 10.1002/jmv.21933 [DOI] [PubMed] [Google Scholar]
- Killian M. S., Teque F., Walker R. L., Meltzer P. S., Killian J. K.(2013). CD8+ lymphocytes suppress human immunodeficiency virus 1 replication by secreting type I interferons. J Interferon Cytokine Res 33632–645. 10.1089/jir.2012.0067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. J., Romero R., Kim C. J., Tarca A. L., Chhauy S., LaJeunesse C., Lee D. C., Draghici S., Gotsch F., et al. (2009). Villitis of unknown etiology is associated with a distinct pattern of chemokine up-regulation in the feto-maternal and placental compartments: implications for conjoint maternal allograft rejection and maternal anti-fetal graft-versus-host disease. J Immunol 1823919–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. N., Ye C., Villani A. C., Raj T., Li W., Eisenhaure T. M., Imboywa S. H., Chipendo P., Ran F. A., et al. (2014). Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 3431246980. 10.1126/science.1246980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy J. A.(2003). The search for the CD8+ cell anti-HIV factor (CAF). Trends Immunol 24628–632. [DOI] [PubMed] [Google Scholar]
- Lim S. G., Condez A., Lee C. A., Johnson M. A., Elia C., Poulter L. W.(1993). Loss of mucosal CD4 lymphocytes is an early feature of HIV infection. Clin Exp Immunol 92448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Chen S., Lai C., Lai M., Fang H., Dao H., Kang J., Fan J., Guo W., et al. (2016). Suppression of HIV replication by CD8+ regulatory T-cells in elite controllers. Front Immunol 7134. 10.3389/fimmu.2016.00134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackewicz C. E., Ortega H. W., Levy J. A.(1991). CD8+ cell anti-HIV activity correlates with the clinical state of the infected individual. J Clin Invest 871462–1466. 10.1172/JCI115153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackewicz C. E., Craik C. S., Levy J. A.(2003). The CD8+ cell noncytotoxic anti-HIV response can be blocked by protease inhibitors. Proc Natl Acad Sci U S A 1003433–3438. 10.1073/pnas.0630379100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manh T. P., Alexandre Y., Baranek T., Crozat K., Dalod M.(2013). Plasmacytoid, conventional, and monocyte-derived dendritic cells undergo a profound and convergent genetic reprogramming during their maturation. Eur J Immunol 431706–1715. 10.1002/eji.201243106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Mariño B., Foster H., Hao Y., Levy J. A.(2007). Differential gene expression in CD8+ cells from HIV-1-infected subjects showing suppression of HIV replication. Virology 362217–225. 10.1016/j.virol.2006.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumiya M., Harris S. A., Satti I., Stockdale L., Tanner R., O'Shea M. K., Tameris M., Mahomed H., Hatherill M., et al. (2014). Inflammatory and myeloid-associated gene expression before and one day after infant vaccination with MVA85A correlates with induction of a T cell response. BMC Infect Dis 14314. 10.1186/1471-2334-14-314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattapallil J. J., Douek D. C., Hill B., Nishimura Y., Martin M., Roederer M.(2005). Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 4341093–1097. 10.1038/nature03501 [DOI] [PubMed] [Google Scholar]
- Mudd P. A., Watkins D., I(2011). Understanding animal models of elite control: windows on effective immune responses against immunodeficiency viruses. Curr Opin HIV AIDS 6197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhl T., Krawczak M., Ten Haaft P., Hunsmann G., Sauermann U.(2002). MHC class I alleles influence set-point viral load and survival time in simian immunodeficiency virus-infected rhesus monkeys. J Immunol 1693438–3446. [DOI] [PubMed] [Google Scholar]
- Mussil B., Sauermann U., Motzkus D., Stahl-Hennig C., Sopper S.(2011). Increased APOBEC3G and APOBEC3F expression is associated with low viral load and prolonged survival in simian immunodeficiency virus infected rhesus monkeys. Retrovirology 877. 10.1186/1742-4690-8-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri D. R., Baroncelli S., Catone S., Comini A., Michelini Z., Maggiorella M. T., Sernicola L., Crostarosa F., Belli R., et al. (2004). Protective efficacy of a multicomponent vector vaccine in cynomolgus monkeys after intrarectal simian immunodeficiency virus challenge. J Gen Virol 851191–1201. 10.1099/vir.0.79794-0 [DOI] [PubMed] [Google Scholar]
- O'Connor S. L., Becker E. A., Weinfurter J. T., Chin E. N., Budde M. L., Gostick E., Correll M., Gleicher M., Hughes A. L., et al. (2012). Conditional CD8+ T cell escape during acute simian immunodeficiency virus infection. J Virol 86605–609. 10.1128/JVI.05511-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochieng W., Sauermann U., Schulte R., Suh Y. S., Kim K. S., Sung Y. C., Hunsmann G., Stahl-Hennig C., Sopper S.(2009). Susceptibility to simian immunodeficiency virus ex vivo predicts outcome of a prime-boost vaccine after SIVmac239 challenge. J Acquir Immune Defic Syndr 52162–169. 10.1097/QAI.0b013e3181b22f4a [DOI] [PubMed] [Google Scholar]
- Pankla R., Buddhisa S., Berry M., Blankenship D. M., Bancroft G. J., Banchereau J., Lertmemongkolchai G., Chaussabel D.(2009). Genomic transcriptional profiling identifies a candidate blood biomarker signature for the diagnosis of septicemic melioidosis. Genome Biol 10R127. 10.1186/gb-2009-10-11-r127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poropatich K., Sullivan D. J.(2011). Human immunodeficiency virus type 1 long-term non-progressors: the viral, genetic and immunological basis for disease non-progression. J Gen Virol 92247–268. [DOI] [PubMed] [Google Scholar]
- Reiser K., Francois K. O., Schols D., Bergman T., Jornvall H., Balzarini J., Karlsson A., Lundberg M.(2012). Thioredoxin-1 and protein disulfide isomerase catalyze the reduction of similar disulfides in HIV gp120. Int J Biochem Cell Biol 44556–562. 10.1016/j.biocel.2011.12.015 [DOI] [PubMed] [Google Scholar]
- Rotger M., Dalmau J., Rauch A., McLaren P., Bosinger S. E., Martinez R., Sandler N. G., Roque A., Liebner J., et al. (2011). Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J Clin Invest 1212391–2400. 10.1172/JCI45235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauermann U., Siddiqui R., Suh Y. S., Platzer M., Leuchte N., Meyer H., Mätz-Rensing K., Stoiber H., Nürnberg P., et al. (2008). MHC class I haplotypes associated with survival time in simian immunodeficiency virus (SIV)-infected rhesus macaques. Genes Immun 969–80. 10.1038/sj.gene.6364448 [DOI] [PubMed] [Google Scholar]
- Schneider T., Jahn H. U., Schmidt W., Riecken E. O., Zeitz M., Ullrich R.(1995). Loss of CD4 T lymphocytes in patients infected with human immunodeficiency virus type 1 is more pronounced in the duodenal mucosa than in the peripheral blood. Berlin Diarrhea/Wasting Syndrome Study Group. Gut 37524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahzad K., Cadeiras M., Memon S., Zeeberg B., Klingler T., Sinha A., Tabak E. G., Unniachan S., Deng M. C.(2010). Gene expression signatures of peripheral blood mononuclear cells during the early post-transplant period in patients developing cardiac allograft vasculopathy. J Transplant 2010719696. 10.1155/2010/719696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard H. W., Lang W., Ascher M. S., Vittinghoff E., Winkelstein W.(1993). The characterization of non-progressors: long-term HIV-1 infection with stable CD4+ T-cell levels. AIDS 71159–1166. [PubMed] [Google Scholar]
- Songok E. M., Osero B., McKinnon L., Rono M. K., Apidi W., Matey E. J., Meyers A. F., Luo M., Kimani J., et al. (2010). CD26/dipeptidyl peptidase IV (CD26/DPPIV) is highly expressed in peripheral blood of HIV-1 exposed uninfected female sex workers. Virol J 7343. 10.1186/1743-422X-7-343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stantchev T. S., Paciga M., Lankford C. R., Schwartzkopff F., Broder C. C., Clouse K. A.(2012). Cell-type specific requirements for thiol/disulfide exchange during HIV-1 entry and infection. Retrovirology 997. 10.1186/1742-4690-9-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranford S. A., Skurnick J., Louria D., Osmond D., Chang S. Y., Sninsky J., Ferrari G., Weinhold K., Lindquist C., Levy J. A.(1999). Lack of infection in HIV-exposed individuals is associated with a strong CD8+ cell noncytotoxic anti-HIV response. Proc Natl Acad Sci U S A 961030–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenbusch M., Ignatius R., Temchura V., Nabi G., Tippler B., Stewart-Jones G., Salazar A. M., Sauermann Ü., Stahl-Hennig C., Uberla K.(2012). Risk of immunodeficiency virus infection may increase with vaccine-induced immune response. J Virol 8610533–10539. 10.1128/JVI.00796-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa-Montoya F., Louahed J., Dizier B., Gruselle O., Spiessens B., Lehmann F. F., Suciu S., Kruit W. H., Eggermont A. M., et al. (2013). Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J Clin Oncol 312388–2395. [DOI] [PubMed] [Google Scholar]
- Vella C., Daniels R. S.(2003). CD8+ T-cell-mediated non-cytolytic suppression of human immuno-deficiency viruses. Curr Drug Targets Infect Disord 397–113. 10.2174/1568005033481196 [DOI] [PubMed] [Google Scholar]
- Walker C. M., Moody D. J., Stites D. P., Levy J. A.(1986). CD8+ lymphocytes can control HIV infection in vitro by suppressing virus replication. Science 2341563–1566. [DOI] [PubMed] [Google Scholar]
- Walker C. M., Erickson A. L., Hsueh F. C., Levy J. A.(1991). Inhibition of human immunodeficiency virus replication in acutely infected CD4+ cells by CD8+ cells involves a noncytotoxic mechanism. J Virol 655921–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcher A. A., Boedigheimer M., Kivitz A. J., Amoura Z., Buyon J., Rudinskaya A., Latinis K., Chiu K., Oliner K. S., et al. (2015). Blockade of interferon-γ normalizes interferon-regulated gene expresson and serum CXCL10 levels in patients with systemic lupus erythematosus. Arthritis Rheumatol 672713–2722. 10.1002/art.39248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiviott L. D., Walker C. M., Levy J. A.(1990). CD8+ lymphocytes suppress HIV production by autologous CD4+ cells without eliminating the infected cells from culture. Cell Immunol 128628–634. [DOI] [PubMed] [Google Scholar]
- Yuan L., Ma L., Fan X., Feng Y., Peng H., Zhao Q., Ruan Y., Levy J. A., Shao Y.(2009). CD8+ cell noncytotoxic antiviral response in long-term HIV-1 infected former blood donors in China. Curr HIV Res 7437–440. [DOI] [PubMed] [Google Scholar]
- Zhang S., Kim C. C., Batra S., McKerrow J. H., Loke P.(2010). Delineation of diverse macrophage activation programs in response to intracellular parasites and cytokines. PLoS Negl Trop Dis 4e648. 10.1371/journal.pntd.0000648 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.