

Abstract
Background
Targeting overlapping behavioral phenotypes in neurogenetic disorders can help elucidate gene-behavior relationships. Fragile X syndrome (FXS) and autism spectrum disorder (ASD) have been studied as a model for this approach, and important areas of phenotypic overlap and divergence have been documented. However, few studies have examined how the manifestation of ASD-related phenotypes in FXS may change over development, a question which has important implications for conceptualizing shared etiologies of these disorders and their constituent phenotypes. The goal of this study was to characterize ASD phenotypes in boys and girls with FXS across development, as well as to compare individual component phenotypes among boys with FXS and boys with idiopathic ASD (ASD-O) over time.
Methods
Sixty-five boys and girls with FXS and 19 boys with ASD-O completed a battery of diagnostic, cognitive, and language assessments at two time points (mean 2.5 years apart). Nonparametric tests assessed changes in diagnostic classification in FXS over time, and hierarchical linear modeling and repeated measures assessed changes in individual ASD symptoms in FXS over time. Additionally, ANCOVAs compared ASD symptom severity and component phenotypes in boys with FXS-O, FXS-ASD, and ASD-O at both time points.
Results
Overall, ASD symptom manifestation for children with FXS significantly increased over time, and developmental predictors varied based on the domain of symptoms assessed. The greatest degree of overlap was observed between boys with FXS-ASD and ASD-O in the domain of reciprocal social communication across time points, whereas boys with ASD-O demonstrated greater impairment in restricted and repetitive behaviors at the later time point.
Conclusions
ASD symptoms increased in FXS with age, and social language impairment emerged as a potential core shared feature of FXS and ASD that may help elucidate underlying molecular genetic variation related to phenotypic variance, and aid intervention planning for subgroups of children showing distinct phenotypes. Results highlight the value of a developmental perspective, and longitudinal data in particular, in evaluating shared behavioral phenotypes across genetic conditions, lending insight into underlying cognitive, neural, and genetic mechanisms associated with key developmental phenotypes in ASD and FXS.
Keywords: Autism spectrum disorder, Fragile X syndrome, Endophenotype, Longitudinal, Social behavior, Language, Pragmatics, FMR1 gene
Background
Since the development of psychiatric nosology, it has been recognized that clinical symptoms are often shared across categorically defined disorders. Consistent with clinical observations, multiple genome-wide analysis studies (GWAS) have produced molecular evidence of shared genetic risk factors in several psychiatric disorders [35, 50]. Clearly defining phenotypes cutting across diagnostic boundaries may help to clarify relationships between behavioral symptoms and underlying biology with long-term implications for individualized treatment; this approach has been proposed as a primary direction for the future of psychiatric research [29]. A critical consideration in such efforts is how psychiatric phenotypes may manifest differently across development. Longitudinal research on disorders such as attention deficit-hyperactivity disorder [52], Down syndrome [44], and autism spectrum disorder (ASD; [14, 62]) demonstrate changes in clinical symptoms and underlying neurobiology across the lifespan, underscoring the importance of examining development when characterizing symptomatology within and across different psychiatric disorders. Yet, few studies have adopted a developmental perspective when examining shared genetic liability across conditions. This paper examines developmental changes in diagnostic classification, symptom expression, and related abilities in fragile X syndrome (FXS) and ASD, two complex neurodevelopmental disorders with considerable phenotypic overlap. At both behavioral and neurobiological levels, FXS and ASD are characterized by atypical developmental trajectories. Therefore, charting the dual paths of phenotypic development in each disorder can help to identify markers of shared etiology throughout the lifespan, with direct clinical, methodological, and theoretical implications.
ASD is characterized by social and communicative impairments and restricted and repetitive behaviors or interests [2]. Hundreds of copy-number variations (CNVs), de-novo mutations, and individual loci conferring elevated risk have been implicated in ASD [56]. Monogenic disorders account for up to 20% of diagnosed cases of ASD, the most common of which is FXS [63]. FXS is caused by a Cytosine-Guanine-Guanine (CGG) repeat expansion of over 200 in the promoter region of the fragile X mental retardation 1 (FMR1) gene on the X chromosome. This expansion leads to methylation of the promoter and effective silencing of the FMR1 gene, resulting in significant reduction or loss of fragile X mental retardation protein (FMRP), an essential protein for brain development and, in particular, the regulation of synaptic function (see [4], for review). Individuals with FXS present with heterogeneous cognitive, language, social, and behavioral deficits [1, 4, 6]. Additionally, over 90% of individuals with FXS display some ASD symptoms [28].
This considerable phenotypic overlap, together with recognition that ASD appears to arise from heterogeneous molecular genetic causes, has prompted investigations of FXS (along with other monogenic conditions overlapping with ASD) as a paradigm for understanding gene-brain-behavior relationships relevant to ASD symptomatology in a simplified genetic context [7, 22, 54]. FMRP is an inhibitor of dendritic translation, involved in the regulation of synaptic development, plasticity, and activity, making it a good candidate for involvement in symptoms associated with ASD given evidence implicating synaptic disruptions in ASD [45, 55]. Specifically, FMRP suppresses group 1 metabotropic (mGluR1, mGluR5) glutamate receptor-regulated translation, thereby regulating numerous genes implicated in ASD (i.e., NLGN3, NRXN, SHANK3, PTEN, TCS2, and NF1; [5, 15, 59]). Of note, inhibition of mGluR5 in the FXS mouse model has been shown to normalize numerous abnormal synaptic, signaling, electrophysiological, and behavioral phenotypes that overlap with ASD [45]. Finally, reduced levels of FMRP in FXS have also been shown to disrupt signaling pathways of neurotransmitters implicated in ASD, such as dopaminergic receptors [43], GABA [12], and 5-HT [30].
Detailed characterization of shared phenotypes in these disorders is necessary to identify specific behaviors that may map more directly onto common underlying biology (i.e., endophenotypes). Family studies of ASD indicate that endophenotypes segregate independently in unaffected relatives, possibly reflecting unique genetic contributions [38, 47]. Therefore, characterizing ASD phenotypes in the context of a monogenic disorder such as FXS offers a unique opportunity to identify endophenotypes related to known genetic variation. Social deficits represent the most consistent area of overlap between idiopathic ASD and individuals with FXS with comorbid ASD (i.e., FXS-ASD) in both severity and quality [8, 31–33, 53]. For instance, Klusek et al. [33] found that boys with idiopathic ASD and FXS-ASD demonstrated highly similar pragmatic (i.e., social) language profiles that were not accounted for by overall cognitive impairment. Further, it has been reported that FMR1 premutation carriers (i.e., CGG repeat length between 55 and 200) may evidence higher rates of ASD [3, 18, 19, 57], though population-based studies are still needed, and clinically unaffected carriers also demonstrate subtle pragmatic difficulties similar to those observed among unaffected relatives of individuals with ASD [40]. Therefore, social language may be a promising candidate ASD endophenotype connected with FMR1-related variation.
Not all ASD phenotypes express similarly among individuals with FXS, and understanding such differences is equally informative in determining common pathways between the two disorders. For instance, within the domain of restricted and repetitive behaviors (RRBs), careful examination of the types of behaviors demonstrated across groups (rather than overall frequency) indicated similar rates of lower level motoric RRBs in FXS and idiopathic ASD but fewer higher order compulsive and repetitive behaviors in individuals with FXS meeting criteria for ASD [61]. A mix of similarities and differences has also been reported in the biophysiological and neuroanatomical profiles of ASD and FXS [27, 41, 46, 62]. Mapping areas of convergence and divergence in FXS and ASD is therefore essential for deconstructing the heterogeneity of ASD, identifying those phenotypes that may relate to FMR1, and guiding clinical interventions for both disorders.
Adopting a developmental perspective in such efforts is critical for accounting for phenotypic changes that occur with children’s growth and maturation. Although pronounced changes are known to occur in such core ASD symptom domains as social communication over time, few studies have examined the role of development in studies of ASD symptom manifestation in FXS, despite the fact that studies conducted at a single time point have included considerable age ranges (5–60 years; [34]). Existing findings from cross-sectional work suggests variation in the expression of phenotypes associated with ASD in children with FXS at different ages and cognitive abilities [25, 34, 42, 53, 58], whereas longitudinal approaches using questionnaires or behavioral ratings suggest more stability of ASD symptoms in FXS [13, 26, 51]. In the one longitudinal study using a gold-standard clinical measure of ASD symptoms, Hernandez et al. [28] found that approximately one third of their FXS sample demonstrated inconsistent ASD classification over time based on the administration of the Autism Diagnostic Interview-Revised (ADI-R; [37]), a comprehensive parent-interview, although this change was not statistically significant. Therefore, further direct-assessment evaluations of ASD phenotypes in FXS are needed to evaluate key phenotypes that overlap and to identify specific developmental factors that impact the manifestation of ASD symptoms in FXS, in order to advance clinical, methodological, and theoretical approaches.
This study examined the developmental manifestation of ASD phenotypes in FXS, drawing from comprehensive longitudinal assessments of children with FXS and idiopathic ASD (i.e., ASD-O) using gold-standard ASD diagnostic measures, and standardized assessments of cognition, structural language (i.e., vocabulary), and pragmatic language. We aimed to (1) characterize trajectories of ASD symptoms in FXS and predictors of ASD symptoms over time and (2) compare the type and severity of symptoms and individual behaviors observed in boys with FXS to a group of boys with ASD-O.
Methods
Participants
Participants included 65 children with FXS (31 male, 34 female) and 19 boys with ASD-O. Because FXS is a rare disorder, 12 pairs of siblings were included in analyses to maximize sample size (see Table 1 for participant characteristics). All participants spoke English as their first language and were screened for use of at least three-word phrases. Inclusion criteria for boys with ASD-O included a previous clinical diagnosis confirmed through direct assessment with the Autism Diagnostic Observation Schedule (ADOS; [36]) and/or the ADI-R [37], and no known ASD-related monogenic disorders.
Table 1.
Overall sample characteristics at time 1 and time 2
| Time one | Group | n | Chronological age M (SD) |
Nonverbal mental age M (SD) |
EVT age equivalent M (SD) |
PPVT age equivalent M (SD) |
ADOS module distribution |
| FXS-girls | 34 | 8.96 (3.39) | 6.18 (1.72) a | 6.95 (2.67) a | 7.40 (3.06) a | 14 M2, 20 M3 | |
| FXS-O | 24 | 8.50 (3.37) | 6.51 (1.87) | 7.32 (2.75) | 7.90 (3.12) | 11 M2, 13 M3 | |
| FXS-ASD | 10 | 9.11 (3.14) | 5.39 (.99) | 6.05 (2.40) | 6.98 (3.81) | 3 M2, 7 M3 | |
| FXS-boys | 31 | 8.97 (2.51) | 4.77 (.69) b | 4.69 (1.23) b | 5.46 (1.53) b | 2 M1, 18 M2, 11 M3 | |
| FXS-O | 14 | 8.47 (2.58) | 4.79 (.75) | 4.51 (2.57) | 5.19 (1.43) | 1 M1, 8 M2, 5 M3 | |
| FXS-ASD | 17 | 9.38 (2.45) | 4.75 (.66) | 4.84 (1.21) | 5.67 (1.62) | 1 M1, 10 M2, 6 M3 | |
| ASD (boys) | 19 | 9.08 (2.31) | 5.82 (1.43) b | 5.79 (1.62) b | 5.85 (1.71) b | 2 M2, 11 M3 | |
| Time two | Group | n | Chronological age M (SD) |
Nonverbal mental age M (SD) |
EVT age equivalent M (SD) |
PPVT age equivalent M (SD) |
ADOS module distribution |
| FXS-girls | 34 | 11.21 (3.31) | 7.31 (3.04) a | 8.50 (2.95) a | 9.48 (3.55) a | 2 M2, 31 M3, 1 M4 | |
| FXS-O | 20 | 11.98 (3.44) | 8.12 (3.60) | 9.61 (2.84) | 10.88 (3.49) | 20 M3 | |
| FXS-ASD | 13 | 10.12 (2.88) | 6.06 (1.18) | 6.88 (2.35) | 7.33 (2.46) | 2 M2, 11 M3, 1 M4 | |
| FXS-boys | 31 | 11.50 | 5.01(.50) b | 5.36(1.27) b | 6.15(1.51) b | 5 M2, 26 M3 | |
| FXS-O | 6 | 10.78 (1.65) | 5.17 (.73) | 6.06 (1.50) | 6.71(.64) | 6 M3 | |
| FXS-ASD | 25 | 11.67 (2.5) | 4.98 (.44) | 5.19 (1.18) | 6.02 (1.64) | 5 M2, 20 M3 | |
| ASD (boys) | 19 | 11.38 (2.63) | 6.93(1.84) b | 6.79 (2.14) b | 7.68(2.11) b | 2 M2, 17 M3 |
Differing superscripts indicate groups which significantly differed overall (p < .05). “M” represents ADOS module type. Italicized data indicate overall means by sex. FXS-O and FXS-ASD group classification changed over time based on behaviors observed during the ADOS. Five participants with FXS had missing covariate data
Procedures
At two time points (mean 2.5 years apart, range 1.15–3.90 years) participants completed a battery of cognitive, language, and ASD diagnostic measures. Because this sample was selected to assess stability over time, only participants with greater than one time point were included in the analyses. Participants were recruited through advertisements at genetic clinics and physicians’ offices, advocacy groups, and participant registries. All participants provided informed consent and the University of North Carolina at Chapel Hill (IRB #07-0044) and Northwestern University Institutional Review Boards (IRB # STU00039816) approved these procedures.
Measures
ASD classification
Participants were administered the ADOS and the ADI-R. The ADOS is a standardized, semi-structured assessment that includes a range of activities designed to elicit social interaction and consists of four different modules that account for differences in developmental level and language abilities. The ADOS determined ASD classification for children with FXS and confirmed ASD classification for boys with ASD-O. Classification was based on revised algorithms of the ADOS, and continuous symptom severity scores were also determined [20, 21]. Consistent with DSM-5 [2], participants with FXS that met either “spectrum” or “autism” criteria on the ADOS were classified as FXS-ASD. The ADOS consists of a range of individual items rated on a scale of 0–3 (absent–severe). In order to compare performance on items across modules, codes that were identical or tapping the same symptoms across modules were identified and used in item-level analyses. For these analyses, items rated an 8 or 9 (i.e., a code was not applicable or there was an administration error) were reduced to 0, consistent with algorithm coding. The ADI-R is a parent interview that assesses early developmental features and provides an algorithm to determine ASD classification. The ADI-R was administered to confirm ASD in participants with ASD-O and to examine classification agreement with the ADOS in individuals with FXS over time.
Cognitive and language abilities
The Leiter International Performance Scale-Revised [49] provided an estimate of nonverbal mental age. The Expressive Vocabulary Test (EVT; [60]) and Peabody Picture Vocabulary Test-3rd or 4th edition (PPVT; [16, 17]) were used to assess expressive and receptive language. To assess pragmatic language, participants completed the Pragmatic Judgment subtest of the Comprehensive Assessment of Spoken Language (CASL; [9]), which evaluates awareness of appropriate language use in various social situations (e.g., how to greet an unfamiliar adult or how to give an appropriate compliment).
Analysis plan
The first aim of this study was to characterize trajectories of ASD symptoms in FXS and predictors of ASD symptoms over time. McNemar’s test of classification assessed whether rates of ASD and agreement between ADOS and ADI-R changed over time. Nonparametric Wilcoxon signed-rank tests were used to assess changes in individual ADOS items across time points. These analyses were run separately by sex and also within the group of boys and girls with FXS who demonstrated a change in ASD classification on the ADOS. Finally, hierarchical linear models, nesting age within participant, were completed to assess the statistical effect of chronological age, sex, mental age, receptive and expressive vocabulary, and pragmatic judgment age equivalence on measures of symptom severity derived from the ADOS across time points. To reduce collinearity among predictors, all predictor variables in these models were mean-centered and expressive and receptive vocabulary measures were combined into a composite by summing the raw score on each measure.
The second aim of this study was to compare the severity and type of ASD symptoms in boys with FXS to a group of boys with ASD-O at each time point. ANCOVAs, controlling for mental age and receptive and expressive vocabulary age equivalence, followed by planned comparisons, compared ASD severity scores as well as individual items at both time points for boys classified as FXS-O, FXS-ASD, and ASD-O. Although item-level scores are ordinal in nature, this parametric statistical approach was chosen in order to include mental age and vocabulary as covariates. Thus, results of item-level comparisons should be interpreted cautiously.
Results
Aim one: characterize trajectories of ASD symptoms in FXS and predictors of ASD symptoms over time
Overall, 41.7% of children with FXS met criteria for ASD at time one (54.8% of boys, 41.5% of girls); at time two, 60% of the sample met criteria (80.6% of boys, 41.2% of girls), a statistically significant change (p = .008), driven by the change in classification in boys with FXS (p = .021). There was a nearly significant increase in ADI-R and ADOS agreement over time (p = .065), driven by a significant increase in agreement for boys with FXS (45.2 to 75%, p = .039), whereas agreement for girls with FXS decreased slightly (73.5 to 64.7%, p = .45).
Repeated measure analyses were then conducted for individual items on the ADOS. Boys with FXS overall demonstrated increased impairments in the area of shared enjoyment (Z = −2.57, p = .010) and social overtures (Z = −2.71, p = .007), and girls with FXS demonstrated an increase in impairments in prosodic features of speech (Z = −2.42, p = .016), facial expressions (Z = −2.04, p = .041), and social overtures (Z = −2.71, p = .05). No other significant changes were noted on any other item in these groups (Zs > −1.90, ps > .05). Item level analyses were replicated within the group of participants who did not meet ASD criteria at time one but did at time two in order to assess symptoms that worsened with time. Within this group, several algorithm items (i.e., items that would contribute to a classification change) increased in impairment: conversation (Z = −2.1, p = .035), facial expressions (Z = −2.31, p = .021), social overtures (Z = −2.95, p = .003), social response (Z = −2.83, p = .005), and quality of rapport (Z = −2.11, p = .035). In addition, impairment on a non-algorithm item, atypical qualities of speech (Z = −1.98, p = .048) also increased. No other significant changes were noted (Zs > −1.83, ps > .068).
Hierarchical linear models revealed a significant main effect of age, in that greater age was associated with greater symptom severity for all measures of severity (see Table 2). Additionally, pragmatic competence was marginally associated with reduced overall severity and severity of social affect (i.e., for every year increase in pragmatic language age equivalence, predicted overall symptom severity decreased by .34 and severity of social affect decreased by .32). In contrast, for restricted and repetitive behaviors, mental age significantly predicted decreases in severity (i.e., for every year increase in age equivalence, severity decreased by .38).
Table 2.
Main effects of hierarchical linear models
| Test | |
|---|---|
| Overall severity | |
| Chronological age | F(1,88.41) = 17.31, p < .001 |
| Sex | F(1,64.60) = .00, p = .98 |
| Chronological age*sex | F(1,104.92) = .404, p = .53 |
| Mental age | F(1,102.26) = 1.59, p = .211 |
| Receptive and expressive vocabulary | F(1,109.50) = .142, p = .71 |
| Pragmatic language | F(1,114.85) = 3.81, p = .054 |
| Severity of social affect | |
| Chronological age | F(1,85.7) = 17.03, p < .001 |
| Sex | F(1,64.21) = .07, p = .80 |
| Chronological age*sex | F(1,101.32) = .43, p = .51 |
| Mental age | F(1,98.02) = .49, p = .49 |
| Receptive and expressive vocabulary | F(1,106.74) = .35, p = .56 |
| Pragmatic language | F(1,115.88) = 3.45, p = .07 |
| Severity of restricted and repetitive behavior | |
| Chronological age | F(1,83.67) = 6.36, p = .014 |
| Sex | F(1,66.56) = .019, p = .87 |
| Chronological age*sex | F(1,96.37) = 1.65, p = .20 |
| Mental age | F(1,92.45) = 4.78, p = .03 |
| Receptive and expressive vocabulary | F(1,102.35) = .02, p = .89 |
| Pragmatic language | F(1,115.17) = 3.06, p = .08 |
* represents the interaction between chronological age and sex
Aim two: comparisons of symptom severity and type of ASD symptoms in boys with FXS and ASD-O over time
At time one, overall ASD symptom severity and severity of social impairment and RRBs in boys with FXS-ASD were nearly indistinguishable from boys with ASD-O (ps > .50; see Figure 1), and significantly greater than boys with FXS-O (F(2,43) = 33.57, p < .001; F(2,43) = 23.81, p < .001; F(2,43) = 7.36, p = .002). At time two, a stepwise pattern was observed in which boys with ASD-O demonstrated significantly greater overall severity (driven by more severe RRBs) than boys with FXS-ASD, who in turn displayed greater severity than boys with FXS-O (F(2,44) = 30.55, p < .001; F(2,44) = 6.86, p = .003). Boys with ASD-O did not differ from boys with FXS-ASD on severity of social impairment at time two (p = .26), and both were significantly greater than boys with FXS-O (F(2,44) = 17.98, p < .001).
Fig. 1.
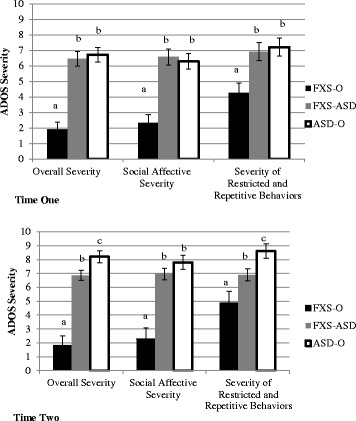
Comparison of ASD symptom severity of boys with FXS-O, FXS-ASD, and ASD-O at each time point. Notes: ADOS severity derived from Gotham et al. [21]. Differing letters convey significant differences at the level of p < .05. Means are adjusted for nonverbal mental age, expressive vocabulary, and receptive vocabulary
Figures 2 and 3 demonstrate profiles of performance on individual ADOS items across boys with FXS-O, FXS-ASD, and ASD-O. At time one, groups did not differ in items tapping prosodic features of speech, shared enjoyment, imagination, sensory seeking behaviors, repetitive motor movements, self-injury, overactivity, negative behavior, or anxiety (F(2,41) < 2, ps > .05). However, several significant group differences emerged (overall model F(2,41) > 3.7, ps < .05). Boys with ASD-O and FXS-ASD demonstrated overlap in conversation, eye contact, quality of social overtures and responses, amount of reciprocal communication, and overall quality of rapport (ps > .05), and significantly greater impairment than boys with FXS-O across these domains (ps < .05). Boys with ASD-O demonstrated greater impairments in restricted interests, gestures, and echolalia relative to both FXS-ASD and FXS-O groups (ps < .05), who did not differ from one another (ps > .07), and boys with ASD also demonstrated greater impairments in facial expressions and stereotyped speech than boys with FXS-O only (ps < .03).
Fig. 2.
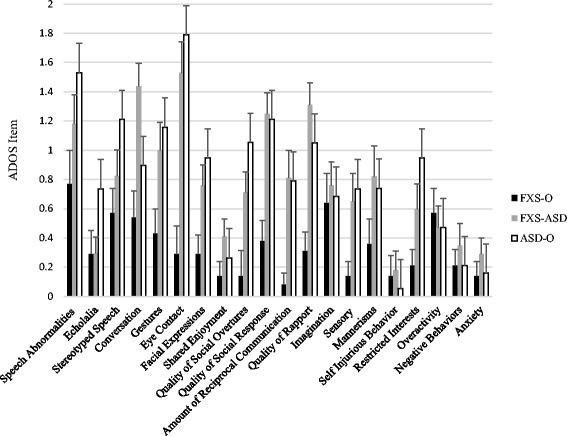
Profile of ASD phenotypic expression on ADOS items at time one. Notes: Significant differences are reported in text. After accounting for mental age, and receptive and expressive vocabulary, males with FXS-ASD and ASD-O demonstrated overlap on several items tapping social communication, whereas individuals with ASD-O demonstrated greater impairment in restricted interests, gestures and echolalia
Fig. 3.
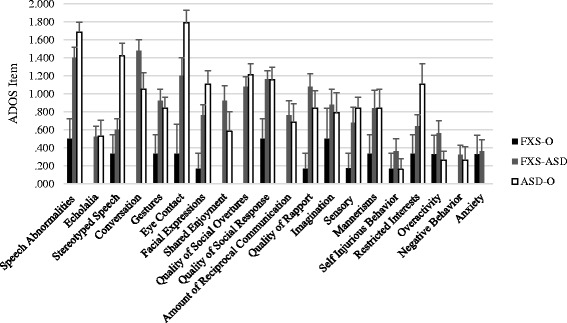
Profile of ASD phenotypic expression on ADOS items at time two. Notes: Significant differences are reported in text. After accounting for mental age, and receptive and expressive vocabulary, males with FXS-ASD and ASD-O demonstrated overlap on several items tapping social communication, whereas individuals with ASD-O demonstrated greater impairment in restricted interests
At the second time point, groups did not differ on items tapping echolalia, gestures, shared enjoyment, imagination, sensory seeking behaviors, repetitive motor movements, self-injurious behavior, overactivity, negativity, or anxiety (F(2,44) < 2.9, ps > .06); group differences emerged on the remaining items (overall F(2,44) > 3.3, ps < .05). Boys with FXS-ASD and ASD-O did not differ from each other (ps > .06), and both demonstrated greater impairment than boys with FXS-O in the domains of prosodic features of speech, conversation, eye contact, quality of social response, amount of reciprocal social interactions, and overall quality of rapport (ps < .05). A stepwise pattern was observed for facial expressions and social overtures in which boys with FXS-O demonstrated less impairment than boys with FXS-ASD, who in turn demonstrated less impairment than boys with ASD-O (ps < .05). Finally, boys with ASD-O demonstrated greater impairments in stereotyped speech and restricted interests than boys with FXS-ASD and FXS-O (ps < .01).
Discussion
This study applied a longitudinal lens to characterize ASD symptom profiles in the context of FXS and identify potential overlapping endophenotypes in ASD and FXS across development. Findings suggest ASD symptoms worsen with age and that this increase is most prominently observed in social communication skills. Substantial phenotypic overlap, as well as key areas of difference, was observed in boys with ASD-O and FXS-ASD at both time points.
In contrast to prior longitudinal reports (e.g., [26, 28]), ASD symptom manifestation in FXS was variable over time, with both classification and symptom severity significantly increasing over time. This study was the first to use the ADOS to examine changes over time; it may be that direct assessments of behavior better capture phenotypic variation contributing to diagnostic status than parent report, in line with Harris et al. [24] finding that the ADOS classifications were most consistent with DSM-IV diagnoses. Of note, Klusek et al. [34] examined ADOS and ADI-R agreement in a cross-sectional sample overlapping with the current study and thus similar agreement rates are not surprising. Rather, findings from the current study build on this prior work by suggesting that agreement increases with age, particularly for males with FXS. Consistent with prior work showing that ASD is more common in males than females with FXS [11, 23, 34], girls with FXS in the current sample continued to demonstrate reduced rates of ASD classification relative to boys over time, although sex did not impact rates of change over time when controlling for mental age and structural language. Whereas studies should continue to investigate the role of assessment approach and sex in observed changes in symptoms over time, findings demonstrating an increase in ASD symptoms with age have important implications for long-term intervention planning for children with FXS, as well as future research examining overlapping profiles of individuals with ASD and FXS.
The longitudinal approach of this study also allowed for analyses of developmental features related to ASD symptoms in individuals with FXS over time. The roles of nonverbal mental age, structural language (i.e., receptive and expressive vocabulary) abilities, and pragmatic competence in ASD symptom presentation in FXS changed as a function of the domain assessed. For example, when examining the significance of mental age in predicting symptom severity, significant relationships emerged for severity of restricted and repetitive behaviors, consistent with prior findings from cross-sectional work [42, 58] and group comparisons at a single time point (e.g., [23]). However, mental age did not predict severity of social-affective symptoms. Thus, while general cognition remains an important consideration when examining the expressions of ASD phenotypes in FXS, lower mental age does not appear to account for ASD symptoms in FXS entirely. Instead, variation in pragmatic competence was the only significant covariate in the model examining predictors of severity of social affect over time, further suggesting the importance of pragmatic development in observed impairments in FXS.
The language demands of the assessment context may also play a role in observed ASD symptoms in FXS. In fact, whereas at baseline approximately 50% of individuals with FXS received a module 2 of the ADOS (a version that is less linguistically demanding), with development of language over 80% of individuals with FXS received a module 3 at the later time point. That is, assessments administered at time one involved limited expressive language requirements whereas assessments at time two entailed a heavier emphasis on expressive language ability and reciprocal conversation (i.e., ADOS module change). Therefore, the development of language abilities may afford more observable manifestations of ASD-related social and communicative deficits in children with FXS.
Interestingly, in the subgroup of children with FXS who demonstrated a change in classification over time, the phenotypes driving this change were primarily related to social interaction, including conversation, amount of reciprocation, and social responses. Indeed, across time points, social impairments emerged as a core shared phenotype across boys with FXS-ASD and ASD-O, with highly similar levels of social affective severity as well as several items linked to social interactions (e.g., social overtures and responses) observed in both children with FXS-ASD and ASD-O across time points. These findings add to a growing body of work demonstrating that communication [31] and reciprocal social behaviors are shared across FXS-ASD and ASD-O [8, 31, 32], predict later ASD classification [48], and align in particular with literature showing a high degree of pragmatic impairment overlap in FXS-ASD and ASD-O [33, 40] as well as similarities in pragmatic language profiles documented among relatives who are genetic carriers [39]. An important next step will be to continue to investigate mechanisms underlying this overlap across groups; for example, Roberts et al. [48] found that differential HPA axis activation (i.e., a measure of the body’s response to stress) impacted social approach behaviors in individuals with FXS with and without comorbid ASD. Such investigations, targeting specific aspects of social behaviors across development, hold promise for continuing to elucidate ASD endophenotypes related to FMR1 variation.
Some important differences between boys with ASD-O and FXS-ASD also emerged across time points. For example, the FXS-ASD group did not differ in severity of RRBs at time one but did differ at the later time point. Important to note is that this difference was driven by the fact that boys with ASD-O demonstrated significantly greater impairments in restricted interests (a higher order RRB) in particular. This difference may therefore have been influenced by a distinct profile of RRBs in FXS-ASD relative to ASD-O, as reported in prior work [61, 62], and that may be more clearly observed with age. Further, males with ASD-O demonstrated significantly greater impairments than those with FXS-ASD on symptom domains tapped by the ADOS that varied across time points, in that at time one, but not at time two, boys with ASD-O demonstrated greater rates of gestures and echolalia. Such findings affirm that ASD in FXS is not identical to that observed in ASD-O, that age is likely to influence manifestation of this overlap, and that specific target behaviors, such as social responses, may best map onto underlying shared genetic etiology.
Together, results suggest that ASD as it presents in FXS is not identical to idiopathic ASD. Given the clinical and etiologic complexities of each disorder, this is not necessarily surprising, nor should it undermine the utility of studying FXS and other monogenic conditions as models for understanding ASD related phenotypes. In line with recent conceptualizations of psychiatric nosology, and complementary findings from psychiatric genomics, current findings strongly suggest that mapping homogeneous sets of phenotypes cutting across disorders (and ultimately, related underlying neural and molecular mechanisms, and their interplay with environmental influences) is crucial to understanding the pathogenesis of complex psychiatric disorders such as ASD [10, 29]. Findings here suggest that adopting a developmental framework in such comparisons can provide necessary sharpened focus to the analysis of complex phenotypes across disorders. In the case of FXS and ASD, it appears that social-communicative behaviors may be particularly fruitful targets for biological study and clinical intervention, where shared phenotypes can serve as a basis for translating new findings and treatments across disorders to inform clinical and research practice.
Conclusions
Current findings highlight the importance of adopting a developmental perspective when investigating shared behavioral features across disorders. Including assessments across multiple time points allowed for the identification of overlapping and divergent phenotypes across two complex neurodevelopmental disorders and clarification for the role of related abilities in observed symptoms over time, with key implications for understanding etiologic mechanisms related to shared phenotypes across ASD and FXS, and implications for clinical intervention. Incorporating a developmental perspective within a shared phenotype approach will continue to inform the search for genetic etiology of psychiatric disorders, with potential for shaping clinical and research practice.
Acknowledgements
Special thanks to all participating families. We would also like to acknowledge the late Joanne Roberts, PhD, who was awarded the NICHD grants that supported the initial phases of this research. Dr. Roberts had a long and distinguished career, including over 125 publications related to language development across multiple clinical populations. We also acknowledge the Research Participant Registry Core of the Carolina Institute for Developmental Disabilities, Grant Award P30 HD03110, as well as the Frank Porter Graham Child Development Institute.
Funding
Funding support was provided by R01 HD038819, R01 HD044935, R01 MH091131-01A1.
Availability of data and materials
The datasets used for the current study are available from the corresponding author upon reasonable request.
Authors’ contributions
All authors had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors contributed to study concept and design and acquisition, analysis or interpretation of data. Ms. Lee was responsible for drafting of the manuscript and statistical analysis. Drs. ML and GM obtained funding and supervised the completion of the study. Drs. ML, GM, and EB-K provided critical edits. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All participants provided informed consent and the University of North Carolina at Chapel Hill (IRB #07-0044) and Northwestern University (IRB # STU00039816) Institutional Review Boards approved these procedures.
Contributor Information
Michelle Lee, Email: michellelee@u.northwestern.edu.
Gary E. Martin, Email: marting@stjohns.edu
Elizabeth Berry-Kravis, Email: Elizabeth_Berry-Kravis@rush.edu.
Molly Losh, Email: M-losh@northwestern.edu.
References
- 1.Abbeduto L, Chapman R. Language development in Down syndrome and fragile X syndrome: current research and implications for theory and practice. In: Fletcher P, Miller JF, editors. Developmental theory and language disorders. Amsterdam: John Benjamins Publishing Co; 2005. pp. 53–72. [Google Scholar]
- 2.American Psychiatric Association . Diagnostic and statistical manual of mental disorders: DSM-5. 5. Arlington: American Psychiatric Association; 2013. [Google Scholar]
- 3.Aziz M, Stathopulu E, Callias M, Taylor C, Turk J, Oostra B, et al. Clinical features of boys with fragile X premutations and intermediate alleles. Am J Med Genet B Neuropsychiatry Geneticts. 2003;121B(1):119–127. doi: 10.1002/ajmg.b.20030. [DOI] [PubMed] [Google Scholar]
- 4.Bagni C, Oostra BA. Fragile X syndrome: from protein function to therapy. Am J Med Genet. 2013;161A(11):2809–2821. doi: 10.1002/ajmg.a.36241. [DOI] [PubMed] [Google Scholar]
- 5.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27(7):370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Bennetto L, Pennington BF, Hagerman RJ, Cronister AJ. The neuropsychology of fragile X syndrome. In: Hagerman RJ, Cronister A, editors. Fragile X synrome: Diagnosis, treatment and research. Balitmore: Johns Hopkins University Press; 2002. pp. 210–248. [Google Scholar]
- 7.Betancur C, Buxbaum JD. SHANK3 haploinsufficiency: a “common” but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Molecular Autism. 2013;4(1):17. doi: 10.1186/2040-2392-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Budimirovic DB, Bukelis I, Cox C, Gray RM, Tierney E, Kaufmann WE. Autism spectrum disorder in fragile X syndrome: differential contribution of adaptive socialization and social withdrawal. Am J Med Genet A. 2006;9999:1–13. doi: 10.1002/ajmg.a.31405. [DOI] [PubMed] [Google Scholar]
- 9.Carrow-Woolfolk E. CASL: comprehensive assessment of spoken language. Circle Pines, MN: American Guidance Services; 1999. [Google Scholar]
- 10.Clementz BA, Sweeney JA, Hamm JP, Ivleva EI, Ethridge LE, Pearlson GD, et al. Identification of distinct psychosis biotypes using brain-based biomarkers. Am J Psychiatr. 2015. [DOI] [PMC free article] [PubMed]
- 11.Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, Loesch DZ. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. Journal of Autism and Developmental Disorders. 2007;37(4):738–47. doi: 10.1007/s10803-006-0205-z. [DOI] [PubMed] [Google Scholar]
- 12.Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ. GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neuroscience and Biobehavior Review. 2012;36(9):2044–2055. doi: 10.1016/j.neubiorev.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cornish K, Cole V, Longhi E, Karmiloff-Smith A, Scerif G. Does attention constrain developmental trajectories in fragile x syndrome? A 3-year prospective longitudinal study. American Journal of Intellectual and Developmental Disabilities. 2012;117(2):103–120. doi: 10.1352/1944-7558-117.2.103. [DOI] [PubMed] [Google Scholar]
- 14.Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP, et al. Mapping early brain development in autism. Neuron. 2007;56(2):399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 15.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunn LM, Dunn DM. Peabody picture vocabulary test-3rd edition. Circle Pines: American Guidance Service; 1997. [Google Scholar]
- 17.Dunn LM, Dunn DM. Peabody picture vocabulary test-4th edition. Circle Pines: American Guidance Service; 2007. [Google Scholar]
- 18.Farzin F, Perry H, Hessl D, Loesch D, Cohen J, Bacalman S, et al. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27(Supplement 2):S137–144. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- 19.Goodlin-Jones BL, Tassone F, Gane LW, Hagerman RJ. Autistic spectrum disorder and the fragile X premutation. J Dev Behav Pediatr. 2004;25(6):392–398. doi: 10.1097/00004703-200412000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Gotham K, Risi S, Pickles A, Lord C. The autism diagnostic observation schedule: revised algorithms for improved diagnostic validitiy. Journal of Autism and Developmetnal Disorders. 2007;37(4):613–627. doi: 10.1007/s10803-006-0280-1. [DOI] [PubMed] [Google Scholar]
- 21.Gotham K, Pickles A, Lord C. Standardizing ADOS scores for a measure of severity in autism spectrum disorders. J Autism Dev Disord. 2009;39:693–705. doi: 10.1007/s10803-008-0674-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagerman R, Hoem G, Hagerman P. Fragile X and autism: intertwined at the molecular level leading to targeted treatments. Molecular Autism. 2010;1(1):12. doi: 10.1186/2040-2392-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hall SS, Lightbody AA, Hirt M, Rezvani A, Reiss AL. Autism in fragile X syndrome: a category mistake? J Am Acad Child Adolesc Psychiatry. 2010;49(9):921–933. doi: 10.1016/j.jaac.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris SW, Hessl D, Goodlin-Jones B, Ferranti J, Bacalman S, Barbato I, Tassone F, Hagerman PJ, Herman H, Hagerman RJ. Autism profiles of males with fragile x syndrome. American Journal of Mental Retardation. 2008;113(6):427–438. doi: 10.1352/2008.113:427-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hartley SL, Wheeler AC, Mailick MR, Raspa M, Mihaila I, Bishop E, et al. Autism symptoms across adulthood in men with fragile X syndrome: a cross-sectional Analysis. Journal of Autismand Developmental Disorders. 2015;45(11):3668–3679. doi: 10.1007/s10803-015-2513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatton DD, Skinner M, Sideris J, Mankowski JB, Bailey DB, Roberts JE, et al. Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am J Med Genet. 2006;140(17):1804–1813. doi: 10.1002/ajmg.a.31286. [DOI] [PubMed] [Google Scholar]
- 27.Hazlett HC, Poe MD, Lightbody AA, Gerig G, Macfall JR, Ross AK, et al. Teasing apart the heterogeneity of autism: Same behavior, different brains in toddlers with fragile X syndrome and autism. Journal of Neurodevelopmental Disorders. 2009;1(1):81–90. doi: 10.1007/s11689-009-9009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hernandez RN, Feinberg RL, Vaurio R, Passanante NM, Thompson RE, Kaufmann WE. Autism spectrum disorder in fragile X syndrome: a longitudinal evaluation. Am J Hum Genet. 2009;149A(6):1125–1137. doi: 10.1002/ajmg.a.32848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Insel TR. The NIMH Research Domain Criteria (RDoC) project: precision medicine for psychiatry. Am J Psychiatry. 2014;171(4):395–397. doi: 10.1176/appi.ajp.2014.14020138. [DOI] [PubMed] [Google Scholar]
- 30.Janusonis S, Anderson GM, Shifrovich I, Rakic P. Ontogeny of brain and blood serotonin levels in 5-HT receptor knockout mice: potential relevance to the neurobiology of autism. J Neurochem. 2006;99(3):1019–1031. doi: 10.1111/j.1471-4159.2006.04150.x. [DOI] [PubMed] [Google Scholar]
- 31.Kau AS, Tierney E, Bukelis I, Stump MH, Kater WR, Trescher WH, et al. Social behavior profile in young boys with fragile X syndrome: characteristics and specificity. Am J Med Genet. 2004;126A(1):9–17. doi: 10.1002/ajmg.a.20218. [DOI] [PubMed] [Google Scholar]
- 32.Kaufmann WE, Cortell R, Kau AS, Bukelis I, Tierney E, Gray RM, et al. Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors. Am J Med Genet. 2004;129(3):225–234. doi: 10.1002/ajmg.a.30229. [DOI] [PubMed] [Google Scholar]
- 33.Klusek J, Martin GE, Losh M. A comparison of pragmatic language in boys with autism and fragile X syndrome. J Speech Lang Hear Res. 2014a;57(5):1692–1707. [DOI] [PMC free article] [PubMed]
- 34.Klusek J, Martin GE, Losh M. Consistency between research and clinical diagnoses of autism among boys and girls with fragile X syndrome. J Intellect Disabil Res. 2014b;58(10):940–52 [DOI] [PMC free article] [PubMed]
- 35.Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45(9):984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lord C, Rutter M, DeLavore PC, Risi S. Autism diagnostic observation schedule. Los Angeles: Western Psychological Services; 2001. [Google Scholar]
- 37.Lord C, Rutter M, Le Couteur A. Autism diagnostic interview-revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24(5):659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 38.Losh M, Childress D, Lam K, Piven J. Defining key features of the broad autism phenotype: A comparison across parents of multiple- and single-incidence autism families. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2008;147B(4). [DOI] [PMC free article] [PubMed]
- 39.Losh M, Klusek J, Martin GE, Sideris J, Parlier M, Piven J. Defining genetically meaningful language and personality traits in relatives of individuals with fragile X syndrome and relatives of individuals with autism. Am J Med Genet B: Neuropsychiatric Genetics. 2012a;159B(6):660–8. [DOI] [PMC free article] [PubMed]
- 40.Losh M, Martin GE, Klusek J, Hogan-Brown AL, Sideris J. Social communication and theory of mind in boys with autism and fragile x syndrome. Fronteiers in Developmental Psychology. 2012b;3:1–12. [DOI] [PMC free article] [PubMed]
- 41.Mazzocco MM, Kates WR, Baumgardner TL, Freund LS, Reiss AL. Autistic behaviors among girls with fragile X syndrome. J Autism Dev Disord. 1997;27(4):415–435. doi: 10.1023/A:1025857422026. [DOI] [PubMed] [Google Scholar]
- 42.McDuffie A, Abbeduto L, Lewis P, Kover S, Kim J, Weber A, et al. Autism Spectrum Disorder in children and adolescents with fragile X syndrome: within-syndrome differences and age-related changes. American Journal of Autism and Developmental Disorders. 2010;115(5):307–326. doi: 10.1352/1944-7558-115.4.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen M, Roth A, Kyzar EJ, Poudel MK, Wong K, Stewart AM, et al. Decoding the contribution of dopaminergic genes and pathways to autism spectrum disorder (ASD) Neurochem Int. 2014;66:15–26. doi: 10.1016/j.neuint.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 44.Paterson SJ, Brown JH, Gsödl MK, Johnson MH, Karmiloff-Smith A. Cognitive modularity and genetic disorders. Science. 1999;286(5448):2355–2358. doi: 10.1126/science.286.5448.2355. [DOI] [PubMed] [Google Scholar]
- 45.Persico AM, Napolioni V. Autism genetics. Behavior and Brain Research. 2013;251:95–112. doi: 10.1016/j.bbr.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 46.Pierce K, Courchesne E. Evidence for a cerebellar role in reduced exploration and stereotyped behavior in autism. Biology and Psychiatry. 2001;49(8):655–664. doi: 10.1016/S0006-3223(00)01008-8. [DOI] [PubMed] [Google Scholar]
- 47.Piven J, Palmer P, Jacobi D, Childress D, Arndt S. Broader autism phenotype: evidence from a family history study of multiple-incidence autism families. Am J Psychiatry. 1997;154(2):185–190. doi: 10.1176/ajp.154.2.185. [DOI] [PubMed] [Google Scholar]
- 48.Roberts JE, Clarke MA, Alcorn K, Carter JC, Long AC, Kaufmann WE. Autistic behavior in boys with fragile X syndrome: social approach and HPA-axis dysfunction. Journal of Neurodevelopmental Disorders. 2009;1:281–291. doi: 10.1007/s11689-009-9028-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roid GH, Miller LJ. Leiter International Performance Scale-Revised. Wood Dale: Stoelting; 1997. [Google Scholar]
- 50.Ruderfer DM, Fanous AH, Ripke S, McQuillin A, Amdur RL, Gejman PV, et al. Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and schizophrenia. Mol Psychiatry. 2014;19(9):1017–1024. doi: 10.1038/mp.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sabaratnam M, Murthy NV, Wijeratne A, Buckingham A, Payne S. Autistic-like behaviour profile and psychiatric morbidity in fragile X sydnrome: a prospective ten-year follow-up study. European Child and Adolescent Psychiatry. 2003;12:172–177. doi: 10.1007/s00787-003-0333-3. [DOI] [PubMed] [Google Scholar]
- 52.Shaw P, Eckstrand K, Sharp W, Blumenthal J, Lerch JP, Greenstein D, et al. Attention-deficit/hyperactivity disorder is characterized by a delay in cortical maturation. Proceedings of the Naional Academy of Sciences. 2007;104(49):19649–19654. doi: 10.1073/pnas.0707741104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith LE, Barker ET, Seltzer MM, Abbeduto L, Greenberg JS. Behavioral phenotype of fragile X syndrome in adolescence and adulthood. Am J Intellect Dev Disabil. 2012;117(1):1–17. doi: 10.1352/1944-7558-117.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soorya L, Kolevzon A, Zweifach J, Lim T, Dobry Y, Schwartz L, et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Molecular Autism. 2013;4(1):18. doi: 10.1186/2040-2392-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spooren W, Lindemann L, Ghosh A, Santarelli L. Synapse dysfunction in autism: a molecular medicine approach to drug discovery in neurodevelopmental disorders. Trends in Pharmacolical Sciences. 2012;33(12):669–684. doi: 10.1016/j.tips.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 56.Talkowski ME, Minikel EV, Gusella JF. Autism spectrum disorder genetics: diverse genes with diverse clinical outcomes. Harv Rev Psychiatry. 2014;22(2):65–75. doi: 10.1097/HRP.0000000000000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tassone F, Hagerman RJ, Taylor AK, Mills JB, Harris SW, Gane LW, et al. Clinical involvement and protein expression in individuals with the FMR1 premutation. Am J Med Genet. 2000;91(2):144–152. doi: 10.1002/(SICI)1096-8628(20000313)91:2<144::AID-AJMG14>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 58.Thurman AJ, McDuffie A, Kover ST, Hagerman RJ, Abbeduto L. Autism symptomatology in boys with fragile X syndrome: a cross sectional developmental trajectories comparison with nonsyndromic autism spectrum disorder. J Autism Dev Disord. 2015;45(9):2816–32. doi: 10.1007/s10803-015-2443-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang LW, Berry-Kravis E, Hagerman RJ. Fragile X: leading the way for targeted treatments in autism. Neurotherapeutics. 2010;7(3):264–274. doi: 10.1016/j.nurt.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams KT. Expressive vocabulary test. Circle Pines: American Guidance Service; 1997. [Google Scholar]
- 61.Wolff JJ, Bodfish JW, Hazlett HC, Lightbody AA, Reiss AL, Piven J. Evidence of a distinct behavioral phenotype in young boys with fragile X syndrome and autism. J Am Acad Child Adolesc Psychiatr. 2012;51(12):1324–1332. doi: 10.1016/j.jaac.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wolff JJ, Hazlett HC, Lightbody AA, Reiss AL, Piven J. Repetitive and self-injurious behaviors: associations with caudate volume in autism and fragile X syndrome. J Neurodevelopmental Disorders. 2013;5(1):12. doi: 10.1186/1866-1955-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zafeiriou DI, Ververi A, Dafoulis V, Kalyva E, Vargiami E. Autism spectrum disorders: the quest for genetic syndromes. Am J Med Genet B: Neuropsychiatric Genetics. 2013;162B(4):327–366. doi: 10.1002/ajmg.b.32152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used for the current study are available from the corresponding author upon reasonable request.