

Abstract
Aim
To investigate the effects and mechanisms of docetaxel and atorvastatin administered individually or in combination on prostate cancer cells.
Materialsandmethods
Cell growth and apoptosis were determined by the trypan blue exclusion assay and morphological assessment of cells stained with propidium iodide. NF-κB activity was determined by luciferase reporter gene assay and the Western blot assay was used to determine the levels of Bcl-2, phosphor-Akt, VEGF, and phosphor-Erk1/2.
Results
Results showed that following pre-treatment with cholesterol, resistance of PC-3 prostate cancer cellsto docetaxel was increased. The combination of docetaxel with atorvastatin potently inhibited growth and induced apoptosis in PC-3 cells. Mechanistic studies indicated that induction of apoptosis in PC-3 cells was associated with significant decreases in the levels of Bcl-2, VEGF, phosphor-Akt, and phosphor-Erk1/2.
Conclusion
Treatment with cholesterol decreased the sensitivity of prostate cancer cells to docetaxel. Docetaxel in combination with cholesterol-lowering drugs such as atorvastatin may be an effective strategy for inhibiting the growth of prostate cancer.
Keywords: Combination treatment, Prostate cancer, Docetaxel, Atorvastatin
Introduction
Prostate cancer (PCa) is the fourth most common cancer in both sexes collectively and the second most common cancer in men. In 2012, an estimated 1.1 million cases were diagnosed worldwide, accounting for 15% of the cancers diagnosed in menand almost 70% of them (759,000) occurring in more developed regions(11). According to Cancer Statistics, PCa accounts highest for 27% of the newly diagnosed cancers in 2014(11). Consequently, it is urgent to gain a better understanding on the mechanisms driving PCa in developed countries and to develop new agents(28).
PCa incidences have been linked to western diet, which includes high levels of red meat, saturated fat, and dairy products(6,23). Epidemiological studies have associated a fat/cholesterol enriched diet with hypercholesterolemia and an increased risk of PCa by causing intratumoral steroidogenesis, increased inflammation, and increased proliferation. For example, preclinical models involving feeding mice a high cholesterol diet have been shown to significantly increase the volume of human PCa xenografts(46). Meanwhile, cholesterol-lowering drugs reduce the risk of advanced PCa. Therefore, cholesterol has an important role on PCa progression.
Furthermore, normal prostate epithelial cells have abnormally high cholesterol content and cholesterol accumulates in solid tumors, with cholesterol levels further increasing during progression of PCa (4, 22, 26, 31, 45). Older studies also demonstrated that cholesterol homeostasis breaks down in the prostate during aging and the transition to malignant state. Since cholesterol is a precursor to androgen production, a decrease in cholesterol levels can reduce PCa risk by decreasing the levels of circulating testosterone. Thisreduction of cholesterol reduces the levels of interprostatic dihydrotestosterone, which is a strong ligand for the androgen receptor and a target to PCa therapy(13, 20, 33). There is a certain potential relationship between cholesterol and PCa. Cholesterol is also shown to accumulate in lipid rafts and regulate the activation of the phosphatidylinositol3-kinase/Akt pathway(33). We investigated whether alterations of cholesterol in PCa will affect treatment refractory in vitro.
Docetaxel, one of the promising chemotherapeutic treatments for carcinomatosis is a member of the taxoid drug class, which is semi-synthetically produced from the needles of the Pacific yew tree (Taxus brevifolia). As an antineoplastic agent, docetaxel has a more beneficial affect against progressive human prostate cancers than that of conventional anti-cancer agents(7, 17, 19, 38, 42). The principal chemotherapeutic targets of docetaxel are microtubules, which cause cell-cycle arrest and apoptosis by increasing tubulin polymerisation, promoting microtubule assembly, and inhibiting tubulin depolymerisation. It has been assumed that docetaxelis able to induce the phosphorylation of Bcl-XL/Bcl-2 members and thus inactivate their anti-apoptotic capacities. The down-regulation of Bcl-2 and/or the upregulation of p53 are certainly one of the important modes of apoptosis induction by docetaxel(14, 25). High concentrations of docetaxel greatly induce the formation of extensive bundles of microtubules and inhibit cell proliferation. However, the use of high doses of docetaxel induces toxic reactions. In cancer clinical trials, tolerability and toxicityare concerns, particularly since most prostate cancer patients are elderly or have other medical problems and so the use of docetaxel as a monotherapy for cancer needs improvement(34, 39).
In an initial experiment, we tested the anti-proliferative effects of docetaxel on PCa cell line PC-3 and discovered that pre-treatment with cholesterol decreased the sensitivity of docetaxel. Treatments with agents that specifically target the biochemical synthesis of cholesterol may increase the sensitivity of docetaxel. Atorvastatin, a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, has been approved for cholesterol reduction and considered to be among the safest drugs (2, 33, 41). Present study was therefore designed to explore the potential synergistic effect of docetaxelin combination with atorvastatin on growth and apoptosis in human prostate cancer cells.
2. Materials and Methods
2.1 Cell culture and reagents
The human prostate carcinoma cell lines PC-3 (AR negative) and LNCaP (ARsensitive) were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). PC-3 cells are human prostatic carcinoma cell lines derived from a bone metastasis of an androgen-independent prostatic adenocarcinoma that has a greatly reduced dependence upon serum for growth when compared to that of normal prostatic epithelial cells. The cells do not respond to androgens, glucocorticoids, epidermal, or fibroblast growth factors[25]. LNCaP cells are androgen-sensitive human prostate adenocarcinoma cells derived from the left supraclavicular lymph node metastasis from a 50-year-old caucasian male in 1977. The PC-3 and LNCaP cells sustained exponential growth by being treated twice a week in RPMI 1640 tissue culture medium with 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA), penicillin (100 units/ml)-streptomycin (100mg/ml), and L-glutamine(300μg/ml). Atorvastatin and Docetaxel were provided by the National Cancer Institute. Cultured cells were placed into tissue culture flasks and dishes and grown at 37°C in a humidified atmosphere of 5% CO2. Atorvastatin and docetaxel were dissolved in DMSO, and the final concentration of DMSO in all the experiments was 0.1%.
2.2 Determination of the number of viable cells
The number of viable cells after each treatment was determined by the trypan blue exclusion assay. After single cells were dissociated, they were seeded in 35-mm dishes containing growth mediums at a final cell concentration of approximately 2.5×104cells/ml and incubated at 37°Cin an atmosphere of 5% CO2. We used a hemocytometer under a light microscope (Nikon Optiphot, Nikon, Tokyo, Japan) to complete the measurement of cell viability, which was performed by mixing 80μl of cell suspension and 20μl of 0.4% trypan blue solution for 2min. Blue cells were marked dead and the cells that did not absorb dye were marked alive.
2.3 Assessment of apoptotic cells by morphology and by activation of caspase-3
Apoptosis was determined by morphological assessment in cells stained with propidium iodide (PI). Apoptotic cells were identified by their classical morphological features such as nuclear condensation, cell shrinkage, and formation of apoptotic bodies. Cytospin slides were prepared after each experiment and cells were fixed with acetone/methanol(1:1) for 10 min at room temperature, followed by 10 min with PI staining (1 μg/ml in PBS: Phosphate Buffered Saline), and finally analyzed using a fluorescence microscope (Nikon Eclipse TE200, Nikon). At least 200 cells were counted in each sample and the percentage of apoptotic cells was presented.
2.4 Western Blotting
After treatment, the cell lysates were prepared as described earlier. Proteins were subjected to sodium dodecyl sulfatepolyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. After blocking nonspecific binding sites with blocking buffer, the membranes were incubated overnight at 4°C with primary antibodies (#2870 for Bcl-2, #4501 for phosphor-Akt, #4376 for phosphor-Erk1/2, and #9272for total-Akt, all from Cell Signaling Co., Beverly, MA; ab46154 for VEGF from Abcam, Cambridge, MA, USA). β-actin was used as a loading control. Following the removal of the primary antibodies, the membranes were then washed three times with TBS (PBS containing 0.05% tween 20) buffer at room temperature and later incubated with fluorochrome-conjugated secondary antibody (Santa Cruz Biotechnology Inc., CA, USA). The membrane was then washed with TBS three times. Final detection was done with an Odyssey infrared imaging system (Li-Cor Biotechnology, Lincoln, NE, USA).
2.5 NF-κB-dependent reporter gene expression assay
NF-κB transcriptional activity was measured by the NF-κB-luciferase reporter gene expression assay. An NF-κB luciferase construct was stably transfected into PC-3 cells and a single stable clone of PC-3 cells that were stably transfected with the NF-κB luciferase reporter gene, became the PC-3/N cell line, which was used in the present study. PC-3/N cells were treated with docetaxel and atorvastatin individually or in combination for 24 h, and the NF-κB-luciferase activities were measured using the luciferase assay kits from Promega (Madison WI, USA). After treatments, the cells were washed with PBS and harvested in 1 x reporter lysis buffer. After centrifugation, 10 μl aliquots of the supernatants were measured for luciferase activity by using a Luminometer from Turner Designs Instrument (Sunnyvale, CA, USA). The luciferase activity was normalized against known protein concentrations and expressed as a percentage of luciferase activity in the control cells, which were treated with DMSO solvent. The protein level was determined by Bio-Rad protein assay kits (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions.
2.6 Statistical analyses
The potential synergistic effect of docetaxel or atorvastatin was assessed by the isobole method(44), using the equation Ac/Ae + Bc/Be = combination index (CI). Ac and Bc represent the concentration of drug A and drug B used in the combination, and Ae and Be represent the concentration of drug A and B that produced the same magnitude of effect when administered alone. If CI is <1, then the drugs are considered to act synergistically. If the CI is >1 or =1, then the drugs act in an antagonistic or additive manner, respectively. The analysis of variance (ANOVA) model with Tukey-Kramer adjustment was used for the comparison of growth inhibition of NF-κB activity and apoptosis ratios among different treatment groups at the end of the treatment.
3. Results
3.1 Pre-treatment with cholesterol affects the inhibitory effect of docetaxel on cultured prostate cancer cells
To determine whether pre-treatment with cholesterol affects the inhibitory effects of docetaxel on prostate cancer cells, we treated PC-3 cells with cholesterol for 5 days, then treated with docetaxel for 72h (Figure 1A), and finally determined the cell viability by trypan blue exclusion assay. As presented in Figure 1B, treatment with cholesterol reduced the inhibitory effects of docetaxel on cultured PC-3 cells, especially in the 5μM cholesterol concentrations. Cholesterol decreased docetaxel inhibition activity by 72.7% and 22.4% in the treatment of docetaxel 2nM and 5nM. The results indicate that cholesterol pre-treatment may suppress the inhibitory effects of docetaxel on prostate cancer PC-3 cells.
Figure 1.

Effects of cholesterol on docetaxel-induced growth inhibition in PC-3 cells. Human prostate cancer cells PC-3 were seeded at a density of 0.25×105 cells/ml in 10 mm tissue culture dishes and incubated for 24 h. Cells were then treated with various concentrations of cholesterol continually for 5 days shown in flow path (A). On the 7th day, cells were harvestedand each group was separated seed into 35mm tissue culture dishes and incubated for 24 h. Cells were then treated with various concentrations of docetaxel. Viable cells weredetermined by the trypan blue exclusion assay and expressed as percentages of solvent-treated control. Each value represents mean ± S.E. from 3separate experiments. Significant numbers of viable cells between a cholesterol treated group and a single agent treated group were analyzed by ANOVA with Tukey-Kramer multiple comparison test (*p<0.05,**p<0.01,***p<0.001).
3.2 Effects of docetaxel and atorvastatin in combination on the growth and apoptosis of human prostate cancer
The growth-inhibitory activities of docetaxel and atorvastatin alone or in combination were assessed in two prostate cancer cell lines: PC-3 (androgen-independent) and LNCaP (androgen-dependent). he effects of different concentrations of docetaxel and atorvastatin on the growth and death of PC-3 and LNCaP cells were determined by using trypan blue dye exclusion assay. As shown in Figure 2, nanomole concentrations (1–5 nM) docetaxel alone and in combination with atorvastatin caused a dose-dependent inhibition of the proliferation of both cells lines, with 5nM docetaxel combined with 5μM atorvastatin as the most effective treatment in both cell lines. Treatments with docetaxel (2nM) or atorvastatin (5μM) alone had small effects on the growth and death of PC-3 and LNCaP cells. The combination caused a momentous increase in the percentage of growth inhibition. Shown in Figure 2, potent additional inhibition occurred when docetaxel was combined with atorvastatin on the two prostate cancer cell lines, particularly PC-3 cells, which increased 35.25% and 29.76% of growth inhibition, compared to that of docetaxel alone in concentration of 2nM and 5nM. The increased number of dead cells suggests similar potency. With concentrations of 5nM docetaxel combined with 5μM atorvastatin, there are more potent increases in inhibitory effect of the combined treatment in comparison to the treatments with single agent. This increase is 29.76% in PC-3 cells and 45.54% in LNCaP cells. As shown in Figure 2, lower concentrations of docetaxel (5nM) in combination with atorvastatin (5μM) produced a better inhibitory effect than docetaxel at higher concentrations (10nM).
Figure 2.

Effects of docetaxel and atorvastatin alone or in combination on the growth of cultured prostate cancer cells. Human prostate cancer cells PC-3 were seeded at a density of 0.25×105 cells/ml in 35 mm tissue culture dishes and incubated for 24 h. The cells were then treated with various concentrations of docetaxel and atorvastatin alone or in combination for 72 h. 1: Control, 2: Atorvastatin 5μM, 3: Docetaxel 1nM, 4: Docetaxel 2nM, 5:Docetaxel 5nM, 6: Docetaxel 1nM+Atorvastatin 5μM, 7: Docetaxel 2nM+Atorvastatin 5μM, 8: Docetaxel 5nM+ Atorvastatin 5μM. Viable cells weredetermined by the trypan blue exclusion assay and expressed as percentages of solvent-treated control. Each value represents mean ± S.E. from 3separate experiments. Significant numbers of viable cells between a combination group and a single agent treated group were analyzed by ANOVA with Tukey-Kramer multiple comparison test (*p<0.05,**p<0.01,***p<0.001).
Morphological assessments of PC-3 and LNCaP cells after their exposure to either docetaxel (1–5nM), atorvastatin (5μM), or a combination of both drugs were performed by staining with propidium iodide, which stains the nuclei of cells. As shown in Table 1 and Figure 3 after combination treatment, more PC-3 cells and LNCaP cells (Staining image not shown is Figure 3) showed morphological changes characteristic of apoptosis in contrast to that treated with each agent alone, indicating a more potent stimulatory effect on apoptosis.
Table 1.
Effect of docetaxel and atorvastatin on apoptosis of cultured prostate cancer cells. LNCaP and PC-3 cells were seeded at a density of 0.25× 105 cells/ml and incubated for 24 h. Cells were then treated with docetaxel (Doc; 1,2 or 5nM) and atorvastatin (Ator; 5 μM) alone or in combination for 48 h. Apoptosis was determined by morphological assessment. Each value is the mean ± S.E. from 3 different experiments. Differences in the number of apoptotic cells between a combination group and a single agent-treated group were analyzed by ANOVA with the Tukey-Kramer multiple comparison test.
| Treatment | % apoptotic cells
|
|
|---|---|---|
| LNCaP | PC-3 | |
| Control | 1.83 ± 1.52 | 2.17 ± 3.05 |
| Doc(1 nM) | 9.66 ± 2.51 | 8.50 ± 5.57 |
| Doc(2nM) | 20.00 ± 5.29 | 17.83 ± 3.05 |
| Doc(5 nM) | 31.83 ± 3.78 | 34.17 ± 4.04 |
| Ator(5 uM) | 8.17 ± 4.04 | 8.33 ± 5.03 |
| Doc(1 nM) + Ator(5 uM) | 18.67 ± 6.50 * | 21.17 ± 1.53 * |
| Doc(2nM) + Ator(5 uM) | 30.83 ± 6.80 * | 35.33 ± 2.51 *** |
| Doc(5nM) + Ator(5 uM) | 39.67 ± 3.21 * | 57.17 ± 4.51 *** |
Figure 3.

The apoptosis nuclear morphology changes in PC-3 cells. The nuclear morphology changes were analyzed by fluorescence microscopy in ×200 magnification using the propidium iodide nuclear fluorescent dye staining. These experiments were performed 48 h after treatment. A: Control, B: Docetaxel 1nM, C: Docetaxel 2nM, D: Docetaxel 5nM, E: Atorvastatin 5μM, F: Docetaxel 1nM+ Atorvastatin 5μM, G: Docetaxel 2nM+ Atorvastatin 5μM, H: Docetaxel 5nM+ Atorvastatin 5μM.
The combination index (CI) for IC50 were calculated as 0.48 forPC-3 cells and0.67 for LNCaP cells, respectively The results (Figure 2, Figure 3 and Table 1) indicate that the combination of docetaxel and atorvastatin synergistically inhibits the growth of cultured prostate cancer cells and induces cell apoptosis.
3.3 Effect of docetaxel and atorvastatin on NF-κB transcriptional activity
A luciferase reporter gene expression assay was used to determine the effect of docetaxel and atorvastatin when used alone or in combination on the activation of NF-κB. In initial experiments, PC-3/N cells were treated with docetaxel (1–5 nM) and atorvastatin (5μM) individually or in combination for 24h, and the activity of NF-κB in prostate cancer cells was reflected by luciferase activity. As shown in Figure 4, Treatment of PC-3/N cells with docetaxel (1, 2 or 5nM) or atorvastatin (5μM) alone caused a modest decrease in luciferase activity in PC-3/N cells, and the combinations of docetaxel (2–5nM) and atorvastatin (5μM) had much stronger effects than either agent alone (Figure 4). Combined treatment of PC-3/N cells with docetaxel (5nM) and atorvastatin (5μM)showed that NF-κB activity was significantly inhibited (84%) (Figure 4). A lower concentration of docetaxel (2nM) combined with atorvastatin (5μM) caused a greater luciferase activity inhibition than docetaxel alone at the higher concentration of 5nM (Figure 4).
Figure 4.

Inhibitory effect of docetaxel and atorvastatin alone or in combination on NF-κB activation in PC-3 cells. PC-3/N cells were seeded at a density of 0.25×105 cells/ml of medium in 12-well plates and incubated for 24 h. Cells were then treated with docetaxel alone or in combination with atorvastatin for 24 h. 1: Control, 2: Atorvastatin 5μM, 3: Docetaxel 1nM, 4: Docetaxel 2nM, 5:Docetaxel 5nM, 6: Docetaxel 1nM+ Atorvastatin 5μM, 7: Docetaxel 2nM+ Atorvastatin 5μM, 8: Docetaxel 5nM+ Atorvastatin 5μM. The NF-κB transcriptional activity was measured by a luciferase activity assay. Analyzed by ANOVA with Tukey-Kramer multiple comparison test, *p<0.05, **p<0.01, *** p<0.001 as compared to docetaxel or atorvastatin alone.
3.4 Effects of docetaxel and atorvastatin on the level of Bcl-2, phospho-Akt, phospho-Erk1/2 and VEGF
The levels of Bcl-2, phospho-Akt, phospho-Erk1/2 and VEGF inPC-3 cells treated with atorvastatin and docetaxel were determined by western blot. As shown in Figure 5, compared to docetaxel or atorvastatin were used individually, ocetaxel (2–5nM) combined with atorvastatin (5μM) treatment generated a significant decrease in the expression of Bcl-2, an apoptosis-related protein. When docetaxel was used alone at low concentrations(1–2nM), Bcl-2 expression levels slightly decreased; at higher docetaxel concentrations (5nM), Bcl-2 expression decreased significantly. However, when docetaxel was used in combination with atorvastatin, even with low concentrations of docetaxel(1–2nM) and atorvastatin (5μM), there was a more potent effect and the decrease in Bcl-2 levels was even greater. This unique oncogene results in cell division by overriding programmed cell death mechanisms(24). The activity of p-Akt and p-Erk1/2 was decreased during combined treatments of docetaxel and atorvastatin compared to the activity expressed when either agent was employed alone While the activity of T-Akt have no significant change. Combinations of atorvastatin and docetaxel inhibited the growth and induced apoptosis of prostate cancer cells via inhibition of p-Akt and p-Erk1/2. Treatment of PC-3 cells with docetaxel (5 nM) and atorvastatin (5 μM) also resulted in a decrease in the level of VEGF expression level.
Figure 5.
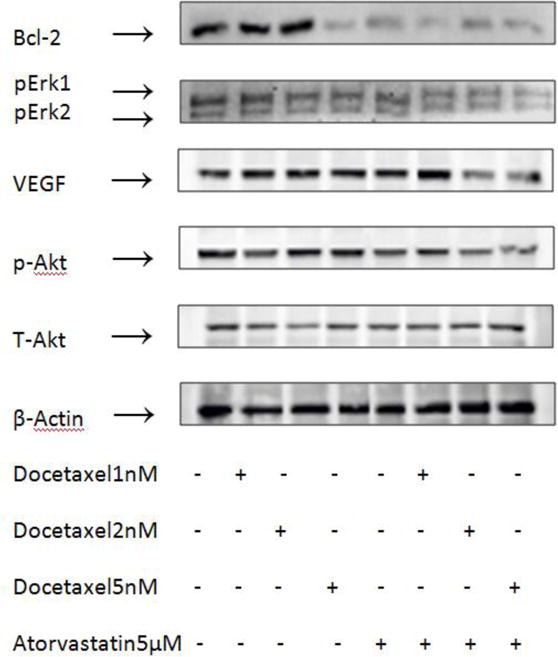
Effects of docetaxel and atorvastatin on the levels of Bcl-2, phospho-Akt, total-Akt, phospho-Erk1/2 and VEGF in PC-3 cells. Cells were seeded at a density of 1×105 cells/ml of medium in 100 mm culture dishes and incubated for 24 h. Cells were then treated with docetaxelandatorvastatin alone and in combination for 24 h (for analysis of phospho-Akt, total-Akt, phospho-Erk1/2 and VEGF) and 48 h (for analysis of Bcl-2). The levels of phospho-Akt, total-Akt, phospho-Erk1/2 and VEGF were determined by the Western blot analysis.
4. Discussion
Earlier studies suggested a link between cholesterol and PCa progress. In the present study, we found that pre-treatment with cholesterol decreased the inhibitory effects of docetaxel on prostate cancer PC-3 cells, and demonstrated for the first time that the cholesterol-lowing drug atorvastatin combined with docetaxel at lower concentrations had even more potent effects on growth inhibition and apoptosis induction than either agent alone at higher concentrations. Previous studies showed that a higher concentration of docetaxel was required to inhibit the growth of prostate cancer cells especially for androgen-independent prostate cancer. Combination of low doses of anticancer agents that work by different mechanisms may be more effective with less toxicity than individual compounds at higher dose levels. We found that a low concentration of docetaxel (2nM) in combination with a low concentration of atorvastatin (5μM), synergistically inhibited the growth of cultured prostate cancer cells. As such we suggest a strategy for the first time that there is a strong combined effect of low concentrations of docetaxel and atorvastatin on prostate cancer cells.
The proposed strategy is beneficial to the treatment of PCa because it inhibits proliferation and induces apoptosis in human prostate cancer cells. The observed potent inhibitory effect in vitro suggests a direction to reduce docetaxel resistance and foster a more beneficial therapy for treating PCa in future clinical trials(37).
Initial treatments for PCa are generally androgen-ablation therapy, prostatectomy, radiation therapy, and cytotoxic chemotherapy(8). However, many patients are not cured by these therapy treatments and the cancer reoccurs and develops from androgen-dependent to androgen-independent prostate cancer(AIPC)(10). At present, treatment of AIPC remains futile. Docetaxel is one of the most promising chemotherapeutic treatments; however, the disease response, survival percentage, and toxicity effects of this anti-prostate cancer drug are not promising (3). Evidence from several studies suggest that hypercholesterolemiais associated with increased risk of aggressive prostate cancer. Cholesterol is shown to accumulate in solid prostate tumors(13, 43). Studies have shown that docetaxel exhibits chemo resistance with cholesterol stimulatory effect (Figure 1). This suggests that decreasing cholesterol levels may be a novel strategy to increase the sensitivity of AIPC to docetaxel treatment. A large number of studies have shown that the combination of different anticancer agents at lower concentrations had even more potent effects on growth inhibition and apoptosis induction than the effects produced when either agent was used alone at higher concentrations.
In the present study, we found that combination of docetaxel and atorvastatin increased growth inhibition and apoptosis induction compared to that of either drug alone on cultured PC-3 (androgen-independent)and LNCaP (androgen-dependent) prostate cancer cells. As shown in Figure 2, potent inhibition occurred when docetaxel was combined with atorvastatin on the two prostate cancer cell lines, particularly PC-3 cells, compared to that of docetaxel alone at concentrations of 2nM and 5nM. The increased number of dead cells suggests similar potency. Moreover, our data demonstrated that the apoptotic cell rate of combined treatment of docetaxel and atorvastatin significantly increased both in PC-3 (p<0.001) and LNCaP (p<0.05) cell lines in comparison to the apoptotic cell rates of each agent used alone. Apoptotic cells were identified by general morphological features, including nuclear condensation, DNA breakdown, plasma and nuclear membrane intense convolution, cell shrinkage, and formation of apoptotic bodies(1, 18). Docetaxel combined with atorvastatin treatment cells showed the morphological changes typical of cells undergoing apoptosis such as shrinkage, blebbing and DNA fragmentation (Figure 3).
To help determine whether the strong effects on PC-3 cells following combination treatment were mediated through apoptosis-related signaling pathways, we evaluated the transcriptional activity of NF-κB, which activates genes associated with limitless replicative potential, angiogenesis, tissue invasion, metastasis, and the suppression of apoptosis in prostate cancer cells(29, 36). Previous studies have demonstrated that constitutive deactivation of NF-κB in human prostate cancer exhibits increased levels of apoptosis(27, 29, 30, 36). Combined treatment of PC-3/N cells with docetaxel (5nM) and atorvastatin (5μM)showed that NF-κB activity was significantly inhibited (Figure 4) while no striking effect was observed with docetaxel or docetaxel used alone(p<0.001). Besides, proteins encoded by Bcl-2 family genes are important regulators of programmed cell death and apoptosis and there is evidence that links NF-κB survival pathway with the upregulation of Bcl-2(5, 16, 21). Bcl-2 expression was remarkably down regulated in the combined treatment of docetaxel and atorvastatin (Figure 5). Overall, data from the trypan blue exclusion assay, along with the NF-κB luciferase reporter gene expression assay and western blot, provide strong evidence that docetaxel combined with atorvastatin effectively inhibits the growth and induces apoptosis in human prostate cancer cells.
Earlier studies have shown that the regulation of Akt pathway plays a central role in growth, proliferation, motility, survival, and angiogenesis in tumor cells(9, 12, 35), and both normal and pathologic angiogenesis are regulated predominantly by the vascular endothelial growth factors(32). Akt activation and VEGF over expression prompt PCa progression(9, 40). Results of the present study suggest that the combination of low concentrations of docetaxel and atorvastatin apparently decreased the level of phosphor-Akt and VEG while each agent used alone even in higher concentration had very slight effects on the level of phospho-Akt and VEGF (Figure 5). Human prostate cancer cells contain cholesterol-rich lipid rafts that mediate epidermal growth factor (EGF)-induced and constitutive signaling through the Akt serine-threonine kinase, when the prostate cancer cells were treated by docetaxel combined with atorvastatin, down-regulated cholesterol, lipid rafts may be disrupted, VEGF and Akt phosphorylation were inhibited and autonomous cell survival was reduce (46). Another signaling pathway that is associated with prostate cancer growth and progression is the mitogen activation protein kinase (MAPK). Erk1/2 belongs to a subfamily of MAPK. The level of activated Erk1/2 increased with increasing Gleason score and tumor stage(15). In the present study, we found that the combination of docetaxel and atorvastatin had a potent effect on decreasing the level of Erk1/2 in the cells (Figure 5). Taken together, considering the molecular level results, simultaneous inhibition of Akt and Erk pathways and downregulation of VEGF and Bcl-2 may initiate a strong inhibitory effect on proliferation and a strong stimulatory effect on apoptosis in prostate cancer cellswith the combined treatment of docetaxel and atorvastatin.
In conclusion, the present study demonstrated that docetaxel and atorvastatin in combination strongly inhibited growth and induced apoptosis in human prostate cancer cells. The effects of docetaxel and atorvastatin on growth inhibition and apoptosis in prostate cancer cells were associated with inhibition of NF-κB activation, increased caspase-3 activation, and decreased levels of Bcl-2, VEGF, phospho-Akt, and phospho-Erk1/2. The combination of docetaxel and atorvastatin may be an effective adjuvant therapy for inhibiting the growth of prostate cancer.
Acknowledgments
The present study was supported by grants from the Rutgers Cancer Institute of New Jersey (CCSG P30-CA072720 RSD), the Chinese National Science Foundation (81272452, 21102020, 21272043), the Guangdong Province Leadership Grant China, the PhD Start-up Fund of Natural Science Foundation of Guangdong Province (2014A030310329), and by Medical Scientific Research Foundation of Guangdong Province (B2014072).
References
- 1.Allen RT, Hunter WJ, Agrawal DK. Morphological and biochemical characterization and analysis of apoptosis. Journal of pharmacological and toxicological methods. 1997;37:215–228. doi: 10.1016/s1056-8719(97)00033-6. [DOI] [PubMed] [Google Scholar]
- 2.Bakker-Arkema RG, Best J, Fayyad R, Heinonen TM, Marais AD, Nawrocki JW, Black DM. A brief review paper of the efficacy and safety of atorvastatin in early clinical trials. Atherosclerosis. 1997;131:17–23. doi: 10.1016/s0021-9150(97)06066-8. [DOI] [PubMed] [Google Scholar]
- 3.Beer T, Pierce W, Lowe B, Henner W. Phase II study of weekly docetaxel in symptomatic androgen-independent prostate cancer. Annals of oncology. 2001;12:1273–1279. doi: 10.1023/a:1012258723075. [DOI] [PubMed] [Google Scholar]
- 4.Bravi F, Scotti L, Bosetti C, Talamini R, Negri E, Montella M, Franceschi S, La Vecchia C. Self-reported history of hypercholesterolaemia and gallstones and the risk of prostate cancer. Annals of Oncology. 2006;17:1014–1017. doi: 10.1093/annonc/mdl080. [DOI] [PubMed] [Google Scholar]
- 5.Catz S, Johnson J. BCL-2 in prostate cancer: a minireview. Apoptosis. 2003;8:29–37. doi: 10.1023/a:1021692801278. [DOI] [PubMed] [Google Scholar]
- 6.Coffey DS. Similarities of prostate and breast cancer: evolution, diet, and estrogens. Urology. 2001;57:31–38. doi: 10.1016/s0090-4295(00)00938-9. [DOI] [PubMed] [Google Scholar]
- 7.Cortes JE, Pazdur R. Docetaxel. Journal of Clinical Oncology. 1995;13:2643–2655. doi: 10.1200/JCO.1995.13.10.2643. [DOI] [PubMed] [Google Scholar]
- 8.Denmeade SR, Isaacs JT. A history of prostate cancer treatment. Nature Reviews Cancer. 2002;2:389–396. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feitelson MA, Arzumanyan A, Kulathinal RJ, Blain SW, Holcombe RF, Mahajna J, Marino M, Martinez-Chantar ML, Nawroth R, Sanchez-Garcia I. Seminars in cancer biology. Elsevier; 2015. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nature Reviews Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 11.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International Journal of Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 12.Foubert E, De Craene B, Berx G. Key signalling nodes in mammary gland development and cancer. The Snail1-Twist1 conspiracy in malignant breast cancer progression. Breast Cancer Res. 2010;12:206. doi: 10.1186/bcr2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freeman MR, Solomon KR. Cholesterol and prostate cancer. Journal of cellular biochemistry. 2004;91:54–69. doi: 10.1002/jcb.10724. [DOI] [PubMed] [Google Scholar]
- 14.Ganansia-Leymarie V, Bischoff P, Bergerat J-P, Holl V. Signal transduction pathways of taxanes-induced apoptosis. Current Medicinal Chemistry-Anti-Cancer Agents. 2003;3:291–306. doi: 10.2174/1568011033482422. [DOI] [PubMed] [Google Scholar]
- 15.Gioeli D, Mandell JW, Petroni GR, Frierson HF, Weber MJ. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer research. 1999;59:279–284. [PubMed] [Google Scholar]
- 16.Hockenbery DM, Oltvai ZN, Yin X-M, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 17.Ikezoe T, Hisatake Y, Takeuchi T, Ohtsuki Y, Yang Y, Said JW, Taguchi H, Koeffler HP. HIV-1 Protease Inhibitor, Ritonavir A Potent Inhibitor of CYP3A4, Enhanced the Anticancer Effects of Docetaxel in Androgen-Independent Prostate Cancer Cells In vitro and In vivo. Cancer research. 2004;64:7426–7431. doi: 10.1158/0008-5472.CAN-03-2677. [DOI] [PubMed] [Google Scholar]
- 18.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British journal of cancer. 1972;26:239. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim SM, Lee SY, Cho JS, Son SM, Choi SS, Yun YP, Yoo HS, Oh K-W, Han SB, Hong JT. Combination of ginsenoside Rg3 with docetaxel enhances the susceptibility of prostate cancer cells via inhibition of NF-κB. European journal of pharmacology. 2010;631:1–9. doi: 10.1016/j.ejphar.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 20.Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR. Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. Journal of Pharmacology and Experimental Therapeutics. 2011;336:496–505. doi: 10.1124/jpet.110.174870. [DOI] [PubMed] [Google Scholar]
- 21.Krajewska M, Krajewski S, Epstein JI, Shabaik A, Sauvageot J, Song K, Kitada S, Reed JC. Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. The American journal of pathology. 1996;148:1567. [PMC free article] [PubMed] [Google Scholar]
- 22.Krycer JR, Brown AJ. Cholesterol accumulation in prostate cancer: a classic observation from a modern perspective. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2013;1835:219–229. doi: 10.1016/j.bbcan.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 23.Llaverias G, Danilo C, Wang Y, Witkiewicz AK, Daumer K, Lisanti MP, Frank PG. A Western-type diet accelerates tumor progression in an autochthonous mouse model of prostate cancer. The American journal of pathology. 2010;177:3180–3191. doi: 10.2353/ajpath.2010.100568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDonnell TJ, Troncoso P, Brisbay SM, Logothetis C, Chung LW, Hsieh J-T, Tu S-M, Campbell ML. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer research. 1992;52:6940–6944. [PubMed] [Google Scholar]
- 25.Montero A, Fossella F, Hortobagyi G, Valero V. Docetaxel for treatment of solid tumours: a systematic review of clinical data. The lancet oncology. 2005;6:229–239. doi: 10.1016/S1470-2045(05)70094-2. [DOI] [PubMed] [Google Scholar]
- 26.Murtola TJ, Tammela TL, Lahtela J, Auvinen A. Cholesterol-lowering drugs and prostate cancer risk: a population-based case-control study. Cancer Epidemiology Biomarkers & Prevention. 2007;16:2226–2232. doi: 10.1158/1055-9965.EPI-07-0599. [DOI] [PubMed] [Google Scholar]
- 27.Naugler WE, Karin M. NF-κB and cancer—identifying targets and mechanisms. Current opinion in genetics & development. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Network NCC. Prostate cancer. NCCN clinical practice guidelines in oncology. Journal of the National Comprehensive Cancer Network: JNCCN. 2004;2:224. doi: 10.6004/jnccn.2004.0021. [DOI] [PubMed] [Google Scholar]
- 29.Orlowski RZ, Baldwin AS. NF-κB as a therapeutic target in cancer. Trends in molecular medicine. 2002;8:385–389. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- 30.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 31.Platz EA, Leitzmann MF, Visvanathan K, Rimm EB, Stampfer MJ, Willett WC, Giovannucci E. Statin drugs and risk of advanced prostate cancer. Journal of the National Cancer Institute. 2006;98:1819–1825. doi: 10.1093/jnci/djj499. [DOI] [PubMed] [Google Scholar]
- 32.Roskoski R. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Critical reviews in oncology/hematology. 2007;62:179–213. doi: 10.1016/j.critrevonc.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 33.Roy M, Kung H-J, Ghosh PM. Statins and prostate cancer: role of cholesterol inhibition vs. prevention of small GTP-binding proteins. American journal of cancer research. 2011;1:542. [PMC free article] [PubMed] [Google Scholar]
- 34.Ryan DP, Kulke MH, Fuchs CS, Grossbard ML, Grossman SR, Morgan JA, Earle CC, Shivdasani R, Kim H, Mayer RJ. A phase II study of gemcitabine and docetaxel in patients with metastatic pancreatic carcinoma. Cancer. 2002;94:97–103. doi: 10.1002/cncr.10202. [DOI] [PubMed] [Google Scholar]
- 35.Steelman LS, Stadelman KM, Chappell WH, Horn S, Bäsecke J, Cervello M, Nicoletti F, Libra M, Stivala F, Martelli AM. Akt as a therapeutic target in cancer. 2008 doi: 10.1517/14728222.12.9.1139. [DOI] [PubMed] [Google Scholar]
- 36.Suh J, Rabson AB. NF‐κB activation in human prostate cancer: Important mediator or epiphenomenon? Journal of cellular biochemistry. 2004;91:100–117. doi: 10.1002/jcb.10729. [DOI] [PubMed] [Google Scholar]
- 37.Tabernero J. The role of VEGF and EGFR inhibition: implications for combining anti–VEGF and anti–EGFR agents. Molecular Cancer Research. 2007;5:203–220. doi: 10.1158/1541-7786.MCR-06-0404. [DOI] [PubMed] [Google Scholar]
- 38.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Théodore C, James ND, Turesson I. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. New England Journal of Medicine. 2004;351:1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 39.Tedesco K, Thor A, Johnson D, Shyr Y, Blum K, Goldstein L, Gradishar W, Nicholson B, Merkel D, Murrey D. Docetaxel combined with trastuzumab is an active regimen in HER-2 3+ overexpressing and fluorescent in situ hybridization–positive metastatic breast cancer: a multi-institutional phase II trial. Journal of clinical oncology. 2004;22:1071–1077. doi: 10.1200/JCO.2004.10.046. [DOI] [PubMed] [Google Scholar]
- 40.Vara JÁF, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. PI3K/Akt signalling pathway and cancer. Cancer treatment reviews. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 41.Youssef S, Stüve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, Bravo M, Mitchell DJ, Sobel RA, Steinman L. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- 42.Yvon A-MC, Wadsworth P, Jordan MA. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Molecular biology of the cell. 1999;10:947–959. doi: 10.1091/mbc.10.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2013;1831:1518–1532. doi: 10.1016/j.bbalip.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao L, Wientjes MG, Au JL. Evaluation of combination chemotherapy integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clinical Cancer Research. 2004;10:7994–8004. doi: 10.1158/1078-0432.CCR-04-1087. [DOI] [PubMed] [Google Scholar]
- 45.Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. The Journal of clinical investigation. 2005;115:959–968. doi: 10.1172/JCI200519935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhuang L, Lin J, Lu ML, Solomon KR, Freeman MR. Cholesterol-rich lipid rafts mediate akt-regulated survival in prostate cancer cells. Cancer research. 2002;62:2227–2231. [PubMed] [Google Scholar]