

Abstract
Host–pathogen interactions may result in either directional selection or in pressure for the maintenance of polymorphism at the molecular level. Hence signatures of both positive and balancing selection are expected in immune genes. Because both overall selective pressure and specific targets may differ between species, large-scale population genomic studies are useful in detecting functionally important immune genes and comparing selective landscapes between taxa. Such studies are of particular interest in amphibians, a group threatened worldwide by emerging infectious diseases. Here, we present an analysis of polymorphism and divergence of 634 immune genes in two lineages of Lissotriton newts: L. montandoni and L. vulgaris graecus. Variation in newt immune genes has been shaped predominantly by widespread purifying selection and strong evolutionary constraint, implying long-term importance of these genes for functioning of the immune system. The two evolutionary lineages differ in the overall strength of purifying selection which can partially be explained by demographic history but may also signal differences in long-term pathogen pressure. The prevalent constraint notwithstanding, 23 putative targets of positive selection and 11 putative targets of balancing selection were identified. The latter were detected by composite tests involving the demographic model and further validated in independent population samples. Putative targets of balancing selection encode proteins which may interact closely with pathogens but include also regulators of immune response. The identified candidates will be useful for testing whether genes affected by balancing selection are more prone to interspecific introgression than other genes in the genome.
Keywords: balancing selection, positive selection, immune genes, demographic model, Lissotriton, newts
Introduction
Pressures exerted by diverse pathogens are the source of selection resulting in rapid evolutionary changes of host molecules, components of both innate and adaptive immunity (Boehm 2012; Quintana-Murci and Clark 2013). Evidence for adaptive evolution of immune genes is ample (Obbard et al. 2009; Spurgin and Richardson 2010) and both experimental (Eizaguirre et al. 2009) and correlational (Fumagalli et al. 2011) studies point to pathogens as causal factors. However, establishing direct and robust links between genetic variation and immunity to pathogens is challenging and requires laborious experiments. Promising candidates for such in-depth studies may be identified by examining patterns of variation within and divergence between species in many genes. A clear advantage of such genomic scans is their ability to screen a large number of potential targets, even if their functions are incompletely understood. Two other advantages of genomic scans are equally important. First, because signatures generated by selection are persistent, targets of historical selection can also be identified providing insights over evolutionary timescales. Second, genomic scans allow meaningful comparison of selection targets and overall selective landscapes between species.
The mode of natural selection can be inferred from patterns of sequence variation and the inferred mode of selection correlates with functional relevance of genes (Quintana-Murci and Clark 2013). Strong purifying selection characterizes molecules indispensable for functioning of the immune system, whereas weak purifying selection is a signature of functional redundancy (Barreiro et al. 2009; Harpur and Zayed 2013). Pathogens can exert directional pressures on host resistance molecules leaving signatures of positive selection in the genome. Most evidence comes from studies on primates and other mammals, where positive selection has been detected, e.g., in Toll-like receptor (TLR) genes (Barreiro et al. 2009; Wlasiuk and Nachman 2010), chemokine receptor genes (Metzger and Thomas 2010), defensins (Das et al. 2010) or genes involved in resistance to malaria (Hedrick 2011). However, the influence of pathogens can be more complex if adaptations are costly, pathogen genes respond to host evolution and vice versa, leading to coevolutionary arms-races, or if interactions with the host are temporary. Hence also balancing selection (BS) is expected to operate and maintain high variation in some immune genes through mechanisms of overdominance, negative frequency-dependent selection or temporally and spatially fluctuating selection. Some studies suggest a major role of BS in the evolution of immunity-related genes (reviewed in Azevedo et al. 2015; Těšický and Vinkler 2015) and good examples of genes under BS exist in a wide spectrum of taxa (Cagliani et al. 2008; Hedrick 2011; Linnenbrink et al. 2011; Tarazona-Santos et al. 2013). However, genome-wide scans, e.g., in humans identify relatively few examples (Leffler et al. 2013; DeGiorgio et al. 2014; Teixeira et al. 2015), and most evidence comes from MHC genes which are somewhat atypical with extreme and unambiguous signatures of long-term BS. It is thus possible that in any given species BS affects only a handful of genes.
In spite of a wide interest in adaptive evolution of immune genes, population genomic studies are underrepresented in nonmodel organisms including vertebrates (Boehm 2012). Studies looking at the impact of selection on vertebrate immune genes have focused so far on expression changes after pathogen infection (Savage et al. 2014), comparison of immune repertoires between vertebrate taxa (You et al. 2014) or inferences of selection based on sequence divergence between taxa (Ekblom et al. 2010). Despite the importance of the clawed frogs of genus Xenopus as a model for evolutionary and comparative immunobiology (Robert and Cohen 2011), patterns of variation and selective landscapes in amphibian immune genes are poorly known. This is unfortunate as amphibians are undergoing a huge biodiversity crisis caused in no small part by emerging infectious diseases, in particular chytridiomycosis (Fisher et al. 2012; Martel et al. 2014; Berger et al. 2016). Not all species (Martel et al. 2014) or even regional amphibian faunas (Bataille et al. 2013) appear equally vulnerable and variation in susceptibility clearly has a genetic component (Savage and Zamudio 2011; Bataille et al. 2015). As a consequence, there has been an interest in identifying targets of selection by experimental and comparative studies (Bataille et al. 2015; Ellison et al. 2015). However, population genomic analyses of selection acting on immune genes, which are invaluable for setting such efforts in a broader context, have lagged behind. The few studies performed so far have been limited in scope and focused on MHC genes (Nadachowska-Brzyska et al. 2012), antimicrobial peptides (Tennessen and Blouin 2008) and Toll-like receptor (TLR) genes (Babik et al. 2015).
Here, we examine sequence polymorphism and divergence in hundreds of genes involved in immune response and infer patterns of selection in two evolutionary lineages of Lissotriton newts. Lissotriton montandoni (Lm) and L. vulgaris graecus (Lvg) have allopatric distributions in the Carpathians and southern Balkans, respectively. There is little evidence for genetic exchange between them after the divergence dated to the Pliocene (Nadachowska-Brzyska et al. 2012; Pabijan et al. 2015). Lissotriton montandoni has however exchanged genes with other evolutionary lineages of L. vulgaris (Lv, Babik et al. 2005; Nadachowska-Brzyska et al. 2012; Zieliński et al. 2013, 2014, 2016). The higher MHC class II variation in Lm has been attributed to introgression from Lv (Nadachowska-Brzyska et al. 2012). Lm and Lvg show similar variation in TLR genes but selective regimes in these genes differ between the species with both purifying and positive selection stronger in Lvg (Babik et al. 2015). If TLRs are representative of other immune genes, we expect to find evidence for stronger overall selective pressures in Lvg than in Lm. We also predict that, because host–pathogen interactions may lead to the maintenance of polymorphism, targets of BS would be found among the analyzed genes. We aimed specifically to: (i) identify immune genes in Lissotriton transcriptomes with emphasis on single copy genes; (ii) examine levels of polymorphism within and divergence between Lm and Lvg using targeted resequencing; (iii) characterize patterns of selection and constraint in these genes within and between species; and (iv) identify targets of positive and balancing selection, whereas controlling for the confounding effects of demography.
Material and Methods
Identification of Immune Genes in the newt Transcriptome
Lissotriton Reference Transcriptome
Coding sequences (ORFs) of newt immune genes were identified in de novo assembled Lissotriton vulgaris reference transcriptome. Spleen (two individuals, 108 million (M) pairs of 100-bp Illumina reads) and liver (six individuals, 195 M pairs of 100 bp reads) transcriptomes were assembled separately with Trinity (release 2012-10-05). Both assemblies were merged and redundancy was removed following the approach of Stuglik et al. (2014). This reference transcriptome contained transcriptome-based gene models, i.e., ideally a single sequence per gene containing all its exons. The most recent assembly of the Lissotriton liver and spleen transcriptome is available at http://newtbase.eko.uj.edu.pl; last accessed October 6, 2016.
Database of Tetrapod Immune Proteins
A nonredundant set of tetrapod immune proteins was compiled as follows. The names and IDs of human genes involved in immune response (GO:000695 “Immune Response”) were obtained from Ensembl (release 69). This set was complemented with additional genes from the Immunome Knowledge Base (Ortutay and Vihinen 2009, http://structure.bmc.lu.se/idbase/IKB/; last accessed October 6, 2016) and InnateDB (Breuer et al. 2013, http://www.innatedb.com; last accessed October 6, 2016) databases giving the total of 2,037 genes. Xenopus tropicalis orthologs of these genes were identified in Ensembl yielding a list of 1,438 X. tropicalis gene IDs, which was augmented with 56 X. tropicalis genes from the ImmunomeKB and Ensembl. Additional 22 immune genes identified in the chicken genome and 5 genes identified in the Anolis lizard genome were also included. For each species, the longest protein per gene was retrieved from Ensembl and within species self BLASTp was used to remove the remaining redundancy: only one of the proteins exhibiting > 90% amino acid identity was retained. Datasets from the four species were merged so that a given set of putative orthologs was represented by X. tropicalis when available, otherwise by the human protein, and in a few cases, when no ortholog was found in human or X. tropicalis, by chicken or the Anolis lizard proteins. The final set, used as the database of tetrapod immune genes against which the newt reference transcriptome was searched, comprised 1,990 protein sequences. We used rather inclusive criteria for classifying genes as “immune”, as some genes assigned to the broad GO category “Immune response” may affect immune system only indirectly. However, such approach minimized the risk of missing genes important for the function of salamander immune system, which is poorly understood.
Identification of Putative Immune Genes in the Lissotriton Reference Transcriptome
Putative newt orthologs of the tetrapod immune genes were identified in the newt transcriptome as best reciprocal BLAST hits. BLASTx searches used newt transcripts as queries and tBLASTn searches used tetrapod immune proteins as queries, both at the E value threshold of 10−20. Only queries having a unique best hit in the database were retained. The hit was defined as unique if the bitscore (summed across all hsps) of the second best hit was not higher than 0.75 × bitscore value of the best hit. In such approach, most immune gene families comprising recently diverged members were probably excluded from the analysis. In principle, this is undesirable as many genes important for the immune response do form families characterized by a considerable rate of gene duplication. However, given limitations of the currently available technologies, large scale analysis of such gene families would not be possible without a high quality newt genome which is not available. Therefore, we focused on putatively single copy immune genes; 775 complete ORFs of the total length of 1.39 Mb were identified (supplementary table S1, Supplementary Material online). To reduce the target length <1 Mb, 141 genes were removed, yielding the final set of 634 putative newt immune genes which were analyzed via targeted resequencing. The probes for sequence capture were 80-bp biotinylated RNA oligonucleotides (MYcroarray) tiled every 48 bp on the ORF sequences.
Sequence Capture and Estimation of Polymorphism and Divergence
Samples
Sequence capture was performed on 25 individuals of L. montandoni (Lm) and 25 individuals of L. vulgaris graecus (Lvg); L. helveticus (Lh) was used as an outgroup (supplementary table S2, Supplementary Material online). Adult newts were sampled by dip-netting during breeding season. Animals were released after tailtips were collected. Tissue samples were stored in 95% ethanol until DNA extraction. DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega).
Intraspecific genetic structure may confound population genetic inferences if not taken into account. To minimize adverse effects of genetic structure sampling scheme should be carefully adjusted, because various sampling schemes may substantially affect measures of variation and shape of the site frequency spectrum (Stadler et al. 2009). Newts form discrete demes corresponding to breeding ponds, such demes undergo extinction and recolonization, and thus the regional population can be considered a metapopulation (Smith and Green 2005). It has been shown (Wakeley 2004) that if one gene copy per locus is sampled per deme in a metapopulation consisting of many demes, the ancestral process producing such sample is identical to the unstructured coalescent process, if time is rescaled appropriately. We approximated this optimal strategy by sampling each newt from a separate locality (supplementary table S2, Supplementary Material online).
Sequence Capture and Sequencing
Genomic DNA (5–6 μg) was sheared to ca. 200-bp fragments using Bioruptor Plus (Diagenode). Indexed genomic libraries were prepared with NEBNext DNA Library Prep Master Mix Set for Illumina (NEB) following the manufacturer’s protocol with modifications at the steps of size selection and library amplification (details in Supplementary Material online) and 10 PCR cycles. Sequence enrichment was performed using MYbaits Custom Target Capture Kit according to the manufacturer’s protocol with some modifications (details in Supplementary Material online). Capture products were sequenced on the Illumina MiSeq platform, producing 2 × 75 bp paired-end reads.
Mapping
Lm and Lvg reads were mapped separately to the reference ORFs (http://newtbase.eko.uj.edu.pl/; last accessed October 6, 2016) using Bowtie 2 (Langmead and Salzberg 2012). Local alignment was performed to exclude fragments of the reads derived from captured intronic sequences flanking the targeted exons. The following mapping settings were used: very-sensitive-local –no-unal –score-min G,20,10 -p 10. PCR duplicates were removed with Picard Tools and the remaining reads were realigned around indels with SAMtools (Li et al. 2009). For Lh, we mapped jointly liver RNAseq reads from a single individual (9.8 mln 2 × 100 bp Illumina reads) and sequence capture reads from two individuals. The Lh alignment was thus a composite of three individuals and comprised both cDNA and gDNA data. Coverage for each sample was calculated using BEDtools (Quinlan and Hall 2010). For Lm and Lvg samples, capture efficiency was calculated as the percentage of reads mapped to the target, specificity as the percentage of unique mapped reads and sensitivity as the percentage of target covered by at least one mapped read.
Sequence Variation and Divergence
Most measures of variation and divergence were estimated directly from genotype likelihoods bypassing the genotype calling step. Such approach is preferred when coverage is limited (Nielsen et al. 2012). Site frequency spectra (SFS) and various statistics described below were calculated with ANGSD (Korneliussen et al. 2014) and ngsTools (Fumagalli et al. 2014).
To identify putative paralogs each polymorphic position was tested, separately for Lm and Lvg, for an excess of heterozygotes compared with the Hardy–Weinberg expectations. To perform this test, we did call genotypes using ANGSD, but these genotypes were not used in any other analysis. Allele frequencies were estimated from SFS in ANGSD. Observed and expected heterozygosities were compared using the Fisher’s exact test. Genes showing an excess of heterozygotes at any position in any species at FDR 0.05 were discarded as paralogs.
Reference sequences of Lm, Lvg, and Lh were constructed to estimate sequence divergence, calculate mutation rates and identify ancestral variants. For each species, the reference sequence was built including in each position the major allele as identified by ANGSD. Divergence (dxy) was estimated as the per site number of differences between reference sequences. Per site mutation rate was estimated for each gene using the average divergence from the outgroup and Lh versus Lm/Lvg divergence time of 4.6 × 106 generations (Pabijan et al. 2015). Mutation rate was estimated for all sites as well as separately for 0- (ZFD) and 4-fold degenerate sites (FFD).
Genotype likelihoods were calculated with the Empirical Bayes approach and the SYK method in ANGSD as described in Kim et al. 2011, and the global, unfolded SFS was estimated using Lh reference sequence to represent the ancestral states. The only filtering options were minimum mapping quality of 1, and minimum base quality of 20. Per locus estimates of the following statistics were obtained: nucleotide diversity (π), total number of segregating sites within species (S), segregating sites private in each species (Sx), shared (Ss), fixed (Sf) sites, Tajima’s D, Fu and Li’s F, Fu and Li’s D, and Fst between species. This dataset, further referred to as ungenotyped, formed the basis of most analyses.
Identification of genes under BS (see below) involved the McDonald–Kreitman (MK) test. This test is normally performed on counts of synonymous and nonsynonymous polymorphisms and fixed differences obtained from called genotypes. A reasonable genotype quality requires substantial coverage so a large amount of potentially useable data is discarded if coverage is variable as was in our case. More precise information about polymorphism is available for ZFD and FFD sites but these are just approximation for synonymous and nonsynonymous sites. As neither of these approaches is ideal, we present results from both to extract as much information as possible from the MK test. Genotypes for each site were inferred using SAMtools mpileup and VCFtools, positions with the minimum genotype quality of 20 and minimum coverage 8× were retained and exported to fasta alignments. Numbers of segregating polymorphisms and fixed differences at synonymous and nonsynonymous positions were calculated using mstatspop (http://bioinformatics.cragenomica.es/numgenomics/people/sebas/software/software.html; last accessed October 6, 2016). We refer to this dataset as genotyped data. All statistics in this study were calculated for genes with at least 30 called bp in the category of interest (all sites, ZFD, FFD, synonymous and nonsynonymous sites).
Tests of Selection
Null Distributions of Statistics Used in Tests of Selection
Null distributions of statistics used in tests of selection were calculated from datasets simulated with ms (Hudson 2002) under the estimated demographic model (Supplementary Material online). A third population (Lh, of constant size 105) was added to our two-population divergence model to simulate divergence from an outgroup. We used the time of Lh divergence of 18.4 Ma (Pabijan et al. 2015) or 4.6 × 106 generations ago, assuming generation time of 4 years (Nadachowska and Babik 2009). For each locus, 104 datasets were simulated, separately for ZFD and FFD sites. In each simulation, mutation rate for each class of sites was randomly sampled from gamma distribution with mean and variance equal to the mean and variance of mutation rates across loci. All statistics were calculated with mstatspop.
Selection and Constraint in newt Immune Genes
Selection and constraint were assessed within species and compared between species using SnIPRE (Eilertson et al. 2012). This method estimates mutation rates, divergence, constraint and selection effects simultaneously using genome-wide data without referring to any specific demographic model. The method models parameters using counts from the MK table, i.e., PN, DS, PS and DN, where P and D denote the numbers of polymorphisms within species and fixed differences between species, whereas N and S refer to nonsynonymous and synonymous mutations, respectively. For this analysis, we used numbers of segregating sites and fixed differences from Lh estimated per gene at ZFD and FFD sites. The program was run in the fully Bayesian mode, with 105 iterations of the MCMC sampler after 2 × 104 iterations of burn-in and thinning parameter set to 4.
We also estimated the distribution of fitness effects of nonsynonymous mutations using the method of Eyre-Walker et al. (2006), which explores information from synonymous and nonsynonymous allele frequency spectra. In this approach, all nonsynonymous mutations are assumed to be deleterious, and the strength of purifying selection has gamma distribution. No underlying demographic model is assumed, any effect of demographic change on frequency spectra influences synonymous and nonsynonymous sites identically. We used estimates of site frequency spectra in ZFD and FFD sites across all genes. The analysis was run for 106 steps with a burn-in of 105.
Identification of Genes under Balancing Selection
We devoted special attention to identification of putative targets of BS because this mode of selection plays an important role in shaping variation of immune genes. Signatures left in genetic data by BS are difficult to identify unambiguously as they can be confounded with other forms of selection, nonequilibrium demography or can result from stochasticity of the genealogical process (Fijarczyk and Babik 2015). Scans for BS should therefore rely on statistics or composite tests which simultaneously consider different lines of evidence. We performed two composite tests to identify candidate BS genes, each separately for Lm and Lvg. The final set of candidates is the sum of those identified by test 1 and test 2. We tested for enrichment of GO categories among candidate genes using GOrilla (Eden et al. 2009).
Test 1 identified genes showing an excess of nonsynonymous polymorphisms segregating at intermediate frequencies which is expected under BS. Hence two conditions had to be met simultaneously: (i) an excess of nonsynonymous polymorphisms and (ii) no excess of rare nonsynonymous polymorphisms (this removes the effect of slightly deleterious variants segregating at low frequencies). The MK test was performed to evaluate the first condition. Its result can be summarized with a neutrality index, NI = PN/PS× DS/DN. A significant MK test with NI > 1 indicates an excess of nonsynonymous polymorphisms and thus either balancing or purifying selection. The MK test was conducted on genotyped and ungenotyped data and its statistical significance was assessed with the Fisher’s exact test. The second condition was imposed by filtering out genes with significantly negative Tajima’s D in ZFD sites. Results from test 1 on ungenotyped data were compared with an alternative approach in which low-frequency variants (singletons and doubletons) were removed prior to the MK test. We observed large overlap between the approaches and thus present only the results of test 1. Test 2 identified genes with a high ratio of polymorphism to divergence, showing at the same time an excess of polymorphisms segregating at high frequencies. Thus, candidates identified in test 2 were those genes which showed significantly high π/dxy ratio and significantly positive values of any of the three statistics: Tajima’s D, Fu and Li’s F or Fu and Li’s D. P values of all statistics were determined with 104 coalescent simulations as described above. Test 2 was conducted separately for ZFD and FFD sites.
We further evaluated evidence for BS in candidate genes by analyzing patterns of variation in high quality resequencing data from population samples and comparing them with control genes (CR) randomly selected from the transcriptome. One Lm and one Lvg population sample (19 individuals each) were analyzed. Resequencing was performed with Molecular Inversion Probes (MIP, Niedzicka et al. 2016, supplementary table S3, Supplementary Material online). MIPs were designed from transcripts with exon boundaries identified through BLAST searches against X. tropicalis gene models (Niedzicka et al. 2016). Because many vertebrate exons are short, to maximize the length of the target available for MIP design in candidate BS genes, sequence capture reads were assembled into contigs using Trinity (release v2.0.4). Such contigs contained exons and flanking intron fragments (see Supplementary Material online). One to three nonoverlapping MIPs (112–336 bp) per gene were resequenced in 88 CR genes (175 MIPs, 19.6 kb). We aimed to cover as much ORFs of BS genes as possible and finally resequenced 270 MIPs (18.4 kb) in 32 candidate genes. Reads were mapped to their respective references (i.e., transcripts or sequence capture contigs) using bwa-mips (Pedersen 2014). Reads mapped to sequence capture contigs were extracted and mapped back to the ORFs using local alignment in Bowtie 2. Within-gene alignments were subsequently merged. MIPs with coverage ≥ 12× in at least 85% of individuals in a population were considered successfully resequenced. Genotypes were called using GATK UnifiedGenotyper (McKenna et al. 2010) with the following settings: -T UnifiedGenotyper -dfrac 1 -pcr_error 5.0E-3 -mbq 20 -stand_call_conf 20.0 -out_mode EMIT_ALL_SITES -gt_mode DISCOVERY. Haplotypes were inferred with PHASE (Stephens and Donnelly 2003) and unphased positions were resolved randomly. Positions with > 25% missing data per population were removed. We applied stringent filters to remove paralogs which might have escaped primary quality filters in sequence capture data. Firstly, the number of heterozygotes within populations was compared with the Hardy–Weinberg expectation using the Fisher’s exact test, and genes with excess of heterozygotes significant at FDR 0.05 were flagged as putative paralogs. Secondly, because all positions within a single MIP target have very similar coverage, presence of more than two alleles per individual could be detected by looking at differences in allele counts between polymorphic positions located in a single MIP. Significant differences in allele counts indicate the difference in the number of gene copies between positions and thus paralogy. Thirdly, MIPs were also resequenced in two laboratory hybrid families (L. montandoni/L. vulgaris F1 hybrids and 111/114 F2 progeny in two families) to test for Mendelian segregation. The presence of positions departing from Mendelian expectations at FDR 0.05 and showing an excess of heterozygotes was considered a sign of paralogy. Taken together, these paralog filters were very strict with a potential to remove, e.g., genes showing even modest copy number variation. These strict filters were necessary to exclude as many putative paralogs as possible, as they are likely to generate false signals of BS.
Tajima’s D, π, dxy, number of segregating and shared polymorphisms were calculated in mstatspop. Sliding window estimates of Tajima’s D along genes and number of haplotypes were obtained in DnaSPv5 (Librado and Rozas 2009). To compare selective pressures between species in BS genes, we used a population genetics-phylogenetics gammamap method which relies on information about polymorphism within and divergence between species (Wilson et al. 2011). The method infers distribution of fitness effects (DFE) of nonsynonymous mutations within species as well as variation in selection coefficients along genes. Priors of model parameters were set as in Wilson et al. (2011). Each lineage (Lm, Lvg and Lh) was assigned a separate prior for sliding window smoothing parameter (p), transition:transversion ratio (κ), branch length (T) and DFE. κ and T had improper log-uniform priors and p had uniform prior within interval (0, 1). Uninformative, symmetric Dirichlet prior was set on DFE, with 12 equally likely classes corresponding to different levels of fitness, from inviable to highly beneficial (γ = −500, −100, −50, −10, −5, −1, 0, 1, 5, 10, 50, 100). Log-normal priors were set on population-scaled mutation rate (θ) for each lineage and each gene, with hyperparameters mean μ (improper uniform prior) and standard deviation σ (log-normal prior with mean 0 and standard deviation 2). Posterior probabilities of population-scaled selection coefficient γ (γ =P*Ne*s, where P is ploidy, Ne is effective population size and s is selective advantage of a derived nonsynonymous mutation) were collected from 4 MCMC runs, each 2 × 106 steps long excluding 5 × 105 steps of burn-in. Separate chains were investigated for convergence, and final estimates were derived from merging all runs together. Codons in which posterior probability of γ ≥ 1 was ≥ 0.75 were considered under positive selection.
Results
Sequence Capture and Patterns of Variation
The ORFs of 634 immunes genes used for sequence capture ranged from 300 to 10,671 bp, averaging 1,560 ± (SD) 1,055 bp; the total target length was 989.3 kb (supplementary table S1, Supplementary Material online). The average capture efficiency was 46% ± 8.2 (supplementary table S2, Supplementary Material online). Capture specificity, i.e., the percentage of unique reads on target was 21 ± 3.1%, and capture sensitivity, i.e., the fraction of the target covered by at least one read was 93 ± 1.8% (supplementary table S2, Supplementary Material online). Median coverage after discarding duplicates was as high as 11 ± 3.3× (mean 38.5 ± 8.9×), we noted however a substantial variance in coverage along the reference, probably due to variation between probes in capture efficiency (supplementary table S4, Supplementary Material online). Lh alignment contained reads from three individuals and included RNAseq data, thus the total coverage was high, with a median of 39× and 99% the target covered. SNPs with an excess of heterozygotes were found in 109 Lvg and 120 Lm genes, mostly common to both species. These 135 putative paralogs were excluded from further analyses. Two additional genes were excluded due to the lack of outgroup information.
Patterns of diversity were thus examined in 497 putative single copy genes (fig. 1). Average nucleotide diversity (π) was comparable in the two species at all sites (πLm = 0.0061 ± 0.0049, πLvg 0.0060 ± 0.0044) as well as at ZFD and FFD sites (fig. 1a). This agreed with similar long-term effective population sizes of both species inferred from the demographic model (fig. 2 and supplementary tables S5a and c, Supplementary Material online). The average divergence (dxy) from the outgroup was ca. 2% (fig. 1b). Tajima’s D was negative in both species at all site classes (fig. 1c), which may be attributed to population expansions. Tajima’s D was 3 times lower in Lvg (DLvg = −0.67 ± 0.74, DLm = −0.22 ± 0.83) and private polymorphisms (Sx) were more numerous in this species (fig. 1d). The inspection of SFS indicates that the surplus of polymorphisms in Lvg can be attributed solely to singletons (fig. 2). Moreover, interspecific differences in Tajima’s D and in the number of Sx were most pronounced at ZFD sites (fig. 1c and d). These patterns are consistent with stronger demographic expansion in Lvg suggested by the demographic model and resulting differences in intensity of purifying selection. At all site classes, shared polymorphisms (Ss) were over 4 times more abundant than fixed differences (Sf) (fig. 1d). Since we found no evidence of post-divergence gene flow between Lvg and Lm (supplementary fig. S2 and supplementary tables S5a and c, Supplementary Material online), Ss are likely ancestral polymorphisms or independent, recurrent mutations. The fraction of shared polymorphisms was a little lower in ZFD than in FFD sites, consistent with purifying selection on nonsynonymous variants. No significant differences in diversity were found between genes involved in innate (GO:0045087) and adaptive (GO:0002250) immune response (supplementary table S6, Supplementary Material online).
Fig. 1.—

Diversity and divergence of 497 newt immune genes estimated for all, 4-fold (FFD) and 0-fold degenerate sites (ZFD). (a) Nucleotide diversity (π), (b) divergence from the outgroup (dxy), (c) Tajima’s D, (d) number of segregating sites (S, gray) and fixed substitutions (Sf, black) between Lissotriton montandoni (Lm) and L. vulgaris graecus (Lvg). Boxplots show medians with upper and lower quartiles. Whiskers indicate the most extreme values which do not exceed the quartile value plus the 1.5 times the interquartile. The outliers are marked as empty dots.
Fig. 2.—
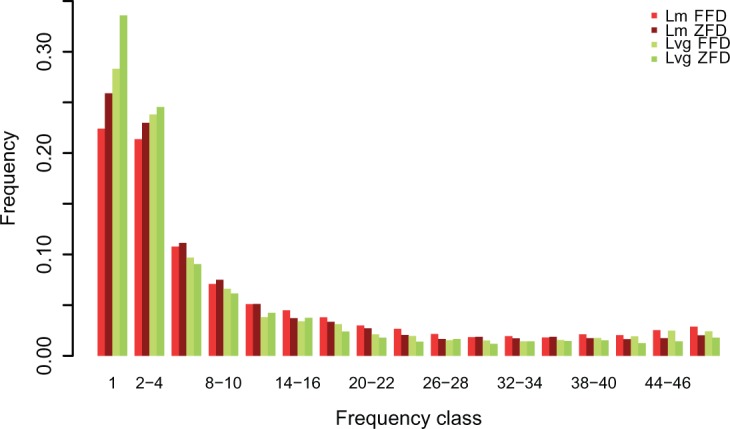
Maximum likelihood estimates of site frequency spectra at 4-fold (FFD) and 0-fold (ZFD) degenerate sites in Lm and Lvg. Each bin, except the first (singletons), sums up three consecutive frequency classes.
Selection Landscapes in newt Immune Genes
Using the MK-like approach in SnIPRE to estimate selection and constraint, we found evidence for the dominant role of purifying selection in newt immune genes. The selection effect allows to detect positive selection or negative selection acting on mildly deleterious alleles. Most genes were classified as neutral while the remaining 20% in Lm and 40% in Lvg appeared to evolve under negative selection, suggesting that selection on mildly deleterious mutations affects twice as many genes in Lvg as in Lm (fig. 3a and b). Consequently, genome-wide estimates of population selection coefficient were significantly more negative in Lvg (γLvg = −0.48 ± (SD) 0.28, γLm = −0.33 ± 0.27; Mann–Whitney paired test, P < 2.2 × 10−16). When the distribution of selection effect was taken at the face value, no gene appeared under positive selection. However, the power to detect such genes can be significantly reduced due to genome-wide effects of negative selection. It is therefore justified to compare gene-specific selection effects relative to the genome-wide average (Eilertson et al. 2012). Lower bounds of selection effect were above the genome-wide average for 12 Lm and 16 Lvg genes, which are thus plausible candidates for positive selection (fig. 3a and b and supplementary table S7, Supplementary Material online). The constraint effect, a function of the fraction of nonlethal nonsynonymous mutations (f) and the population selection coefficient, is useful to identify cases of strong purifying selection. In both species, the constraint effect was negative in almost all genes, suggesting widespread purifying selection against strongly deleterious mutations (fig. 3c and d). The average proportion of nonlethal mutations was slightly but significantly lower in Lm (fLm = 0.23 ± (SD) 0.13, fLvg = 0.26 ± 0.13; Mann–Whitney paired test, P = 1.2×10−14) and did not differ between genes involved in innate and adaptive immune response (supplementary table S6, Supplementary Material online).
Fig. 3.—

Selection and constraint effects in 497 newt immune genes. (a) and (b) sizes of selection effect in Lm and Lvg, respectively, sorted in the increasing order. Black line follows the average, vertical lines span the Bayesian credibility intervals. Dashed line indicates neutrality, red line indicates genome-wide average. Genes with negative selection effect are marked in blue. Genes with selection effect significantly larger than the genome-wide average are shown in orange. (c) and (d) selection versus constraint effect in Lm and Lvg, respectively.
The distributions of fitness effects estimated for both species were very similar, as shown by the almost identical shape parameters of the gamma distribution (Lm: 0.12 [CI: 0.11–0.13], Lvg: 0.11 [0.11–0.13]). The most abundant class comprised sites under very strong negative selection (Nes > 100, fig. 4) reflecting high constraint identified in the SniPRE analysis (fig. 4c and d). Although the 95% credibility intervals for the two species overlap in all selection classes, we note an apparent shift towards sites under very strong selection (Nes > 100) in Lm, and towards weakly and moderately selected sites in Lvg (fig. 4). This is consistent with more widespread weak purifying selection in Lvg, but higher constraint in Lm inferred by SnIPRE.
Fig. 4.—
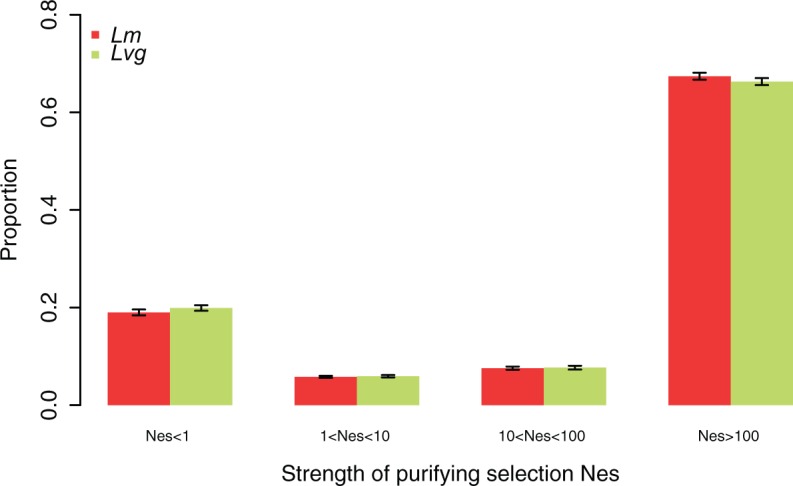
The distribution of fitness effects of deleterious mutations in Lm and Lvg estimated using the method of Eyre-Walker et al. (2006); 95% credible intervals are shown.
Targets of Balancing Selection
Putative targets of BS were identified using two composite tests. Test 1 identified genes with an excess of nonsynonymous polymorphisms segregating at appreciable frequencies. Test 2 identified those with an excess of polymorphism compared with divergence and the SFS skewed towards more frequent variants. Test 1 revealed 15 candidates in Lm and 19 in Lvg, 5 of them were common to both species (table 1 and supplementary table S8, Supplementary Material online). Test 2 detected 7 candidate genes in Lm and only one in Lvg, all species-specific. In total, we found 35 candidates for BS, 20 genes in each species, but only 5 common to both species (table 1).
Table 1.
Putative Targets of Balancing Selection
| Test 1 |
Test 2—ZFD |
Test 2—FFD |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Species | MK1 + TD | MK2 + TD | TajD0 | FuF0 | FuD0 | π/dxy0 | TajD4 | FuF4 | FuD4 | π/dxy4 |
| AQP9 | Lm | 0.28 | NA | 0.05 | 0.02* | 0.11 | <1E-4* | 0.31 | 0.69 | 0.8 | 0.11 |
| BCL3 | Lm | 0.03* | 0.76 | 0.83 | 0.95 | 0.95 | 0.32 | 0.69 | 0.82 | 0.82 | 0.87 |
| CYLD | Lm | 0.02* | 0.06 | 0.75 | 0.79 | 0.75 | 0.02* | 0.46 | 0.8 | 0.85 | 0.94 |
| ELK1 | Lm | NA | NA | 0.66 | 0.47 | 0.42 | 0.3 | 0.62 | 0.15 | 0.02* | 0.02* |
| EPS8 | Lm | 0.25 | 0.03* | 0.39 | 0.38 | 0.42 | 0.02* | 0.90 | 0.84 | 0.73 | 0.57 |
| IL17RD | Lm | 0.02* | 0.21 | 0.63 | 0.68 | 0.68 | 0.02* | 0.50 | 0.09 | 0.02* | 0.63 |
| IL28RA | Lm | NA | NA | 0.22 | 0.03* | 0.02* | 0.22 | 0.20 | 0.05* | 0.05* | 0.03* |
| NBN | Lm | 0.1 | 0.03* | 0.56 | 0.52 | 0.49 | 0.48 | 0.51 | 0.52 | 0.52 | 0.88 |
| NFE2L2 | Lm | 0.17 | 0.01* | 0.83 | 0.97 | 0.98 | 0.18 | 0.43 | 0.4 | 0.43 | 0.7 |
| PAFAH1B1 | Lm | 4E-3* | 0.04* | 0.84 | 0.87 | 0.83 | <1E-4* | 0.14 | 0.21 | 0.38 | 0.24 |
| PRDX6 | Lm | 0.04* | 4E-4* | 0.67 | 0.68 | 0.63 | 0.08 | 0.40 | 0.29 | 0.3 | 0.46 |
| PRELID1 | Lm | 0.23 | 0.45 | 0.93 | 0.39 | 0.04* | 0.01* | 0.95 | 0.83 | 0.7 | 0.45 |
| RNF41 | Lm | 5E-3* | 0.22 | 0.52 | 0.8 | 0.82 | NA | 0.31 | 0.38 | 0.44 | 0.27 |
| SEC61A1 | Lm | 0.01* | 1 | 0.43 | 0.11 | 0.04* | <1E-4* | 0.27 | 0.06 | 0.04* | 0.01* |
| SIGLEC1 | Lm | 0.01* | 0.51 | 0.88 | 0.76 | 0.63 | 0.02* | 0.67 | 0.7 | 0.68 | 0.31 |
| SOCS3 | Lm | 0.12 | 0.03* | 0.64 | 0.68 | 0.68 | 0.65 | 0.26 | 0.18 | 0.21 | 0.96 |
| SOD1 | Lm | 0.01* | 0.11 | 0.71 | 0.72 | 0.66 | 0.71 | 0.81 | 0.65 | 0.5 | 0.96 |
| TAX1BP1 | Lm | 0.82 | 0.35 | 0.10 | 0.03* | 0.06 | 0.04* | 0.23 | 0.24 | 0.35 | 0.07 |
| UBA52 | Lm | 0.02* | 1 | 0.33 | 0.06 | 0.01* | <1E-4* | 0.26 | 0.02* | 1E-3* | 0.33 |
| USP7 | Lm | 1E-3* | 1 | 0.62 | 0.55 | 0.50* | 0.01* | 0.68 | 0.41 | 0.28 | 0.78 |
| AAMP | Lvg | 0.04* | NA | 0.44 | 0.58 | 0.59 | <1E-4* | 0.73 | 0.38 | 0.21 | 0.7 |
| AKIRIN2 | Lvg | 0.6 | NA | 0.05* | 0.03* | 0.14 | <1E-4* | 0.28 | 0.22 | 0.23 | 0.09 |
| APP | Lvg | 0.02* | 1 | 0.75 | 0.7 | 0.63 | NA | 0.54 | 0.69 | 0.71 | 0.66 |
| CANX | Lvg | 0.35 | 0.05* | 0.38 | 0.5 | 0.56 | 0.04* | 0.25 | 0.11 | 0.1 | 0.22 |
| CD40LG | Lvg | 0.09 | 0.04* | 0.65 | 0.66 | 0.64 | 0.11 | 0.71 | 0.37 | 0.25 | 0.86 |
| CEBPG | Lvg | 0.04* | 0.56 | 0.94 | 0.97 | 0.96 | NA | 0.70 | 0.85 | 0.85 | 0.3 |
| GP5 | Lvg | 0.02* | 0.12 | 0.33 | 0.16 | 0.15 | 0.11 | 0.31 | 0.2 | 0.2 | 0.74 |
| HSF1 | Lvg | 0.03* | 0.07 | 0.70 | 0.62 | 0.54 | 0.07 | 0.85 | 0.54 | 0.31 | 0.86 |
| ITFG1 | Lvg | 0.01* | NA | 0.62 | 0.65 | 0.63 | <1E-4* | 0.52 | 0.52 | 0.51 | 0.72 |
| KLHL6 | Lvg | 0.03* | 0.3 | 0.85 | 0.66 | 0.51 | NA | 0.23 | 0.08 | 0.09 | 0.02* |
| OTUD5 | Lvg | 0.05* | 0.01* | 0.46 | 0.48 | 0.46 | 0.36 | 0.22 | 0.39 | 0.5 | 0.83 |
| PAMR1 | Lvg | 0.02* | 0.45 | 0.80 | 0.96 | 0.96 | 0.06 | 0.65 | 0.51 | 0.43 | 0.78 |
| PRDX6 | Lvg | 0.22 | 0.02* | 0.29 | 0.58 | 0.67 | 0.07 | 0.35 | 0.27 | 0.29 | 0.22 |
| SEC61A1 | Lvg | 1E-3* | 0.51 | 0.76 | 0.28 | 0.11 | <1E-4* | 0.73 | 0.42 | 0.25 | 0.1 |
| SIGLEC1 | Lvg | 4E-3* | 0.13 | 0.93 | 0.96 | 0.95 | 0.05* | 0.68 | 0.55 | 0.46 | 0.62 |
| SQSTM1 | Lvg | 0.57 | 0.02* | 0.16 | 0.23 | 0.32 | 0.31 | 0.62 | 0.68 | 0.65 | 0.83 |
| TAP1 | Lvg | 0.02* | 0.2 | 0.79 | 0.85 | 0.82 | 0.01* | 0.44 | 0.14 | 0.08 | 0.03* |
| TRIM32 | Lvg | 0.01* | 0.03* | 0.89 | 0.96 | 0.95 | 0.01* | 0.68 | 0.71 | 0.67 | 0.3 |
| UBA52 | Lvg | 4E-4* | <1E-4* | 0.74 | 0.96 | 0.96 | <1E-4* | 0.94 | 0.95 | 0.87 | 0.93 |
| USP7 | Lvg | 1E-4* | 1 | 0.76 | 0.79 | 0.74 | <1E-4* | 0.78 | 0.68 | 0.56 | 0.95 |
Note.—List of genes identified with composite tests 1 or 2 as targets of balancing selection in L. montandoni (Lm) and L. v. graecus (Lvg). Test 1 identified genes showing an excess of nonsynonymous polymorphisms segregating at intermediate frequencies: a combination of significant McDonald–Kreitman test conducted on ungenotyped (MK1) or genotyped (MK2) data and not significant or significantly positive Tajima’s D (TD). Test 2 identified genes with high ratio of polymorphism to divergence showing an excess of polymorphisms segregating at high frequencies: a combination of increased polymorphism-to-divergence ratio (π/dxy) and significantly positive Tajima’s D (TajD) or Fu and Li’s F (FuF) or Fu and Li’s D (FuD). Test 2 was performed for 0-fold degenerate (ZFD, 0) and 4-fold degenerate (FFD, 4) sites.
P values in bold indicate test combinations determining inclusion of a particular gene as candidate. Symbols of genes which were confirmed as single-copy are in bold.
NA, not applicable.
To further validate the candidates identified by the composite tests, we resequenced 32 candidate BS and 88 control (CR) genes in population samples of 19 individuals for each species and also checked for Mendelian segregation in families. Balancing selection causes high diversity and excess of variants at intermediate frequencies, the pattern expected also if similar paralogs were lumped together. Hence paralogs overlooked at earlier steps of quality control were likely be overrepresented among BS candidates. Indeed, using stringent paralog filters, we found that as much as 66% candidate BS genes were actually similar paralogs, over twice as many as CR genes (29%). After paralog removal, the dataset included 11 BS candidates (9.1 kb resequenced) and 57 CR genes (11.1 kb resequenced). We compared Tajima’s D and the ratio of polymorphism to divergence between these two gene sets to see whether the results from species-wide sequence capture data hold in resequenced population samples. Seven BS candidates were identified as such only in Lm, three only in Lvg and one in both species, but below we used all 11 BS candidates in each species.
In Lm, Tajima’s D was positive and significantly higher in BS candidates than in CR genes, both at synonymous and nonsynonymous sites (fig. 5a; Mann–Whitney test, P = 0.02). On the contrary, in Lvg Tajima’s D did not differ between BS candidates and CR genes (Mann–Whitney test, P > 0.05). Other tests based on SFS revealed a similar pattern (supplementary fig. S3a and b, Supplementary Material online). The π/dxy ratio at nonsynonymous sites was significantly higher in BS candidates in both species (Mann–Whitney test PLm = 0.02, PLvg = 0.04, fig. 5b). The similar tendency was also seen for synonymous sites but was not significant (fig. 5b). Thus, the candidate genes indeed exhibit on average more even SFS and excess of nonsynonymous polymorphism to divergence. None of the GO categories were enriched in candidate genes.
Fig. 5.—

Comparison of Tajima’s D (a) and π/dxy (b) between control genes (CR) and balancing selection candidates (BS). Medians of statistics are compared between BS and CR genes, separately in synonymous (S), nonsynonymous (NS) sites in Lm and Lvg.
Looking at BS candidates individually (supplementary table S9 and supplementary figs. S4 and S6, Supplementary Material online), we observed segregating nonsynonymous polymorphisms in all but one gene (USP7, 29% of ORF resequenced), but few nonsynonymous polymorphisms were shared between species. In all but three cases, Tajima’s D in nonsynonymous positions was positive with most prominent examples being SQSTM1 (1.72; 17 nonsynonymous polymorphisms—SN), NFE2L (1.64; SN = 11), AQP9 (1.29; SN = 2) and NBN (1.24; SN = 9). If BS maintains nonsynonymous polymorphisms, such codons may be detected as significant but polymorphic by methods searching for targets of positive selection. In NBN, gammamap method inferred three adjacent codons under positive selection segregating in Lm (codon 558 segregates also in Lvg and in codon 553 three nonsynonymous variants segregate), and in SQSTM1 were four such codons segregating in Lvg (supplementary table S10, Supplementary Material online). These codons fall in the regions of positive Tajima’s D (fig. 6a and b) and may be actual targets of BS. An exceptionally high ratio of nonsynonymous to synonymous diversity (2.4) was found in CD40LG (supplementary table S9, Supplementary Material online).
Fig. 6.—

Tajima’s D estimated in 100-bp sliding windows with a step of 25 bp for two genes, NBN (a) and SQSTM1 (b). Asterisks indicate location of segregating codons under selection. Vertical gray lines separate sequenced regions belonging to distinct exons.
Discussion
Selection Landscape and Targets of Positive Selection
In this first population-genomic study of amphibian immune genes, we examined over 600 genes in two evolutionary lineages of Lissotriton newts, describing variation and inferring selective pressures. Variation in newt immune genes has been shaped predominantly by widespread purifying selection and strong evolutionary constraint, implying long-term importance of these genes for functioning of the immune system. This prevalent constraint notwithstanding, we identified 23 candidates for positive selection and 11 genes possibly evolving under BS.
Genome scans use diverse approaches to identify targets of positive selection and often find immune genes among the candidates, but overlap between candidates detected in various studies is limited (Clark et al. 2003; Kosiol et al. 2008; Leffler et al. 2013). In newts, a large proportion of immune genes, similarly to most protein-coding genes in vertebrates, evolve under purifying selection and many genes are strongly constrained. Distribution of fitness effects of nonsynonymous mutations in newt immune genes is skewed towards strongly deleterious and lethal, similarly to protein-coding genes in chimpanzees (Bataillon et al. 2015) and murid rodents (Halligan et al. 2013). In the face of high constraint, positive selection is expected to be limited to a small number of genes and codons, decreasing the power to detect such signatures. Indeed, candidates for positive selection were identified in this study only using the average genome-wide selection effect as the baseline and should not be considered as strongly supported. We identified 16 outliers in Lvg, and 12 outliers in Lm; these are genes which exhibit significantly higher positive selection effects than average genes.
The candidates for positive selection carry out various immune functions and are not restricted to the components of the adaptive immune system. Assuming functions in newts similar to their mammalian counterparts, candidate genes participate in innate pathways, such as the complement cascade (CFI, C4A, C9), Fc epsilon receptor (FCERI) signaling (CSF2RB, FOS, IL2RB), Toll-like receptor cascades (FOS), cytosolic sensing of pathogen-associated DNA (ZBP1), as well as pathways connected with the adaptive immune system (CD226). Several of the candidate genes sense and directly fight pathogens. Two examples include nucleoporins encoded by NUP153 and NUP214. Nucleoporins are part of the nuclear pore complex, which modulates transcript transport between cell and nucleus upon contact with a virus. Another pathogen-interacting protein is the collectin sub-family member 12 (COLEC12). COLEC12 is a cell surface glycoprotein and a scavenger receptor, which binds and directs bacteria and yeast for phagocytosis (Ohtani et al. 2001). Z-DNA binding protein 1 (ZBP1) is a cytosolic sensor of pathogen DNA, triggering cytokine production. Finally, there are complement component 9 proteins (C9) which do not directly identify microbes, but constitute part of the membrane attack complex destroying bacteria. The complement complex is part of the innate immune system, which initiates a cascade of immune response at the site of infection and activates a mechanism for destroying the pathogen. Interestingly, several members of the complement complex have already been recognized as targets of positive selection in mammals (Kosiol et al. 2008; Webb et al. 2015), including two genes that were among candidates also in newts: CFI and C9. Linking specific molecular adaptations with immunity to pathogens is essential for planning conservation actions in amphibians in the face of quickly spreading emerging diseases (Martel et al. 2014). This could be done by looking at associations between allelic variation and prevalence of various pathogen species and also by examining changes in gene expression level following the pathogen challenge in experimental conditions.
A clear advantage of our study lies in examination of a large number of immune genes, which increases the probability of finding targets of positive selection. However, many immune genes form families characterized by high rates of genomic duplication, which are technically challenging to analyze in nonmodel species without sequenced and well-assembled genomes (Larsen et al. 2014). Although we intended to focus on single-copy genes, >20% of studied genes turned out to have closely related paralogs in the newt genome and had to be excluded from the analyses. Gene duplication is an important source of novel or refined functions in the immune system (Huang et al. 2008; Vilcinskas 2013). Novel gene copies released from the constraint of purifying selection, readily acquire molecular changes which can be of evolutionary importance (Hellgren and Ekblom 2010). Therefore, we suspect that the amount of adaptive evolution found in this study could have been underestimated due to exclusion of families of closely related paralogs. Several immune gene families are characterized by high levels of gene duplication, e.g., interferons, defensins, interleukins, chemokine receptor proteins, and their members were shown to follow different evolutionary trajectories, including species-specific adaptations (Metzger and Thomas 2010; Manry et al. 2011; Vasseur et al. 2012). In addition, our analysis of Toll-like receptors in Lissotriton newts found species-specific signatures of positive selection in at least three members of this family (Babik et al. 2015). A group of genes important in amphibian immune defense but missing from this study are antimicrobial peptides (AMPs). The numerous AMPs apparently play an essential role in amphibian immunity against fungi and bacteria and may be important targets of positive selection (Tennessen et al. 2009; Roelants et al. 2013). Unfortunately, there is only very little information about AMPs in urodeles (Meng et al. 2013).
Interaction of Demography and Selection
The two evolutionary lineages differ in the overall strength of purifying selection acting on immune genes. On the one hand, more genes are under purifying selection in Lvg, but on the other, strong purifying selection appears more prevalent in Lm. The former is in line with larger current effective population size in Lvg, which makes selection against mildly deleterious mutations more efficient (Gazave et al. 2013). The latter observation is however difficult to explain by simple differences in Ne between the two lineages unless SnIPRE erroneously interprets patterns of nonsynonymous polymorphism as evidence for stronger constraint in Lm. One possibility is that indeed immune genes are more constrained in Lm, e.g., due to stronger long-term pathogen pressure. It is also possible that nonsynonymous polymorphisms maintained by BS may mislead inferences. There are however other plausible explanations not necessarily involving differences in selection regimes. Inferences of selection from patterns of molecular variation in nonequilibrium populations are complicated by the differences between neutral sites and those under purifying selection in the time needed for reaching an equilibrium (Brandvain and Wright 2016). Moreover, if demographic expansion is accompanied by spatial expansion, as was likely in Lvg (Pabijan et al. 2015), deleterious mutations can increase in frequency forming the so called expansion load (Peischl and Excoffier 2015) further complicating inferences. Clearly joint reconstruction of demography and selection, a subject of intensive ongoing work (Li et al. 2012), would be desirable to disentangle the effect of nonequilibrium demography from actual differences in selection regimes.
Targets of Balancing Selection
We put special emphasis on identification of possible targets of BS among newt immune genes. Variation in such genes, often involved in host–pathogen coevolution, may be of considerable adaptive significance and understanding molecular underpinnings of amphibian resistance is key to planning conservation strategies. Detection of BS is however not straightforward, except in cases when it maintains extreme variation and transspecies polymorphism as in MHC genes. Often genetic signatures left by this mode of selection are subtle and easily confounded by various nonselective processes (Fijarczyk and Babik 2015). Here identification of outliers was based on composite tests employing the reconstructed demographic model and the results were validated by comparing variation in candidate genes with that of control genes in independent population samples. The use of composite tests should decrease the rate of false positives due to multiple tests; unfortunately it is unclear how to calculate false discovery rate in this case.
Altogether 11 candidates for BS were detected, mostly encoding proteins that may interact closely with pathogens. Sequestosome-1 (SQSTM1) is an adaptor protein which recognizes ubiquitin coated proteins and targets them for degradation through autophagy. This mechanism can be used by the cell to eliminate intracellular bacteria or virus RNA (Levine et al. 2011). Nibrin (NBN) forms part of the complex that is responsible for the recognition and repair of DNA double stranded breaks, but some studies show that it can be used for the benefit of a virus for its efficient replication (Anacker et al. 2014). The ubiquitin-specific protease encoded by USP7 interacts with herpes simplex virus type-1 immediate early protein ICP0 through processes of deubiquitinization and ubiquitinization. Expression of ICP0 is necessary for reactivation of viral genome and is stabilized via recruiting USP7 to remove ubiquitin (Antrobus and Boutell 2008). Suppressor of cytokine signaling 3 (SOCS3) is another protein, which can be exploited by pathogens. By boosting expression of SOCS3, viruses downregulate cytokines and inhibit proinflammatory response of the host, facilitating their replication (Okumura et al. 2015). Another interesting gene APP encodes amyloid beta (A4) precursor protein. APP is cleaved to form a number of amyloid peptides, which are known to be associated with many human diseases, however some beta amyloid products exhibit antimicrobial properties similar to antimicrobial peptides (Wang et al. 2012). Interestingly, another candidate gene, ELK1 encodes a transcription factor, which inhibits transcription of genes responsible for processing of APP proteins (Besnard et al. 2011). Although ELK1 and APP were identified as BS candidates in different species (Lm and Lvg, respectively), nevertheless signatures of selection within these two genes suggest that production of amyloid peptides can be influenced by interaction with pathogens. Signatures of BS were also detected in aquaporin 9 (AQP9), a water-selective membrane channel responsible for remodeling of cell shape. Bacteria induce expression of AQP9 leading to changes in size and morphology of macrophages, which affects the outcome of infection (Holm et al. 2015). The Red Queen hypothesis predicts that co-evolutionary arms race between host and pathogen molecules should involve mostly host genes responsible for sensing pathogens. We found examples of genes involved in pathogen recognition among candidates for BS (APP, SQSTM1) and positive selection (e.g., COLEC12, ZBP1). However, among BS candidates, we also found genes which regulate important cell processes and control immune response (NBN, USP7, SOCS3, AQP9). Pathogens may target cell signaling, compromising the host’s ability to defend and increasing their own replication (Alto and Orth 2012). Balancing selection is not confined only to host ability to recognize foreign and harmful factors, but the conflict between host and pathogen interest can also drive molecular evolution of host regulators of immune response or other basic cell processes (Wilson et al. 2006).
Recently, new model-based tests aiming at identification of targets of BS have been proposed (DeGiorgio et al. 2014; Gao et al. 2015). They have good statistical properties and could be used to further validate candidates identified in this study. These tests however require information from continuous genomic regions around the putative targets of BS. Unfortunately, currently obtaining such long range genomic information in urodele amphibians is challenging as their huge genomes are still refractory to sequencing and assembly (Keinath et al. 2015). Until these challenges are overcome, an interesting option would be to identify the regions of interest in large insert (BAC or fosmid) libraries, resequence them and use this data to test for BS.
The BS candidates identified in the current study will be useful for testing whether genes affected by BS are more prone to interspecific introgression than other genes in the genome. Two rationales underlie this hypothesis: (i) theory predicts that, because introgressed alleles are initially rare, BS operating through rare-allele advantage facilitates their establishment in the recipient species (Schierup et al. 2000) and (ii) scant evidence suggests plausibility of the process but its generality and evolutionary importance are unknown (Castric et al. 2008; Abi-Rached et al. 2011; Grossen et al. 2014). Patterns of allele sharing and allele frequency differentiation among populations are consistent with adaptive introgression of MHC class II genes between Lm and Lv in central Europe (Nadachowska-Brzyska et al. 2012). It remains however unknown, whether adaptive introgression is a peculiar feature of MHC genes or is it more widespread among genes evolving under BS. The Lm/Lv system is particularly well suited to test this hypothesis, because patterns of relationships and gene flow in this group allow formulation of predictions that facilitate distinguishing the effect of introgression from incomplete lineage sorting (ILS), expected to be prevalent in BS genes (Nadachowska-Brzyska et al. 2012). Introgression of genes under BS in amphibians may be important for both their adaptive potential and conservation. Amphibians often show old intraspecific genetic structure and reproductive isolation develops relatively slowly in this group (Vences and Wake 2007). Range changes triggered by, e.g., climatic fluctuations, create opportunity for episodic contact and genetic exchange. As long as reproductive isolation is not complete, adaptive variants introgress easily between the differentiating lineages (Barton 1979). Such lineages may thus effectively share a common pool of adaptive variation (Rieseberg et al. 2004). Variants that emerged during periods of isolation would contribute to the pool through episodes of hybridization and this process should be particularly efficient in genes evolving under BS. Secondary contact followed by hybridization would thus boost adaptive potential of interacting lineages and alleviate the effects of past bottlenecks.
In conclusion, this first population-genomic study of amphibian immune genes showed that an overwhelming majority of immune genes in two Lissotriton newts are characterized by high evolutionary constraint. Patterns of synonymous and nonsynonymous variation indicate more widespread selection against mildly deleterious mutations in L. vulgaris graecus. In spite of the overall sequence conservation, we find signatures of positive and balancing selection in genes, which are involved both in pathogen recognition and in regulation of immune response. We provide a list of candidate genes potentially involved in resistance and defense against pathogens, which are worth further exploration in newts and other amphibians.
Supplementary Material
Supplementary tables S1–S10, text and figures S1–S5 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
We thank Michal Stuglik and Marta Niedzicka for bioinformatics support and three anonymous reviewers for insightful comments. We are grateful to the Center for Medical Genomics OMICRON, Jagiellonian University, Medical College in Krakow for access to the sonicator system which greatly facilitated our work. The work was supported by the Polish National Science Center grant UMO-2012/04/A/NZ8/00662 to W.B., Jagiellonian University grant DS/WBiNoZ/INoS/762/15 and in part by PL-Grid infrastructure.
Literature Cited
- Abi-Rached L, et al. 2011. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 334:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alto NM, Orth K. 2012. Subversion of cell signaling by pathogens. Cold Spring Harb Perspect Biol. 4:a006114–a006114.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anacker DC, Gautam D, Gillespie KA, Chappell WH, Moody CA. 2014. Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J Virol. 88:8528–8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antrobus R, Boutell C. 2008. Identification of a novel higher molecular weight isoform of USP7/HAUSP that interacts with the Herpes simplex virus type-1 immediate early protein ICP0. Virus Res. 137:64–71. [DOI] [PubMed] [Google Scholar]
- Azevedo L, Serrano C, Amorim A, Cooper DN. 2015. Trans-species polymorphism in humans and the great apes is generally maintained by balancing selection that modulates the host immune response. Hum Genomics 9:21.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babik W, et al. 2005. Phylogeography of two European newt species-discordance between mtDNA and morphology. Mol Ecol. 14:2475–2491. [DOI] [PubMed] [Google Scholar]
- Babik W, et al. 2015. Constraint and adaptation in newt Toll-like receptor genes. Genome Biol Evol. 7:81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro LB, et al. 2009. Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet. 5:e1000562.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton N. 1979. The dynamics of hybrid zones. Heredity 43:341–359. [Google Scholar]
- Bataille A, et al. 2013. Genetic evidence for a high diversity and wide distribution of endemic strains of the pathogenic chytrid fungus Batrachochytrium dendrobatidis in wild Asian amphibians. Mol Ecol. 22:4196–4209. [DOI] [PubMed] [Google Scholar]
- Bataille A, et al. 2015. Susceptibility of amphibians to chytridiomycosis is associated with MHC class II conformation. Proc Biol Sci. 282:20143127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataillon T, et al. 2015. Inference of purifying and positive selection in three subspecies of chimpanzees (Pan troglodytes) from exome sequencing. Genome Biol Evol. 7:1122–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger L, et al. 2016. History and recent progress on chytridiomycosis in amphibians. Fungal Ecol. 19:89–99. [Google Scholar]
- Besnard A, Galan-Rodriguez B, Vanhoutte P, Caboche J. 2011. Elk-1 a transcription factor with multiple facets in the brain. Front Neurosci. 5:35.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm T. 2012. Evolution of vertebrate immunity. Curr Biol. 22:R722–R732. [DOI] [PubMed] [Google Scholar]
- Brandvain Y, Wright SI. 2016. The limits of natural selection in a nonequilibrium world. Trends Genet. 32:201–210. [DOI] [PubMed] [Google Scholar]
- Breuer K, et al. 2013. InnateDB: systems biology of innate immunity and beyond-recent updates and continuing curation. Nucleic Acids Res. 41:D1228–D1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagliani R, et al. 2008. The signature of long-standing balancing selection at the human defensin β-1 promoter. Genome Biol. 9:R143.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castric V, Bechsgaard J, Schierup MH, Vekemans X. 2008. Repeated adaptive introgression at a gene under multiallelic balancing selection. PLoS Genet. 4:e1000168.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, et al. 2003. Positive selection in the human genome inferred from human-chimp-mouse orthologous gene alignments. Cold Spring Harb Sym. 68:471–477. [DOI] [PubMed] [Google Scholar]
- Das S, et al. 2010. Comparative genomics and evolution of the alpha-defensin multigene family in primates. Mol Biol Evol. 27:2333–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGiorgio M, Lohmueller KE, Nielsen R. 2014. A model-based approach for identifying signatures of ancient balancing selection in genetic data. PLoS Genet. 10:e1004561.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10:48.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilertson KE, Booth JG, Bustamante CD. 2012. SnIPRE: selection inference using a Poisson random effects model. PLoS Comput Biol. 8:e1002806.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizaguirre C, Lenz TL, Traulsen A, Milinski M. 2009. Speciation accelerated and stabilized by pleiotropic major histocompatibility complex immunogenes. Ecol Lett. 12:5–12. [DOI] [PubMed] [Google Scholar]
- Ekblom R, French L, Slate J, Burke T. 2010. Evolutionary analysis and expression profiling of zebra finch immune genes. Genome Biol Evol. 2:781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison AR, et al. 2015. More than skin deep: functional genomic basis for resistance to amphibian chytridiomycosis. Genome Biol Evol. 7:286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A, Woolfit M, Phelps T. 2006. The distribution of fitness effects of new deleterious amino acid mutations in humans. Genetics 173:891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijarczyk A, Babik W. 2015. Detecting balancing selection in genomes: limits and prospects. Mol Ecol. 24:3529–3545. [DOI] [PubMed] [Google Scholar]
- Fisher MC, et al. 2012. Emerging fungal threats to animal, plant and ecosystem health. Nature 484:186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, et al. 2011. Signatures of environmental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution PLoS Genet. 7:e1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Vieira FG, Linderoth T, Nielsen R. 2014. ngsTools: methods for population genetics analyses from next-generation sequencing data. Bioinformatics 30:1486–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Przeworski M, Sella G. 2015. Footprints of ancient-balanced polymorphisms in genetic variation data from closely related species. Evolution 69:431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazave E, Chang D, Clark AG, Keinan A. 2013. Population growth inflates the per-individual number of deleterious mutations and reduces their mean effect. Genetics 195:969–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossen C, Keller L, Biebach I, The International Goat Genome Consortium, Croll D. 2014. Introgression from domestic goat generated variation at the Major Histocompatibility Complex of Alpine ibex. PLoS Genet. 10:e1004438.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halligan DL, et al. 2013. Contributions of protein-coding and regulatory change to adaptive molecular evolution in murid rodents. PLoS Genet. 9:e1003995.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpur BA, Zayed A. 2013. Accelerated evolution of innate immunity proteins in social insects: adaptive evolution or relaxed constraint?. Mol Biol Evol. 30:1665–1674. [DOI] [PubMed] [Google Scholar]
- Hedrick PW. 2011. Population genetics of malaria resistance in humans. Heredity 107:283–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellgren O, Ekblom R. 2010. Evolution of a cluster of innate immune genes (beta-defensins) along the ancestral lines of chicken and zebra finch. Immunome Res. 6:3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm A, Karlsson T, Vikström E. 2015. Pseudomonas aeruginosa lasI/rhlI quorum sensing genes promote phagocytosis and aquaporin 9 redistribution to the leading and trailing regions in macrophages. Front Microbiol. 6:915.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, et al. 2008. Genomic analysis of the immune gene repertoire of amphioxus reveals extraordinary innate complexity and diversity. Genome Res. 18:1112–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18:337–338. [DOI] [PubMed] [Google Scholar]
- Keinath MC, et al. 2015. Initial characterization of the large genome of the salamander Ambystoma mexicanum using shotgun and laser capture chromosome sequencing. Sci Rep. 5:16413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, et al. 2011. Estimation of allele frequency and association mapping using next-generation sequencing data. BMC Bioinformatics 12:231.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneliussen TS, Albrechtsen A, Nielsen R. 2014. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15:356.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosiol C, et al. 2008. Patterns of positive selection in six mammalian genomes. PLoS Genet. 4:e1000144.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PA, Campbell CR, Yoder AD. 2014. Next-generation approaches to advancing eco-immunogenomic research in critically endangered primates. Mol Ecol Resour. 14:1198–1209. [DOI] [PubMed] [Google Scholar]
- Leffler EM, et al. 2013. Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science 339:1578–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Mizushima N, Virgin HW. 2011. Autophagy in immunity and inflammation. Nature 469:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, et al. 2012. Joint analysis of demography and selection in population genetics: where do we stand and where could we go?. Mol Ecol. 21:28–44. [DOI] [PubMed] [Google Scholar]
- Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. [DOI] [PubMed] [Google Scholar]
- Linnenbrink M, et al. 2011. Long-term balancing selection at the blood group-related gene B4galnt2 in the genus Mus (Rodentia; Muridae). Mol Biol Evol. 28:2999–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manry J, et al. 2011. Evolutionary genetic dissection of human interferons. J Exp Med. 208:2747–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel A, et al. 2014. Recent introduction of a chytrid fungus endangers Western Palearctic salamanders. Science 346:630–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng P, et al. 2013. The first salamander defensin antimicrobial peptide. PLoS One 8:e83044.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger KJ, Thomas MA. 2010. Evidence of positive selection at codon sites localized in extracellular domains of mammalian CC motif chemokine receptor proteins. BMC Evol Biol. 10:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadachowska K, Babik W. 2009. Divergence in the face of gene flow: the case of two newts (amphibia: salamandridae). Mol Biol Evol. 26:829–841. [DOI] [PubMed] [Google Scholar]
- Nadachowska-Brzyska K, Zieliński P, Radwan J, Babik W. 2012. Interspecific hybridization increases MHC class II diversity in two sister species of newts. Mol Ecol. 21:887–906. [DOI] [PubMed] [Google Scholar]
- Niedzicka M, Fijarczyk A, Dudek K, Stuglik M, Babik W. 2016. Molecular Inversion Probes for targeted resequencing in non-model organisms. Sci Rep. 6:24051.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Korneliussen T, Albrechtsen A, Li Y, Wang J. 2012. SNP calling, genotype calling, and sample allele frequency estimation from New-Generation Sequencing data. PLoS One 7:e37558.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obbard DJ, Welch JJ, Kim K-W, Jiggins FM. 2009. Quantifying adaptive evolution in the Drosophila immune system. PLoS Genet. 5:e1000698.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, et al. 2001. The membrane-type collectin CL-P1 is a scavenger receptor on vascular endothelial cells. J Biol Chem. 276:44222–44228. [DOI] [PubMed] [Google Scholar]
- Okumura A, et al. 2015. Suppressor of cytokine signaling 3 is an inducible host factor that regulates virus egress during Ebola virus infection. J Virol. 89:10399–10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortutay C, Vihinen M. 2009. Identification of candidate disease genes by integrating Gene Ontologies and protein-interaction networks: case study of primary immunodeficiencies. Nucleic Acids Res. 37:622–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabijan M, et al. 2015. The dissection of a Pleistocene refugium: phylogeography of the smooth newt, Lissotriton vulgaris, in the Balkans. J Biogeogr. 42:671–683. [Google Scholar]
- Pedersen BS. 2014. Aligning sequence from molecular inversion probes. bioRxiv doi: http://dx.doi.org/10.1101/007260. [Google Scholar]
- Peischl S, Excoffier L. 2015. Expansion load: recessive mutations and the role of standing genetic variation. Mol Ecol. 24:2084–2094. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana-Murci L, Clark AG. 2013. Population genetic tools for dissecting innate immunity in humans. Nat Rev Immunol. 13:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH, Church SA, Morjan CL. 2004. Integration of populations and differentiation of species. New Phytol. 161:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert J, Cohen N. 2011. The genus Xenopus as a multispecies model for evolutionary and comparative immunobiology of the 21st century. Dev Comp Immunol. 35:916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelants K, et al. 2013. Origin and functional diversification of an amphibian defense peptide arsenal. PLoS Genet. 9:e1003662.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage AE, Kiemnec-Tyburczy KM, Ellison AR, Fleischer RC, Zamudio KR. 2014. Conservation and divergence in the frog immunome: pyrosequencing and de novo assembly of immune tissue transcriptomes. Gene 542:98–108. [DOI] [PubMed] [Google Scholar]
- Savage AE, Zamudio KR. 2011. MHC genotypes associate with resistance to a frog-killing fungus. Proc Natl Acad Sci U S A. 108:16705–16710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierup MH, Vekemans X, Charlesworth D. 2000. The effect of subdivision on variation at multi-allelic loci under balancing selection. Genet Res. 76:51–62. [DOI] [PubMed] [Google Scholar]
- Smith MA, Green DM. 2005. Dispersal and the metapopulation paradigm in amphibian ecology and conservation: are all amphibian populations metapopulations? Ecography 28:110–128. [Google Scholar]
- Spurgin LG, Richardson DS. 2010. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc Biol Sci. 277:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler T, Haubold B, Merino C, Stephan W, Pfaffelhuber P. 2009. The impact of sampling schemes on the site frequency spectrum in nonequilibrium subdivided populations. Genetics 182:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Donnelly P. 2003. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 73:1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuglik MT, Babik W, Prokop Z, Radwan J. 2014. Alternative reproductive tactics and sex-biased gene expression: the study of the bulb mite transcriptome. Ecol Evol. 4:623–632. [Google Scholar]
- Tarazona-Santos E, et al. 2013. Evolutionary dynamics of the human NADPH oxidase genes CYBB, CYBA, NCF2, and NCF4: functional implications. Mol Biol Evol. 30:2157–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira JC, et al. 2015. Long-term balancing selection in LAD1 maintains a missense trans-species polymorphism in humans, chimpanzees, and bonobos. Mol Biol Evol. 32:1186–1196. [DOI] [PubMed] [Google Scholar]
- Tennessen JA, Blouin MS. 2008. Balancing selection at a frog antimicrobial peptide locus: fluctuating immune effector alleles?. Mol Biol Evol. 25:2669–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, et al. 2009. Variations in the expressed antimicrobial peptide repertoire of northern leopard frog (Rana pipiens) populations suggest intraspecies differences in resistance to pathogens. Dev Comp Immunol. 33:1247–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Těšický M, Vinkler M. 2015. Trans-species polymorphism in immune genes: general pattern or MHC-restricted phenomenon? J Immunol Res. 2015:838035.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasseur E, et al. 2012. The evolutionary landscape of cytosolic microbial sensors in humans. Am J Hum Genet. 91:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vences M, Wake DB. 2007. Speciation, species boundaries and phylogeography of amphibians In: Heatwole H, Tyler MJ, editors. Amphibian biology. Chipping Norton: Surrey Beatty and Sons; p. 2613–2671. [Google Scholar]
- Vilcinskas A. 2013. Evolutionary plasticity of insect immunity. J Insect Physiol. 59:123–129. [DOI] [PubMed] [Google Scholar]
- Wakeley J. 2004. Metapopulation models for historical inference. Mol Ecol. 13:865–875. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. 2012. Antimicrobial activity of human islet amyloid polypeptides: an insight into amyloid peptides’ connection with antimicrobial peptides. Biol Chem. 393:641–646. [DOI] [PubMed] [Google Scholar]
- Webb AE, et al. 2015. Adaptive evolution as a predictor of species-specific innate immune response. Mol Biol Evol. 32:1717–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DJ, Hernandez RD, Andolfatto P, Przeworski M. 2011. A population genetics-phylogenetics approach to inferring natural selection in coding sequences. PLoS Genet. 7:e1002395.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JN, et al. 2006. A hallmark of balancing selection is present at the promoter region of interleukin 10. Genes Immun. 7:680–683. [DOI] [PubMed] [Google Scholar]
- Wlasiuk G, Nachman MW. 2010. Adaptation and constraint at Toll-like receptors in primates. Mol Biol Evol. 27:2172–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You X, et al. 2014. Mudskipper genomes provide insights into the terrestrial adaptation of amphibious fishes. Nat Commun. 5:5594.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieliński P, Dudek K, Stuglik MT, Liana M, Babik W. 2014. Single nucleotide polymorphisms reveal genetic structuring of the Carpathian newt and provide evidence of interspecific gene flow in the nuclear genome. PLoS One 9:e97431.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieliński P, et al. 2013. No evidence for nuclear introgression despite complete mtDNA replacement in the Carpathian newt (Lissotriton montandoni). Mol Ecol. 22:1884–1903. [DOI] [PubMed] [Google Scholar]
- Zieliński P, Nadachowska-Brzyska K, Dudek K, Babik W. 2016. Divergence history of the Carpathian and smooth newts modelled in space and time. Mol Ecol 25:3912–3928. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.