

Abstract
Proglucagon-derived peptides, especially glucagon-like peptide-1 (GLP-1) and its long-acting mimetics, have exhibited neuroprotective effects in animal models of stroke. Several of these peptides are in clinical trials for stroke. Oxyntomodulin (OXM) is a proglucagon-derived peptide that co-activates the GLP-1 receptor (GLP-1R) and the glucagon receptor (GCGR). The neuroprotective action of OXM, however, has not been thoroughly investigated. In this study, the neuroprotective effect of OXM was first examined in human neuroblastoma (SH-SY5Y) cells and rat primary cortical neurons. GLP-1R and GCGR antagonists, and inhibitors of various signaling pathways were used in cell culture to characterize the mechanisms of action of OXM. To evaluate translation in vivo, OXM-mediated neuroprotection was assessed in a 60-minute, transient middle cerebral artery occlusion (MCAo) rat model of stroke. We found that OXM dose- and time-dependently increased cell viability and protected cells from glutamate toxicity and oxidative stress. These neuroprotective actions of OXM were mainly mediated through the GLP-1R. OXM induced intracellular cAMP production and activated cAMP-response element-binding protein (CREB). Furthermore, inhibition of the PKA and MAPK pathways, but not inhibition of the PI3K pathway, significantly attenuated the OXM neuroprotective actions. Intracerebroventricular administration of OXM significantly reduced cerebral infarct size and improved locomotor activities in MCAo stroke rats. Therefore, we conclude that OXM is neuroprotective against ischemic brain injury. The mechanisms of action involve induction of intracellular cAMP, activation of PKA and MAPK pathways and phosphorylation of CREB.
Keywords: Oxyntomodulin, Neuroprotection, Stroke, Glucagon-like peptide-1 receptor, Glucagon receptor, Glutamate excitotoxicity, Oxidative stress
Graphical abstract
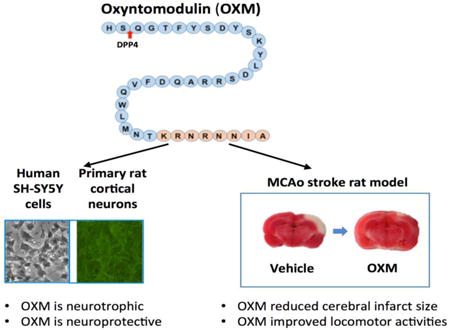
Introduction
Oxyntomodulin (OXM), an endogenous proglucagon-derived intestinal peptide, was isolated from porcine jejunoileum extract in 1981. OXM contains a 29-amino acid sequence of glucagon followed by 8 amino acids at its carboxyl-terminus. It was mainly produced in enteroendocrine L-cells and released together with glucagon-like peptide-1 (GLP-1) in response to food intake. Since OXM modulated gastric acid secretion in gastric oxyntic glands, it was named “oxyntomodulin” (Bataille et al., 1981). OXM is a dual agonist for the GLP-1 receptor (GLP-1R) and the glucagon receptor (GCGR) (Pocai et al., 2009). Limited reports have indicated that OXM activities control nutrition and metabolism in the periphery (Estall and Drucker, 2006; Tan and Bloom, 2013), and reduce pancreatic β-cell apoptosis (Maida et al., 2008). OXM improved glucose tolerance, inhibited food intake, increased energy expenditure, and reduced body weight in both rodents and humans (Dakin et al., 2004 and Wynne et al., 2006). Several long-acting OXM analogues, which avoid rapid inactivation of OXM by dipeptidyl peptidase-4 (DPP4), are currently under investigation in clinical trials for the treatment of type 2 diabetes mellitus (T2DM) and obesity (Santoprete et al., 2011, Bagger et al., 2015 and Muppidi et al., 2016).
OXM can cross the blood brain barrier (BBB) through mechanisms similar to those of GLP-1 (Kastin et al., 2002). We previously demonstrated that the GLP-1R agonists exenatide and liraglutide had neuroprotective and neurotrophic effects in cell and animal models of ischemic stroke, traumatic brain injury, Parkinson's disease (PD), Alzheimer's disease (AD) and ALS (Li et al., 2009, Li et al., 2010a, Li et al., 2012, Salcedo et al., 2012, Eakin et al., 2013, Greig et al., 2014). These data, together with findings by other groups (Bassil et al., 2014; Holscher 2014; Darsalia et al., 2015; Athauda & Foltynie 2016 and Kuroki et al., 2016), suggest that incretin- based anti-diabetic drugs are neuroprotective and hold promise for repurposing as a treatment strategy for neurodegeneration. Positive pre-clinical data on these compounds has led to several ongoing clinical trials, such as a recent successful open label clinical trial of exenatide in Parkinson's disease patients (Aviles-Olmos et al., 2013). Unlike GLP-1 or glucagon, the CNS effect of OXM has not been well examined (Bataille and Dalle, 2014). One recent study has indicated that D-Ser2-oxytomodulin, a protease-resistant OXM analogue, improved locomotor activities and protected dopaminergic neurons in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD (Liu et al., 2015). The neuroprotective actions of OXM, however, have yet to be fully characterized in other neurodegenerative conditions, including stroke that involves both necrotic and apoptotic cell death where the actions of OXM remain unknown.
We hypothesize that OXM is neuroprotective in ischemic stroke based on its structural similarity with other glucagon-derived peptides, its anti-apoptotic actions in pancreatic β-cells, and its features of inducing intracellular cAMP and glucoregulatory effects. Elevations in intracellular cAMP levels, whether the consequence of activation of membrane receptors by neuropeptides, classical neurotransmitters or other protein-ligand interactions, have long been associated with plasticity and cytoprotection within the nervous system (Silveira and Linden, 2006; Sakomoto et al., 2011). Of note, obesity and T2DM, for which OXM analogues are being evaluated as treatment strategies, are major risk factors for stroke (Najarian et al., 2006; Chen et al., 2016). Based on these factors, it is feasible that OXM may be a potentially useful therapy, which targets stroke and metabolism (Shankar et al., 2013). In this study, we characterize the neuroprotective effects of OXM peptide in cellular and animal models of stroke. Our data suggest that OXM is, indeed, a potentially neuroprotective agent against ischemic stroke and warrants further evaluation.
Materials and Methods
Materials
Oxyntomodulin was purchased from the Phoenix Pharmaceuticals (Burkingama, CA). GLP-1R antagonist Exendin 9-39 was obtained from Anaspec (Fremond, CA). GCGR antagonist des-His1-[Glu9]-Glucagon (1-29) amide was purchased from Tocris (Minneapolis, MN). The kinase inhibitors, H89, LY294002 and U0126 were purchased from Calbiochem (Gibbstown, NJ). All other reagents were from Sigma (St. Louis, MO), unless otherwise stated.
Cell culture
SH-SY5Y cells, obtained from American Type Culture Collection (Manassa, VA), and SH-hGLP-1R#9 cells, a human GLP-1R over-expressing cell line derived from SH-SY5Y cells were maintained as described previously (Li et al., 2010b). Primary cultures of cortical neurons (PCN) were prepared from embryonic day 15 Sprague-Dawley rats in accordance with approved procedures by the Animal Care and Usage Committee of the National Health Research Institutes, Taiwan, and were cultured as previously described (Howard et al., 2008; Yu et al., 2016). They were maintained and evaluated from 10 day in vitro (DIV) onward. Briefly, brain cortices from E15 embryos were pooled and digested for 20 min in 0.05% trypsin-ETDA (0.2% (Invitrogen, Carlsbad, CA), 37°C, 1 ml/embryo). Cortices were triturated and then diluted into plating media (Neurobasal media (Invitrogen) containing 2% heat-inactivated fetal bovine serum (Sigma-Aldrich, St Louis, MO), 2% B27 supplement (Invitrogen), 0.5 mM l-glutamate with and without antioxidants (AO) supplement (on DIV 3 and 5 (with AO) and DIV 7 and 10 (without AO)) at 2 ml/embryo. Following evaluation of viability, cells were then plated at 5×104 viable cells/well in 0.2 ml plating media into 96 well plates pre-coated with 0.15-0.2% polyethyleneimine in 150mM sodium borate, pH 8.5 (Sigma-Aldrich). Plated cells were maintained in a 37°C humidified incubator with 5.5% CO2, and fed by 50% media exchange on DIV3, 5, 7 and 10 with feed media.
Cell viability assays
Cell viability was assessed via 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay performed in 96-well plates. Cells were serum-starved (with 0.5% of serum) overnight before pretreatment with various concentrations of OXM for 1 h. Cells were then exposed to different concentrations of glutamate or H2O2 for either 24 h or 48 h, time points selected from prior studies. Glutamate concentrations were selected from concentration-dependent pilot studies focused to provide a statistically significant but submaximal loss of viability. A CellTiter 96 Aqueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI) was utilized to measure a formazan product, which is directly proportional to the cell viability.
cAMP assay
Cells grown in 24-well plates were first serum-starved overnight and then treated with various concentrations of OXM (10-9, 10-8, 10-7 M) in serum-free media for 15 min at 37°C. Cells were then lysed with 0.1 M HCl containing 0.5% Triton x-100 for 10 min at room temperature. Cell lysates were collected and centrifuged at 600g at room temperature to remove cell debris. Supernatants were directly used for cAMP measurement. Intracellular cAMP content was determined using the Direct cAMP ELISA kit (Enzo Life Sciences, Inc., Farmingdale, NY), as per the manufacturer's protocol for the acetylated version.
Immunocytochemistry
At 48 h after drug treatment, PCN cells were fixed with 4% paraformaldehyde (PFA) for 1 h at room temperature. After removing the 4% PFA solution, cells were washed with phosphate-buffered saline (PBS), and the fixed cells were treated with blocking solution (2 % BSA, 0.1 % Triton X-100, and 5 % goat serum in PBS) for 1 h. The cells were then incubated with a mouse monoclonal antibody against MAP2 (1:500; Millipore, Billerica, MA) for 1 day at 4 °C. The cells were next rinsed three times in PBS and treated with a secondary antibody (AlexaFluor 488 goat anti-mouse Ab, 1:500, Invitrogen, Carslbad, CA). Images were acquired using a monochrome camera Qil-mc (Diagnostic Instruments, Inc., Sterling Heights, MI) attached to a NIKON TE2000 inverted microscope (Nikon, Melville, NY). Data were analyzed using NIS Elements AR 3.2 Software (Nikon).
Western Blotting
Cells grown in 100 mm-plates at a density of approximately 5 × 106 cells were used to extract total protein. Standard Western Blotting procedure was used with approximately 50 μg of protein extracts for all samples. cAMP-response element-binding protein (CREB) and phospo-CREB (pCREB) antibodies from Cell Signaling (Danvers, MA) were used at a dilution of 1:1000. Glucagon receptor antibody from Novus Biologicals (Littleton, CO) was used at a dilution of 1:500. α-tubulin antibody from Sigma was used at a dilution of 1:5000. Densitometric quantification of the protein bands was performed using a PC version of NIH IMAGE (ImageJ software).
Animals and drug administration
Adult male Sprague-Dawley rats were used and treated in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) on protocols approved by the Animal Care and Usage Committee of the National Health Research Institutes, Taiwan. Briefly, animals were anesthetized with chloral hydrate (0.4 g/kg, i.p.). As a proof of concept, following randomization of animals into groups, either OXM (20 μg in 20 μl sterile physiological saline per rat) or vehicle (20 μl sterile physiological saline per rat) was given intracerebroventricularly through a 25 μl Hamilton syringe 15 min before a 60 min MCAo. The coordinates for intracerebroventricular injections were: 0.8 mm posterior to the bregma; 1.5 mm lateral to the midline; 3.5 mm below the dura surface. The speed of injection was controlled by a syringe pump at a rate of 2.5 μl per min. The needle was retained in place for 5 min after injection. After injection, a piece of bone wax (W810, Ethicon) was applied to the skull defect to prevent leakage of the solution.
MCA ligation
The right middle cerebral artery (MCA) was transiently occluded as previously described (Luo et al., 2009). In brief, the bilateral common carotids (CCAs) were ligated with nontraumatic arterial clips. A craniotomy of about 2 × 2 mm2 was made in the right squamosal bone. The right MCA was ligated with a 10-O suture, as previously described (Luo et al., 2009), to generate focal infarction in the cerebral cortex. The ligature and clips were removed after 60 min ischemia to generate reperfusional injury. Core body temperature was monitored with a thermistor probe and maintained at 37 °C with a heating pad during anesthesia. After recovery from anesthesia, body temperature was maintained at 37 °C using a temperature-controlled incubator. Immediately after recovery from anesthesia, an elevated body swing test was used to evaluate the success of MCAo surgery, and animals were randomized between treatment vs. vehicle groups.
Behavioral assay
Animals were placed in an Accuscan activity monitor (Columbus, OH) for 24 h at 2 days after MCAo for behavioral recording, as previously described (Shen and Wang, 2010). The monitor contains 16 horizontal infrared sensors spaced 2.5 cm apart. Each animal was placed in a 42 × 42 × 31 cm plexiglass open box. Food and water were provided. Motor activity was calculated using the number of beams broken by the animals.
Triphenyltetrazolium chloride (TTC) staining
Three days after MCAo, animals were culled by decapitation. The brains were removed, immersed in cold saline for 5 minutes, and sliced into 2.0 mm thick sections. The brain slices were incubated in 2% TTC (Sigma), dissolved in normal saline for 10 minutes at room temperature, and then transferred into a 5% formaldehyde solution for fixation. The area of infarction on each brain slice was measured double blind using a digital scanner and the Image Tools program (University of Texas Health Sciences Center, San Antonio, TX).
Statistical Analysis
Data are presented as mean ± standard error mean (SEM) and were analyzed by Prism software. One-way or two-way analysis of variance (ANOVA) tests are used for comparison of multiple samples, followed by post hoc tests (Newman-Keuls test and Dunnetts test). *P <0.05, **P <0.01, ***P <0.001.
Results
OXM induces intracellular cAMP production and activates the CREB pathway
The effect of OXM on intracellular cAMP levels was examined in human neuroblastoma SH-SY5Y and #9 cells (a stable SH-SY5Y cell line that overexpresses the hGLP-1R) (Li et al., 2010b and Li et al., 2015). We previously reported GLP-1Rs were present in SH-SY5Y and #9 cells (Li et al., 2009 and Li et al., 2010b). In this study, we validated this finding and further demonstrated the presence of the GCGR in SH-SY5Y and #9 cells via Western blot. The expression of GLP-1R protein is increased about 2-fold in #9 cells compared to original SH-SY5Y cells, while the expression levels of GCGR are similar in these two cell lines (Figure 1A). Cells were treated with OXM (1, 10, 100 nM) or vehicle for 15 min under serum-free conditions. Intracellular cAMP levels were analyzed via ELISA. We found that OXM at the highest doses marginally increased cAMP levels in the SH-SY5Y cells (p= 0.06, Figure 1B), which were significant at 10 min (p=0.03, not shown). A significant and dose-dependent increase in intracellular cAMP by OXM was found in #9 cells at both time points (p < 0.01, Figure 1C). These data suggest that OXM can induce cAMP response in neuronal cells, and that this response is at least partially mediated through the GLP-1 receptor as #9 cells have enhanced GLP-1R signaling.
Figure 1.
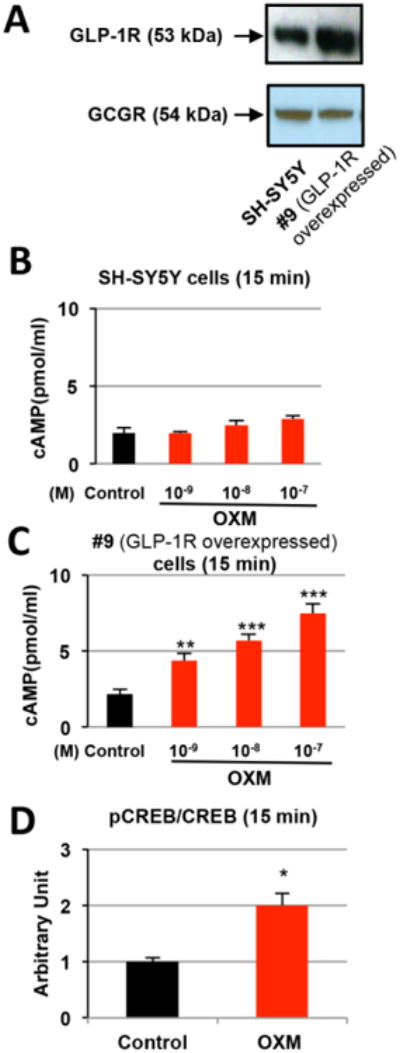
OXM dose-dependently increases intracellular cAMP levels in neural cells. A, The GCGR protein is present in SH-SY5Y and #9 cells as detected by Western blotting; B, Intracellular cAMP levels were marginally elevated and showed a trend of increase in SH-SY5Y cells treated with increasing doses of OXM for 15 min. P=0.06 for the highest concentration of OXM (10-7 M); C, Intracellular cAMP levels were significantly elevated in #9 cells (SH-SY5Y cells that stably over-expresses the hGLP-1R) treated with increasing concentrations (0, 10-9, 10-8, 10-7 M) of OXM for 15 min; D, Western blot analysis indicated that the pCREB/CREB ratio increased 2-fold after 15 min treatment with 10-7M OXM in SH-SY5Y cells (statistical comparison vs. control value, *P < 0.05; **P <0.01; ***P < 0.001).
We next examined whether or not OXM activated the cAMP/CREB signaling pathway, which plays a key role in promoting neuronal growth and survival (Rydel and Greene, 1988; Silveira and Linden, 2006; Sakomoto et al., 2011). pCREB and CREB protein levels in SH-SY5Y cells were determined by Western blot analysis after treatment with 100 nM OXM. The ratio of pCREB/CREB was significantly increased (2-fold) by OXM treatment as compared to control, indicating that OXM activates the CREB pathway in SH-SY5Y cells (Figure 1D). Our results also indicate that OXM is functional in both SH-SY5Y and #9 cells, although the latter are far more sensitive due to elevated expression levels of the GLP-1R, again suggesting importance of the GLP-1R to the actions of OXM in neurons.
OXM dose- and time-dependently promotes cell proliferation
SH-SY5Y and #9 cells were treated with OXM (0, 1, 10, 100, 1000 nM) for 24 or 48 h, and, thereafter, cell viabilities were assessed via MTS assay. There were no changes in SH-SY5Y cell viabilities after 24 h OXM treatment (Figure 2A). However, in hGLP-1R overexpressing #9 cells, cell viabilities were significantly increased at 24 h after OXM treatment (≥10 nM, Figure 2A). At 48 h, OXM (≥ 10 nM) significantly enhanced cell viability in both SH-SY5Y and #9 cells (Figure 2B). Taken together, these data suggest that OXM dose- and time-dependently promoted neural cell proliferation in vitro. Increases in cell viability due to OXM treatment are enhanced by GLP-1R overexpression, which reiterates the important role of the GLP-1R in the actions of OXM in neurons.
Figure 2.
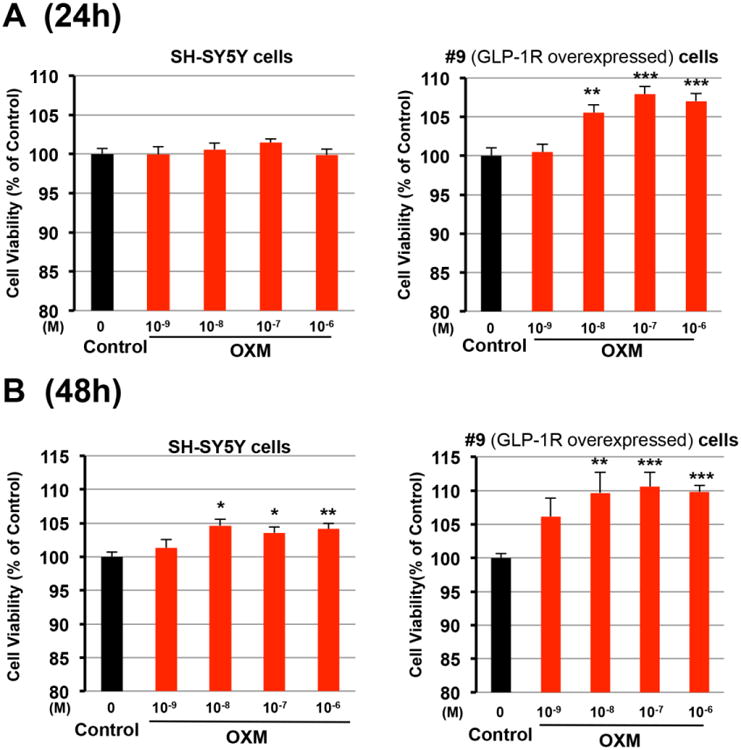
OXM dose- and time-dependently increases cell viabilities in human SH-SY5Y neuroblastoma cells and #9 cells. A and B, Both cells were treated with increasing concentrations (0, 10-9, 10-8, 10-7, 10-6 M) of OXM for either 24 h (A) or 48 h (B). MTS assays were used to assess cell viabilities at 24 h or 48 h after OXM treatment (statistical comparison vs. control value, *P < 0.05; **P <0.01; ***P < 0.001).
OXM dose-dependently protects against glutamate toxicity and oxidative stress in neuronal cells
SH-SY5Y and #9 cells were pretreated with OXM (0, 1, 10, 100, 1000 nM) for 1h before glutamate or H2O2 administration. Cell viability was examined by the MTS assay at 24h after drug treatment. Evaluated at 24 h, glutamate or H2O2 significantly reduced cell survival (Figure 3). In SH-SY5Y cells, OXM (1 to 1000 nM) effectively reduced glutamate (100 mM)–mediated cell death (Figure 3A). A higher concentration (150 mM) of glutamate induced yet greater cell death (70% vs. 20%). In accord with this, a high dose of OXM (100 nM) was required to reduce glutamate (150 mM) toxicity (Figure 3A). It has been reported that #9 cells were relatively resistant to toxins due to enhanced GLP-1R expression (Li et al., 2010b). In line with this, in our experiment glutamate (150 mM) induced 70% cell death in SH-SY5Y cells, and 30% in #9 cells (Figure 3A). We demonstrated that glutamate toxicity was ameliorated by OXM (≥ 10 nM) in #9 cells (Figure 3A, lower panel). Similarly, a 150 μM H2O2 insult significantly reduced #9 cell viabilities, which likewise were protected by OXM ((≥ 10 nM, Figure 3B).
Figure 3.
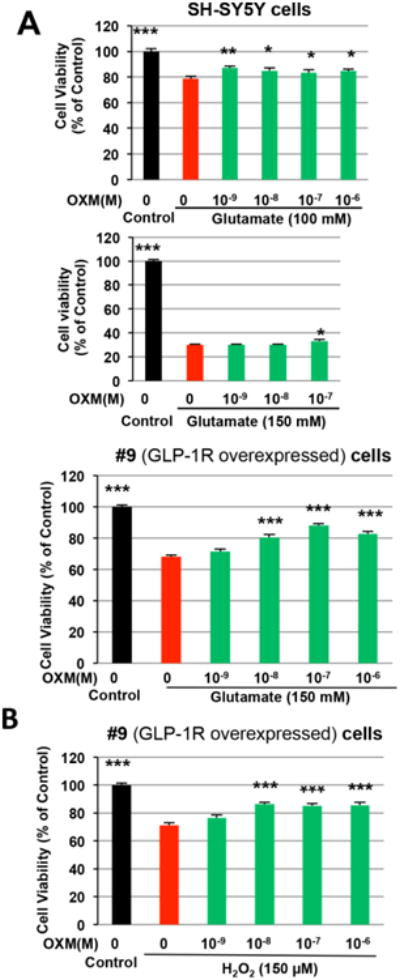
OXM dose-dependently protects neuronal cells from glutamate-induced toxicity and H2O2-induced oxidative stress. A and B, SH-SY5Y and/or #9 cells were pre-treated with increasing concentrations (0, 10-9, 10-8, 10-7, 10-6 M) of OXM for 1 h and then exposed to either glutamate (100 mM or 150 mM) (A) or H2O2 (100 μM) (B). MTS assays were used to assess cell viabilities at 24 h after glutamate and H2O2 treatment (statistical comparison vs. glutamate or H2O2 alone, *P < 0.05; **P <0.01; ***P < 0.001).
Neurotrophic and neuroprotective effects of OXM are mediated through GLP-1R but not GCGR
Using specific antagonists against the GLP-1R and the GCGR, we evaluated the relative influence of the GLP-1R and GCGR on the neurotrophic and neuroprotective effects of OXM. As shown in Figures 2 and 3, OXM treatment increased SH-SY5Y and #9 cell viabilities, as a marker of neurotrophic and neuroprotective actions, respectively (Li et al., 2010b). Blocking the GCGR with a 100-fold greater concentration of GCGR antagonist, des-His1-[Glu9]-Glucagon (1-29) amide, did not alter the neurotrophic effect of OXM in either cell line (Figure 4A). On the other hand, the neurotrophic effect of OXM was totally abolished with the addition of 100-fold greater concentration of GLP-1R antagonist, exendin 9-39, evaluated at two different doses of OXM (Figure 4B). These results show the essential role of the GLP-1R in the neurotrophic effect of OXM. Furthermore, the addition of 100-fold exendin 9-39, but not des-His1-[Glu9]-Glucagon (1-29) amide, attenuated the neuroprotective effect of OXM against glutamate in both cell lines (Figure 4C). Altogether, our data indicate that the neurotrophic and neuroprotective actions of OXM were chiefly mediated through the GLP-1R.
Figure 4.
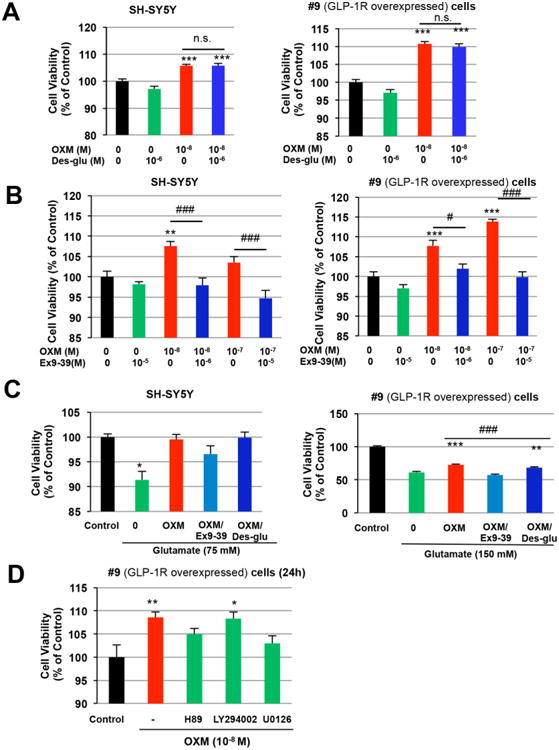
Roles of GLP-1R and GCGR in the neurotrophic and neuroprotective effects of OXM and signaling pathways involved. A, OXM at a concentration of 10-8 M increased cell viabilities in both SH-SY5Y and #9 cells. In the presence of GCGR antagonist des-His1-[Glu9]-Glucagon (1-29) amide (10-6 M, 100-fold of OXM), the effect of OXM on cell viability was maintained in both cell lines. B, In the presence of GLP-1R antagonist exendin 9-39 (10-6 or 10-5 M, 100-fold of OXM at 10-8 or 10-7 M), however, the neurotrophic effect of OXM was abolished in both cell lines. C, Glutamate-induced cell death can be protected by 1 h pretreatment with OXM (10-8 M) in both SH-SY5Y and #9 cells. Addition of 100-fold Ex 9-39, but not des-His1-[Glu9]-Glucagon (1-29) amide, reduced the neuroprotective effect of OXM, indicating a more significant role for the GLP-1R than for the GCGR in this action of OXM. D, Signaling pathways involved in the neurotrophic effect of OXM. #9 cells were incubated with 10 μM H89 (protein kinase A [PKA] inhibitor), 10 μM LY294002 (phosphoinositide 3-kinase [PI3K] inhibitor) or 5 μM U0126 (MEK1/2 inhibitor) for 20 min prior to OXM (10-8 M) treatment. After 24 h incubation with OXM, cell viability was evaluated via MTS assay. H89 and U0126 both decreased OXM-induced increase in cell viability, while LY294002 did not. (Statistical comparison vs. control value, *P < 0.05; **P < 0.01; ***P < 0.001. Comparison between groups, # P <0.05; ## P <0.01; ### P< 0.001).
Signaling pathways involved in the neurotrophic action of OXM
To evaluate relevant signaling pathways underpinning neurotrophic action, serum-starved #9 cells were incubated with the PKA inhibitor H89 (10 μM), PI3K inhibitor LY294002 (10 μM) or MEK inhibitor U0126 (5 μM) (MEK1/2) for 20 min. OXM (10 nM) was thereafter added, and cell viability was assessed via MTS assay at 24 h. As evident in Figure 4D, inhibition of PKA by H89 and of MEK1/2 by U0126 significantly attenuated the neurotrophic effect of OXM, whereas inhibition of PI3K by LY294002 did not significantly effect cell viability. Our data thus suggest that the PKA and MAPK, but not PI3K, pathways play essential roles in the neurotrophic action of OXM.
OXM dose-dependently reduces glutamate-mediated neurotoxicity in primary cortical neurons
To evaluate translation of the neuroprotective action of OXM from an immortal human cell line with neuronal features to cultured neurons, rat primary cortical cultures were exposed to glutamate (100 μM) and then treated with either OXM (1 μM) or vehicle on DIV10. Cell survival was assessed using microtubule-associated protein 2 (MAP2) immunostaining at 48 h after OXM treatment, as MAP2 is a neuron specific protein that plays a role in maintaining dendritic structure through its interaction with microtubules. The timeline of primary neuron maintenance, treatment and immunostaining is shown in Figure 5A. Glutamate reduced MAP2 immunoreactivity, which was significantly antagonized by post-treatment with OXM (p<0.001, Figure 5B). Typical photomicrographs are displayed in Figure 5B. In a separate experiment, PCNs were pre-treated with either vehicle or OXM (10 and 100 nM) for 1 h, and then exposed to glutamate at a concentration of 150 μM. After 24 h, glutamate induced significant cell death in vehicle treated cells. OXM dose-dependently protected the cortical neurons. Cell viability was significantly higher in 100 nM OXM treated cells than in cells treated with glutamate alone (p<0.01, Figure 5C).
Figure 5.
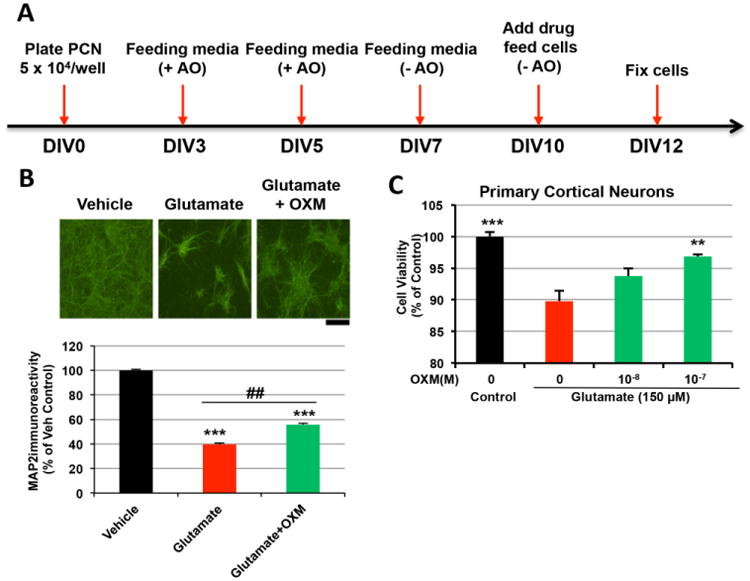
OXM reduces glutamate-induced neurotoxicity in primary cortical neuronal cultures. A, Timeline of in vitro primary cortical culture and immunocytochemistry study. B, Glutamate (100 μM) significantly reduced MAP2 immunoreactivity. This response was significantly antagonized by co-administration of OXM (1 μM) (p<0.001, F2,39=86.209, one-way ANOVA; p=0.002, posthoc Newman-Keuls test). Representative photomicrographs are shown of primary cortical cultures under vehicle, glutamate and glutamate+OXM conditions; calibration mark=200 um. C, Primary cortical neurons were pre-treated with different concentrations (0, 10-8, 10-7 M) of OXM for 1 h, and then exposed to glutamate at a toxic concentration of 150 μM. MTS assays were used to assess cell viabilities at 24 h after glutamate treatment. (statistical comparison vs. glutamate alone, *P < 0.05; **P <0.01; ***P < 0.001. Comparison between groups, ## P <0.01).
Intracerebroventricular administration of OXM improves locomotor activity in stroke rats
The schematic of this in vivo experimental design is shown in Figure 6A. In this proof of principle study, rats received intracerebroventricular injections of either OXM or vehicle 15 min before a 60-min MCAo. This route, rather than systemic administration, was selected to ensure OXM presence within the brain and to thereby safeguard against pharmacokinetic confounds (such as blood-brain barrier permeability and short systemic half-life) impacting potential OXM efficacy. Eleven rats were used for behavioral analysis two days after MCAo. Animals were housed in the activity chamber for 24 h. Pretreatment with OXM (n=5), compared to vehicle (n=6), significantly increased horizontal activity (p=0.005), movement number (p=0.020), movement time (p=0.003), total distance travelled (p=0.002), vertical activity (p=0.002), vertical movement number (p=0.018), and vertical movement time (p=0.007). Conversely, rest time was significantly reduced by OXM (p=0.003). These data suggest that OXM, given intracerebroventricularly, significantly improved locomotor function in the stroke rats (Figure 6B).
Figure 6.
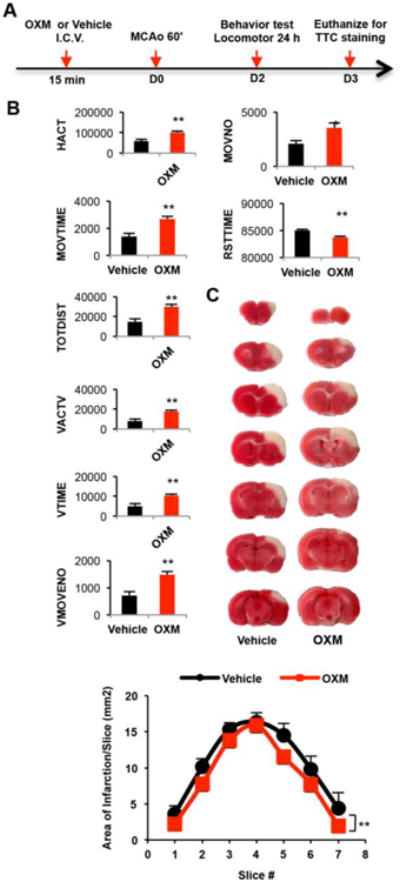
OXM reduces ischemia/reperfusion-induced bradykinesia and cerebral infarction in stroke rats. A, Schematic timeline of in vivo experiment (after randomization of animals into groups, vehicle or OXM was administered intracerebroventricular (I.C.V.) followed by MCAo surgery - with use of an elevated body swing test immediately after recovery from anesthesia to ensure the success of this MCAo surgery. Behavioral analyses were performed over a 24 h period starting 24 h after mCAo surgery, and animal were euthanized, thereafter (on day (D) 3), for evaluation of the brain by TTC staining). B, Pretreatment with OXM significantly increased horizontal activity (HACT, cm), movement number (MOVNO), movement time (MOVTIME, sec), total distance traveled (TOTDIST, cm), vertical activity (VACTV), vertical movement number (VMOVENO), and vertical movement time (VTIME, sec), while it reduced rest time (RSTTIME, sec). All locomotor behaviors were measured 2 days after stroke for 24 hours. (*p<0.05; **P <0.01 t-test). C, Rats received intracerebroventricular injections of OXM or vehicle 15 min before a 60 minute MCAo. Tissue was sectioned (2 mm) and stained with TTC at 3 days post-stroke. TTC staining demonstrating that administration of OXM reduced cortical infarction in stroke animals (A). Area of infarction per 2-mm section (7 sections/brain; anterior to posterior) was significantly reduced for all sections in the oxyntomodulin group, as compared to that in the vehicle control (**p<0.01, F1,63=8.052, two-way ANOVA).
OXM significantly reduces cerebral infarction in rat model of stroke
Stroke rats (n=11) were euthanized 3 days after MCAo. A total of 7 brain slices (each 2 mm) were collected from each brain. The area of infarction was quantified after TTC staining. Typical TTC histological images from animals receiving either OXM or vehicle are shown in Figure 6C. We found that treatment with OXM significantly reduced the extent of cortical infarction compared to vehicle pretreatment (Figure 6C) (p=0.006).
Discussion
In this study, we demonstrate for the first time that native OXM peptide is both neurotrophic and neuroprotective against glutamate toxicity and oxidative stress in cultured human SH-SY5Y neuroblastoma cells and rat primary cortical neurons, two cellular models that are widely used to evaluate neurotrophic and neuroprotective actions of experimental therapeutics in neurological drug development (Li et al., 2009; Melo et al., 2011; Lilja et al., 2013; Inaba-Hasegawa et al., 2013; Sharma et al., 2014). OXM treatment significantly increased cell viabilities as assessed both by MTS assay and immunoreactivities of neuronal marker MAP2. To evaluate in vivo translation, in the rat MCAo stroke model, pre-treatment with OXM through intracerebroventricular administration significantly reduced cortical infarction sizes and improved locomotor activities. Our data hence support the concept that OXM and analogues is a potential neuroprotective agent that may have relevance to the treatment of stroke and other neurological disorders.
Both the GLP-1R and GCGR are G protein-coupled receptors (GPCR) that are members of the secretin receptor-like family of seven transmembrane-spanning domain proteins. These group B receptors additionally include GPCRs that selectively bind glucose-dependent insulinotropic peptide (GIP), vasoactive intestinal peptide (VIP), pituitary adenylyl cyclase-activating peptide (PACAP) and other peptides (Harmar 2001; Nadkarni et al., 2014), several of which have demonstrated neuroprotective actions (Dejda et al., 2005; Brenneman 2007; Li et al., 2016; Yu et al., 2016). Heterotrimeric stimulatory (Gs) GTP-binding proteins are activated in response to GLP-1R and GCGR agonist binding, and they couple agonist occupancy to the stimulation of transmembrane adenylyl cyclases that in neurons, like pancreatic β-cells, catalyze conversion of ATP to cytosolic cAMP, a second messenger that then activates key downstream proteins (Campbell and Drucker, 2013; Nadkarni et al., 2014). With this knowledge and strong evidence indicating that cAMP induces gene transcription through the activation of cAMP-dependent PKA and subsequent phosphorylation of the transcription factor CREB at Ser-133 (Delghandi et al., 2005; Sakomoto et al., 2011), we therefore probed neuronal cells and demonstrated that OXM increased intracellular cAMP levels and activated CREB. With the use of specific pathway inhibitors, we also found that among the downstream signaling pathways that OXM is known to stimulate, the PKA and MAPK pathways are essential to the neuroprotective effect of OXM, while the PI3K pathway is not. This is different from our previous findings on the neuroprotective effect of GLP-1, which relies on the PKA and PI3K pathways more than the MAPK pathway (Li et al., 2010b). Indeed, others have shown distinct stimulation patterns of OXM and GLP-1 in hypothalamic pathways (Chaudhri et al., 2006). These differences could be due to the different affinities of OXM and GLP-1 to the GLP-1R, or to their distinct interactions with the GCGR. The neuroprotective effects of glucagon have been reported in the literature. The mechanism of glucagon's neuroprotection is suggested mediated through stimulation of the cAMP/PKA pathway and reduction of neurotoxic glutamate (Fanne et al., 2011 and Armstead et al., 2011). It is also possible that OXM may stimulate a specific yet to be discovered receptor that explains these differences. OXM signaling in general, and its neuroprotective signaling pathways in particular, are still largely unexamined.
OXM is a natural peptide that interacts with both the GLP-1R and GCGR. With the use of receptor-specific antagonists, our results also suggest that the GLP-1R plays a more important role than does the GCGR in the neurotrophic and neuroprotective actions of OXM. Across a variety of non-neuronal cell lines, OXM is reported to be a low potency full agonist for cAMP accumulation from the GLP-1R (Schepp et al., 1996; Jorgensen et al., 2007) and the GCGR (Jorgensen et al., 2007). It additionally is a full agonist for recruitment of G protein-coupled receptor kinase (GRK) 2, β-arrestin 1 and β-arrestin 2 to the GCGR; whereas, at the GLP-1R, OXM is only a partial agonist for these actions (Jorgensen et al., 2007). Nonetheless, OXM is described to possess a higher affinity for the GLP-1R as compared to the GCGR, and hence OXM is considered to be a ‘biased’ ligand whose action is primarily mediated via the GLP-1R (Pabreja et al., 2014). Our studies in neuronal cells support this view and are additionally in line with previous findings showing that the anorectic effects of OXM were preserved in GCGR knockout mice but abolished in GLP-1R knockout mice (Baggio et al., 2004). It would be helpful to use receptor-specific knockout animals to further study the relative contributions of these two receptors to the neuroprotective action of OXM.
As a GLP-1R/GCGR dual agonist with promising effects on glycemic control and weight, OXM-based drugs have the potential to supersede traditional GLP-1R agonists as anti-diabetes and obesity treatments. Indeed, single agents that target two or more receptors present distinct advantages in pharmacological treatment design and are currently an interesting direction for future research. A series of GLP-1R/GCGR co-agonist peptide chimeras have been designed and tested in animal models (Day et al., 2009). A recently synthesized dual-agonist, nicknamed “twincretin”, was designed as a co-agonist for both GLP-1R and GIPR as a way to lower drug dose and reduce dose-limiting side effects (Finan et al., 2013). Most recently, a unimolecular, triple agonist that targets the GLP-1R, GIPR and GCGR has been reported to be successful in animal models of diabetes and obesity (Finan et al., 2015a and Finan et al., 2015b). Several of these agonists have recently been reported to provide neuroprotective actions (Jalewa et al., 2016; Tamargo et al., 2016).
In light of compelling evidence that glutamate excitotoxicity is a principle mechanism underlying neurodegeneration following cerebral ischemia associated with stroke (Lai et al., 2014) and that oxidative stress plays a critical role in the pathogenesis of ischaemic brain injury (Allen and Bayraktutan 2009; Rodrigo et al., 2013), we evaluated whether OXM neuroprotection in cell culture would translate to in vivo in a well characterized model of stroke, as this neurological disorder involves aspects of both apoptotic and necrotic cell death (Broughton et al., 2009; Yuan 2009; Filichia et al., 2015). As a proof of principle, although OXM, similar to GLP-1 and long-acting analogs, gains brain access (Kastin et al., 2002), it was administered by intracerebroventricular injection, rather than systemically, 15 min prior to 60 min MCAo to ensure that pharmacokinetic confounds did not undermine evaluation of its potential efficacy. Due to its rapid proteolytic inactivation by DPP4, OXM has a short half-life of 12 min in humans (Schjoldager et al., 1988) and 6.4 min in rats (Kervran et al., 1990). For therapeutic purposes, DPP4-resistant long-lasting OXM analogs have been developed using various strategies, such as PEGylation and crosslinking (Santoprete et al., 2011; Muppidi et al., 2016), and are generally administered subcutaneously; alternatively, short-acting OXM could potentially be administered intra-nasally to mitigate its rapid systemic metabolism. TT401 (LY2944876) from Transition Therapeutics Inc. is an OXM analog with dual agonist activity on the GLP-1R and GCGR. It appears to be the most clinically advanced drug candidate among the new class of GLP-1R/GCGR dual agonists. A recently completed phase II clinical trial in 420 T2DM patients showed that once weekly TT401 had superior weight loss effects as compared to the long-acting GLP-1 agonist Bydureon, while holding similar HbA1c reduction effects (Transition Therapeutics media release 2016); although its clinical development path is current uncertain. In addition to the potential benefits of OXM on neurodegeneration due to its anti-diabetes and anti-obesity actions, our work provides evidence that OXM stimulates specific neurotrophic and neuroprotective pathways that can combat neurodegeneration directly. Taken together, our cellular and proof of principle animal study suggest that OXM may be helpful for stroke subjects that are also diabetic and/or obese and add support for the investigation of long-acting OXM analogs as a potential treatment strategy for stroke and other neurodegenerative disorders (Liu et al., 2015) in which clinically translatable doses are administered systemically or intra-nasally after the development of the neurological disorder.
Conclusion
Our data in cellular and animal models indicate that the endogenous peptide OXM is neuroprotective against oxidative stress and glutamate toxicity and can reduce infarction size and improve locomotor activities in MCAo stroke rats. These effects appear to be primarily mediated via the GLP-1R and act through PKA/MAPK signaling pathways. Our results suggest that OXM and, in particular, long acting analogs may mitigate ischemic stroke and are worthy of further investigation for this and other neurodegenerative conditions.
Highlights.
We evaluated the endogenous 37-amino acid peptide hormone oxyntomodulin (OXM) for neurotrophic and neuroprotective actions in neuronal cultures and in a rat model of ischemic stroke.
OXM provided neurotrophic properties in immortal human SH-SY5Y neuronal cells - elevating cAMP levels and pCREB.
OXM provided neuroprotection against glutamate excitotoxicity in human SH-SY5Y cells and against glutamate excitotoxicity and oxidative stress injury in rat primary neuron cultures.
OXM neurotrophic and neuroprotective actions were primarily mediated by the GLP-1 receptor, rather than via the glucagon receptor, and involved the PKA and MAPK pathways, but not PI3K pathway.
In a proof of principle study involving direct intracerebroventricular delivery, OXM significantly reduced cerebral infarct size and improved locomotor activities in a 60-min, transient middle cerebral artery occlusion (MCAo) rat model of stroke.
Acknowledgments
Sources of Funding: This research was supported in part by: (i) the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Baltimore, MD, USA, and, (ii) National Health Research Institutes, Taiwan.
Footnotes
Disclosure/competing interests: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4:461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- Armstead WM, Kiessling JW, Cines DB, Higazi AA. Glucagon protects against impaired NMDA-mediated cerebrovasodilation and cerebral autoregulation during hypotension after brain injury by activating cAMP protein kinase A and inhibiting upregulation of tPA. J Neurotrauma. 2011;28:451–457. doi: 10.1089/neu.2010.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athauda D, Foltynie T. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson's disease: mechanisms of action. Drug Discov Today. 2016;21:802–818. doi: 10.1016/j.drudis.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T. Exenatide and the treatment of patients with Parkinson's disease. J Clin Invest. 2013;123:2730–2736. doi: 10.1172/JCI68295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagger JI, Holst JJ, Hartmann B, Andersen B, Knop FK, Vilsbøll T. Effect of Oxyntomodulin, Glucagon, GLP-1, and Combined Glucagon +GLP-1 Infusion on Food Intake, Appetite, and Resting Energy Expenditure. J Clin Endocrinol Metab. 2015;100:4541–4552. doi: 10.1210/jc.2015-2335. [DOI] [PubMed] [Google Scholar]
- Baggio LL, Huang Q, Brown TJ, Drucker DJ. Oxyntomodulin and glucagon-like peptide-1 differentially regulate murine food intake and energy expenditure. Gastroenterology. 2004;127:546–558. doi: 10.1053/j.gastro.2004.04.063. [DOI] [PubMed] [Google Scholar]
- Bassil F, Fernagut PO, Bezard E, Meissner WG. Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: targets for disease modification? Prog Neurobiol. 2014;118:1–18. doi: 10.1016/j.pneurobio.2014.02.005. [DOI] [PubMed] [Google Scholar]
- Bataille D, Dalle S. The forgotten members of the glucagon family. Diabetes Res Clin Pract. 2014;106:1–10. doi: 10.1016/j.diabres.2014.06.010. [DOI] [PubMed] [Google Scholar]
- Bataille D, Gespach C, Tatemoto K, Marie JC, Coudray AM, Rosselin G, Mutt V. Bioactive enteroglucagon (oxyntomodulin): present knowledge on its chemical structure and its biological activities. Peptides. 1981;2(2):41–44. doi: 10.1016/0196-9781(81)90008-5. [DOI] [PubMed] [Google Scholar]
- Brenneman DE. Neuroprotection: a comparative view of vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide. Peptides. 2007;28:1720–1726. doi: 10.1016/j.peptides.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–9. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013;17:819–837. doi: 10.1016/j.cmet.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Chaudhri OB, Parkinson JR, Kuo YT, Druce MR, Herlihy AH, Bell JD, Dhillo WS, Stanley SA, Ghatei MA, Bloom SR. Differential hypothalamic neuronal activation following peripheral injection of GLP-1 and oxyntomodulin in mice detected by manganese-enhanced magnetic resonance imaging. Biochem Biophys Res Commun. 2006;350:298–306. doi: 10.1016/j.bbrc.2006.09.033. [DOI] [PubMed] [Google Scholar]
- Chen R, Ovbiagele B, Feng W. Diabetes and Stroke: Epidemiology, Pathophysiology, Pharmaceuticals and Outcomes. Am J Med Sci. 2016;351:380–386. doi: 10.1016/j.amjms.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakin CL, Small CJ, Batterham RL, Neary NM, Cohen MA, Patterson M, Ghatei MA, Bloom SR. Peripheral oxyntomodulin reduces food intake and body weight gain in rats. Endocrinology. 2004;145:2687–2695. doi: 10.1210/en.2003-1338. [DOI] [PubMed] [Google Scholar]
- Darsalia V, Larsson M, Nathanson D, Klein T, Nyström T, Patrone C. Glucagon-like receptor 1 agonists and DPP-4 inhibitors: potential therapies for the treatment of stroke. J Cereb Blood Flow Metab. 2015;35:718–723. doi: 10.1038/jcbfm.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, Findeisen H, Bruemmer D, Drucker DJ, Chaudhary N, Holland J, Hembree J, Abplanalp W, Grant E, Ruehl J, Wilson H, Kirchner H, Lockie SH, Hofmann S, Woods SC, Nogueiras R, Pfluger PT, Perez-Tilve D, DiMarchi R, Tschöp MH. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5:749–757. doi: 10.1038/nchembio.209. [DOI] [PubMed] [Google Scholar]
- Dejda A, Sokołowska P, Nowak JZ. Neuroprotective potential of three neuropeptides PACAP, VIP and PHI. Pharmacol Rep. 2005;57:307–320. [PubMed] [Google Scholar]
- Delghandi MP, Johannessen M, Moens U. The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell Signal. 2005;17:1343–1351. doi: 10.1016/j.cellsig.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Eakin K, Li Y, Chiang YH, Hoffer BJ, Rosenheim H, Greig NH, Miller JP. Exendin-4 ameliorates traumatic brain injury-induced cognitive impairment in rats. PLoS One. 2013;8:e82016. doi: 10.1371/journal.pone.0082016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estall JL, Drucker DJ. Glucagon and glucagon-like peptide receptors as drug targets. Curr Pharm Des. 2006;12:1731–50. doi: 10.2174/138161206776873671. [DOI] [PubMed] [Google Scholar]
- Fanne RA, Nassar T, Heyman SN, Hijazi N, Higazi AA. Insulin and glucagon share the same mechanism of neuroprotection in diabetic rats: role of glutamate. Am J Physiol Regul Integr Comp Physiol. 2011;301:R668–673. doi: 10.1152/ajpregu.00058.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filichia E, Shen H, Zhou X, Qi X, Jin K, Greig NH, Hoffer B, Luo Y. Forebrain neuronal specific ablation of p53 gene provides protection in a cortical ischemic stroke model. Neuroscience. 2015;295:1–10. doi: 10.1016/j.neuroscience.2015.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finan B, Clemmensen C, Müller TD. Emerging opportunities for the treatment of metabolic diseases: Glucagon-like peptide-1 based multi-agonists. Mol Cell Endocrinol. 2015a;418:42–54. doi: 10.1016/j.mce.2015.07.003. [DOI] [PubMed] [Google Scholar]
- Finan B, Ma T, Ottaway N, Müller TD, Habegger KM, Heppner KM, Kirchner H, Holland J, Hembree J, Raver C, Lockie SH, Smiley DL, Gelfanov V, Yang B, Hofmann S, Bruemmer D, Drucker DJ, Pfluger PT, Perez-Tilve D, Gidda J, Vignati L, Zhang L, Hauptman JB, Lau M, Brecheisen M, Uhles S, Riboulet W, Hainaut E, Sebokova E, Conde-Knape K, Konkar A, DiMarchi RD, Tschöp MH. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. 2013;5:209ra151. doi: 10.1126/scitranslmed.3007218. [DOI] [PubMed] [Google Scholar]
- Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, Chabenne J, Zhang L, Habegger KM, Fischer K, Campbell JE, Sandoval D, Seeley RJ, Bleicher K, Uhles S, Riboulet W, Funk J, Hertel C, Belli S, Sebokova E, Conde-Knape K, Konkar A, Drucker DJ, Gelfanov V, Pfluger PT, Müller TD, Perez-Tilve D, DiMarchi RD, Tschöp MH. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. 2015b;21:27–36. doi: 10.1038/nm.3761. [DOI] [PubMed] [Google Scholar]
- Greig NH, Tweedie D, Rachmany L, Li Y, Rubovitch V, Schreiber S, Chiang YH, Hoffer BJ, Miller J, Lahiri DK, Sambamurti K, Becker RE, Pick CG. Incretin mimetics as pharmacologic tools to elucidate and as a new drug strategy to treat traumatic brain injury. Alzheimers Dement. 2014;10:S62–75. doi: 10.1016/j.jalz.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmar AJ. Family-B G-protein-coupled receptors. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-12-reviews3013. REVIEWS3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holscher C. Central effects of GLP-1: new opportunities for treatments of neurodegenerative diseases. J Endocrinol. 2014;221:T31–41. doi: 10.1530/JOE-13-0221. [DOI] [PubMed] [Google Scholar]
- Howard DB, Powers K, Wang Y, Harvey BK. Tropism and toxicity of adeno-associated viral vector serotypes 1, 2, 5, 6, 7, 8, and 9 in rat neurons and glia in vitro. Virology. 2008;372:24–34. doi: 10.1016/j.virol.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba-Hasegawa K, Akao Y, Maruyama W, Naoi M. Rasagiline and selegiline, inhibitors of type B monoamine oxidase, induce type A monoamine oxidase in human SH-SY5Y cells. J Neural Transm (Vienna) 2013;120:435–444. doi: 10.1007/s00702-012-0899-3. [DOI] [PubMed] [Google Scholar]
- Jalewa J, Sharma MK, Hölscher C. Novel incretin analogues improve autophagy and protect from mitochondrial stress induced by rotenone in SH-SY5Y cells. J Neurochem. 2016 doi: 10.1111/jnc.13736. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Jorgensen R, Kubale V, Vrecl M, Schwartz TW, Elling CE. Oxyntomodulin differentially affects glucagon-like peptide-1 receptor beta-arrestin recruitment and signaling through Galpha(s) J Pharmacol Exp Ther. 2007;322:148–154. doi: 10.1124/jpet.107.120006. [DOI] [PubMed] [Google Scholar]
- Kastin AJ, Akerstrom V, Pan W. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neurosci. 2002;18:7–14. doi: 10.1385/JMN:18:1-2:07. [DOI] [PubMed] [Google Scholar]
- Kervran A, Dubrasquet M, Blache P, Martinez J, Bataille D. Metabolic clearance rates of oxyntomodulin and glucagon in the rat: contribution of the kidney. Regul Pept. 1990;31:41–52. doi: 10.1016/0167-0115(90)90194-2. [DOI] [PubMed] [Google Scholar]
- Kuroki T, Tanaka R, Shimada Y, Yamashiro K, Ueno Y, Shimura H, Urabe T, Hattori N. Exendin-4 Inhibits Matrix Metalloproteinase-9 Activation and Reduces Infarct Growth After Focal Cerebral Ischemia in Hyperglycemic Mice. Stroke. 2016;47:1328–1335. doi: 10.1161/STROKEAHA.116.012934. [DOI] [PubMed] [Google Scholar]
- Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157–188. doi: 10.1016/j.pneurobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Li Y, Bader M, Tamargo I, Rubovitch V, Tweedie D, Pick CG, Greig NH. Liraglutide is neurotrophic and neuroprotective in neuronal cultures and mitigates mild traumatic brain injury in mice. J Neurochem. 2015;135:1203–17. doi: 10.1111/jnc.13169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chigurupati S, Holloway HW, Mughal M, Tweedie D, Bruestle DA, Mattson MP, Wang Y, Harvey BK, Ray B, Lahiri DK, Greig NH. Exendin-4 ameliorates motor neuron degeneration in cellular and animal models of amyotrophic lateral sclerosis. PLoS One. 2012;7:e32008. doi: 10.1371/journal.pone.0032008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, Tweedie D, Perry T, Mattson MP, Kapogiannis D, Sambamurti K, Lahiri DK, Greig NH. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer's disease. J Alzheimers Dis. 2010a;19:1205–1219. doi: 10.3233/JAD-2010-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu W, Li L, Hölscher C. Neuroprotective effects of a GIP analogue in the MPTP Parkinson's disease mouse model. Neuropharmacology. 2016;101:255–263. doi: 10.1016/j.neuropharm.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, Powers K, Shen H, Egan JM, Sambamurti K, Brossi A, Lahiri DK, Mattson MP, Hoffer BJ, Wang Y, Greig NH. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A. 2009;106:1285–1290. doi: 10.1073/pnas.0806720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tweedie D, Mattson M, Holloway H, Greig N. Enhancing the GLP-1 receptor signaling pathway leads to proliferation and neuroprotection in human neuroblastoma cells. J Neurochem. 2010b;113:1621–1631. doi: 10.1111/j.1471-4159.2010.06731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilja AM, Luo Y, Yu QS, Röjdner J, Li Y, Marini AM, Marutle A, Nordberg A, Greig NH. Neurotrophic and neuroprotective actions of (-)- and (+)-phenserine, candidate drugs for Alzheimer's disease. PLoS One. 2013;8:e54887. doi: 10.1371/journal.pone.0054887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Li Y, Jalewa J, Saunders-Wood T, Li L, Hölscher C. Neuroprotective effects of an oxyntomodulin analogue in the MPTP mouse model of Parkinson's disease. Eur J Pharmacol. 2015;765:284–90. doi: 10.1016/j.ejphar.2015.08.038. [DOI] [PubMed] [Google Scholar]
- Luo Y, Kuo CC, Shen H, Chou J, Greig NH, Hoffer BJ, Wang Y. Delayed treatment with a p53 inhibitor enhances recovery in stroke brain. Ann Neurol. 2009;65:520–530. doi: 10.1002/ana.21592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maida A, Lovshin JA, Baggio LL, Drucker DJ. The glucagon-like peptide-1 receptor agonist oxyntomodulin enhances beta-cell function but does not inhibit gastric emptying in mice. Endocrinology. 2008;149:5670–8. doi: 10.1210/en.2008-0336. [DOI] [PubMed] [Google Scholar]
- Melo A, Monteiro L, Lima RM, Oliveira DM, Cerqueira MD, El-Bachá RS. Oxidative stress in neurodegenerative diseases: mechanisms and therapeutic perspectives. Oxid Med Cell Longev. 2011;2011:467180. doi: 10.1155/2011/467180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muppidi A, Zou H, Yang PY, Chao E, Sherwood L, Nunez V, Woods AK, Schultz PG, Lin Q, Shen W. Design of Potent and Proteolytically Stable Oxyntomodulin Analogs. ACS Chem Biol. 2016;11:324–328. doi: 10.1021/acschembio.5b00787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadkarni P, Chepurny OG, Holz GG. Regulation of glucose homeostasis by GLP-1. Prog Mol Biol Transl Sci. 2014;121:23–65. doi: 10.1016/B978-0-12-800101-1.00002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najarian RM, Sullivan LM, Kannel WB, Wilson PW, D'Agostino RB, Wolf PA. Metabolic syndrome compared with type 2 diabetes mellitus as a risk factor for stroke: the Framingham Offspring Study. Arch Intern Med. 2006;166:106–111. doi: 10.1001/archinte.166.1.106. [DOI] [PubMed] [Google Scholar]
- Pabreja K, Mohd MA, Koole C, Wootten D, Furness SG. Molecular mechanisms underlying physiological and receptor pleiotropic effects mediated by GLP-1R activation. Br J Pharmacol. 2014;171:1114–1128. doi: 10.1111/bph.12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L, Du X, Petrov A, Lassman ME, Jiang G, Liu F, Miller C, Tota LM, Zhou G, Zhang X, Sountis MM, Santoprete A, Capito' E, Chicchi GG, Thornberry N, Bianchi E, Pessi A, Marsh DJ, SinhaRoy R. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes. 2009;58:2258–2266. doi: 10.2337/db09-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachmany L, Tweedie D, Li Y, Rubovitch V, Holloway HW, Miller J, Hoffer BJ, Greig NH, Pick CG. Exendin-4 induced glucagon-like peptide-1 receptor activation reverses behavioral impairments of mild traumatic brain injury in mice. Age (Dordr) 2013;35:1621–1636. doi: 10.1007/s11357-012-9464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo R, Fernández-Gajardo R, Gutiérrez R, Matamala JM, Carrasco R, Miranda-Merchak A, Feuerhake W. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 2013;12:698–714. doi: 10.2174/1871527311312050015. [DOI] [PubMed] [Google Scholar]
- Rydel RE, Greene LA. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci U S A. 1988;85:1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Karelina K, Obrietan K. CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem. 2011;116:1–9. doi: 10.1111/j.1471-4159.2010.07080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo I, Tweedie D, Li Y, Greig NH. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: an emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br J Pharmacol. 2012;166:1586–1599. doi: 10.1111/j.1476-5381.2012.01971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoprete A, Capitò E, Carrington PE, Pocai A, Finotto M, Langella A, Ingallinella P, Zytko K, Bufali S, Cianetti S, Veneziano M, Bonelli F, Zhu L, Monteagudo E, Marsh DJ, Sinharoy R, Bianchi E, Pessi A. DPP-IV-resistant, long-acting oxyntomodulin derivatives. J Pept Sci. 2011;17:270–280. doi: 10.1002/psc.1328. [DOI] [PubMed] [Google Scholar]
- Schepp W, Dehne K, Riedel T, Schmidtler J, Schaffer K, Classen M. Oxyntomodulin: a cAMP-dependent stimulus of rat parietal cell function via the receptor for glucagon-like peptide-1 (7-36)NH2. Digestion. 1996;57:398–405. doi: 10.1159/000201367. [DOI] [PubMed] [Google Scholar]
- Schjoldager BT, Baldissera FG, Mortensen PE, Holst JJ, Christiansen J. Oxyntomodulin: a potential hormone from the distal gut. Pharmacokinetics and effects on gastric acid and insulin secretion in man. Eur J Clin Invest. 1988;18:499–503. doi: 10.1111/j.1365-2362.1988.tb01046.x. [DOI] [PubMed] [Google Scholar]
- Shankar SS, Shankar RR, Mixson L, Pramanik BS, Stoch A, Stainberg HO, et al. Oxyntomodulin has significant acute glucoregulatory effects comparable to liraglutide in subjects with type 2 diabetes. Diabetes. 2013;62(Suppl.1):A48. [Google Scholar]
- Sharma MK, Jalewa J, Hölscher C. Neuroprotective and anti-apoptotic effects of liraglutide on SH-SY5Y cells exposed to methylglyoxal stress. J Neurochem. 2014;128:459–471. doi: 10.1111/jnc.12469. [DOI] [PubMed] [Google Scholar]
- Shen H, Wang Y. Correlation of locomotor activity and brain infarction in rats with transient focal ischemia. J Neurosci Methods. 2010;186:150–154. doi: 10.1016/j.jneumeth.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira MS, Linden R. Neuroprotection by cAMP: Another brick in the wall. Adv Exp Med Biol. 2006a;557:164–176. doi: 10.1007/0-387-30128-3_10. [DOI] [PubMed] [Google Scholar]
- Tamargo IA, Bader M, Li Y, Yu SJ, Wang Y, Talbot K, DiMarchi RD, Pick CG, Greig NH. Novel GLP-1R/GIPR co-agonist “twincretin” is neuroprotective in cell and rodent models of mild traumatic brain injury. Exp Neurol. 2016 doi: 10.1016/j.expneurol.2016.11.005. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T, Bloom SR. Gut hormones as therapeutic agents in treatment of diabetes and obesity. Curr Opin Pharmacol. 2013;13:996–1001. doi: 10.1016/j.coph.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Transition Therapeutics Inc. media release. 2016 http://www.transitiontherapeutics.com/media/press_releases/20160418.pdf, viewed Nov. 11, 2016.
- Wynne K, Bloom SR. The role of oxyntomodulin and peptide tyrosine-tyrosine (PYY) in appetite control. Nat Clin Pract Endocrinol Metab. 2006;2:612–620. doi: 10.1038/ncpendmet0318. [DOI] [PubMed] [Google Scholar]
- Yuan J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis. 2009;14:469–77. doi: 10.1007/s10495-008-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SJ, Wu KJ, Bae EK, Hsu MJ, Richie CT, Harvey BK, Wang Y. Methamphetamine induces a rapid increase of intracellular Ca(++) levels in neurons overexpressing GCaMP5. Addict Biol. 2016a;21:255–266. doi: 10.1111/adb.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YW, Hsieh TH, Chen KY, Wu JC, Hoffer BJ, Greig NH, Li Y, Lai JH, Chang CF, Lin JW, Chen YH, Yang LY, Chiang YH. Glucose-Dependent Insulinotropic Polypeptide Ameliorates Mild Traumatic Brain Injury-Induced Cognitive and Sensorimotor Deficits and Neuroinflammation in Rats. J Neurotrauma. 2016b doi: 10.1089/neu.2015.4229. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]