

Abstract
Brain extracellular matrix (ECM) is a highly organized system that consists of collagens, non-collagenous proteins, glycoproteins, hyaluronan and proteoglycans (PGs). Recognized physiological roles of ECM include developmental regulation, tissue homeostasis, cell migration, proliferation, differentiation, neuronal plasticity, and neurite outgrowth. Aberrant ECM structure is associated with brain neurodegenerative conditions. This review focuses on two neurodegenerative conditions, schizophrenia (Sz) and Alzheimer's disease (AD), and summarizes recent findings of altered ECM components, including PGs, glycosaminoglycans (GAGs), proteins and glycoproteins, and proteins and genes related to other brain components. The scope includes immunohistochemical, genomics, transcriptomics, proteomics and glycomics studies, and a critical assessment of current state of proteomics studies for neurodegenerative disorders. The intent is to summarize the ECM molecular alterations associated with neurodegenerative pathophysiology.
Keywords: brain extracellular matrix, proteoglycans, glycosaminoglycans, perineuronal nets, schizophrenia, Alzheimer's disease
Graphical abstract

Introduction
Extracellular matrix (ECM), constitutes approximately 20% of the adult brain volume [1] and has become a focus for neurological research. Functional roles of ECM include developmental regulation, tissue homeostasis, cell migration, proliferation, and differentiation [2]. The constituents of ECM include collagenous and non-collagenous proteins, glycoproteins, hyaluronan, and proteoglycans (PGs) [2]. Proteoglycans are proteins glycosylated with glycosaminoglycan (GAG) chains. GAGs play essential roles in all areas of physiology through their biophysical properties and capacities to bind growth factors and growth factor receptors. Thus, there is a keen interest in the roles of PG neuronal functions including cell adhesion, migration, regeneration, ECM assembly, neurite outgrowth and plasticity [3]. Proteoglycan structure is regulated in a spatial and temporal manner. As a result, structures and functions of proteoglycans in ECM depend on expression of core proteins via the endoplasmic reticulum and Golgi apparatus-mediated assembly of GAG chains attached to the core protein. Because the biosynthetic reactions do not go to completion, the GAG chains on PGs are heterogeneous with respect to chain length and modifications.
Neurodegenerative diseases, including schizophrenia (Sz), Alzheimer's disease (AD), multiple sclerosis (MS), Parkinson's disease (PD), and human immunodeficiency virus dementia (HIVD), result from gradual and progressive loss of neural cells and changes in ECM components that lead to brain dysfunction [4-7]. This review focuses on changes in ECM components specifically, proteoglycans and GAGs in SZ and AD. A better understanding of these changes in ECM will help unravel the biomolecular mechanisms related to complex neurodegenerative conditions in the human brain.
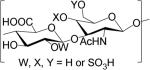
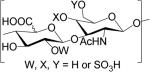
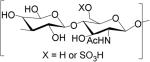
GAGs are classified into five major types based on the disaccharide unit; heparan sulfate (HS), chondroitin sulfate (CS), dermatan sulfate (DS), keratan sulfate (KS), and hyaluronan (HA). HS and CS/DS are unbranched polymers composed of ~20-200 repeating disaccharide units attached to serine or threonine residue of core protein through a characteristic tetrasaccharide linker [8]. The disaccharide units contain either of two modified sugars, N-acetylgalactosamine (GalNAc) or N-acetylglucosamine (GlcNAc) and a uronic acid (either glucuronate (GlcA) or iduronate (IdoA)). The structures of the GAG classes are shown in Table 1.
Table 1.
Glycosaminoglycan repeating unit structures: hyaluronan (HA), heparan sulfate (HS, including heparin), chondroitin sulfate (CS), dermatan sulfate (DS), and keratan sulfate (KS).
| GAG | Repeating unit | Modifications |
|---|---|---|
| Hyaluronan (HA) | GlcAβ3GlcNAcβ4 |

|
| Heparan sulfate (HS) and Heparin | GlcA/IdoAβ4/α4GlcNα4 |

|
| Chondroitin Sulfate (CS) | GlcAβ3GalNAcβ4 |
|
| Dermatan Sulfate (DS) | GlcA/IdoAβ/α3GalNAcβ4 |
|
| Keratan Sulfate (KS) | Galβ4GlcNAcβ3 |
|
The four PG classes include interstitial PGs, which are present in the ECM, basement membrane PGs, secretory granule PGs, and membrane-bound PGs [9]. Interstitial PGs are a critical component of ECM involved in stabilization and organization of collagen fibers. The interstitial PGs include small leucine-rich PGs (SLRPs) such as decorin, asporin, lumican, and biglycan [9, 10], and hyalectans, including aggrecan, versican, neurocan, and brevican [11]. The major proteoglycan found in cytoplasmic secretory granules is serglycin. It has heparin and/or CS chains attached to the core protein. It binds to several chemokines, growth factors, and inflammatory mediators, and modulates their bioactivity. Basement membrane PGs include HSPGs perlecan, agrin, collagen type XVIII, and leprecan. They mainly have HS chains attached to the core protein, except CSPG leprecan [12]. These PGs play important roles in cell adhesion, proliferation, growth factor signaling, embryogenesis, tissue morphogenesis, and cartilage development [12-14]. The membrane-bound syndecans and glypicans are involved in processes such as cell growth and angiogenesis [15, 16]. The syndecan family consists four members, syndecans 1, 2, 3, and 4, which carry HS and/or CS chains. On the other hand, glypicans consist of six members, GPC-1, 2, 3, 4, 5, and 6 attached to the plasma membrane via a glycosyl phosphatidyl inositol anchor and carrying only HS chains. CSPG4 is a single-pass, type-1 transmembrane proteoglycan that carry one CS chain [9].
Hyalectans functions in brain ECM are related to modulation of cell adhesion and migration [17]. The C-type lectin domain of hyalectans mediates binding to other ECM proteins [18], while the EGF-like repeats make hyalectans right candidates for growth control [19]. The roles of hyalectans in neurite outgrowth are also well-recognized [20]. Studies have shown that all four hyalectans inhibit neurite outgrowth in various neuronal cell types, through the CS chains [21-23]. A recent study showed a significant reduction in neurite outgrowth in vitro, in the presence of ECM components, including aggrecan, brevican, and tenascin-R [24].
HA is an important component of brain ECM. It binds to growth factors and cytokines including TGF-β, TNFα, epidermal growth factor (EGF), and keratinocytes [25]. Exogenous HA induces mesenchymal properties of epithelial cells, and digestion of HA by hyaluronidase disrupts epithelial-mesenchymal transition [26, 27]. Both increase and a decrease of hyaluronidase have been observed during tumorigenesis [28].
Function and Composition of Brain ECM
As shown in Fig. 1, brain ECM consists of relatively small amounts of fibrous proteins such as collagens and fibronectin and relatively high proportion of proteoglycans and hyaluronan. Brain hyalectans include aggrecan (210 kDa protein chain, CS and KS GAGs), neurocan (136 kDa protein chain, CS and DS GAGs), brevican (145 kDa protein chain, CS GAGs) and versican (265 kDa protein chain, CS and DS GAGs) [29]. Brain SLRPs include asporin (39 kDa protein chain, CS and DS GAGs), fibromodulin (42 kDa protein chain, KS GAGs), lumican (40 kDa protein chain, KS GAGs), biglycan (42 kDa, CS and DS GAGs) and decorin (40 kDa, CS and DS GAGs) [2]. Glycoproteins including collagens, laminins, tenascin, reelin, and fibronectin are among other components of the brain ECM.
Fig. 1.

Brain extracellular matrix showing HSPGs, CSPGs, HA, collagens, and other glycoproteins
ECM proteoglycans interact with brain ligands including growth factors, cell adhesion molecules, matrix components, and enzymes. This binding becomes considerably affected by variation in the GAG chains that influences the accessibility of ligands to cell surface receptors. The activity of growth factors may be either potentiated or inhibited by their interactions with ECM molecules. For example, vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) bind heparin / HS, which in turn modulates their biological activity [30, 31]. The CSPG phosphacan binds to FGFs and potentiates their mitogenic activities [32].
Among brain ECM structures, perineuronal nets (PNNs) are differentially distributed, highlighting their importance in brain physiology [24]. First described by Camillo Golgi and Santiago Ramon y Cajal in 1890, PNNs are highly organized reticular networks composed of ECM molecules surrounding neuronal cell bodies and proximal dendrites [33]. PNNs consist of hyalectican PGs, HA, tenascin-C, tenascin-R, hyaluronan and PG binding link proteins (Haplns, Hapln1/ Crtl1, and Hapln4/ Bral2). PNNs surrounding neurons carry three main components: a hyaluronan synthase (HAS), a link protein (usually cartilage link protein-1; crtl1) and a chondroitin sulfate proteoglycan (usually aggrecan). The physiological role of PNNs are not yet well-established; however, their postnatal appearance suggests a role in restricting the development of new synaptic contacts: synapse formation and synaptic plasticity [11]. In line with this, a study showed the development of synapses in parallel with the emergence of PNNs [34]. PNNs are observed ensheathing neurons, suggesting a supportive role in providing a microenvironment for neurons [35].
Changes in Brain ECM in Neurodegenerative conditions
Schizophrenia
Schizophrenia is a serious brain disorder affecting approximately 1% of the population worldwide. The clinical symptoms of Sz appear during late adolescence or early adulthood when cognitive functions, including reasoning, thinking, and planning, achieve maturation [36]. Characterized by defects in thinking and decision-making ability, Sz is defined by positive (hallucinations and delusions), and negative (affective flattening and cognitive deficits) symptoms [37]. Currently, antipsychotic medications are the only treatment available for Sz. Treatment provides relief to a subset of Sz patients but does not appear to impact the disease outcome significantly. Thus, lack of appropriate and effective treatment demands a better understanding of neuropathophysiology of the disease, paving the way for better treatment efficacy. It has been speculated that a better understanding of critical clinical signatures that occur before the onset of symptoms and timely intervention of these early signatures may reduce or prevent the onset of severe symptoms related to Sz [38]. However, significant progress towards these ends has not been reported. Maturation of prefrontal cortex (PFC) via extensive synaptic pruning occurs during late adolescence and early adulthood, and thus, it has been hypothesized that synaptic pruning that takes place in the PFC may play a critical role in the onset of Sz [39]. Interestingly, ECM molecules regulate processes occurring during late adolescence and early adulthood, including altered GABAergic function, synaptic pruning and plasticity, axonal guidance, neuronal differentiation, and migration [5].
A number of immunohistochemical (IHC) studies indicated patterns of ECM molecular expression associated with Sz, [40-45] (Table 2A). A marked decrease of CSPG-labeled PNNs in the amygdala and entorhinal cortex of subjects with Sz was reported [41, 46-49]. The PNNs were detected using Wisteria floribunda agglutinin (WFA) lectin which is believed to bind N-acetylgalactosamine residue of CS in the PNNs [50]. Interestingly, treatment with chondroitinase ABC, an enzyme known to degrade CS chains, abolished the WFA staining of PNNs, indicating the specificity of WFA for CSPGs [41]. Another study reported a decrease in WFA-labeled PNNs in layer 3 and 5 of the PFC of subjects with Sz compared to normal controls [42]. Intriguingly, an increase in the number of PNNs in the PFC during postnatal development through early adulthood has been reported [42], suggesting a parallel relationship of PNN development with the onset of Sz symptoms and indicating the contribution of altered PNN expression in the onset of the disease. A similar decrease in PNNs using antibodies against aggrecan, one of the main CSPG in the amygdala was observed for Sz subjects [44]. Another study quantified densities and fluorescence intensities of parvalbumin (PV) neurons and PNNs labeled with WFA versus an immunoreactive against major PNN protein, aggrecan in Sz and normal controls. They observed a decrease in the fluorescence intensities for both PV and PNNs labeled with WFA and aggrecan immunoreactive. However, densities of PV and PNNs were unchanged, suggesting that normal complements of PV and PNNs are preserved in Sz, but levels of PV and PNN proteins and carbohydrate moieties attached to it are dysregulated [51]. A significant reduction in numerical densities of cytoplasmic-CSPG and olfactory receptor neurons (ORNs) in Sz has been reported, suggesting a reduction of CSPG expression in mature ORNs, that may contribute to the ORN lineage dysregulation and olfactory identification abnormalities, observed in Sz [42].
Table 2.
Expression of ECM related components, including PNNs, PGs, glycoproteins, proteins and other ECM unrelated proteins in Schizophrenia (Sz) as reported in A) immunohistochemical studies B) Genomics studies, C) Proteomics studies, and D) Glycomics or glycosylation based studies.
| A. Immunohistochemical studies | ECM component | Brain region | Change in Sz | References |
|---|---|---|---|---|
| CSPG-labeled PNN (WFA labeling) | Amygdala and Entorhinal cortex | ↓ | [41] | |
| WFA-labeled PNNs | Layer 3 and 5 of the PFC | ↓ | [42] | |
| Aggrecan positive PNNs | Amygdala | ↓ | [44] | |
| Aggrecan positive PNNs and WFA labeled PNNs | Dorsolateral Prefrontal Cortex | ↓ | [51] | |
| Numerical densities of cytoplasmic-CSPG | Olfactory receptor neurons (ORNs) | ↓ | [42] |
| B. Genomics / Transcriptomics | Gene | Symbol | Change in Sz | References |
|---|---|---|---|---|
| ECM-related genes | ||||
| Aggrecan | ACAN | ↓ | [71] | |
| Versican | VCAN | ↓ | [71] | |
| Neurocan | NCAN | ↓ | [71] | |
| Lumican | LUM | ↓ | [71] | |
| Hyaluronan and proteoglycan link protein 1 | HAPLN1 | ↓ | [71] | |
| Reelin | RELN | ↑, ↓ | [52, 53], [164] | |
| Tenascin XB | TNXB | ↑ | [79] | |
| Fibronectin | FN | ↓ | [58, 59] | |
| Integrin α2bβ3a | ITGα2bβ3a | ↓ | [63] | |
| Matrix metalloproteinase-9 | MMP-9 | ↓, ↑ | [72], [69, 70] | |
| Matrix metalloproteinase-16 | MMP-16 | ↑ | [71] | |
| Matrix metalloproteinase-24 | MMP-24 | ↓ | [71] | |
| Matrix metalloproteinase-25 | MMP-25 | ↓ | [71] | |
| A disintegrin and metalloproteinase with thrombospondin motifs 1 | ADAMTS 1 | ↑ | [71] | |
| A disintegrin and metalloproteinase with thrombospondin motifs 6 | ADAMTS 6 | ↑ | [71] | |
| Phosphotyrosine phosphatase beta/zeta | PTPRZ1 | ↓ | [40, 73] | |
| Other proteins | ||||
| Neuregulin-1 isoform 1 | NRG1 | ↑ | [165, 166] | |
| Neuregulin-1 isoform 2 | NRG1 | ↓ | [165, 166] | |
| Receptor tyrosine-protein kinase erbB-4 | ERBB4 | ↑ | [167] | |
| C. Proteomics | Protein | Symbol | Change in Sz | References |
|---|---|---|---|---|
| ECM-related proteins | ||||
| Versican core protein | VCAN | ↓ | [78] | |
| Hyaluronan and proteoglycan link protein 2 | HAPLN2 | ↓ | [78] | |
| Galectin-1 | LGALS1 | ↓ | [81, 82] | |
| Vimentin | VIM | ↑ | [78, 81, 82] | |
| Other proteins | ||||
| Enolase 1 | ENO1 | ↑ | [78, 83, 84] | |
| Enolase 2 | ENO2 | ↑ | [78, 79, 83] | |
| Peroxiredoxin-1, 5 | PRDX1, PRDX5 | ↓ | [78-81, 85],[84] | |
| Glyceraldehyde-3 -phosphate dehydrogenase | GAPDH | ↓ | [78-81, 85] | |
| Superoxide dismutase 1 | SOD1 | ↓,↑ | [78-81, 85], [84] | |
| Heat shock 70 kDa protein | HSPA8 | ↑,↓ | [79, 80, 82], [84] | |
| Transferrin | TF | ↓,↑ | [79, 80], [82] | |
| 14-3-3 protein zeta/delta | YWHAZ | ↓ | [78, 80, 82] | |
| Protein S100-B | S100B | ↓ | [86] | |
| Septin-11 | SEPT11 | ↓ | [83, 84] | |
| Annexin A5, 6 | ANXA5, ANXA6 | ↓ | [83, 84] | |
| D. Glycomics / Glycosylation based studies | Glycans or glycosylated genes | Symbol | Change in Sz | References |
|---|---|---|---|---|
| Bisecting N-glycans | - | ↓ | [91] | |
| Sialylated N-glycans | - | ↓ | [91] | |
| UDP-GlcNAc:BetaGal Beta-1,3 GlcNAcT 8 | B3GNT8 | ↓ | [92] | |
| Mannosyl (alpha-1,3-)-glycoprotein beta-1,4 GlcNAcT | MGAT4A | ↓ | [92] | |
| 2-hydroxyacylsphingosine 1-beta-galactosyltransferase | UGT8 | ↓ | [90] | |
| Sphingosine-1-phosphate phosphatase 1 | SGPP1 | ↓ | [90] | |
| Galactocerebrosidase | GALC | ↓ | [90] | |
| Beta-1, 4-galactosyltransferase 6 | B4GALT6 | ↓ | [90] | |
| Serine palmitoyltransferase 2 | SPTLC2 | ↓ | [90] | |
| Acid ceramidase | ASAH1 | ↓ | [90] | |
| Galactosylceramide sulfotransferase | GAL3ST1 | ↓ | [90] |
Reelin, tenascin-X, fibronectin, and integrins have been extensively investigated in Sz, Table 2B. Reelin is an extracellular glycoprotein that performs diverse roles in the developing and adult brain, including regulation of neuronal migration, synapse development, synaptic plasticity, and learning and memory. In subjects with Sz, a reduced expression of both mRNA and protein levels of reelin was reported [52, 53]. Tenascin-X is the largest member of the tenascin family of ECM glycoproteins. It is involved in behavioral functions including anxiety, emotional learning and memory [54], and has been associated with Sz [55-57]. Fibronectin is a high molecular weight ECM glycoprotein that binds to integrin receptors and plays important roles in neuro- and synaptogenesis. Dysregulation of fibronectin may result in neurodevelopmental abnormalities related to the pathogenesis of Sz [58, 59]. In contrast, a study showed no significant association of fibronectin with Sz [60].
Integrins assist in cell-cell and cell-ECM interactions and play a significant role in the maintenance and modulation of neuronal synaptic activity, development of the central nervous system, and have been associated with the pathophysiology of Sz. For example, altered expression of ITGA8 and ITGB3 have been observed in Sz [61, 62]. In addition, a flow cytometry study analyzed surface expression of several glycoproteins and found a significant increase in integrin α2bβ3a expression in first episode Sz patients compared with healthy controls [63].
Levels of proteolytic enzymes including matrix metalloproteases (MMPs), a disintegrin and metalloproteases (ADAMs) and a disintegrin and metalloproteases with a thrombospondin motif (ADAMTS) have been associated with Sz, Table 2B [64-68]. These enzymes mediate proteolytic ECM remodeling by interaction with cell adhesion molecules, ECM receptors, and CSPGs, and thus, play important roles in synaptic plasticity [24]. Elevated levels of MMP-9 have been reported in previous Sz studies, including those of treatment-resistant Sz patients [69, 70]. Additionally, altered mRNA expression of MMPs (MMP 16, MMP 24, MMP 25), and ADAMTS-1 and -6 has also been reported in Sz [71]. In contrast, a recent study showed no difference in the level of MMP-9 in Sz compared to healthy controls. However, this expression level was demonstrated to be sensitive to electroconvulsive therapy, where MMP-9 levels were significantly decreased in Sz patients compared to healthy controls [72].
Several genetic studies indicate abnormal CSPG expression in Sz, Table 2B. For example, increased expression of receptor phosphotyrosine phosphatase beta/zeta (RPTPbeta), a transmembrane CSPG involved in critical processes such as synaptic plasticity and learning, was observed in patients with Sz. The signaling of neuregulin-1 (NRG1), a multifunctional protein that performs an important role in the development of the central nervous system, was reduced in mice overexpressing RPTPbeta [40]. Further, an increased expression of PTPRZ1, the gene encoding RPTPbeta has been reported in the amygdala and PFC of Sz subjects [40, 73]. Altered expression of NCAN, a gene encoding CSPG neurocan, have also been reported in Sz [74, 75]. Chondroitin sulfate proteoglycan 5 has been indicated as the potential susceptibility gene for Sz, along with other integrating partners, including NR1, ERRB4, and ERBB3 [76]. A recent study combining laser capture microdissection with gene expression profiling extracted RNA from laser-captured pyramidal neurons from postmortem brains from Sz and normal control subjects. The number of genes encoding CSPGs and associated with ECM, including ACAN, VCAN, LUM, and HAPLN1 were downregulated in Sz. In addition, altered expression of genes belonging to ECM related pathways, including transforming growth factor beta (TGF β) signaling, focal adhesion and ECM-receptor interaction pathways were observed [71]. These data strongly suggest a decrease in ECM structural components and enzymes that regulate ECM-related molecules and pathways in Sz.
Proteomics studies of Sz
Proteomics studies using different methods including label-free LC-MS/MS, 2DE-MS, 2D-DIGE-MS, have revealed several common altered proteins and pathways in Sz, compared to normal control samples [77], Table 2C. Altered proteins included enolase proteins (ENO1, ENO2), heat shock proteins (HSP60, HSPA8, HSPA5), vimentin (VIM), tubulin beta chain, 14-3-3 protein zeta/delta (YWHAZ), 14-3-3 protein epsilon (YWHAE), septin 11 (SEPT11), annexin 5, 6 (ANAX5, ANAX6), protein-L-isoaspartate(D-aspartate) O-methyltransferase (PCMT1), galectin-1 (LGALS1), superoxide dismutase (SOD1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), peroxiredoxin-1, 5 (PRDX1, PRDX5), and protein S100B, and pathways such as energy metabolism, oxidative stress response, cell growth and maintenance, cell signaling and interaction, cellular assembly and organization, oligodendrocyte dysfunction, and cytoskeleton structure assembly [78-86]. Some serum and blood-based biomarker studies have indicated sets of proteins that correlate with Sz. Examples of these are growth factors; VEGF, EGF, FGFs and cell adhesion molecule ICAM1 [87-89].
With respect to ECM, a recent study of the cytoplasmic proteome of a corpus callosum portion of the human brain from Sz and normal control subjects illustrated 65 differentially expressed proteins, including ECM-related proteins, versican, and HAPLN2 which were observed to be downregulated in Sz patients compared to normal controls [78]. Additionally, an extensive multi- (gen-, prote-, metabolo-) omics study indicated significant upregulation of ECM related tenascin XB, but only at the gene level [79].
Glycomics studies of Sz
Given to the inherent complexity of the human brain, studying only genetic or proteomic variation may not deliver a complete picture of the pathophysiology of Sz and other neurodegenerative disorders. Since most of the ECM molecules are either proteoglycans containing GAG side chains or glycoproteins containing N- and/or O-linked glycans, understanding glycosylation changes in Sz (and other disorders) is essential, Table 2D. Previous reports have suggested aberrant expression of genes (UGT8, SGPP1, GALC, B4GALT6, SPTLC2, ASAH1) particularly related to glycosphingolipid metabolism and N- and O-linked glycan biosynthesis in subjects with schizophrenia, compared to controls [90]. Moreover, a study on blood and cerebrospinal fluid (CSF) from Sz and control subjects identified decreases in bisecting and sialylated N-glycosylation in Sz patients. In addition, gender-based variations of N-glycans, including triantennary trisialylated glycan and tetraantennary tetrasialylated glycan with a polylactosamine extension, were observed between male and female Sz patients [91]. Some studies have measured protein expression of distinct N-acetylglucosamine transferases (GlcNAcTs) and found decreased protein expression of UDP-GlcNAc: BetaGal Beta-1,3 GlcNAcT 8 (B3GNT8) and mannosyl (alpha-1,3-)-glycoprotein beta-1,4 GlcNAcT (MGAT4A) expression in Sz, providing further evidence for dysregulated glycosylation in Sz [92].
Alzheimer's disease
Alzheimer's disease (AD) is the leading cause of dementia and cognitive decline in aged populations. Amyloid-beta (Aβ) precursor protein (APP) is associated with the formation of amyloid plaques. Tau protein is involved in the formation of neurofibrillary tangles [93]. Studies have indicated roles of HSPGs and CSPGs in Alzheimer's disease [94-97]. HS has been shown to bind and affect aggregation, intracellular internalization and clearance of Tau and APP [98]. It also regulates APP processing by BACE1, an enzyme that cleaves β site of the amyloid protein [99]. In addition, HS enhances and stabilizes Aβ fibril formation [100], dependent on the degree of sulfation of the HS [101, 102]. Several HSPGs including perlecan, glypican, syndecan, and agrin are associated with amyloid plaques in AD [100, 103-107]. Moreover, several reports have shown perlecan in senile plaques, neurofibrillary tangles, and amyloid-laden vessels [108-110].
Among CSPGs, CS-A, CS-C, and unsulfated chondroitin are associated with lesions of AD [96], and the DSPG decorin has been associated with amyloid plaques and neurofibrillary tangles in AD [111]. Intriguingly, appican, a CSPG modified alternatively spliced isoform of APP, have been observed to express in astrocytes, but not neurons, in AD. Appican-enriched ECM serves as a better substrate for attachment of N2a neuroblastomas, pheochromocytoma PC12 cells, and primary astrocytes, than APP enriched ECM [97, 112].
With respect to ECM, the distribution patterns CSPG in cortical areas of AD and normal human brains suggest that cortical neurons associated with PNNs and large proportions of hyalectan CSPGs were largely absent from neurofibrillary changes associated with AD [113]. Another study indicated decreases in the densities of WFA labeled PNNs in AD compared to normal brain samples [114]. Contrastingly, aggrecan-based PNN's were shown to be unaffected in a transgenic (Tg2576) mouse model of AD [115]. Loss of CSPGs was reported in cells stained with lectin Vicia villosa, which binds to terminal N-acetyl galactosamine residues present in CSPGs and/or glycoproteins, in AD compared to normal brains [116]. Taken together, ECM components, including CSPGs, and PNNs decrease in AD, and appear to play a neuroprotective role against tau pathology [117, 118]. Among other ECM components, hyaluronan positive neurons were observed to remain unchanged in AD brains, suggesting resistance of such neurons to neuropathological processes related to AD [119].
Because plaque formation characterizes AD, expression of Aβ degrading enzymes or proteases that degrade Aβ peptides are attractive therapeutic options. Examples of such enzymes include MMP-2, 3, 9, plasmin, neprilysin (NEP), and ADAM-10. ADAMs are increasingly recognized as targets for AD therapy [120]. Because it is an α-secretase involved in non-amyloidogenic cleavage of APP, upregulation of ADAM-10 mediated proteolysis could be a promising strategy to treat a neurodegenerative disorder such as AD [120]. In addition, upregulation of ADAM-10 mRNA expression in severe AD cases has been reported [121], and mutations in ADAM10 have been associated with a reduction of α-secretase in familial late-onset AD [122].
Among ECM glycoproteins, altered expression of reelin and its glycosylation patterns have been reported in CSF from AD patients [123]. A recent study showed perturbed expression of reelin and its downstream signaling members APOER2, VLDLR, and DAB1 in AD. In addition, it was demonstrated that depletion of reelin is an early event of AD pathology, occurring long before the onset of Aβ pathology, suggesting a possible role of decreased reelin expression in precipitation of AD pathology and supporting it as a potential pre-clinical marker of AD [124]. Congruently, reduced expression of reelin in the entorhinal cortex of transgenic mice and humans with AD has been reported [125]. Additionally, reelin and its signaling members have been shown to inhibit Aβ generation, promote Aβ clearance, or prevent tau hyperphosphorylation in AD pathology [126-128].
The αV integrin plays an important role in mediating synaptic dysfunction before neurodegeneration in AD [129]. Also, α2β1 and αVβ1 integrin signaling pathways mediate Aβ induced neurotoxicity in AD and may be a critical component of neurodegeneration [130]. LFA-1 integrin mediates infiltration of neutrophils to amyloid deposits in the brain. Inhibition of neutrophils or blockage of LFA-1 integrin-mediated neutrophil trafficking in a transgenic AD mouse model was shown to reduce AD neuropathology and improve memory in mice, suggesting a direct contribution of integrin-mediated neutrophil trafficking in AD pathogenesis and cognitive impairment [131]. Interestingly, APP regulates the activity of integrin β1 by binding to it. On shedding APP by treatment with alpha-secretase, the inhibitory activity of APP is removed and neurite outgrowth is triggered via integrins [132].
Proteomics studies of AD
In contrast to immunohistochemical studies that showed a reduction of ECM components and PNNs in AD, a recent proteomics study on a mouse model of AD, demonstrated significant increases in several protein components of the extracellular matrix including HAPLN1, neurocan, brevican, and tenascin-R (TNR), with increases in Aβ levels in the hippocampus. These increases in expression occurred with impairment of long-term potentiation and contextual memory, before the onset of plaque formation. Interestingly, chondroitinase ABC injection into the hippocampus restored both long-term potentiation and contextual memory performance [133]. Another recent study used laser capture microdissection (LCM) to excise hippocampal subareas CA1 and subiculum of 40 AD brains with subsequent LC-MS/MS-based proteomics. Among 372 altered proteins, several ECM components were increased in AD, including tenascin, versican, laminin subunit beta-2, and galectin-1. In addition, increases in adhesion molecules that interact with ECM, including CD44, integrin β1, and integrin αV were observed in AD [134]. In contrast, versican and tenascin-R were significantly decreased in frontal cortex AD brain tissue [135]. Table 3, summarizes the changes in ECM and related components in AD.
Table 3.
Expression pattern of ECM and associated components in AD.
| ECM and related component | Change | References |
|---|---|---|
| WFA-labeled PNNs | ↓ | [114] |
| N-acetyl galactosamine residues stained by lectin Vicia villosa | ↓ | [116] |
| ADAM-10 | ↑ | [121] |
| Reelin | ↓ | [125], [124] |
| Hyaluronan and proteoglycan link protein 1 | ↑ | [133] |
| Neurocan | ↑ | [133] |
| Brevican | ↑ | [133] |
| Tenascin-R | ↑,↓ | [133] ,[134], [135] |
| Versican | ↑, ↓ | [134], [135] |
| Laminin subunit beta-2 | ↑ | [134] |
| Galectin-1 | ↑ | [134] |
| CD44 antigen | ↑ | [134] |
| Integrin beta-1 | ↑ | [134] |
| Integrin alpha-V | ↑ | [134] |
Other LC-MS/MS-based proteomics studies on AD brain have identified several proteins, including microtubule-associated protein tau (MAPT), Aβ A4 protein precursor (APP), apolipoprotein E (apoE), ubiquitin carboxy-terminal hydrolase 1 (UCHL1), syntaxin-binding protein 1 (Munc-18), malate dehydrogenase (MDH2), protein piccolo (PCLO), transformation/transcription domain-associated protein (TRRAP), 14-3-3 protein zeta/delta (YWHAZ), and mucin 19 (MUC19), and pathways including signal transduction, immune response, cytoskeleton organization, lipid metabolism, energy metabolism, glycolysis, apoptosis, oxidative stress, oxidative phosphorylation and synaptic functioning, significantly altered in AD [135-141]. Further, blood plasma and serum proteomics studies from AD and control samples have been utilized to identify AD diagnostic biomarker candidates [142, 143]. For details on neuroproteomics in AD, refer to a recent review [144].
Glycomics studies of AD
Both tau and APP carry N- and O-glycosylation. Further, tau protein N-glycosylation occurs only in AD, and not in controls [145]. A recent study indicated GnT-III, an enzyme responsible for the formation of bisecting-GlcNAc type N-glycans, as a promising target for AD therapeutics. It was also shown that BACE1, an enzyme required to cleave amyloid-β (Aβ) and generate Aβ plaques in AD, is post-translationally modified with bisecting-GlcNAc type N-glycans in AD patients. Using knockout mouse mutants lacking GnT-III, decreased cleavage of APP by BACE1 was observed, resulting in reduced amyloid plaques and improved cognitive functions. It was suggested that due to lack of this sugar modification, BACE1 is directed towards late lysosomes, subjected to lysosomal degradation and thus less co-localized with APP [146]. Consistent with the above observation, an N-glycome profiling study indicated an increase in bisecting-GlcNAc type N-glycans and a decrease in overall sialylation in CSF from AD patients compared to normal controls [147]. The decrease in sialylated N-glycans in AD could be attributed to decreased sialyltransferase activity, which has been reported in serum as well as membrane and soluble fractions from postmortem brain from AD patients [148, 149]. In addition, lectin studies have shown decreased binding of WGA (wheat germ agglutinin), a lectin, that binds to sialic acid residues, in CSF of AD patients [150, 151]. Other than sialylated and bisecting N-glycans, reduction of NA2F, a core-fucosylated biantennary terminal galactose N-glycan structure, has been reported in AD compared to healthy controls, and it has been suggested to serve as suitable serum N-glycan marker for AD [152].
Current state of proteomics in studies of neurodegenerative disorders
Genomics and immunohistochemical analysis are widely employed to study neurodegenerative disorders [41, 42, 69, 71, 113, 114, 118]. They have provided a base of information for understanding the molecular deregulation associated with neurodegenerative disorders; however, genomics analysis does not sufficiently define the alterations at the protein level and associated biological functions. Expression and function of proteins can be modulated during transcription, translation and subsequently by post-translational modifications (PTMs), including phosphorylation, glycosylation, and acetylation. Proteomics analysis allows identification and quantification of thousands of proteins from complex mixtures, together with PTM and protein-protein interaction information, that are important to understand complete neuropathophysiology of these disorders.
In recent years, significant advances have been made in neuroproteomics, with a large number of well-executed proteomics studies providing some common altered proteins and pathways. However, the lack of consistent results and follow-up studies remains a limiting factor. For example, a recent Sz proteomics study [78], compared their findings with previous studies [79-85], and found consistent alteration of only 30 proteins, including 14-3-3 protein zeta/delta (YWHAZ), 14-3-3 protein epsilon (YWHAE), ENO1, ENO2, SOD1, GAPDH, PRDX1, VIM, and pathways, including energy and metabolism, cytoskeletal structure and function oxidative stress response, and oligodendrocyte dysfunction; however, the expression patterns of some of the proteins were observed to be in the opposite direction.
Similarly, for AD, a review [153], compared 43 2-D proteomics studies published between 1999-2010. A total of 93 proteins were differentially expressed in 13 different brain regions. Among these, only 42 altered proteins, were found in more than one study, regardless of brain region, while only 11 proteins (APOA1, apoE, PTGDS, TTR, ALDOA, FABP3, GAPDH, ENO1, GFAP, P1N1, UCHL1) were found to alter in a similar direction, in more than one study within the same brain region. For LC-MS/MS proteomics, a study [136], compared its 197 differentially expressed proteins with previous work [140, 141, 153-156] and found only 19 common proteins, including APP, YWHAZ, PRDX1, ANXA1, and HSPA12A, altered in the same direction.
Interestingly, low concordance was observed for differential proteins, while high concordance was observed for non-differential proteins. The low concordance rate of the altered proteome among different studies leads to inconsistent conclusions regarding pathological patterns related to these disorders. It is difficult to compare proteomics studies because several points of variation can emerge based on the type of sample used, sample handling techniques, and mass spectrometry methods. As shown in Table 4, several recent proteomics studies for Sz and AD employed different brain regions, a wide range of sample handling techniques and mass spectrometry instrumentation, leading to variable data, and only a few similarities in the findings, as indicated above. Increased use of standardized methods will improve the ability to compare protein expression in brain proteomics.
Table 4.
Examples of proteomics studies for Sz and AD, in recent years, employing different brain regions, a wide range of sample handling techniques and mass spectrometry instruments.
| Brain Region | Sample handling techniques |
Mass Spectrometry |
% Sequence coverage range for differential expressed proteins |
Modifications Identified |
Orthogonal Validation |
Differential expression of ECM-related proteins observed? |
References |
|---|---|---|---|---|---|---|---|
| Schizophrenia (Sz) | |||||||
| Corpus Callosum (CC) | Shotgun proteomics (Label-free) | LTQ-Orbitrap XL | No information | None | None | Yes (VCAN, HAPLN2) | Saia-Cereda et al., 2015 [78] |
| Dorsolateral Prefrontal Cortex | 2DE | QSTAR XL | 14-78% | None | None | No | Sivagnanasundaram et al., 2007 [85] |
| Dorsolateral Prefrontal Cortex | 2DE | MALDI-TOF/TOF | No information | None | Western blotting | No | Martins-de-Souza et al., 2009 [80] |
| Mediodorsal Thalamus | 2DE, iTRAQ-IEF | MALDI-TOF/TOF | No | None | Western blotting | Yes (LGALS1, VIM) | Martins-de-Souza et al., 2010 [81] |
| Prefrontal Cortex | 2D-DIGE | QTOF/TOF | 2-56% | None | Metabolomics, genomics | No | Prabakaran et al., 2004 [79] |
| Dorsolateral Prefrontal Cortex | 2D-DIGE | LTQ | 1-55% | None | ELISA | Yes (LGALS1, VIM) | English et al., 2009 [82] |
| Hippocampus | 2D-DIGE | Q-TOF | No information | None | Western blotting and ELISA | No | Focking et al., 2011 [83] |
| Hippocampus | 2D-DIGE | Q-TOF | No information | None | None | No | Schubert et al., 2015 [84] |
| Alzheimer's disease (AD) | |||||||
| Whole Brain | Shotgun proteomics (AMT tag approach) | LTQ Orbitrap | No information | None | Western blotting | No | Andreev et al., 2012 [136] |
| Frontal Cortex | Resolution shotgun proteomics (Label-free) | Orbitrap | Yesa | None | IHC, Western blotting | None | Gozal et al., 2009 [141] |
| Frontal Cortex | Resolution shotgun proteomics (Label-free) | LTQ Orbitrap | 8-68% | None | Immunoblotting | Yes (VCAN, TNR) | Donovan et al., 2012 [135] |
| Hippocampus | Laser capture microdissection (LCM), In-gel shotgun proteomics (Label-free) | LTQ Orbitrap | Yesa | None | IHC | Yes (VCAN, TNC, LGALS1, LAMB2) | Hondius et al., 2016 [134] |
| Frontal Cortex | 2D-DIGE | MALDI-TOF | 8-59% | None | None | No | Muller et al., 2008 [168] |
| Entorhinal Cortex | 1DE, 2DE | MALDI-TOF | No | Increased ubiquitination | IHC, Western blotting | No | Riederer et al., 2009 [169] |
| Inferior Parietal Lobule | 2DE | MALDI-TOF | 39-56% (for glutathionylated proteins) | Increased S-glutathionylation | None | No | Newman et al., 2007 [170] |
Percent not included, peptide sequence information provided
The human brain is a complex structure, with cellular and ECM heterogeneity based on the brain region. Thus, studies covering different brain regions make proteomics analysis much more challenging. In addition, studies on the human brain may have been affected by post-mortem interval and differences in tissue processing procedures [157]. Nevertheless, there remains an overwhelming need to characterize protein expression in both postmortem brains and animal models [158]. A recent study indicated a significant variability in quantitative LC-MS proteomics of human brain tissue samples arise from extraction methods (72%), and other variables, including instrumental variance (16%), instrumental stability (8.4%) and proteolytic digestion (3.1%) [159]. Because of this variability, it is necessary to analyze a large cohort of biological samples to validate the findings with enough statistical significance. In addition, differences in results could also be attributed to the method of separation and fractionation. Two-dimensional gel electrophoresis (2-DE), later modified to 2-D difference in-gel electrophoresis (2-D DIGE), were two methods of choice over the past two decades. However, these gel-based techniques involve certain limitations, including low sensitivity, underrepresentation of some low abundance and low solubility membrane proteins [160-162]. Gel-free approaches using liquid chromatography directly coupled to a mass spectrometer have been employed for label-free or label-based quantitative proteomics, to overcome such limitations. Although label-based quantitative proteomics allows accurate protein profiling, the limitations of such approaches include additional time-consuming sample preparation, the requirement of higher sample amounts, the risk of incomplete labeling, and the cost of the (commercial) reagents. Label-free quantitative proteomics is, therefore, an attractive alternative since it uses a robust, reasonable accurate, straightforward and time-effective experimental workflow.
For neuroproteomics, the majority of studies have taken a discovery approach, and not all have performed orthogonal validation of the findings, Table 4. Minimal attention has been paid to ECM-related proteins and PTMs, with very few studies indicating modifications of the proteins, and ECM proteins among differentially expressed proteins. Importantly, many of these proteomics studies were lacking relevant information including complete proteome lists, peptide lists, sequence coverage, and modifications. Such information is of high relevance to the neuroproteomics community, for cross-validation and comparison of the data, to understand neuropathophysiology.
ECM-related proteins were observed to be differentially expressed in only six studies, Table 4. Intriguingly, versican, one of the hyalectans, was differentially expressed in three out of six studies [78, 134, 135], reporting differential expression of ECM proteins. Notably, all three studies were performed as shotgun proteomics experiments, with label-free LC-MS/MS analysis, using an LTQ-Orbitrap instrument. The peptide sequence coverage for versican identified by two out of the three studies (one study did not provide sequence coverage information), is shown in Fig. 2. A study [135] identified 8% sequence coverage (yellow), while another study [134], identified 17% sequence coverage (red). Major domains covered by these studies were Ig-like V-type (21-146), link 1(150-245), link 2 (251-347), EGF-like 2, calcium binding (3176-3290) and c-type lectin (3294-3354) domains. Some peptides from GAG regions (GAG-alpha; 348-1335, GAG beta; 1336-3089) were also covered, Fig.2.
Fig. 2.

Peptide sequence coverage for Versican protein, identified by previous studies [135] (yellow) and [134] (red). Domain (blue numbers), Ig-like V-type; 21-146; link 1; 150-245, link 2; 251-347, EGF-like 1; 3089-3125, EGF-like 2, calcium binding; 3127-3163, c-type lectin; 3176-3290, Sushi; 3294-3354. GAG region (green numbers), GAG-alpha; 348-1335, GAG beta; 1336-3089.
Future directions
Previous proteomics studies of neurodegenerative disorders, including Sz and AD, show a lack of concordance with respect to differentially expressed proteins and associated pathways, which could be due to technical variability among different studies, including sample (number of samples, different brain regions), sample handling (2D, 2D-DIGE, LC-MS/MS), and MS (Orbitrap, Q-TOF, TOF/TOF), Table 4. Several steps can be taken to rule out such technical limitations and to obtain reliable and confident proteome profiles such as use of larger cohort of samples, robust and appropriate biological models with the correct controls, validation of protein expression levels by using a sufficient number of biological and technical replicate experiments, stringent data analysis, cross-validation of regulated proteins using orthogonal molecular techniques, e.g. western blotting, MRM, and ELISA, and use of ultra-performance high-end mass spectrometers.
Furthermore, although PTMs play important roles in regulating protein expression and related biological pathways [163], they have received very little attention in neuroproteomics. There is a need to perform in-depth analysis of global proteome data, since proteomics experiments deliver thousands of peptide fragments, and thus, there are substantial chances of finding modified peptides. Since modified peptides are present in low abundances compared to non-modified peptides, the use of enrichment methods will likely increase the relative abundances of PTM-peptides in the protein mixtures. Some examples of enrichment techniques include immunoprecipitation, affinity chromatography, and chemical derivatisation.
A plethora of altered proteins and biological pathways has been identified in neuroproteomics; but, disappointingly, only a handful of proteomics studies have identified altered ECM components. Nonetheless, some IHC and genomics studies have indicated changes in neural ECM associated with neurodegenerative conditions. Thus, given the importance of ECM components in neurodegenerative disorders, future neuroproteomics studies should target such ECM proteins for a better understanding of neuropathophysiology. At present, despite low concordance, and lack of additional information, studies have provided a large amount of information and datasets contributing to the knowledge base for understanding complex neuropathophysiology of neurodegenerative disorders.
Acknowledgements
This work was supported by NIH grants P41GM104602, R21CA177479, and R01MH105608.
Abbreviations
- ECM
extracellular matrix
- PGs
proteoglycans
- GAGs
glycosaminoglycans
- CS
chondroitin sulfate
- HS
heparan sulfate
- DS
dermatan sulfate
- KS
keratan sulfate
- HA
hyaluronic acid
- Sz
schizophrenia
- AD
Alzheimer's disease
- PNNs
perineuronal nets
- GalNAc
N-acetylgalactosamine
- GlcNAc
N-acetylglucosamine
- GlcA
glucuronic acid
- IdoA
iduronic acid
- 1-DE
one-dimensional gel electrophoresis
- 2-DE
two-dimensional gel electrophoresis
- 2-D DIGE
two-dimensional difference in-gel electrophoresis
- LC
liquid chromatography
- MS
mass spectrometer
- MALDI
matrix-assisted laser desorption/ionization
- TOF
time-of-flight
- PTMs
post-translational modifications
Footnotes
Published in the topical collection Glycomics, Glycoproteomics and Allied Topics with guest editors Yehia Mechref and David Muddiman.
Compliance with Ethical Standards
The authors declare no conflict of interest.
References
- 1.Nicholson C, Sykova E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998;21:207–215. doi: 10.1016/s0166-2236(98)01261-2. [DOI] [PubMed] [Google Scholar]
- 2.Novak U, Kaye AH. Extracellular matrix and the brain: components and function. J clin Neurosci. 2000;7:280–290. doi: 10.1054/jocn.1999.0212. [DOI] [PubMed] [Google Scholar]
- 3.Smith PD, Coulson-Thomas VJ, Foscarin S, Kwok JC, Fawcett JW. “GAG-ing with the neuron”: The role of glycosaminoglycan patterning in the central nervous system. Exp Neurol. 2015;274:100–114. doi: 10.1016/j.expneurol.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Bonneh-Barkay D, Wiley CA. Brain extracellular matrix in neurodegeneration. Brain Pathol. 2009;19:573–585. doi: 10.1111/j.1750-3639.2008.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berretta S. Extracellular matrix abnormalities in schizophrenia. Neuropharmacology. 2012;62:1584–1597. doi: 10.1016/j.neuropharm.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Busch SA, Silver J. The role of extracellular matrix in CNS regeneration. Curr Opin Neurobiol. 2007;17:120–127. doi: 10.1016/j.conb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 7.Green REA. Editorial on Brain Injury as a Neurodegenerative Disorder. Front Hum Neurosci. 2016 doi: 10.3389/fnhum.2015.00615. doi:10.3389/fnhum.2015.00615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kjellen L, Lindahl U. Proteoglycans: structures and interactions. Annu Rev Biochem. 1991;60:443–475. doi: 10.1146/annurev.bi.60.070191.002303. [DOI] [PubMed] [Google Scholar]
- 9.Iozzo RV, Schaefer L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015;42:11–55. doi: 10.1016/j.matbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schaefer L, Iozzo RV. Biological functions of the small leucine-rich proteoglycans: from genetics to signal transduction. J Biol Chem. 2008;283:21305–21309. doi: 10.1074/jbc.R800020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaguchi Y. Lecticans: organizers of the brain extracellular matrix. Cell Mol Life Sci. 2000;57:276–289. doi: 10.1007/PL00000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iozzo RV. Basement membrane proteoglycans: from cellar to ceiling. Nat Rev Mol Cell Biol. 2005;6:646–656. doi: 10.1038/nrm1702. [DOI] [PubMed] [Google Scholar]
- 13.Lord MS, Chuang CY, Melrose J, Davies MJ, Iozzo RV, Whitelock JM. The role of vascular-derived perlecan in modulating cell adhesion, proliferation and growth factor signaling. Matrix Biol. 2014;35:112–122. doi: 10.1016/j.matbio.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilusz RE, Sanchez-Adams J, Guilak F. The structure and function of the pericellular matrix of articular cartilage. Matrix Biol. 2014;39:25–32. doi: 10.1016/j.matbio.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Rossi G, Evans AR, Kay E, Woodfin A, McKay TR, Nourshargh S, Whiteford JR. Shed syndecan-2 inhibits angiogenesis. J Cell Sci. 2014;127:4788–4799. doi: 10.1242/jcs.153015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Filmus J, Capurro M, Rast J. Glypicans. Genome Biol. 2008;9:224. doi: 10.1186/gb-2008-9-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang BL, Zhang Y, Cao L, Yang BB. Cell adhesion and proliferation mediated through the G1 domain of versican. J Cell Biochem. 1999;72:210–220. doi: 10.1002/(sici)1097-4644(19990201)72:2<210::aid-jcb5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 18.Olin AI, Morgelin M, Sasaki T, Timpl R, Heinegard D, Aspberg A. The proteoglycans aggrecan and Versican form networks with fibulin-2 through their lectin domain binding. J Biol Chem. 2001;276:1253–1261. doi: 10.1074/jbc.M006783200. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Cao L, Yang BL, Yang BB. The G3 domain of versican enhances cell proliferation via epidermial growth factor-like motifs. J Biol Chem. 1998;273:21342–21351. doi: 10.1074/jbc.273.33.21342. [DOI] [PubMed] [Google Scholar]
- 20.Beller JA, Snow DM. Proteoglycans: road signs for neurite outgrowth. Neural Regen Res. 2014;9:343–355. doi: 10.4103/1673-5374.128235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedlander DR, Milev P, Karthikeyan L, Margolis RK, Margolis RU, Grumet M. The neuronal chondroitin sulfate proteoglycan neurocan binds to the neural cell adhesion molecules Ng-CAM/L1/NILE and N-CAM, and inhibits neuronal adhesion and neurite outgrowth. J Cell Biol. 1994;125:669–680. doi: 10.1083/jcb.125.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamada H, Fredette B, Shitara K, Hagihara K, Miura R, Ranscht B, Stallcup WB, Yamaguchi Y. The brain chondroitin sulfate proteoglycan brevican associates with astrocytes ensheathing cerebellar glomeruli and inhibits neurite outgrowth from granule neurons. J Neuroscience. 1997;17:7784–7795. doi: 10.1523/JNEUROSCI.17-20-07784.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snow DM, Lemmon V, Carrino DA, Caplan AI, Silver J. Sulfated proteoglycans in astroglial barriers inhibit neurite outgrowth in vitro. Exp Neurol. 1990;109:111–130. doi: 10.1016/s0014-4886(05)80013-5. [DOI] [PubMed] [Google Scholar]
- 24.Dauth S, Grevesse T, Pantazopoulos H, Campbell PH, Maoz BM, Berretta S, Parker KK. Extracellular matrix protein expression is brain region dependent. J Comp Neurol. 2016;524:1309–1336. doi: 10.1002/cne.23965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwok JC, Dick G, Wang D, Fawcett JW. Extracellular matrix and perineuronal nets in CNS repair. Dev Neurobiol. 2011;71:1073–1089. doi: 10.1002/dneu.20974. [DOI] [PubMed] [Google Scholar]
- 26.Zoltan-Jones A, Huang L, Ghatak S, Toole BP. Elevated hyaluronan production induces mesenchymal and transformed properties in epithelial cells. J Biol Chem. 2003;278:45801–45810. doi: 10.1074/jbc.M308168200. [DOI] [PubMed] [Google Scholar]
- 27.Spicer AP, Tien JY. Hyaluronan and morphogenesis. Birth Defects Res C Embryo Today. 2004;72:89–108. doi: 10.1002/bdrc.20006. [DOI] [PubMed] [Google Scholar]
- 28.McAtee CO, Barycki JJ, Simpson MA. Emerging roles for hyaluronidase in cancer metastasis and therapy. Adv Cancer Res. 2014;123:1–34. doi: 10.1016/B978-0-12-800092-2.00001-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Margolis RK, Margolis RU. Nervous tissue proteoglycans. EXS. 1993;49:429–446. doi: 10.1007/BF01923587. [DOI] [PubMed] [Google Scholar]
- 30.Schlessinger J, Lax I, Lemmon M. Regulation of growth factor activation by proteoglycans: what is the role of the low affinity receptors? Cell. 1995;83:357–360. doi: 10.1016/0092-8674(95)90112-4. [DOI] [PubMed] [Google Scholar]
- 31.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 32.Milev P, Monnerie H, Popp S, Margolis RK, Margolis RU. The core protein of the chondroitin sulfate proteoglycan phosphacan is a high-affinity ligand of fibroblast growth factor-2 and potentiates its mitogenic activity. J Biol Chem. 1998;273:21439–21442. doi: 10.1074/jbc.273.34.21439. [DOI] [PubMed] [Google Scholar]
- 33.Celio MR, Spreafico R, De Biasi S, Vitellaro-Zuccarello L. Perineuronal nets: past and present. Trends Neurosci. 1998;21:510–515. doi: 10.1016/s0166-2236(98)01298-3. [DOI] [PubMed] [Google Scholar]
- 34.Pyka M, Wetzel C, Aguado A, Geissler M, Hatt H, Faissner A. Chondroitin sulfate proteoglycans regulate astrocyte-dependent synaptogenesis and modulate synaptic activity in primary embryonic hippocampal neurons. Eur J Neurosci. 2011;33:2187–2202. doi: 10.1111/j.1460-9568.2011.07690.x. [DOI] [PubMed] [Google Scholar]
- 35.Reimers S, Hartlage-Rubsamen M, Bruckner G, Rossner S. Formation of perineuronal nets in organotypic mouse brain slice cultures is independent of neuronal glutamatergic activity. Eur J Neurosci. 2007;25:2640–2648. doi: 10.1111/j.1460-9568.2007.05514.x. [DOI] [PubMed] [Google Scholar]
- 36.Andersen SL. Trajectories of brain development: point of vulnerability or window of opportunity? Neurosci Biobehav Rev. 2003;27:3–18. doi: 10.1016/s0149-7634(03)00005-8. [DOI] [PubMed] [Google Scholar]
- 37.Woo TU. Neurobiology of schizophrenia onset. Curr Top Behav Neurosci. 2014;16:267–295. doi: 10.1007/7854_2013_243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGorry PD. Early intervention in psychotic disorders: beyond debate to solving problems. Br J Psychiatry Suppl. 2005;48:108–110. doi: 10.1192/bjp.187.48.s108. [DOI] [PubMed] [Google Scholar]
- 39.McGlashan TH, Hoffman RE. Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch Gen Psychiatry. 2000;57:637–648. doi: 10.1001/archpsyc.57.7.637. [DOI] [PubMed] [Google Scholar]
- 40.Takahashi N, Sakurai T, Bozdagi-Gunal O, Dorr NP, Moy J, Krug L, Gama-Sosa M, Elder GA, Koch RJ, Walker RH, Hof PR, Davis KL, Buxbaum JD. Increased expression of receptor phosphotyrosine phosphatase-beta/zeta is associated with molecular, cellular, behavioral and cognitive schizophrenia phenotypes. Transl Psychiatry. 2011 doi: 10.1038/tp.2011.8. doi: 10.1038/tp.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pantazopoulos H, Woo TU, Lim MP, Lange N, Berretta S. Extracellular matrix-glial abnormalities in the amygdala and entorhinal cortex of subjects diagnosed with schizophrenia. Arch Gen Psychiatry. 2010;67:155–166. doi: 10.1001/archgenpsychiatry.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mauney SA, Athanas KM, Pantazopoulos H, Shaskan N, Passeri E, Berretta S, Woo TU. Developmental pattern of perineuronal nets in the human prefrontal cortex and their deficit in schizophrenia. Biol Psychiatry. 2013;74:427–435. doi: 10.1016/j.biopsych.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shah A, Lodge DJ. A loss of hippocampal perineuronal nets produces deficits in dopamine system function: relevance to the positive symptoms of schizophrenia. Transl Psychiatry. 2013 doi: 10.1038/tp.2012.145. doi:10.1038/tp.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pantazopoulos H, Berretta S. Sickness and in Health: Perineuronal Nets and Synaptic Plasticity in Psychiatric Disorders. Neural Plast. 2016 doi: 10.1155/2016/9847696. doi: 10.1155/2016/9847696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Habl G, Schmitt A, Zink M, von Wilmsdorff M, Yeganeh-Doost P, Jatzko A, Schneider-Axmann T, Bauer M, Falkai P. Decreased reelin expression in the left prefrontal cortex (BA9) in chronic schizophrenia patients. Neuropsychobiology. 2012;66:57–62. doi: 10.1159/000337129. [DOI] [PubMed] [Google Scholar]
- 46.Niu L, Matsui M, Zhou SY, Hagino H, Takahashi T, Yoneyama E, Kawasaki Y, Suzuki M, Seto H, Ono T, Kurachi M. Volume reduction of the amygdala in patients with schizophrenia: a magnetic resonance imaging study. Psychiatry Res. 2004;132:41–51. doi: 10.1016/j.pscychresns.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 47.Aleman A, Kahn RS. Strange feelings: do amygdala abnormalities dysregulate the emotional brain in schizophrenia? Prog Neurobiol. 2005;77:283–298. doi: 10.1016/j.pneurobio.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 48.Baiano M, Perlini C, Rambaldelli G, Cerini R, Dusi N, Bellani M, Spezzapria G, Versace A, Balestrieri M, Mucelli RP, Tansella M, Brambilla P. Decreased entorhinal cortex volumes in schizophrenia. Schizophr Res. 2008;102:171–180. doi: 10.1016/j.schres.2007.11.035. [DOI] [PubMed] [Google Scholar]
- 49.Harrison PJ. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain. 1999;122:593–624. doi: 10.1093/brain/122.4.593. [DOI] [PubMed] [Google Scholar]
- 50.Nakagawa F, Schulte BA, Spicer SS. Selective cytochemical demonstration of glycoconjugate-containing terminal N-acetylgalactosamine on some brain neurons. J Comp Neurol. 1986;243:280–290. doi: 10.1002/cne.902430210. [DOI] [PubMed] [Google Scholar]
- 51.Enwright JF, Sanapala S, Foglio A, Berry R, Fish KN, Lewis DA. Reduced Labeling of Parvalbumin Neurons and Perineuronal Nets in the Dorsolateral Prefrontal Cortex of Subjects with Schizophrenia. Neuropsychopharmacology. 2016 doi: 10.1038/npp.2016.24. doi: 10.1038/npp.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Impagnatiello F, Guidotti AR, Pesold C, Dwivedi Y, Caruncho H, Pisu MG, Uzunov DP, Smalheiser NR, Davis JM, Pandey GN, Pappas GD, Tueting P. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc Natl Acad Sci U.S.A. 1998;95:15718–15723. doi: 10.1073/pnas.95.26.15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, Impagnatiello F, Pandey G, Pesold C, Sharma R, Uzunov D, Costa E. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- 54.Kawakami K, Matsumoto K. Behavioral alterations in mice lacking the gene for tenascin-x. Biol Pharm Bull. 2011;34:590–593. doi: 10.1248/bpb.34.590. [DOI] [PubMed] [Google Scholar]
- 55.Liu LL, Wei J, Zhang X, Li XY, Shen Y, Liu SZ, Ju GZ, Shi JP, Yu YQ, Xu Q, Hemmings GP. Lack of a genetic association between the TNXB locus and schizophrenia in a Chinese population. Neurosci Lett. 2004;355:149–151. doi: 10.1016/j.neulet.2003.10.059. [DOI] [PubMed] [Google Scholar]
- 56.Tochigi M, Zhang X, Ohashi J, Hibino H, Otowa T, Rogers M, Kato T, Okazaki Y, Kato N, Tokunaga K, Sasaki T. Association study between the TNXB locus and schizophrenia in a Japanese population. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:305–309. doi: 10.1002/ajmg.b.30441. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Sun S, Zhang L, Wang Z, Ye L, Liu L, Wu N, Li H, Zhang X, Wu J. Further study of genetic association between the TNXB locus and schizophrenia. Psychiatr Genet. 2011 doi: 10.1097/YPG.0b013e3283413398. doi: 101097/YPG.0b013e3283413398. [DOI] [PubMed] [Google Scholar]
- 58.Mahadik SP, Mukherjee S, Wakade CG, Laev H, Reddy RR, Schnur DB. Decreased adhesiveness and altered cellular distribution of fibronectin in fibroblasts from schizophrenic patients. Psychiatry Res. 1994;53:87–97. doi: 10.1016/0165-1781(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 59.Miyamae Y, Nakamura Y, Kashiwagi Y, Tanaka T, Kudo T, Takeda M. Altered adhesion efficiency and fibronectin content in fibroblasts from schizophrenic patients. Psychiatry Clin Neurosci. 1998;52:345–352. doi: 10.1046/j.1440-1819.1998.00386.x. [DOI] [PubMed] [Google Scholar]
- 60.Nakata K, Ujike H, Sakai A, Takaki M, Imamura T, Tanaka Y, Kuroda S. Association study between the fibronectin gene and schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2003;116B:41–44. doi: 10.1002/ajmg.b.10796. [DOI] [PubMed] [Google Scholar]
- 61.Supriyanto I, Watanabe Y, Mouri K, Shiroiwa K, Ratta-Apha W, Yoshida M, Tamiya G, Sasada T, Eguchi N, Okazaki K, Shirakawa O, Someya T, Hishimoto A. A missense mutation in the ITGA8 gene, a cell adhesion molecule gene, is associated with schizophrenia in Japanese female patients. Progr Neuropsychopharmacol Biol Psychiatry. 2013;40:347–352. doi: 10.1016/j.pnpbp.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 62.Wang KS, Liu X, Arana TB, Thompson N, Weisman H, Devargas C, Mao C, Su BB, Camarillo C, Escamilla MA, Xu C. Genetic association analysis of ITGB3 polymorphisms with age at onset of schizophrenia. J Mol Neurosci. 2013;51:446–453. doi: 10.1007/s12031-013-0059-8. [DOI] [PubMed] [Google Scholar]
- 63.Walsh MT, Ryan M, Hillmann A, Condren R, Kenny D, Dinan T, Thakore JH. Elevated expression of integrin alpha(IIb) beta(IIIa) in drug-naive, first-episode schizophrenic patients. Biol Psychiatry. 2002;52:874–879. doi: 10.1016/s0006-3223(02)01400-2. [DOI] [PubMed] [Google Scholar]
- 64.Dow DJ, Huxley-Jones J, Hall JM, Francks C, Maycox PR, Kew JN, Gloger IS, Mehta NA, Kelly FM, Muglia P, Breen G, Jugumauth S, Pederoso I, St Clair D, Rujescu D, Barnes MR. ADAMTSL3 as a candidate gene for schizophrenia: gene sequencing and ultra-high density association analysis by imputation. Schizophr Res. 2011;127:28–34. doi: 10.1016/j.schres.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 65.Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chopra K, Baveja A, Kuhad A. MMPs: a novel drug target for schizophrenia. Exp Opin Ther Targets. 2015;19:77–85. doi: 10.1517/14728222.2014.957672. [DOI] [PubMed] [Google Scholar]
- 67.Wei J, Richbourgh B, Jia T, Liu C. ADAMTS-12: a multifaced metalloproteinase in arthritis and inflammation. Mediators Inflamm. 2014 doi: 10.1155/2014/649718. doi: 10.1155/2014/649718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Farkas N, Lendeckel U, Dobrowolny H, Funke S, Steiner J, Keilhoff G, Schmitt A, Bogerts B, Bernstein HG. Reduced density of ADAM 12-immunoreactive oligodendrocytes in the anterior cingulate white matter of patients with schizophrenia. World J Biol Psychiatry. 2010;11:556–566. doi: 10.3109/15622970903497936. [DOI] [PubMed] [Google Scholar]
- 69.Domenici E, Wille DR, Tozzi F, Prokopenko I, Miller S, McKeown A, Brittain C, Rujescu D, Giegling I, Turck CW, Holsboer F, Bullmore ET, Middleton L, Merlo-Pich E, Alexander RC, Muglia P. Plasma protein biomarkers for depression and schizophrenia by multi analyte profiling of case-control collections. PLos One. 2010 doi: 10.1371/journal.pone.0009166. doi: 10.1371/journal.pone.0009166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamamori H, Hashimoto R, Ishima T, Kishi F, Yasuda Y, Ohi K, Fujimoto M, Umeda-Yano S, Ito A, Hashimoto K, Takeda M. Plasma levels of mature brain-derived neurotrophic factor (BDNF) and matrix metalloproteinase-9 (MMP-9) in treatment-resistant schizophrenia treated with clozapine. Neurosci Lett. 2013;556:37–41. doi: 10.1016/j.neulet.2013.09.059. [DOI] [PubMed] [Google Scholar]
- 71.Pietersen CY, Mauney SA, Kim SS, Lim MP, Rooney RJ, Goldstein JM, Petryshen TL, Seidman LJ, Shenton ME, McCarley RW, Sonntag KC, Woo TU. Molecular profiles of pyramidal neurons in the superior temporal cortex in schizophrenia. J Neurogenet. 2014;28:53–69. doi: 10.3109/01677063.2014.882918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shibasaki C, Takebayashi M, Itagaki K, Abe H, Kajitani N, Okada-Tsuchioka M, Yamawaki S. Altered Serum Levels of Matrix Metalloproteinase-2, -9 in Response to Electroconvulsive Therapy for Mood Disorders. Int J Neuropsychopharmacol. 2016 doi: 10.1093/ijnp/pyw019. doi:10.1093/ijnp/pyw019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buxbaum JD, Georgieva L, Young JJ, Plescia C, Kajiwara Y, Jiang Y, Moskvina V, Norton N, Peirce T, Williams H, Craddock NJ, Carroll L, Corfas G, Davis KL, Owen MJ, Harroch S, Sakurai T, O'Donovan MC. Molecular dissection of NRG1-ERBB4 signaling implicates PTPRZ1 as a potential schizophrenia susceptibility gene. Mol Psychiatry. 2008;13:162–172. doi: 10.1038/sj.mp.4001991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Muhleisen TW, Mattheisen M, Strohmaier J, Degenhardt F, Priebe L, Schultz CC, Breuer R, Meier S, Hoffmann P; Group Investigators. Rivandeneira F, Hofman A, Uitterlinden AG, Moebus S, Gieger C, Emeny R, Ladwig KH, Wichmann HE, Schwarz M, Kammerer-Ciernioch J, Schlösser RG, Nenadic I, Sauer H, Mössner R, Maier W, Rujescu D, Lange C, Ophoff RA, Schulze TG, Rietschel M, Nöthen MM, Cichon S. Association between schizophrenia and common variation in neurocan (NCAN), a genetic risk factor for bipolar disorder. Schizophr Res. 2012;138:69–73. doi: 10.1016/j.schres.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 75.Schultz CC, Muhleisen TW, Nenadic I, Koch K, Wagner G, Schachtzabel C, Siedek F, Nothen MM, Rietschel M, Deufel T, Kiehntopf M, Cichon S, Reichenbach JR, Sauer H, Schlösser RG. Common variation in NCAN, a risk factor for bipolar disorder and schizophrenia, influences local cortical folding in schizophrenia. Psychol Med. 2014;44:811–820. doi: 10.1017/S0033291713001414. [DOI] [PubMed] [Google Scholar]
- 76.So HC, Fong PY, Chen RY, Hui TC, Ng MY, Cherny SS, Mak WW, Cheung EF, Chan RC, Chen EY, Li T, Sham PC. Identification of neuroglycan C and interacting partners as potential susceptibility genes for schizophrenia in a Southern Chinese population. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:103–113. doi: 10.1002/ajmg.b.30961. [DOI] [PubMed] [Google Scholar]
- 77.Davalieva K, Maleva Kostovska I, Dwork AJ. Proteomics Research in Schizophrenia. FronT Cell Neurosci. 2016 doi: 10.3389/fncel.2016.00018. doi:10.3389/fncel.2016.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saia-Cereda VM, Cassoli JS, Schmitt A, Falkai P, Nascimento JM, Martins-de-Souza D. Proteomics of the corpus callosum unravel pivotal players in the dysfunction of cell signaling, structure, and myelination in schizophrenia brains. Eur Arch Psychiatry Clin Neurosci. 2015;265:601–612. doi: 10.1007/s00406-015-0621-1. [DOI] [PubMed] [Google Scholar]
- 79.Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL, Wayland M, Freeman T, Dudbridge F, Lilley KS, Karp NA, Hester S, Tkachev D, Mimmack ML, Yolken RH, Webster MJ, Torrey EF, Bahn S. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry. 2004;9:684–697, 643. doi: 10.1038/sj.mp.4001511. [DOI] [PubMed] [Google Scholar]
- 80.Martins-de-Souza D, Gattaz WF, Schmitt A, Maccarrone G, Hunyadi-Gulyas E, Eberlin MN, Souza GH, Marangoni S, Novello JC, Turck CW, Dias-Neto E. Proteomic analysis of dorsolateral prefrontal cortex indicates the involvement of cytoskeleton, oligodendrocyte, energy metabolism and new potential markers in schizophrenia. J Psychiatr Res. 2009;43:978–986. doi: 10.1016/j.jpsychires.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 81.Martins-de-Souza D, Maccarrone G, Wobrock T, Zerr I, Gormanns P, Reckow S, Falkai P, Schmitt A, Turck CW. Proteome analysis of the thalamus and cerebrospinal fluid reveals glycolysis dysfunction and potential biomarkers candidates for schizophrenia. J Psychiatr Res. 2010;44:1176–1189. doi: 10.1016/j.jpsychires.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 82.English JA, Dicker P, Focking M, Dunn MJ, Cotter DR. 2-D DIGE analysis implicates cytoskeletal abnormalities in psychiatric disease. Proteomics. 2009;9:3368–3382. doi: 10.1002/pmic.200900015. [DOI] [PubMed] [Google Scholar]
- 83.Focking M, Dicker P, English JA, Schubert KO, Dunn MJ, Cotter DR. Common proteomic changes in the hippocampus in schizophrenia and bipolar disorder and particular evidence for involvement of cornu ammonis regions 2 and 3. Arch Gen Psychiatry. 2011;68:477–488. doi: 10.1001/archgenpsychiatry.2011.43. [DOI] [PubMed] [Google Scholar]
- 84.Schubert KO, Focking M, Cotter DR. Proteomic pathway analysis of the hippocampus in schizophrenia and bipolar affective disorder implicates 14-3-3 signaling, aryl hydrocarbon receptor signaling, and glucose metabolism: potential roles in GABAergic interneuron pathology. Schizophr Res. 2015;167:64–72. doi: 10.1016/j.schres.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 85.Sivagnanasundaram S, Crossett B, Dedova I, Cordwell S, Matsumoto I. Abnormal pathways in the genu of the corpus callosum in schizophrenia pathogenesis: a proteome study. Proteomics Clin Appl. 2007;1:1291–1305. doi: 10.1002/prca.200700230. [DOI] [PubMed] [Google Scholar]
- 86.Steiner J, Schmitt A, Schroeter ML, Bogerts B, Falkai P, Turck CW, Martins-de-Souza D. S100B is downregulated in the nuclear proteome of schizophrenia corpus callosum. Eur Arch Psychiatry Clin Neurosci. 2014;264:311–316. doi: 10.1007/s00406-014-0490-z. [DOI] [PubMed] [Google Scholar]
- 87.Schwarz E, Izmailov R, Spain M, Barnes A, Mapes JP, Guest PC, Rahmoune H, Pietsch S, Leweke FM, Rothermundt M, Steiner J, Koethe D, Kranaster L, Ohrmann P, Suslow T, Levin Y, Bogerts B, van Beveren NJ, McAllister G, Weber N, Niebuhr D, Cowan D, Yolken RH, Bahn S. Validation of a blood-based laboratory test to aid in the confirmation of a diagnosis of schizophrenia. Biomark Insights. 2010;5:39–47. doi: 10.4137/bmi.s4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schwarz E, Guest PC, Rahmoune H, Harris LW, Wang L, Leweke FM, Rothermundt M, Bogerts B, Koethe D, Kranaster L, Ohrmann P, Suslow T, McAllister G, Spain M, Barnes A, van Beveren NJ, Baron-Cohen S, Steiner J, Torrey FE, Yolken RH, Bahn S. Identification of a biological signature for schizophrenia in serum. Mol Psychiatry. 2012;17:494–502. doi: 10.1038/mp.2011.42. [DOI] [PubMed] [Google Scholar]
- 89.Schwarz E, van Beveren NJ, Ramsey J, Leweke FM, Rothermundt M, Bogerts B, Steiner J, Guest PC, Bahn S. Identification of subgroups of schizophrenia patients with changes in either immune or growth factor and hormonal pathways. Schizophr Bull. 2014;40:787–795. doi: 10.1093/schbul/sbt105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Narayan S, Head SR, Gilmartin TJ, Dean B, Thomas EA. Evidence for disruption of sphingolipid metabolism in schizophrenia. J Neurosci Res. 2009;87:278–288. doi: 10.1002/jnr.21822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stanta JL, Saldova R, Struwe WB, Byrne JC, Leweke FM, Rothermund M, Rahmoune H, Levin Y, Guest PC, Bahn S, Rudd PM. Identification of N-glycosylation changes in the CSF and serum in patients with schizophrenia. J Proteome Res. 2010;9:4476–4489. doi: 10.1021/pr1002356. [DOI] [PubMed] [Google Scholar]
- 92.Kippe JM, Mueller TM, Haroutunian V, Meador-Woodruff JH. Abnormal N-acetylglucosaminyltransferase expression in prefrontal cortex in schizophrenia. Schizophr Res. 2015;166:219–224. doi: 10.1016/j.schres.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Selkoe DJ. Alzheimer's disease: genotypes, phenotypes, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 94.Schworer R, Zubkova OV, Turnbull JE, Tyler PC. Synthesis of a targeted library of heparan sulfate hexa to dodecasaccharides as inhibitors of beta-secretase: potential therapeutics for Alzheimer's disease. Chemistry. 2013;19:6817–6823. doi: 10.1002/chem.201204519. [DOI] [PubMed] [Google Scholar]
- 95.Patey SJ, Edwards EA, Yates EA, Turnbull JE. Engineered heparins: novel beta-secretase inhibitors as potential Alzheimer's disease therapeutics. Neurodegener Dis. 2008;5:197–199. doi: 10.1159/000113701. [DOI] [PubMed] [Google Scholar]
- 96.DeWitt DA, Silver J, Canning DR, Perry G. Chondroitin sulfate proteoglycans are associated with the lesions of Alzheimer's disease. Exp Neurol. 1993;121:149–152. doi: 10.1006/exnr.1993.1081. [DOI] [PubMed] [Google Scholar]
- 97.Pangalos MN, Shioi J, Efthimiopoulos S, Wu A, Robakis NK. Characterization of appican, the chondroitin sulfate proteoglycan form of the Alzheimer amyloid precursor protein. Neurodegeneration. 1996;5:445–451. doi: 10.1006/neur.1996.0061. [DOI] [PubMed] [Google Scholar]
- 98.Cui H, Freeman C, Jacobson GA, Small DH. Proteoglycans in the central nervous system: role in development, neural repair, and Alzheimer's disease. IUBMB Life. 2013;65:108–120. doi: 10.1002/iub.1118. [DOI] [PubMed] [Google Scholar]
- 99.Scholefield Z, Yates EA, Wayne G, Amour A, McDowell W, Turnbull JE. Heparan sulfate regulates amyloid precursor protein processing by BACE1, the Alzheimer's beta-secretase. J Cell Biol. 2003;163:97–107. doi: 10.1083/jcb.200303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Castillo GM, Ngo C, Cummings J, Wight TN, Snow AD. Perlecan binds to the beta-amyloid proteins (A beta) of Alzheimer's disease, accelerates A beta fibril formation, and maintains A beta fibril stability. J Neurochem. 1997;69:2452–2465. doi: 10.1046/j.1471-4159.1997.69062452.x. [DOI] [PubMed] [Google Scholar]
- 101.Castillo GM, Lukito W, Wight TN, Snow AD. The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta-amyloid protein fibril formation. J Neurochem. 1999;72:1681–1687. doi: 10.1046/j.1471-4159.1999.721681.x. [DOI] [PubMed] [Google Scholar]
- 102.Bruinsma IB, te Riet L, Gevers T, ten Dam GB, van Kuppevelt TH, David G, Kusters B, de Waal RM, Verbeek MM. Sulfation of heparan sulfate associated with amyloid-beta plaques in patients with Alzheimer's disease. Acta Neuropathol. 2010;119:211–220. doi: 10.1007/s00401-009-0577-1. [DOI] [PubMed] [Google Scholar]
- 103.Laikko I, Larmas M. Adenosine triphosphate in normal and carious human dentine. Arch Oral Biol. 1979;24:15–20. doi: 10.1016/0003-9969(79)90169-9. [DOI] [PubMed] [Google Scholar]
- 104.Watanabe N, Araki W, Chui DH, Makifuchi T, Ihara Y, Tabira T. Glypican-1 as an Abeta binding HSPG in the human brain: its localization in DIG domains and possible roles in the pathogenesis of Alzheimer's disease. FASEB J. 2004;18:1013–1015. doi: 10.1096/fj.03-1040fje. [DOI] [PubMed] [Google Scholar]
- 105.Leonova EI, Galzitskaia OV. Role of syndecan-2 in amyloid plaque formation. Mol Biol (Mosk) 2015;49:89–98. [PubMed] [Google Scholar]
- 106.Verbeek MM, Otte-Holler I, van den Born J, van den Heuvel LP, David G, Wesseling P, de Waal RM. Agrin is a major heparan sulfate proteoglycan accumulating in Alzheimer's disease brain. Am J Pathol. 1999;155:2115–2125. doi: 10.1016/S0002-9440(10)65529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Snow AD, Sekiguchi R, Nochlin D, Fraser P, Kimata K, Mizutani A, Arai M, Schreier WA, Morgan DG. An important role of heparan sulfate proteoglycan (Perlecan) in a model system for the deposition and persistence of fibrillar A beta-amyloid in rat brain. Neuron. 1994;12:219–234. doi: 10.1016/0896-6273(94)90165-1. [DOI] [PubMed] [Google Scholar]
- 108.Snow AD, Kinsella MG, Parks E, Sekiguchi RT, Miller JD, Kimata K, Wight TN. Differential binding of vascular cell-derived proteoglycans (perlecan, biglycan, decorin, and versican) to the beta-amyloid protein of Alzheimer's disease. Arch Biochem Biophys. 1995;320:84–95. doi: 10.1006/abbi.1995.1345. [DOI] [PubMed] [Google Scholar]
- 109.Snow AD, Mar H, Nochlin D, Sekiguchi RT, Kimata K, Koike Y, Wight TN. Early accumulation of heparan sulfate in neurons and in the beta-amyloid protein-containing lesions of Alzheimer's disease and Down's syndrome. Am J Pathol. 1990;137:1253–1270. [PMC free article] [PubMed] [Google Scholar]
- 110.Snow AD, Sekiguchi RT, Nochlin D, Kalaria RN, Kimata K. Heparan sulfate proteoglycan in diffuse plaques of hippocampus but not of cerebellum in Alzheimer's disease brain. Am J Pathol. 1994;144:337–347. [PMC free article] [PubMed] [Google Scholar]
- 111.Snow AD, Mar H, Nochlin D, Kresse H, Wight TN. Peripheral distribution of dermatan sulfate proteoglycans (decorin) in amyloid-containing plaques and their presence in neurofibrillary tangles of Alzheimer's disease. J Histochem Cytochem. 1992;40:105–113. doi: 10.1177/40.1.1370306. [DOI] [PubMed] [Google Scholar]
- 112.Pangalos MN, Shioi J, Robakis NK. Expression of the chondroitin sulfate proteoglycans of amyloid precursor (appican) and amyloid precursor-like protein 2. J Neurochem. 1995;65:762–769. doi: 10.1046/j.1471-4159.1995.65020762.x. [DOI] [PubMed] [Google Scholar]
- 113.Bruckner G, Hausen D, Hartig W, Drlicek M, Arendt T, Brauer K. Cortical areas abundant in extracellular matrix chondroitin sulphate proteoglycans are less affected by cytoskeletal changes in Alzheimer's disease. Neuroscience. 1999;92:791–805. doi: 10.1016/s0306-4522(99)00071-8. [DOI] [PubMed] [Google Scholar]
- 114.Baig S, Wilcock GK, Love S. Loss of perineuronal net N-acetylgalactosamine in Alzheimer's disease. Acta Neuropathol. 2005;110:393–401. doi: 10.1007/s00401-005-1060-2. [DOI] [PubMed] [Google Scholar]
- 115.Morawski M, Pavlica S, Seeger G, Grosche J, Kouznetsova E, Schliebs R, Bruckner G, Arendt T. Perineuronal nets are largely unaffected in Alzheimer model Tg2576 mice. Neurobiol Aging. 2010;31:1254–1256. doi: 10.1016/j.neurobiolaging.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 116.Kobayashi K, Emson PC, Mountjoy CQ. Vicia villosa lectin-positive neurones in human cerebral cortex. Loss in Alzheimer-type dementia. Brain Res. 1989;498:170–174. doi: 10.1016/0006-8993(89)90416-2. [DOI] [PubMed] [Google Scholar]
- 117.Suttkus A, Rohn S, Weigel S, Glockner P, Arendt T, Morawski M. Aggrecan, link protein and tenascin-R are essential components of the perineuronal net to protect neurons against iron-induced oxidative stress. Cell Death Dis. 2014 doi: 10.1038/cddis.2014.25. doi: 10.1038/cddis.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Morawski M, Bruckner G, Jager C, Seeger G, Arendt T. Neurons associated with aggrecan-based perineuronal nets are protected against tau pathology in subcortical regions in Alzheimer's disease. Neuroscience. 2010;169:1347–1363. doi: 10.1016/j.neuroscience.2010.05.022. [DOI] [PubMed] [Google Scholar]
- 119.Yasuhara O, Akiyama H, McGeer EG, McGeer PL. Immunohistochemical localization of hyaluronic acid in rat and human brain. Brain Res. 1994;635:269–282. doi: 10.1016/0006-8993(94)91448-6. [DOI] [PubMed] [Google Scholar]
- 120.Saftig P, Reiss K. The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: novel drug targets with therapeutic potential? European journal of cell biology. 2011;90(6-7):527–535. doi: 10.1016/j.ejcb.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 121.Gatta LB, Albertini A, Ravid R, Finazzi D. Levels of beta-secretase BACE and alpha-secretase ADAM10 mRNAs in Alzheimer hippocampus. Neuroreport. 2002;13:2031–2033. doi: 10.1097/00001756-200211150-00008. [DOI] [PubMed] [Google Scholar]
- 122.Kim M, Suh J, Romano D, Truong MH, Mullin K, Hooli B, Norton D, Tesco G, Elliott K, Wagner SL, Moir RD, Becker KD, Tanzi RE. Potential late-onset Alzheimer's disease-associated mutations in the ADAM10 gene attenuate {alpha}-secretase activity. Hum Mol Genet. 2009;18:3987–3996. doi: 10.1093/hmg/ddp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Botella-Lopez A, Burgaya F, Gavin R, Garcia-Ayllon MS, Gomez-Tortosa E, Pena-Casanova J, Urena JM, Del Rio JA, Blesa R, Soriano E, Sáez-Valero J. Reelin expression and glycosylation patterns are altered in Alzheimer's disease. Proc Natl Acad Sci U.S.A. 2006;103:5573–5578. doi: 10.1073/pnas.0601279103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Herring A, Donath A, Steiner KM, Widera MP, Hamzehian S, Kanakis D, Kolble K, ElAli A, Hermann DM, Paulus W, Keyvani K. Reelin depletion is an early phenomenon of Alzheimer's pathology. Journal of Alzheimer's disease. J Alzheimers Dis. 2012;30:963–979. doi: 10.3233/JAD-2012-112069. [DOI] [PubMed] [Google Scholar]
- 125.Chin J, Massaro CM, Palop JJ, Thwin MT, Yu GQ, Bien-Ly N, Bender A, Mucke L. Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer's disease. J Neurosci. 2007;27:2727–2733. doi: 10.1523/JNEUROSCI.3758-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, Anton ES. Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron. 2000;27:33–44. doi: 10.1016/s0896-6273(00)00007-6. [DOI] [PubMed] [Google Scholar]
- 127.Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brich J, Shie FS, Howell BW, Li R, Tus K, Wakeland EK, Jin LW, Mumby M, Churchill G, Herz J, Cooper JA. Genetic modulation of tau phosphorylation in the mouse. J Neurosci. 2003;23:187–192. doi: 10.1523/JNEUROSCI.23-01-00187.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang Q, Klyubin I, Wright S, Griswold-Prenner I, Rowan MJ, Anwyl R. Alpha v integrins mediate beta-amyloid induced inhibition of long-term potentiation. Neurobiol Aging. 2008;29:1485–1493. doi: 10.1016/j.neurobiolaging.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 130.Wright S, Malinin NL, Powell KA, Yednock T, Rydel RE, Griswold-Prenner I. Alpha2beta1 and alphaVbeta1 integrin signaling pathways mediate amyloid-beta-induced neurotoxicity. Neurobiol Aging. 2007;28:226–237. doi: 10.1016/j.neurobiolaging.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 131.Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, Turano E, Rossi B, Angiari S, Dusi S, Montresor A, Carlucci T, Nanì S, Tosadori G, Calciano L, Catalucci D, Berton G, Bonetti B, Constantin G. Neutrophils promote Alzheimer's disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. 2015;21:880–886. doi: 10.1038/nm.3913. [DOI] [PubMed] [Google Scholar]
- 132.Young-Pearse TL, Chen AC, Chang R, Marquez C, Selkoe DJ. Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev. 2008 doi: 10.1186/1749-8104-3-15. doi: 101186/1749-8104-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vegh MJ, Heldring CM, Kamphuis W, Hijazi S, Timmerman AJ, Li KW, van Nierop P, Mansvelder HD, Hol EM, Smit AB,van, Kesteren RE. Reducing hippocampal extracellular matrix reverses early memory deficits in a mouse model of Alzheimer's disease. Acta Neuropathol Commun. 2014 doi: 10.1186/s40478-014-0076-z. doi: 10.1186/s40478-014-0076-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hondius DC, van Nierop P, Li KW, Hoozemans JJ, van der Schors RC, van Haastert ES, van der Vies SM, Rozemuller AJ, Smit AB. Profiling the human hippocampal proteome at all pathologic stages of Alzheimer's disease. Alzheimers Dement. 2016;12:654–668. doi: 10.1016/j.jalz.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 135.Donovan LE, Higginbotham L, Dammer EB, Gearing M, Rees HD, Xia Q, Duong DM, Seyfried NT, Lah JJ, Levey AI. Analysis of a membrane-enriched proteome from postmortem human brain tissue in Alzheimer's disease. Proteomics Clin Appl. 2012;6:201–211. doi: 10.1002/prca.201100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Andreev VP, Petyuk VA, Brewer HM, Karpievitch YV, Xie F, Clarke J, Camp D, Smith RD, Lieberman AP, Albin RL, Nawaz Z, El Hokayem J, Myers AJ. Label-free quantitative LC-MS proteomics of Alzheimer's disease and normally aged human brains. J Proteome Res. 2012;11:3053–3067. doi: 10.1021/pr3001546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Musunuri S, Wetterhall M, Ingelsson M, Lannfelt L, Artemenko K, Bergquist J, Kultima K, Shevchenko G. Quantification of the brain proteome in Alzheimer's disease using multiplexed mass spectrometry. J Proteome Res. 2014;13:2056–2068. doi: 10.1021/pr401202d. [DOI] [PubMed] [Google Scholar]
- 138.Ho Kim J, Franck J, Kang T, Heinsen H, Ravid R, Ferrer I, Hee Cheon M, Lee JY, Shin Yoo J, Steinbusch HW, Salzet M, Fournier I, Mok Park Y. Proteome-wide characterization of signalling interactions in the hippocampal CA4/DG subfield of patients with Alzheimer's disease. Sci Rep. 2015 doi: 10.1038/srep11138. doi: 101038/srep11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Minjarez B, Calderon-Gonzalez KG, Rustarazo ML, Herrera-Aguirre ME, Labra-Barrios ML, Rincon-Limas DE, Del Pino MM, Mena R, Luna-Arias JP. Identification of proteins that are differentially expressed in brains with Alzheimer's disease using iTRAQ labeling and tandem mass spectrometry. J Proteomics. 2016;139:103–121. doi: 10.1016/j.jprot.2016.03.022. [DOI] [PubMed] [Google Scholar]
- 140.Sultana R, Boyd-Kimball D, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA. Proteomics analysis of the Alzheimer's disease hippocampal proteome. J Alzheimers Dis. 2007;11:153–164. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- 141.Gozal YM, Duong DM, Gearing M, Cheng D, Hanfelt JJ, Funderburk C, Peng J, Lah JJ, Levey AI. Proteomics analysis reveals novel components in the detergent-insoluble subproteome in Alzheimer's disease. J Proteome Res. 2009;8:5069–5079. doi: 10.1021/pr900474t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shah DJ, Rohlfing F, Anand S, Johnson WE, Alvarez MT, Cobell J, King J, Young SA, Kauwe JS, Graves SW. Discovery and Subsequent Confirmation of Novel Serum Biomarkers Diagnosing Alzheimer's Disease. J Alzheimers Dis. 2015;49(2):317–327. doi: 10.3233/JAD-150498. [DOI] [PubMed] [Google Scholar]
- 143.Perneczky R, Guo LH. Plasma Proteomics Biomarkers in Alzheimer's Disease: Latest Advances and Challenges. Methods Mol Biol. 2016;1303:521–529. doi: 10.1007/978-1-4939-2627-5_32. [DOI] [PubMed] [Google Scholar]
- 144.Brinkmalm A, Portelius E, Ohrfelt A, Brinkmalm G, Andreasson U, Gobom J, Blennow K, Zetterberg H. Explorative and targeted neuroproteomics in Alzheimer's disease. Biochim Biophys Acta. 2015;1854:769–778. doi: 10.1016/j.bbapap.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 145.Schedin-Weiss S, Winblad B, Tjernberg LO. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014;281:46–62. doi: 10.1111/febs.12590. [DOI] [PubMed] [Google Scholar]
- 146.Kizuka Y, Kitazume S, Fujinawa R, Saito T, Iwata N, Saido TC, Nakano M, Yamaguchi Y, Hashimoto Y, Staufenbiel M, Hatsuta H, Murayama S, Manya H, Endo T, Taniguchi N. An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer's disease. EMBO Mol Med. 2015;7:175–189. doi: 10.15252/emmm.201404438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Palmigiano A, Barone R, Sturiale L, Sanfilippo C, Bua RO, Romeo DA, Messina A, Capuana ML, Maci T, Le Pira F, Zappia M, Garozzo D. CSF N-glycoproteomics for early diagnosis in Alzheimer's disease. J Proteomics. 2016;131:29–37. doi: 10.1016/j.jprot.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 148.Maguire TM, Gillian AM, O'Mahony D, Coughlan CM, Dennihan A, Breen KC. A decrease in serum sialyltransferase levels in Alzheimer's disease. Neurobiol Aging. 1994;15:99–102. doi: 10.1016/0197-4580(94)90149-x. [DOI] [PubMed] [Google Scholar]
- 149.Maguire TM, Breen KC. A decrease in neural sialyltransferase activity in Alzheimer's disease. Dementia. 1995;6:185–190. doi: 10.1159/000106944. [DOI] [PubMed] [Google Scholar]
- 150.Fodero LR, Saez-Valero J, Barquero MS, Marcos A, McLean CA, Small DH. Wheat germ agglutinin-binding glycoproteins are decreased in Alzheimer's disease cerebrospinal fluid. J Neurochem. 2001;79:1022–1026. doi: 10.1046/j.1471-4159.2001.00640.x. [DOI] [PubMed] [Google Scholar]
- 151.Taniguchi M, Okayama Y, Hashimoto Y, Kitaura M, Jimbo D, Wakutani Y, Wada-Isoe K, Nakashima K, Akatsu H, Furukawa K, Arai H, Urakami K. Sugar chains of cerebrospinal fluid transferrin as a new biological marker of Alzheimer's disease. Dement Geriatr Cogn Disord. 2008;26:117–122. doi: 10.1159/000147479. [DOI] [PubMed] [Google Scholar]
- 152.Chen CC, Engelborghs S, Dewaele S, Le Bastard N, Martin JJ, Vanhooren V, Libert C, De Deyn PP. Altered serum glycomics in Alzheimer disease: a potential blood biomarker? Rejuvenation Res. 2010;13:439–444. doi: 10.1089/rej.2009.0992. [DOI] [PubMed] [Google Scholar]
- 153.Korolainen MA, Nyman TA, Aittokallio T, Pirttila T. An update on clinical proteomics in Alzheimer's research. J Neurochem. 2010;112:1386–1414. doi: 10.1111/j.1471-4159.2009.06558.x. [DOI] [PubMed] [Google Scholar]
- 154.Papassotiropoulos A, Fountoulakis M, Dunckley T, Stephan DA, Reiman EM. Genetics, transcriptomics, and proteomics of Alzheimer's disease. J Clin Psychiatry. 2006;67:652–670. doi: 10.4088/jcp.v67n0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Di Domenico F, Sultana R, Barone E, Perluigi M, Cini C, Mancuso C, Cai J, Pierce WM, Butterfield DA. Quantitative proteomics analysis of phosphorylated proteins in the hippocampus of Alzheimer's disease subjects. J Proteomics. 2011;74:1091–1103. doi: 10.1016/j.jprot.2011.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang J, Goodlett DR, Peskind ER, Quinn JF, Zhou Y, Wang Q, Pan C, Yi E, Eng J, Aebersold RH, Montine TJ. Quantitative proteomic analysis of age-related changes in human cerebrospinal fluid. Neurobiol Aging. 2005;26:207–227. doi: 10.1016/j.neurobiolaging.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 157.Hynd MR, Lewohl JM, Scott HL, Dodd PR. Biochemical and molecular studies using human autopsy brain tissue. J Neurochem. 2003;85:543–562. doi: 10.1046/j.1471-4159.2003.01747.x. [DOI] [PubMed] [Google Scholar]
- 158.McCullumsmith RE, Hammond JH, Shan D, Meador-Woodruff JH. Postmortem brain: an underutilized substrate for studying severe mental illness. Neuropsychopharmacology. 2014;39:65–87. doi: 10.1038/npp.2013.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Piehowski PD, Petyuk VA, Orton DJ, Xie F, Moore RJ, Ramirez-Restrepo M, Engel A, Lieberman AP, Albin RL, Camp DG, Smith RD, Myers AJ. Sources of technical variability in quantitative LC-MS proteomics: human brain tissue sample analysis. J Proteome Res. 2013;12:2128–2137. doi: 10.1021/pr301146m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chiou S-H, Wu S-H. Evaluation of commonly used electrophoretic methods for the analysis of proteins and peptides and their application to biotechnology. Anal Chim Acta. 1999;383:47–60. [Google Scholar]
- 161.Rabilloud T, Chevallet M, Luche S, Lelong C. Two-dimensional gel electrophoresis in proteomics: past, present and future. J Proteomics. 2010;73:2064–2077. doi: 10.1016/j.jprot.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 162.Cellulaire B. Two-dimensional gel electrophoresis in proteomics: old, old fashioned, but it still climbs up the mountains. Proteomics. 2002;2:3–10. [PubMed] [Google Scholar]
- 163.Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003;21:255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- 164.Eastwood SL, Harrison PJ. Cellular basis of reduced cortical reelin expression in schizophrenia. Am J Psychiatry. 2006;163:540–542. doi: 10.1176/appi.ajp.163.3.540. [DOI] [PubMed] [Google Scholar]
- 165.Parlapani E, Schmitt A, Wirths O, Bauer M, Sommer C, Rueb U, Skowronek MH, Treutlein J, Petroianu GA, Rietschel M, Falkai P. Gene expression of neuregulin-1 isoforms in different brain regions of elderly schizophrenia patients. World J Biol Psychiatry. 2010;11:243–250. doi: 10.3109/15622970802022376. [DOI] [PubMed] [Google Scholar]
- 166.Keri S, Beniczky S, Kelemen O. Suppression of the P50 evoked response and neuregulin 1-induced AKT phosphorylation in first-episode schizophrenia. Am J Psychiatry. 2010;67:444–450. doi: 10.1176/appi.ajp.2009.09050723. [DOI] [PubMed] [Google Scholar]
- 167.Law AJ, Kleinman JE, Weinberger DR, Weickert CS. Disease-associated intronic variants in the ErbB4 gene are related to altered ErbB4 splice-variant expression in the brain in schizophrenia. Hum Mol Genet. 2007;16:129–141. doi: 10.1093/hmg/ddl449. [DOI] [PubMed] [Google Scholar]
- 168.Muller T, Jung K, Ullrich A, Schrotter A, Meyer HE, Stephan C, Egensperger R, Marcus K. Disease state, age, sex, and post-mortem time-dependent expression of proteins in AD vs. control frontal cortex brain samples. Curr Alzheimer Res. 2008;5:562–571. doi: 10.2174/156720508786898488. [DOI] [PubMed] [Google Scholar]
- 169.Riederer IM, Schiffrin M, Kovari E, Bouras C, Riederer BM. Ubiquitination and cysteine nitrosylation during aging and Alzheimer's disease. Brain Res Bull. 2009;80:233–241. doi: 10.1016/j.brainresbull.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 170.Newman SF, Sultana R, Perluigi M, Coccia R, Cai J, Pierce WM, Klein JB, Turner DM, Butterfield DA. An increase in S-glutathionylated proteins in the Alzheimer's disease inferior parietal lobule, a proteomics approach. J Neurosci Res. 2007;85:1506–1514. doi: 10.1002/jnr.21275. [DOI] [PubMed] [Google Scholar]