

Abstract
We report a new method to quantify the affinity of small molecules for proteins. This method is based on Förster resonance energy transfer (FRET) between endogenous tryptophan (Trp) residues and the coumarin-derived fluorophore Pacific Blue (PB). Tryptophan residues are frequently found in proteins near ligand-binding sites, making this approach potentially applicable to a wide range of systems. To improve access to PB, we developed a scalable multigram synthesis of this fluorophore, starting with inexpensive 2,3,4,5-tetrafluorobenzoic acid. This route was used to synthesize fluorescent derivatives of biotin, as well as lower affinity thiobiotin, iminobiotin, and imidazolidinethione analogues that bind the protein streptavidin. Compared with previously published FRET acceptors for tryptophan, PB proved to be superior in both sensitivity and efficiency. These unique properties of PB enabled direct quantification of dissociation constants (Kd) as well as competitive inhibition constants (Ki) in the micromolar to nanomolar range. In comparison to analogous binding studies using fluorescence polarization, fluorescence quenching, or fluorescence enhancement, affinities determined using Trp-FRET were more precise and accurate as validated using independent isothermal titration calorimetry studies. FRET between tryptophan and PB represents a new tool for the characterization of protein–ligand complexes.
Introduction
To quantify the affinity of small molecules for proteins, a wide variety of biophysical techniques have been developed. Homogeneous methods include isothermal titration calorimetry (ITC),1 fluorescence quenching or enhancement,2 fluorescence polarization (FP),3 and Förster resonance energy transfer (FRET).4 Although these techniques are powerful, each has limitations. ITC, considered the gold standard for affinity determination, is material-intensive and low-throughput and requires an appreciable change in heat upon binding. In the widely employed FP (or the related fluorescence anisotropy),3,5 small molecules linked to fluorophores report changes in the polarization of emitted photons when a rapidly rotating small molecule binds a slowly tumbling protein. However, probe size, solvent viscosity, fluorescence quenching, and linker flexibility can limit the applicability.3
FRET has the potential to overcome some of these limitations. This method involves nonradiative transfer of excitation (Ex.) energy from a donor fluorophore to a proximal acceptor fluorophore.4 This transfer is highly distance-dependent, with an efficiency of 50% at the Förster distance (R0, typically <∼5 nm). FRET-based binding assays generally involve one fluorophore attached to a receptor of interest and another fluorophore conjugated to a ligand. However, this requirement for two exogenous fluorophores poses challenges, given that conjugation of fluorophores to proteins can result in heterogeneity, or requires site-directed labeling reactions.
As an alternative method for detection of small molecule–protein interactions, FRET initiated by the excitation of intrinsically fluorescent tryptophan (Trp, 1, Figure 1) residues is of increasing interest.6−8 Beneficially, these residues are commonly found in or near protein–ligand binding sites and at interfaces between proteins and other biomolecules.9,10 Although maximally excited at 280 nm, tryptophan can be selectively excited over tyrosine at 295 nm. Fluorescent photons emitted by tryptophan exhibit λmax values that range from ∼308 to ∼355 nm, roughly correlated with exposure to an aqueous solution,11 typically with a modest quantum yield (Φ ≈ 0.2). A previously reported11 comparison of 19 tryptophan residues from 17 proteins with experimentally determined structures revealed an average λmax of tryptophan emission of 333 nm. Because the emission λmax and the quantum yield of tryptophan are affected by the polarity of the local environment,12 its environmental sensitivity and quenching by exogenous factors have been widely used to study changes in protein conformation and binding of ligands. The intrinsic emission of tryptophan can also participate in FRET with other fluorophores,7 and this approach has been extensively used to study protein folding and dynamics.13−15 Fluorophores known to participate in FRET with tryptophan include derivatives of dansyl (2), 7-hydroxycoumarin (7HC, 3), and 7-dimethylaminocoumarin (DMACA, 4).4 Coumarins 3 and 4 have recently been reported6 by Chung to be the best FRET acceptors for tryptophan identified to date. However, the photophysical properties of coumarin 3 are highly sensitive to changes in pH in the physiological range (phenol pKa = 7.8),16,17 and the absorbance18 and emission19 of coumarin 4 shift substantially when transitioning from an aqueous to a hydrophobic environment. Moreover, a biotin-linked derivative of coumarin 4 was reported to have a low quantum yield (Φ = 0.06),6 which increases the concentration of the probe required for detection. These environmental effects, limited brightness, and low FRET efficiencies continue to hinder sensitivity for Trp-FRET applications. A few noncompetitive binding assays using Trp-FRET have been reported,20−23 but they generally involve quenching of tryptophan fluorescence or require high concentrations of fluorescent probes for detection. These high concentrations can prevent quantitative measurements of low dissociation constants (Kd) and competitive inhibition constants (Ki), which are frequently observed between small molecules and proteins.
Figure 1.
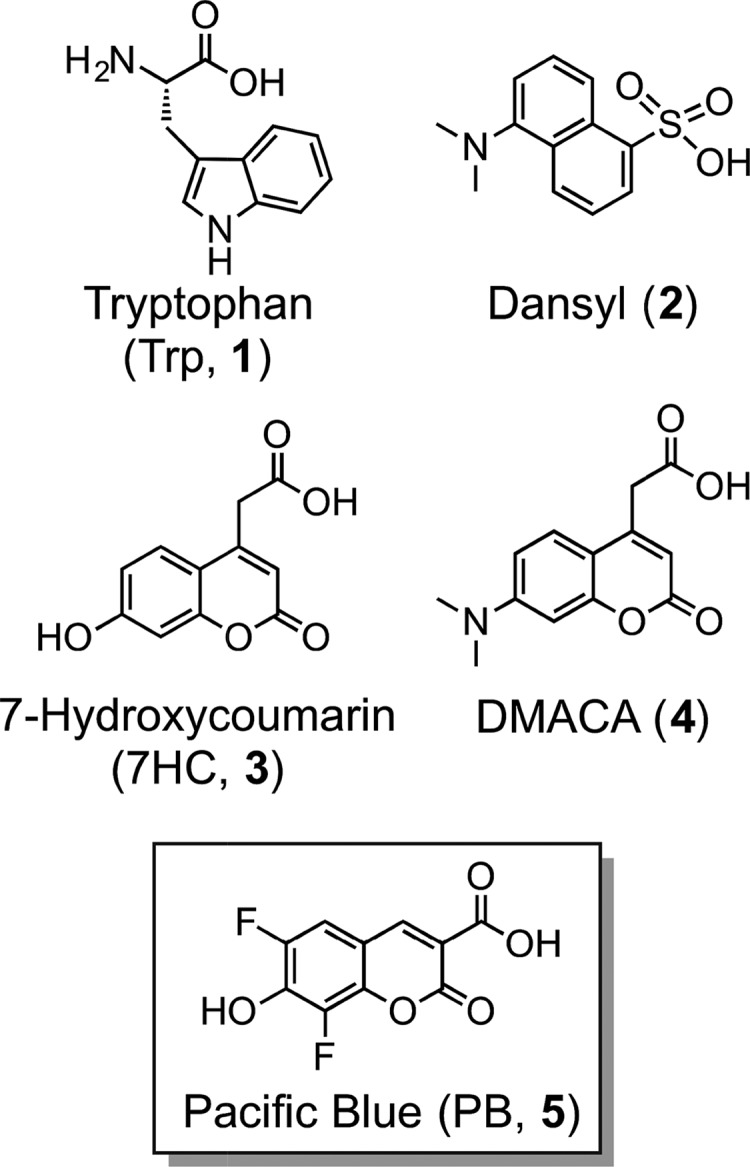
Structures of tryptophan (Trp, 1) and other fluorophores (2–5).
Results and Discussion
To identify a more sensitive FRET acceptor for tryptophan, we examined the overlap between the emission spectrum of this amino acid (Ex. 280 nm, Em. ∼ 350 nm, ε = 5600 M–1 cm–1) and the absorbance spectrum of 6,8-difluoro-7-hydroxycoumarin, a fluorophore known as Pacific Blue (PB, 5).17 Because of its favorable photophysical properties (Ex. 400 nm, Em. 447 nm, ε = 29 500 M–1 cm–1, Φ = 0.75, and phenol pKa = 3.7), PB is of substantial interest for labeling of proteins and other biomolecules.24,25 As shown in Figure 2, the large overlap between the emission of tryptophan and the absorption of PB suggested that this fluorophore might be a particularly sensitive FRET partner. To investigate this hypothesis, we developed an improved synthesis of PB that readily allows access to gram quantities of this compound (Scheme 1). In contrast to previously reported routes based on expensive 2,4-difluororesorcinol,17,26 we found that the relatively inexpensive 2,3,4,5-tetrafluorobenzoic acid can be converted to the common intermediate aldehyde 10(17,26) in 66% overall yield in a four-pot process.
Figure 2.
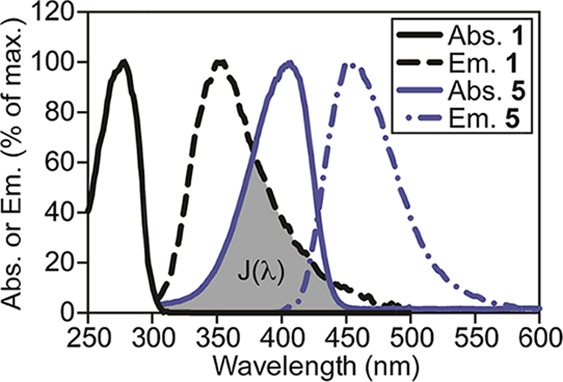
Absorbance (Abs., solid lines) and emission (Em., dotted lines) spectra of tryptophan (1, black lines, 32 μM for Abs. and 2 μM for Em.) and PB (5, blue lines, 32 μM for Abs. and 50 nM for Em.) in phosphate-buffered saline (PBS, pH 7.4). The spectral overlap integral, J(λ), critical for FRET, is shaded gray.
Scheme 1. Improved Gram-Scale Synthesis of the PB Fluorophore.

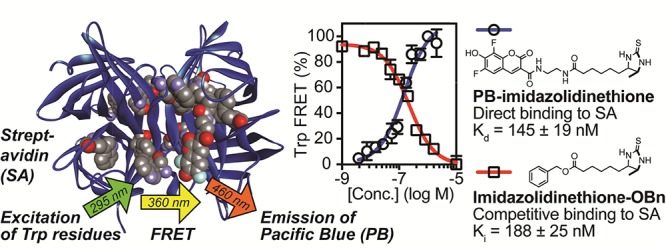
As a model small molecule–protein interaction, we investigated binding of the bacterial protein streptavidin (SA) to biotin and analogues. This tetrameric protein binds biotin with exceptionally high affinity (Kd = 10–14 M–1),27 and X-ray crystallography28 has revealed six tryptophan residues in close proximity to each of its four noncooperative29 biotin-binding sites. Additionally, Trp79, Trp108, and Trp120 are critical for binding of SA to biotin via van der Waals and hydrophobic interactions.28,30 Moreover, in a buffered aqueous solution, excitation of these tryptophan residues leads to an average emission λmax of 336 nm that can initiate FRET with biotinylated fluorophores.20,31,32 Correspondingly, docking33 of this X-ray structure (Figure 3) to PB-biotin derivative 12 (Figure 4, panel A) supported the notion that binding of SA would favorably position the fluorophore in close proximity to endogenous tryptophan residues that make noncovalent contacts to biotin and tryptophan residues in other subunits of the SA tetramer.
Figure 3.
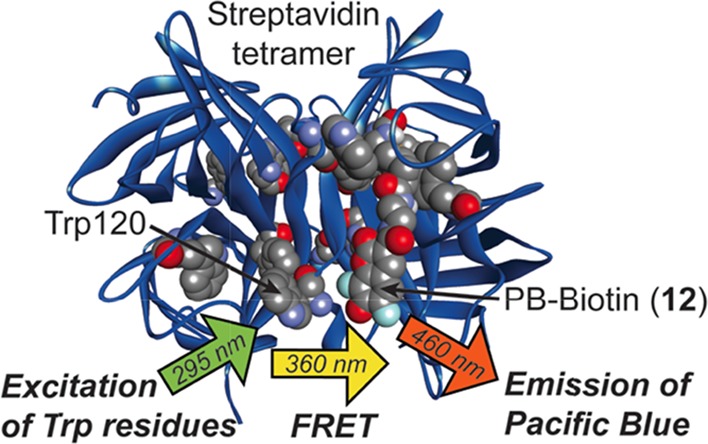
Model of SA (PDB 3RY2) docked to the PB-biotin derivative 12 (CPK model) with AutoDock Vina. In each monomer, residues Trp79, Trp108, and Trp120 are shown as CPK models. The distance between the most proximal Trp120 (marked) and the PB moiety of 12 is ∼11 Å. For clarity, only one bound ligand is shown.
Figure 4.
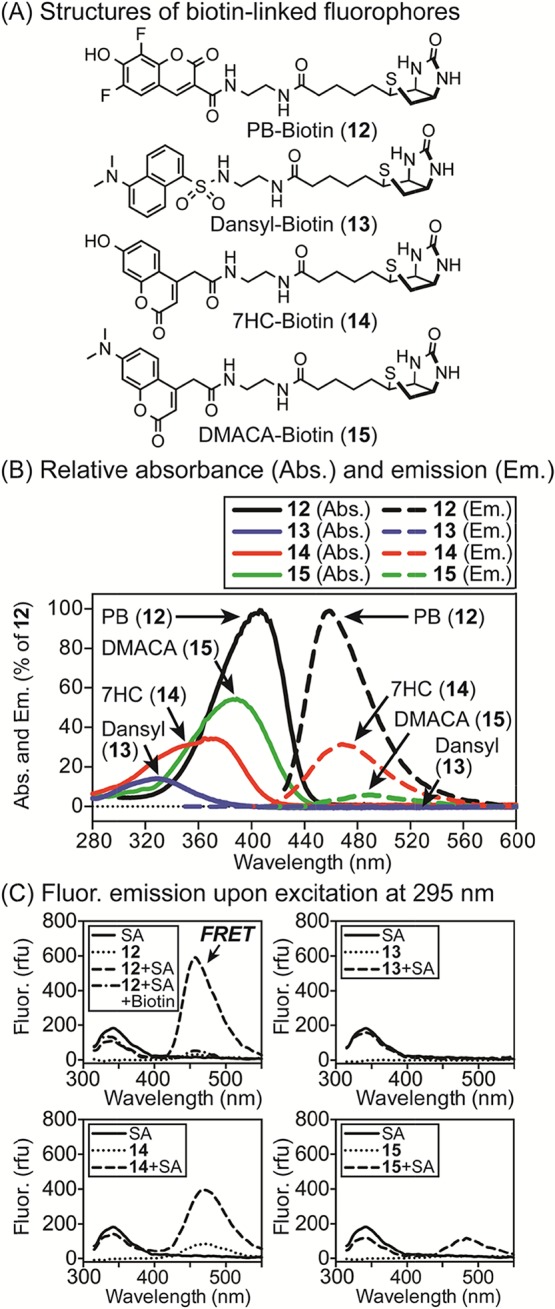
(A) Structures of fluorophores linked to biotin (12–15). (B) Normalized absorbance (Abs., solid lines) and fluorescence (Fluor.) emission (Em., dashed lines) spectra of 12–15 (32 μM for Abs., 50 nM for Fluor.) in PBS (pH 7.4). Fluorophores 12–15 were excited at their λmax [405 nm (12), 330 nm (13), 375 nm (14), and 390 nm (15)]. Intensities are % of λmax of 12. (C) Analysis of Trp-FRET upon binding of 12–15 (100 nM) to SA ([monomer] = 400 nM). Ex. of SA (295 nm) results in maximal Em. at 340 nm. After binding of 12–15, FRET Em. was observed at 457 (12), 532 (13), 467 (14), and 475 nm (15). Addition of biotin (10 μM) blocked FRET from 12, confirming specificity.
To compare PB (5) with known Trp-FRET acceptors (2–4), we synthesized biotin derivatives 12–15 (Figure 4, panel A). Comparison of normalized absorbance and emission spectra of these compounds (Figure 4, panel B) revealed that the PB-derived probe 12 was ∼3-fold brighter than 14, ∼17-fold brighter than 15, and more than 100-fold brighter than 13 in aqueous PBS (pH 7.4). FRET can be measured as the relative fluorescence intensity of the donor in the absence (Id) and presence (Ida) of the acceptor.4 When probes 12–15 were added to SA, excitation of tryptophan revealed substantial differences in emission because of FRET (Figure 4, panel C). As listed in Table 1, values from these studies were used to analyze the sensitivity (Iad), efficiency (E), and other properties of these fluorophore pairs. Compared with PB (12), dansyl (13) was 33-fold less sensitive, 7HC (14) was 1.5-fold less sensitive, and DMACA (15) was 6-fold less sensitive. Additionally, the blue-shifted absorbance of 7HC (14) increased its excitation at 295 nm, reducing its FRET fold effect (FF) by a factor of 4. The specificity of the FRET signal of 12 upon binding to SA was confirmed by addition of biotin as a competitor (Figure 4, panel C). Consequently, PB proved to be the most efficient and sensitive FRET acceptor for tryptophan under these conditions.
Table 1. Photophysical Properties of Fluorescent Probesa.
| probe | Iad | E | FF | R0 (nm) | Φf | ε (M–1cm–1) |
|---|---|---|---|---|---|---|
| 12 | 3.3 | 0.40 | 19.2 | 3.0 | 0.74 | 24 200 |
| 13 | 0.1 | 0.13 | 6.8 | 2.1 | 0.068 | 3500 |
| 14 | 2.2 | 0.21 | 4.8 | 2.7 | 0.476 | 7800 |
| 15 | 0.6 | 0.35 | 8.0 | 2.8 | 0.066 | 11 600 |
The intensity (Iad) of FRET acceptors (100 nM) was measured at Em. λmax when bound to SA (400 nM, Ex. 295 nm) and normalized to Id. The efficiency (E) of FRET with tryptophan when bound to SA was calculated as E = 1 – Ida/Id. FF was calculated as FF = Iad/Ia. R0 is the theoretical Förster distance calculated for each Trp-acceptor pair as described in the Experimental Section. Ida = intensity of Em. of tryptophan (340 nm) in the presence of the acceptor. Id = intensity of Em. of tryptophan (340 nm) in the absence of the acceptor. Iad and Ia are the intensities of Em. of the acceptor [457 (12), 532 (13), 467 (14), and 475 nm (15)] in the presence and absence of the donor (SA), respectively. Values for Φf and molar extinction coefficients (ε) were measured as described in the Experimental Section and Supporting Information.
Biotin binds SA with such high affinity that its association is essentially irreversible. To investigate fluorescent analogues with lower affinity that might be suitable for equilibrium binding measurements, we synthesized probes 16–18 (Figure 5, panel A).34,35 To generate equilibrium binding curves, SA was titrated into a low fixed concentration of the fluorescent probe (below Kd), followed by excitation of tryptophan at 295 nm to trigger FRET (measured at 460 nm). Curve fitting was used to calculate the Kd values of 11 ± 2 nM for thiobiotin-PB derivative 17, 145 ± 19 nM for imidazolidinethione-PB 18, and 2730 ± 340 nM for iminobiotin-PB 16 in PBS (Figure 5, panel B). These values were in excellent agreement with the independent assessments of Kd using ITC under the same conditions (12 ± 4 nM for 17 and 125 ± 45 nM for 18, Figure 5, panels C and D). For these high affinity probes, attempts to measure affinities by FP were unsuccessful because of fluorescence quenching, which affects the lifetime of the fluorophore.3 However, using higher concentrations of the lower affinity probe 16, FP yielded a comparable Kd value of 2950 ± 270 nM (data shown in Supporting Information).
Figure 5.
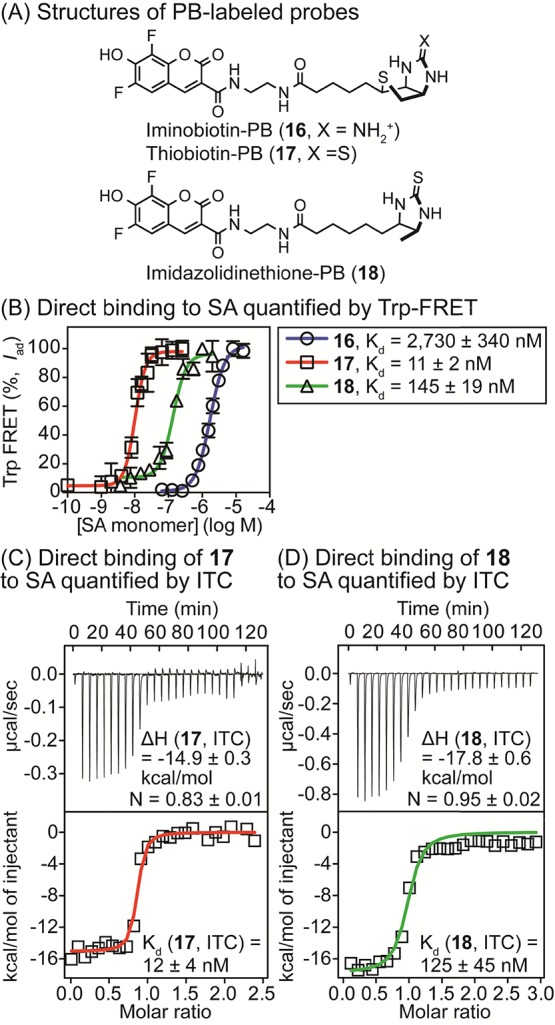
(A) Structures of analogues of biotin (16–18) used in direct binding assays. (B) Quantification of the affinity (Kd) of SA for probes 16 (25 nM), 17 (5 nM), and 18 (25 nM) in PBS (pH 7.4) by Trp-FRET. Tryptophan residues were excited at 295 nM and FRET was measured at 460 nm. Values were corrected to account for fluorescence quenching or enhancement upon binding, as described in Experimental Section. [SA] was based on monomeric protein. Dissociation constants (Kd) were calculated using a one-site binding model (GraphPad Prism 6.0). (C,D) Quantification of the binding of 17 (C) and 18 (D) to SA in PBS (pH 7.4) using ITC. Thermodynamic parameters and Kd values were calculated with Origin software. For 17, [SA in sample cell] = 4 μM and [17 in syringe] = 50 μM. For 18, [SA in sample cell] = 10 μM and [18 in syringe] = 150 μM.
We hypothesized that the high sensitivity of Trp-FRET with PB might be particularly valuable for quantifying small molecule–protein interactions in a competition assay format. To test this approach, we measured competitive inhibition constants (Ki) of the unlabeled biotin analogues 19–22 (Figure 6, panel A) using PB-imidazolidinethione 18 as a fluorescent probe. The unlabeled ligands 19–22 were added to SA (held at a concentration near the Kd) bound to 18 (held below Kd, 25 nM). After incubation for 1 h at room temperature to achieve equilibrium, excitation of tryptophan at 295 nm was used to trigger FRET (measured at 460 nm). This approach allowed the calculation of Ki values of 188 ± 25 nM for the benzyl ester imidazolidinethione derivative 22, 693 ± 91 nM for the methyl ester imidazolidinethione 21, 1350 ± 180 nM for imidazolidinethione 20, and 44 600 ± 5800 nM for iminobiotin 19 in PBS (Figure 6, panel B) by nonlinear curve fitting. Moreover, we independently measured the affinity of the nonfluorescent imidazolidinethione analogue 20 for SA using ITC (Kd = 1550 ± 270 nM, Figure 6, panel C), which was in excellent agreement with the Ki quantified by Trp-FRET. Other previously published studies36,37 of 19 have reported values similar to the Ki value measured by Trp-FRET.
Figure 6.
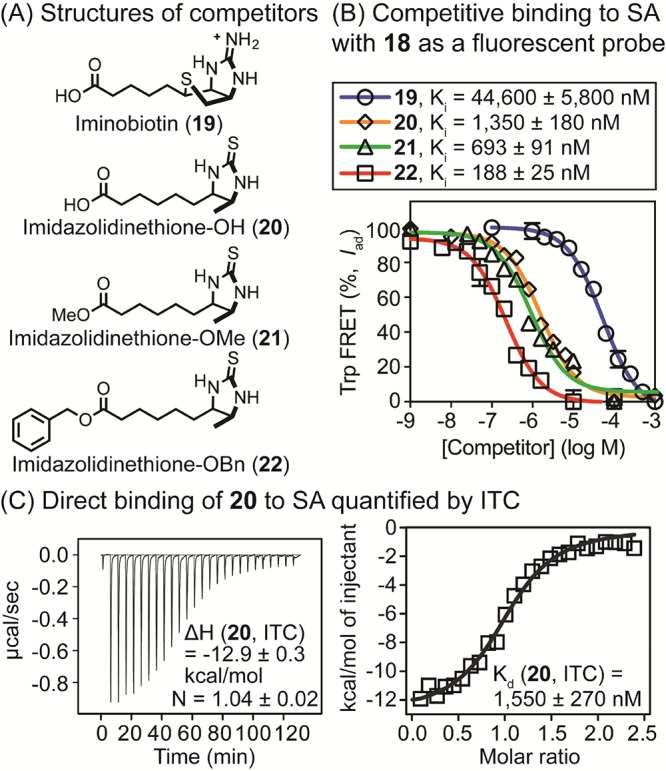
(A) Structures of nonfluorescent analogues of biotin (19–22) used in competition binding assays. (B) Quantification of competitive inhibitory constants (Ki) of 19–22 for SA complexed with 18 (175 nM for SA and 25 nM for 18) by Trp-FRET. Tryptophan residues were excited at 295 nM, and FRET was measured at 460 nm. Values were corrected to account for fluorescence quenching upon binding as described in Experimental Section. [SA] was based on monomeric protein. Half-maximal inhibitory concentrations (IC50) were calculated using a log(inhibitor) vs response model (GraphPad Prism 6.0), and IC50 values were converted to Ki values. (C) Evaluation of direct binding of 20 to SA using ITC. Compound 20 was titrated into [SA] in PBS (pH 7.4), and thermodynamic parameters and Kd values were calculated using the Origin software. [SA in sample cell] = 20 μM and [20 in syringe] = 250 μM.
When PB-linked ligands bind to SA, their fluorescence is partially quenched (16, 17) or enhanced (18). Hence, simple changes in fluorescence could potentially allow determination of their affinities for this protein. To examine the merits of this approach, we quantified the Kd of 16–18 for SA using only fluorescence quenching or enhancement, and we examined changes in fluorescence upon addition of the nonfluorescent competitor 20 to SA-18. As shown in Figure 7, we found that simple fluorescence quenching or enhancement could be used to estimate the affinity of 16–18 for SA, but Kd values obtained using this method differed by twofold (18) to sixfold (17) from gold-standard measurements using ITC. Moreover, examination of competitive binding of 20 to SA-18 revealed substantial deviation from the binding model at high concentrations (Figure 7, panel D), which prevented accurate determination of Ki. This lower accuracy and precision associated with the sole use of the fluorescence intensity presumably arises from its high sensitivity to aggregation of fluorophores and other environmental factors. In contrast, the dependence of FRET on the distance between fluorophores presumably reduces its susceptibility to these confounding factors. On the basis of these results, we conclude that Trp-FRET with PB is superior to fluorescence intensity measurements for quantifying equilibrium binding in this system.
Figure 7.
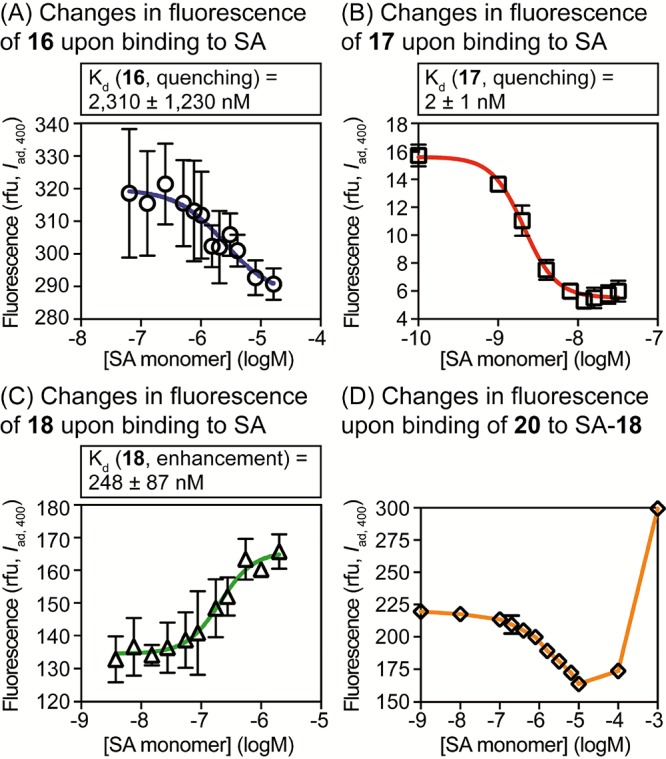
Analysis of interactions with SA by quenching or enhancement of fluorescence. PB was excited at 400 nm, and the intensity of fluorescence emission at 460 nm was measured. (A–C) Quantification of Kd values for direct binding. (D) Fluorescence resulting from competition of 20 for SA bound to probe 18 (25 nM). In (D), because of the poor fit to the binding model, the Ki value was not calculated.
In conclusion, we developed an improved synthesis of the PB fluorophore starting from inexpensive 2,3,4,5-tetrafluorobenzoic acid. This synthesis was used to prepare fluorescent molecular probes that bind the Trp-containing protein SA. Comparison with other fluorescent probes that bind SA revealed that PB is a highly sensitive and efficient FRET acceptor for tryptophan. This high sensitivity enabled quantification of Kd and competitive Ki values into the nM range. As validated using independent ITC measurements, this method proved to be more precise and accurate than analogous binding studies using fluorescence polarization, fluorescence quenching, or fluorescence enhancement for this system. FRET between tryptophan and PB offers a new method for quantification of small molecule–protein interactions.
Experimental Section
Synthesis
Chemicals were purchased from Sigma Aldrich, Acros Organics, Alfa Aesar, Oakwood Chemical, or Chem-Impex International. All nonaqueous reactions were carried out using flame- or oven-dried glassware under an atmosphere of dry argon or nitrogen. Tetrahydrofuran (THF), dichloromethane (CH2Cl2), N,N-dimethylformamide (DMF), and methanol (CH3OH) were purified via filtration through two columns of activated basic alumina under an atmosphere of Ar using a solvent purification system from Pure Process Technology (GlassContour). Other commercial reagents were used as received unless otherwise noted. 1H nuclear magnetic resonance (NMR), 13C NMR, and 19F NMR spectra were acquired on a Bruker DRX-400 or an Avance AVIII 500 MHz instrument. For 1H and 13C, chemical shifts (δ) are reported in ppm referenced to CDCl3 (7.26 ppm for 1H and 77.2 ppm for 13C), CD3OD (3.31 ppm for 1H, 49.0 ppm for 13C), or dimethyl sulfoxide (DMSO)-d6 (2.50 ppm for 1H, 39.5 ppm for 13C). For 19F, chemical shifts (δ) are reported in ppm referenced to trifluoroethanol (−77.0 ppm for 19F). 1H coupling constants (JHH, Hz), 13C coupling constants (JCF, Hz), and 19F coupling constants (JFF, Hz) are reported as chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet), coupling constant, and integration. High-resolution mass spectra were obtained at the Mass Spectrometry Laboratory at the University of Kansas on a Micromass LCT Premier. Thin layer chromatography (TLC) was performed using EMD aluminum-backed (0.20 mm) silica plates (60 F-254), and flash chromatography used ICN silica gel (200–400 mesh). TLC plates were visualized with a UV lamp or by staining with I2. Preparative high-performance liquid chromatography (HPLC) was performed with an Agilent 1200 instrument equipped with a Hamilton PRP-1 reverse phase column (250 mm length, 21.2 mm ID, and 7 μm particle size) with detection by absorbance at 215, 254, and 370 nm.
2,3,4,5-Tetrafluorobenzonitrile (7)
To a solution of 2,3,4,5-tetrafluorobenzoic acid (6, 10 g, 51 mmol) in CH2Cl2 (50 mL) was added oxalyl chloride (5.4 mL, 62.9 mmol) and DMF (ca. 2 drops), and the reaction mixture was stirred at 22 °C for 16 h while venting to the atmosphere to allow escape of evolved gases. The solvent was removed under reduced pressure, and the vessel was placed on high vacuum for 2 h to afford the acid chloride as a viscous oil. This oil was dissolved in chloroform (40 mL) and cooled to 4 °C. Aqueous ammonia (28%, 55 mL) was slowly added and the reaction was stirred at 4 °C for 30 min. The mixture was extracted with chloroform, and the organic layer was dried, filtered, and concentrated under reduced pressure to afford a white solid. To this solid was added phosphoryl chloride (32 mL), and the mixture was stirred at 80 °C for 3 h. This mixture was treated with diethyl ether (150 mL) and ice water (100 mL), followed by sat. aqueous NaHCO3 (100 mL) for 1 h. This mixture was extracted with diethyl ether, and the organic layer was washed with sat. aqueous NaHCO3 (3 × 100 mL). The organic layer was dried, filtered, and concentrated under reduced pressure to afford 7 (7.9 g, 88%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.33 (dddd, J = 8.8, 7.6, 5.3, 2.6 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 149.5 (dddd, J = 269.0, 12.1, 4.0, 2.0 Hz), 147.4 (dddd, J = 259.0, 10.8, 3.9, 2.2 Hz), 144.4 (dddd, J = 265.6, 15.9, 12.2, 3.2 Hz), 143.4 (ddd, J = 16.0, 12.1, 3.2 Hz), 141.3 (dddd, J = 258.6, 15.0, 12.7, 4.0 Hz), 115.0 (dd, J = 21.9, 4.1 Hz), 111.2 (m), 97.8 (dddd, J = 14.2, 9.3, 4.7, 1.7 Hz); 19F NMR (376 MHz, CDCl3) δ −129.99 (dddt, J = 31.8, 19.9, 11.9, 6.9 Hz), −134.49 (dddt, J = 32.4, 20.6, 11.8, 5.3 Hz), −143.33 (tdd, J = 20.5, 12.9, 8.3 Hz), −150.62 (dddd, J = 39.3, 24.4, 17.3, 6.4 Hz); HRMS (ESI−) m/z 173.9993 (M – H+, C7F4N requires 173.9967). Note that this compound is commercially available (e.g. Alfa Aesar, 25 g/$173).
2,4-Bis(benzyloxy)-3,5-difluorobenzonitrile (8)
To a solution of 7 (7.8 g, 45 mmol) in DMF (5 mL) was added benzyl alcohol (23 mL, 223 mmol) and potassium carbonate (37 g, 267 mmol). The vessel was heated to 105 °C for 16 h, subsequently placed under high vacuum for 2 h to remove DMF, followed by purification using column chromatography on silica gel (eluent: hexanes/ethyl acetate (17:1)) to afford 8 (13.5 g, 86%) as a viscous colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.46–7.33 (m, 11H), 7.04 (dd, J = 10.0, 2.3 Hz, 1H), 5.29 (s, 2H), 5.2251 (d, J = 1.0 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 151.1 (dd, J = 247.5, 4.8 Hz), 149.4 (dd, J = 251.5, 6.3 Hz), 146.3 (dd, J = 11.9, 3.3 Hz), 140.7 (dd, J = 14.7, 12.1 Hz), 135.5, 135.2, 129.1, 129.0, 128.84, 128.80, 128.77, 128.4, 114.9 (dd, J = 23.6, 3.7 Hz), 114.8 (d, J = 3.0 Hz), 101.0 (dd, J = 10.5, 5.0 Hz), 76.8 (d, J = 5.6 Hz), 76.2 (t, J = 3.9 Hz); 19F NMR (376 MHz, CDCl3) δ −129.79 (d, J = 6.9 Hz), −138.96 (d, J = 6.9 Hz); HRMS (ESI+) m/z 374.0979 (M + Na+, C21H15F2NO2Na requires 374.0969).
2,4-Bis(benzyloxy)-3,5-difluorobenzaldehyde (9)
To a solution of 8 (13.4 g, 38 mmol) in CH2Cl2 (20 mL) at −78 °C was added diisobutylaluminum hydride (Acros Organics, 1 M in cyclohexane, 45.9 mL). The reaction mixture was stirred at −78 °C for 3.5 h and was then warmed to 22 °C. The reaction was quenched with aqueous HCl (0.5 M, 150 mL) by stirring for 1 h. The mixture was extracted with ethyl acetate, dried, filtered, and evaporated under reduced pressure. The resultant solid was recrystallized from ethanol to afford 9 (11.6 g, 87%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 10.01 (d, J = 3.3 Hz, 1H), 7.49–7.27 (m, 11H), 5.34 (s, 2H), 5.17 (d, J = 1.0 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 187.1 (dd, J = 3.2, 1.8 Hz), 151.8 (dd, J = 247.3, 4.0 Hz), 149.1 (dd, J = 250.8, 5.5 Hz), 146.6 (dd, J = 10.1, 2.9 Hz), 141.6 (dd, J = 15.4, 12.2 Hz), 135.8, 135.3, 129.2, 128.94, 128.91, 128.90, 128.8, 128.4, 124.1 (dd, J = 6.3, 1.6 Hz), 109.2 (dd, J = 21.2, 3.1 Hz), 77.4 (d, J = 6.5 Hz), 76.0 (t, J = 4.0 Hz); 19F NMR (376 MHz, CDCl3) δ −130.48 (d, J = 5.7 Hz), −140.71 (dd, J = 6.0, 2.5 Hz); HRMS (ESI+) m/z 377.0983 (M + Na+, C21H16F2O3Na requires 377.0965).
3,5-Difluoro-2,4-dihydroxybenzaldehyde (10)
To a solution of 9 (11.0 g, 31 mmol) in CH3OH/THF (100 mL, 7:3) was added Pd/C (10%, 1.67 g), and the mixture was stirred under an atmosphere of hydrogen (1 atm) at 22 °C for 8 h. After removing the catalyst by filtration over celite, the filtrate was concentrated under reduced pressure and purified using column chromatography over silica gel (eluent: hexanes/ethyl acetate/acetic acid (82:18:1)) to afford 10 (4.6 g, 83%) as a light pink solid. 1H NMR (500 MHz, DMSO-d6) δ 11.58 (s, 1H), 10.86 (s, 1H), 10.0439 (d, J = 2.4 Hz, 1H), 7.29 (dd, J = 10.8, 2.1 Hz, 1H); 13C NMR (126 MHz, DMSO-d6) δ 189.5, 146.8 (d, J = 11.8 Hz), 145.6 (dd, J = 236.6, 4.5 Hz), 141.5 (dd, J = 18.0, 13.5 Hz), 141.2 (dd, J = 238.9, 5.7 Hz), 113.9 (d, J = 5.8 Hz), 109. 4 (dd, J = 19.4, 2.9 Hz); 19F NMR (376 MHz, DMSO-d6) δ −144.25, −156.25 (d, J = 6.4 Hz); HRMS (ESI−) m/z 173.0038 (M – H+, C7H3F2O3 requires 173.0050). Note: This product can be carried forward without further purification. Following simple filtration through celite and concentration, this crude material has yielded PB (5) in high purity (79% yield over two steps, 8.5 mmol scale).
6,8-Difluoro-7-hydroxy-2-oxo-2H-chromene-3-carboxylic Acid (PB, 5)
To a suspension of 10 (4.2 g, 24.1 mmol) in water (110 mL) was added Meldrum’s acid (3.8 g, 26.6 mmol) and ammonium acetate (550 mg, 7.2 mmol). The suspension was stirred at 22 °C for 3.5 h. Aqueous HCl (2 M, 75 mL) was added, and the mixture was placed at 4 °C for 1 h. A precipitate that formed was filtered, washed with cold water (2 × 25 mL), and dried under high vacuum to afford PB (5, 5.4 g, 92%) as a light yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.67 (d, J = 1.4 Hz, 1H), 7.66 (dd, J = 10.5, 2.0 Hz, 1H); 13C NMR (126 MHz, DMSO-d6) δ 163.9, 155.6, 148.6 (dd, J = 240.8, 4.7 Hz), 148.5 (t, J = 2.9 Hz), 141.3 (d, J = 9.6 Hz), 140.3 (dd, J = 18.1, 12.6 Hz), 138.7 (dd, J = 244.7, 6.4 Hz), 115.3, 110.5 (dd, J = 20.9, 3.0 Hz), 109.0 (d, J = 10.2 Hz). 19F NMR (376 MHz, DMSO-d6) δ −137.33 (d, J = 9.3 Hz), −155.68 (d, J = 9.8 Hz); HRMS (ESI−) m/z 240.9944 (M – H+, C10H3F2O5 requires 240.9949).
2,5-Dioxopyrrolidin-1-yl 6,8-Difluoro-7-hydroxy-2-oxo-2H-chromene-3-carboxylate (PB NHS Ester, 11)
To a solution of 5 (0.75 g, 3.1 mmol) in DMF (5 mL) was added N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, 1.2 g, 6.2 mmol) and N-hydroxysuccinimide (0.89 g, 7.8 mmol). This mixture was stirred at 22 °C for 16 h and was subsequently added dropwise to cold aq HCl (1 N, 75 mL). A precipitate formed, which was filtered, washed with cold aq HCl (1 N, 25 mL), and dried under high vacuum to give 11 (920 mg, 87%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 9.01 (d, J = 1.4 Hz, 1H), 7.78 (dd, J = 10.2, 1.9 Hz, 1H), 2.89 (s, 4H); 13C NMR (126 MHz, DMSO-d6) δ 170.3, 158.4, 154.4, 152.1 (t, J = 2.9 Hz), 148.8 (dd, J = 241.3, 4.9 Hz), 142.4 (d, J = 9.0 Hz), 138.7 (dd, J = 244.8, 6.6 Hz), 111.5 (dd, J = 21.1, 2.8 Hz), 108.8, 108.4 (d, J = 10.3 Hz), 25.6; 19F NMR (376 MHz, DMSO-d6) δ −136.84, −155.95 (d, J = 13.4 Hz); HRMS (ESI−) m/z 338.0129 (M – H+, C14H6F2NO7 requires 338.0112).
General Procedure for the Synthesis of Fluorescent Derivatives of Biotin
General Procedure A (12, 13)
N-Boc-ethylenediamine-d-biotin (64–87 mg, 1.5 equiv), prepared as previously reported,38 was treated with a solution of trifluoroacetic acid (TFA) in CH2Cl2 (2 mL, 30:70) for 20 min. The mixture was concentrated under vacuum and washed with CH2Cl2 (5 mL) and diethyl ether (5 mL × 2) to remove excess TFA. The activated fluorophore (1 equiv), N,N-diisopropylethylamine (DIEA, 5 equiv), and DMF (1–3 mL) were added, and the reaction mixture was stirred at 22 °C for 16 h. The solvent was removed under vacuum, the residue was dissolved in DMSO (1.5 mL), and the product was purified by preparative reversed-phase (RP)-HPLC [gradient: H2O:CH3CN (9:1) to (0:100) with added TFA (0.1%) over 20 min; elution time = 6–10 min]. Pure fractions were collected and combined, and the solvent was removed using lyophilization.
General Procedure B (14, 15)
N-Boc-ethylenediamine-d-biotin (92–120 mg, 1.5 equiv), prepared as previously reported,38 was treated with a solution of TFA/CH2Cl2 (2 mL, 30:70) for 20 min. The mixture was concentrated under vacuum and washed with CH2Cl2 (5 mL) and ether (5 mL × 2) to remove excess TFA. The fluorophore (1 equiv), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 1.1 equiv), 4-dimethylaminopyridine (DMAP, 0.5 equiv), and DMF (2 mL) were added, and the reaction mixture was stirred at 22 °C for 16 h. The solvent was removed under vacuum, the residue was dissolved in DMSO (1.5 mL), and the product was purified by preparative RP-HPLC (gradient: H2O:CH3CN (9:1) to (0:100) with added TFA (0.1%) over 20 min; elution time = 6–10 min). Pure fractions were collected, combined, and dried using lyophilization.
6,8-Difluoro-7-hydroxy-2-oxo-N-(2-(5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamido)ethyl)-2H-chromene-3-carboxamide (PB-Biotin, 12)
Following the general procedure A, PB NHS ester (11, 50 mg, 0.15 mmol) yielded compound 12 (45 mg, 60%) as a pale yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.82 (d, J = 1.4 Hz, 1H), 8.67 (t, J = 5.8 Hz, 1H), 7.93 (t, J = 5.6 Hz, 1H), 7.78 (dd, J = 10.5, 1.9 Hz, 1H), 6.44 (s, 1H), 6.36 (s, 1H), 4.29 (dd, J = 7.7, 4.9 Hz, 1H), 4.11 (dd, J = 7.8, 4.4 Hz, 1H), 3.39 (q, J = 6.2 Hz, 2H), 3.22 (q, J = 6.1 Hz, 2H), 3.07 (ddd, J = 8.5, 6.3, 4.4 Hz, 1H), 2.78 (dd, J = 12.4, 5.1 Hz, 1H), 2.55 (d, J = 12.4 Hz, 1H), 2.06 (t, J = 7.4 Hz, 2H), 1.64–1.26 (m, 6H); 13C NMR (126 MHz, DMSO-d6) δ 172.3, 162.7, 161.3, 159.5, 148.8 (dd, J = 241.2, 4.6 Hz), 147.3 (t, J = 2.9 Hz), 140.5 (d, J = 9.2 Hz), 140.0 (d, J = 13.2 Hz), 138.8 (dd, J = 245.1, 6.6 Hz), 116.2, 110.6 (dd, J = 21.0, 3.0 Hz), 109.6 (d, J = 10.2 Hz), 61.0, 59.2, 55.4, 40.1, 39.4, 38.5, 38.1, 35.2, 28.2, 28.1, 28.0, 25.3; HRMS (ESI−) m/z 509.1298 (M – H+, C22H23F2N4O6S requires 509.1306).
N-(2-((5-(Dimethylamino)naphthalene)-1-sulfonamido)ethyl)-5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamide (Dansyl–Biotin, 13)
Following the general procedure A, dansyl chloride (30 mg, 0.11 mmol) yielded compound 13 (25 mg, 42%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.47 (dt, J = 8.6, 1.1 Hz, 1H), 8.28 (dt, J = 8.8, 0.9 Hz, 1H), 8.10 (dd, J = 7.3, 1.2 Hz, 1H), 7.99 (t, J = 5.9 Hz, 1H), 7.73 (t, J = 5.8 Hz, 1H), 7.62 (m, 2H), 7.28 (d, J = 7.4 Hz, 1H), 6.42 (s, 1H), 4.30 (dd, J = 7.4, 4.7 Hz, 1H), 4.11 (dd, J = 7.7, 4.4 Hz, 1H), 3.13–3.05 (m, 1H), 3.03 (dt, J = 7.5, 5.9 Hz, 2H), 2.85 (s, 6H), 2.83–2.75 (m, 3H), 2.57 (d, J = 12.5 Hz, 1H), 1.93 (t, J = 7.4 Hz, 2H), 1.66–1.15 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 172.2, 162.7, 158.3, 158.0, 151.2, 135.8, 129.5, 129.0, 128.4, 128.3, 127.9, 123.7, 117.0, 115.2, 114.7, 61.0, 59.2, 55.4, 45.1, 42.0, 39.5, 35.1, 28.2, 25.1; HRMS (ESI+) m/z 542.1900 (M + Na+, C24H33N5O4S2Na requires 542.1872).
N-(2-(2-(7-Hydroxy-2-oxo-2H-chromen-4-yl)acetamido)ethyl)-5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamide (7-Hydroxycoumarin–Biotin, 7HC–Biotin, 14)
2-(7-Hydroxy-2-oxo-2H-chromen-4-yl)acetic acid was prepared as previously reported.39 Following the general procedure B, 2-(7-hydroxy-2-oxo-2H-chromen-4-yl)acetic acid (45 mg, 0.21 mmol) yielded compound 14 (57 mg, 57%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 10.57 (s, 1H), 8.27–8.15 (m, 1H), 7.93–7.75 (m, 1H), 7.58 (d, J = 8.7 Hz, 1H), 6.79 (dd, J = 8.7, 2.4 Hz, 1H), 6.71 (d, J = 2.4 Hz, 1H), 6.40 (d, J = 27.8 Hz, 2H), 6.16 (s, 1H), 4.30 (dd, J = 7.7, 5.0 Hz, 1H), 4.12 (ddd, J = 7.7, 4.4, 1.8 Hz, 1H), 3.74–3.50 (m, 2H), 3.09 (m, 5H), 2.81 (dd, J = 12.4, 5.1 Hz, 1H), 2.57 (d, J = 12.5 Hz, 2H), 2.02 (t, J = 7.4 Hz, 2H), 1.67–1.18 (m, 6H); 13C NMR (126 MHz, DMSO) δ 172.3, 167.8, 162.7, 161.2, 160.3, 155.0, 151.1, 126.8, 112.9, 111.9, 111.5, 102.3, 61.0, 59.2, 55.4, 40.4, 40.1, 40.0, 39.94, 39.86, 39.78, 39.69, 39.61, 39.5, 39.4, 39.2, 39.0, 38.9, 38.7, 38.1, 35.2, 28.2, 28.1, 25.2; HRMS (ESI−) m/z 487.1647 (M – H+, C23H27N4O6S requires 487.1651).
N-(2-(2-(7-(Dimethylamino)-2-oxo-2H-chromen-4-yl)acetamido)ethyl)-5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamide (DMACA–Biotin, 15)
2-(7-(Dimethylamino)-2-oxo-2H-chromen-4-yl)acetic acid was prepared as previously reported.39 Following the general procedure B, 2-(7-(dimethylamino)-2-oxo-2H-chromen-4-yl)acetic acid (DMACA, 40 mg, 0.16 mmol) yielded compound 15 (36 mg, 43%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.25–8.18 (m, 1H), 7.82 (d, J = 5.4 Hz, 1H), 7.53 (d, J = 9.0 Hz, 1H), 6.73 (dd, J = 9.0, 2.6 Hz, 1H), 6.56 (d, J = 2.5 Hz, 1H), 6.46–6.41 (m, 1H), 6.38 (s, 1H), 6.00 (s, 1H), 4.30 (ddd, J = 7.7, 5.2, 1.0 Hz, 1H), 4.16–4.10 (m, 1H), 3.61–3.57 (m, 2H), 3.10 (m, 5H), 3.02 (s, 6H), 2.82 (dd, J = 12.4, 5.1 Hz, 1H), 2.58 (d, J = 12.5 Hz, 1H), 2.03 (t, J = 7.4 Hz, 2H), 1.69–1.18 (m, 6H); 13C NMR (126 MHz, DMSO) δ 172.7, 168.4, 163.2, 161.2, 155.8, 153.2, 151.6, 126.5, 109.9, 109.5, 108.7, 97.9, 61.5, 59.6, 55.9, 40.9, 39.3, 39.1, 38.6, 35.7, 28.7, 28.5, 25.6; HRMS (ESI+) m/z 516.2291 (M + H+, C25H34N5O5S requires 516.2281).
tert-Butyl(2-(6,8-difluoro-7-hydroxy-2-oxo-2H-chromene-3-carboxamido)ethyl)carbamate (N-Boc-ethylenediamine-PB, 23)
To a solution of 11 (200 mg, 0.59 mmol) in DMF (5 mL) was added N-Boc-ethylenediamine (114 mg, 0.71 mmol). The reaction mixture was stirred at 22 °C for 16 h. The vessel was placed under high vacuum for 1 h to remove DMF (4 mL). The remaining reaction mixture was added dropwise to cold aq HCl (1 N, 40 mL). A precipitate formed, which was filtered, washed with cold aq HCl (1 N, 10 mL), and dried under high vacuum to give 23 (200 mg, 88%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.81 (d, J = 1.4 Hz, 1H), 8.67 (t, J = 5.8 Hz, 1H), 7.77 (dd, J = 10.4, 1.9 Hz, 1H), 6.95 (t, J = 5.6 Hz, 1H), 3.36 (q, J = 6.1 Hz, 2H), 3.10 (q, J = 6.0 Hz, 2H), 1.37 (s, 11H), 1.45–1.39 (m, 1H); 13C NMR (126 MHz, DMSO-d6) δ 161.3, 159.5, 155.7, 148.9 (dd, J = 240.9, 4.8 Hz), 147.3, 140.5 (d, J = 9.0 Hz), 140.1, 138.8 (dd, J = 245.0, 6.4 Hz), 116.2, 110.7, 110.5, 109.5, 109.5, 77.7, 28.2; HRMS (ESI−) m/z 383.1043 (M – H+, C17H17F2N2O6 requires 383.1055).
General Procedure for the Synthesis of Analogues of Biotin Linked to PB
General Procedure C (16–18)
N-Boc-ethylenediamine-PB (23, 23–29 mg, 1 equiv) was treated with a solution of TFA in CH2Cl2 (2 mL, 30:70) for 20 min. The mixture was concentrated and washed with CH2Cl2 (5 mL) and MeOH (5 mL × 2) to remove excess TFA. The biotin analogue (1.3 equiv), HBTU (1.5 equiv), hydroxybenzotriazole (HOBt, 1.5 equiv), DIEA (5 equiv), and DMF (1 mL) were added. The reaction mixture was stirred at 22 °C for 16 h. The solvent was removed under vacuum, the residue was dissolved in DMSO (1.5 mL), and the product was purified by preparative RP-HPLC (gradient: H2O:CH3CN (9:1) to (0:100) with added TFA (0.1%) over 20 min; elution time = 6–10 min). Pure fractions were collected, combined, and dried using lyophilization.
(3aS,4S,6aR)-4-(5-((2-(6,8-Difluoro-7-hydroxy-2-oxo-2H-chromene-3-carboxamido)ethyl)amino)-5-oxopentyl)tetrahydro-1H-thieno[3,4-d]imidazol-2(3H)-iminium (PB–Iminobiotin, 16)
Following the general procedure C, 2-iminobiotin (19 mg, 0.077 mmol) yielded compound 16 (12 mg, 40%) as a pale yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.80 (d, J = 1.4 Hz, 1H), 8.67 (t, J = 5.8 Hz, 1H), 8.22 (d, J = 6.5 Hz, 1H), 8.11–8.01 (m, 1H), 7.94 (t, J = 5.6 Hz, 1H), 7.82–7.70 (m, 1H), 7.62 (s, 2H), 6.55 (s, 1H), 4.63 (dd, J = 7.9, 4.8 Hz, 1H), 4.44 (ddd, J = 7.9, 4.5, 1.7 Hz, 1H), 3.38 (d, J = 6.3 Hz, 2H), 3.22 (dd, J = 7.7, 3.7 Hz, 2H), 3.01–2.82 (m, 2H), 2.77 (d, J = 12.9 Hz, 1H), 2.07 (t, J = 7.3 Hz, 2H), 1.75–1.23 (m, 6H); 13C NMR (126 MHz, DMSO-d6) δ 172.3, 161.3, 159.5 (d, J = 3.0 Hz), 159.4, 148.9 (dd, J = 240.8, 4.8 Hz), 147.4 (t, J = 3.0 Hz), 140.6 (d, J = 8.8 Hz), 140.2 (d, J = 17.4 Hz), 138.8 (dd, J = 245.3, 6.6 Hz), 116.1, 110. 6 (dd, J = 21.1, 2.9 Hz), 109.5 (d, J = 10.2 Hz), 64.4, 63.0, 55.1, 40.1, 39.7, 38.1, 35.2, 28.2, 27.9, 25.2; HRMS (ESI−) m/z 508.1471 (M – H+, C22H24F2N5O5S requires 508.1466).
Methyl 5-((3aS,4S,6aR)-2-thioxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanoate (24)
d-Biotin methyl ester (387 mg, 1.50 mmol), prepared as previously reported,40 was dissolved in xylenes (6 mL). Lawesson’s reagent (607 mg, 1.50 mmol) was then added, and the reaction mixture was heated to 95 °C for 1 h. The solvent was removed under reduced pressure, and the resultant residue was extracted with ethyl acetate, dried, filtered, and evaporated under reduced pressure. The residue was purified using column chromatography over silica gel (eluent: CH2Cl2/CH3OH (50:1)) to afford 24 (330 mg, 80%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.24 (m, 1H), 8.17 (s, 1H), 4.54 (dd, J = 8.4, 5.0 Hz, 1H), 4.36 (ddd, J = 8.4, 4.5, 1.5 Hz, 1H), 3.58 (s, 3H), 3.17 (ddd, J = 8.6, 6.0, 4.5 Hz, 1H), 2.86 (dd, J = 12.6, 5.1 Hz, 1H), 2.67 (d, J = 12.7 Hz, 1H), 2.30 (t, J = 7.5 Hz, 2H), 1.77–1.24 (m, 6H); 13C NMR (126 MHz, CDCl3) δ 182.4, 173.3, 66.0, 64.1, 55.7, 39.8, 33.1, 28.1, 27.9, 24.5; HRMS (ESI+) m/z 297.0707 (M + Na+, C11H18N2O2S2Na requires 297.0707).
5-((3aS,4S,6aR)-2-Thioxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanoic Acid (25)
To a solution of 24 (125 mg, 0.46 mmol) in CH3OH (4 mL) was added aq NaOH (1 N, 2 mL). The reaction mixture was heated to 35 °C for 0.5 h, followed by acidification with aq HCl (1 N, 10 mL). A precipitate formed, which was filtered, washed with cold aq HCl (1 N, 5 mL), and dried under high vacuum to give 25 (90 mg, 76%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 12.00 (s, 1H), 8.24 (s, 1H), 8.17 (s, 1H), 4.54 (dd, J = 8.4, 4.9 Hz, 1H), 4.36 (ddd, J = 8.5, 4.7, 1.4 Hz, 1H), 3.17 (ddd, J = 8.8, 5.9, 4.4 Hz, 1H), 2.86 (dd, J = 12.6, 5.0 Hz, 1H), 2.67 (d, J = 12.6 Hz, 1H), 2.20 (t, J = 7.4 Hz, 2H), 1.75–1.20 (m, 6H); 13C NMR (126 MHz, DMSO-d6) δ 182.4, 174.5, 66.0, 64.1, 55.7, 40.1, 33.5, 28.2, 28.0, 24.6; HRMS (ESI−) m/z 259.0572 (M – H+, C10H15N2O2S2 requires 259.0575).
6,8-Difluoro-7-hydroxy-2-oxo-N-(2-(5-((3aS,4S,6aR)-2-thioxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamido)ethyl)-2H-chromene-3-carboxamide (PB–Thiobiotin, 17)
Following the general procedure C, 25 (24 mg, 0.091 mmol) yielded compound 17 (18 mg, 49%) as a pale yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.79 (d, J = 1.4 Hz, 1H), 8.66 (t, J = 5.8 Hz, 1H), 8.23 (s, 1H), 8.15 (s, 1H), 7.92 (t, J = 5.6 Hz, 1H), 7.73 (d, J = 10.5 Hz 1H), 4.69–4.46 (m, 1H), 4.34 (ddd, J = 8.5, 4.6, 1.6 Hz, 1H), 3.54–3.28 (m, 10H), 3.28–3.10 (m, 4H), 2.82 (dd, J = 12.7, 5.1 Hz, 1H), 2.65 (d, J = 12.7 Hz, 1H), 2.15–1.97 (m, 2H), 1.74–1.16 (m, 6H); 13C NMR (126 MHz, DMSO-d6) δ 182.4, 172.4, 161.5, 159.7, 149.3 (d, J = 239.7 Hz), 147.4 (t, J = 2.8 Hz), 140.8, 140.7, 139.0 (dd, J = 244.6, 6.8 Hz), 115.3, 110.6, 108.8, 66.0, 64.2, 55.8, 40.4, 40.1, 38.9, 35.3, 30.8, 28.3, 28.0, 25.3; HRMS (ESI−) m/z 525.1050 (M – H+, C22H23F2N4O5S2 requires 525.1078).
Methyl 6-((4R,5S)-5-methyl-2-oxoimidazolidin-4-yl)hexanoate (26)
To a solution of d-desthiobiotin (215 mg, 1.0 mmol) in CH3OH (5 mL), thionyl chloride (218 μL, 3.0 mmol) was added dropwise. The reaction mixture was stirred at 22 °C for 3 h. The solvent was removed under reduced pressure followed by purification using column chromatography on silica gel (eluent: CH2Cl2/CH3OH (20:1)) to afford 26 (217 mg, 95%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 5.34 (s, 1H), 5.07 (s, 1H), 3.89–3.78 (m, 1H), 3.70 (ddd, J = 9.2, 7.8, 4.9 Hz, 1H), 3.66 (s, 3H), 2.31 (t, J = 7.4 Hz, 2H), 1.77–1.17 (m, 8H), 1.12 (d, J = 6.5 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 174.3, 164.0, 77.41, 77.36, 77.2, 76.9, 56.2, 51.7, 51.6, 34.0, 29.6, 29.1, 26.3, 24.8, 15.8; HRMS (ESI+) m/z 251.1374 (M + H+, C11H20N2O3Na requires 251.1372).
Methyl 6-((4R,5S)-5-methyl-2-thioxoimidazolidin-4-yl)hexanoate (21)
To a solution of 26 (100 mg, 0.44 mmol) in xylenes (3 mL) was added Lawesson’s reagent (178 mg, 0.44 mmol). The reaction mixture was heated to 95 °C for 1 h. The solvent was removed under reduced pressure, and the resultant residue was extracted with ethyl acetate, dried, filtered, and evaporated under reduced pressure. The residue was purified using column chromatography over silica gel (eluent: CH2Cl2/CH3OH (50:1)) to afford 21 (91 mg, 85%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 6.29 (br, 2H), 4.10 (t, J = 7.2 Hz, 1H), 4.01–3.87 (m, 1H), 3.67 (s, 3H), 2.32 (t, J = 7.3 Hz, 2H), 1.81–1.23 (m, 8H), 1.18 (d, J = 6.1 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 182.8, 174.1, 60.5, 55.9, 51.7, 34.0, 29.0, 28.9, 26.4, 24.8, 15.2; HRMS (ESI+) m/z 267.1146 (M + Na+, C11H20N2O2SNa requires 267.1143).
6-((4R,5S)-5-Methyl-2-thioxoimidazolidin-4-yl)hexanoic Acid (Imidazolidinethione, 20)
To a solution of 21 (63 mg, 0.26 mmol) in CH3OH (2 mL) was added aq NaOH (1 N, 1 mL). The reaction mixture was heated to 35 °C for 0.5 h followed by acidification with aq HCl (1 N, 5 mL). The resultant mixture was extracted with ethyl acetate, dried, filtered, and evaporated under reduced pressure. The residue was purified using column chromatography over silica gel (eluent: CH2Cl2/CH3OH (25:1)) to afford 20 (55 mg, 93%) as an off-white solid. 1H NMR (500 MHz, CD3OD) δ 4.11–3.96 (m, 1H), 3.87 (dt, J = 8.8, 6.8 Hz, 1H), 2.30 (t, J = 7.4 Hz, 2H), 1.71–1.25 (m, 8H), 1.13 (d, J = 6.6 Hz, 3H); 13C NMR (126 MHz, CD3OD) δ 183.5, 177.7, 61.4, 56.7, 34.8, 30.2, 30.0, 27.1, 25.9, 14.8; HRMS (ESI−) m/z 229.1006 (M – H+, C10H17N2O2S requires 229.1011).
6,8-Difluoro-7-hydroxy-N-(2-(6-((4R,5S)-5-methyl-2-thioxoimidazolidin-4-yl)hexanamido)ethyl)-2-oxo-2H-chromene-3-carboxamide (PB–Imidazolidinethione, 18)
Following the general procedure C, 20 (23 mg, 0.1 mmol) yielded compound 18 (26 mg, 68%) as a pale yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.81 (br, 1H), 8.66 (t, J = 5.9 Hz, 1H), 8.18 (s, 1H), 8.02 (s, 1H), 7.91 (t, J = 5.7 Hz, 1H), 7.77 (dd, J = 10.6, 2.0 Hz, 1H), 3.92–3.77 (m, 1H), 3.68 (td, J = 8.2, 4.7 Hz, 1H), 3.37 (m, 2H), 3.21 (q, J = 6.1 Hz, 2H), 2.05 (t, J = 7.4 Hz, 2H), 1.58–1.08 (m, 8H), 0.96 (d, J = 6.4 Hz, 3H); 13C NMR (126 MHz, DMSO-d6) δ 181.8, 172.4, 161.3, 159.5, 148.9 (dd, J = 240.1, 4.1 Hz), 147.3 (d, J = 3.3 Hz), 140.6 (d, J = 8.6 Hz), 138.8 (dd, J = 245.1, 6.7 Hz), 116.1, 110.6 (dd, J = 21.6, 2.2 Hz), 109.4, 59.1, 54.4, 38.1, 35.4, 28.7, 28.6, 25.5, 25.2, 20.7, 14.6; HRMS (ESI−) m/z 495.1534 (M – H+, C22H25F2N4O5S requires 495.1514).
Benzyl 6-((5S)-5-methyl-2-oxoimidazolidin-4-yl)hexanoate (27)
To a solution of d-desthiobiotin (100 mg, 0.46 mmol) in DMF (2 mL) was added benzyl alcohol (95 μL, 0.92 mmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, 56 mg, 0.6 mmol), HOBt (92 mg, 0.6 mmol), and DMAP (56 mg, 0.46 mmol). The reaction mixture was stirred at 22 °C for 16 h. The solvent was removed under reduced pressure, and the residue was purified using column chromatography over silica gel (eluent: CH2Cl2/CH3OH (50:2)) to afford 27 (137 mg, 96%) as an off-white solid. 1H NMR (500 MHz, CDCl3) δ 7.34–7.22 (m, 5H), 5.05 (s, 2H), 3.87–3.76 (m, 1H), 3.65 (td, J = 8.3, 5.0 Hz, 1H), 2.30 (t, J = 7.4 Hz, 2H), 1.68–1.13 (m, 8H), 1.07 (d, J = 6.4 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 173.6, 163.4, 136.1, 128.7, 128.4, 66.3, 56.4, 51.8, 34.2, 29.5, 29.0, 26.3, 24.8, 15.8; HRMS (ESI+) m/z 305.1889 (M + H+, C17H24N2O3 requires 305.1865).
Benzyl 6-((5S)-5-methyl-2-thioxoimidazolidin-4-yl)hexanoate (Imidazolidinethione–OBn, 22)
To a solution of 27 (100 mg, 0.33 mmol) in xylenes (2.5 mL) was added Lawesson’s reagent (133 mg, 0.33 mmol). The reaction mixture was heated to 95 °C for 1.5 h. The solvent was removed under reduced pressure, and the resultant residue was extracted with ethyl acetate, dried, filtered, and evaporated under reduced pressure. The residue was purified using column chromatography over silica gel (eluent: CH2Cl2/CH3OH (50:1)) to afford 22 (86 mg, 82%) as an off-white solid. 1H NMR (500 MHz, CDCl3) δ 7.45–7.27 (m, 5H), 5.11 (s, 2H), 4.07 (dq, J = 8.8, 6.5 Hz, 1H), 3.90 (td, J = 9.0, 4.8 Hz, 1H), 2.36 (t, J = 7.4 Hz, 2H), 1.73–1.21 (m, 8H), 1.16 (d, J = 6.6 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 182.5, 173.4, 136.0, 128.6, 128.3, 66.2, 60.3, 55.8, 34.1, 28.8, 28.7, 26.2, 24.6, 15.0; HRMS (ESI+) m/z 321.1652 (M + H+, C17H24N2O2S requires 321.1637).
Assays of Binding of Small Molecules to SA
SA protein from Streptomyces avidinii(lyophilized powder) was purchased from Alfa Aesar. Absorbance spectra and measurements of molar extinction coefficients (ε) were generated using semimicro (1.4 mL) UV quartz cuvettes (Sigma-Aldrich, Z27667-7) on an Agilent 8452A diode array spectrometer. All optical spectroscopy and protein-binding assays were conducted in PBS (10 mM Na2HPO4, 137 mM NaCl, 2.7 mM KCl, and 1.8 mM KH2PO4, pH 7.4), unless otherwise noted. Molar extinction coefficients were determined in PBS (0.5% DMSO) and were calculated from Beer’s Law plots of absorbance λmax versus concentration, as shown in Figure S1. Linear least-squares fitting of the data (including a zero intercept) was used to determine the slope (corresponding to ε). Values (M–1 cm–1) were calculated as follows: absorbance = ε[concentration(M)]L, where L = 1 cm. SA concentrations were quantified using UV absorbance at 280 nm based on its calculated molar extinction coefficient (εmonomer = 41 326 M–1 cm–1) using a Thermo Scientific NanoDrop 1000 spectrophotometer. All fluorescence spectra were acquired using a SUPRASIL ultra-micro quartz cuvette (PerkinElmer, B0631079) on a Perkin-Elmer LS55 fluorescence spectrometer (10 nm excitation and emission slit widths). Relative quantum yields (Φ) in PBS were determined by the Williams’ method.41 In brief, fluorophores were excited at 396 nm and the integrated fluorescence emission (415–700 nm) was quantified (concentrations of 5–160 nM). Coumarin 102 (Φ = 0.66 in water) provided the standard.42 The integrated fluorescence emission at a given concentration was plotted against the maximum absorbance of the sample at that concentration, determined by extrapolation based on absorbance measurements at higher concentrations, as shown in Figure S1. Linear least-squares fitting of the data (including a zero intercept) was used to calculate the slope, which is proportional to the quantum yield. Quantum yields were calculated as follows: Φx = Φst(Gradx/Gradst), where Φst represents the quantum yield of the standard, Φx represents the quantum yield of the unknown, and Grad is the slope of the best linear fit. Theoretical Förster distances were calculated using a previously described protocol.43,44 The following parameters and equations were used: ΦD is the quantum yield of the donor, η is the refractive index of the solvent, κ is the orientation factor, J is the degree of spectral overlap between the donor and the acceptor, FD(λ) is the normalized donor fluorescence intensity, and εA(λ) is the absorbance spectrum of the acceptor normalized to its maximum molar extinction coefficient.
Theoretical Förster distances were calculated using the PhotoChemCAD software, and the measured extinction coefficient for each probe were Φtryptophan = 0.2, ηphosphate buffer, pH 7.4 = 1.33, and κ = 2/3.
Determination of Kd Values Using FRET, Fluorescence Enhancement, and Fluorescence Quenching
Different concentrations of SA protein, chosen to span a range of at least 20–80% complexation, were incubated with fixed concentrations of 16 (25 nM), 17 (5 nM), or 18 (25 nM) in PBS (pH 7.4) at room temperature with shaking for 1 h. These fixed probe concentrations were chosen to be substantially below the predicted Kd values to assure equilibrium binding measurements. Averages of three measurements of raw FRET values (I295, λex = 295 nm, and λem = 460 nm) and fluorescence intensity values (I400, λex = 400 nm, λem = 460 nm) were recorded for each sample using a Perkin-Elmer LS55 fluorescence spectrometer. These raw FRET values were corrected (Iad), and changes in fluorescence were corrected (Iad,400) by subtracting background signals and factoring in quenching or enhancement of the fluorescence of PB upon binding as follows: averages of raw FRET (Id,295) values and averages of the fluorescence (Id,400) intensity of SA alone (background fluorescence) were calculated for each concentration of SA. Additionally, the average emission at 460 nm from three measurements of the free PB ligand (16–18 for direct binding and 18 for competition binding) in the PBS buffer upon excitation at 295 (Ia,295) and 400 nm (Ia,400) was calculated. Background-subtracted FRET (IFRET), fluorescence (Iad,400), and the quenching ratio (Qr) were calculated as
Background-subtracted FRET values (IFRET) were processed to directly factor in quenching or enhancement of fluorescence using the following equation, where Iad is the corrected FRET signal
To calculate the dissociation constants (Kd) using FRET (Figure 5), corrected FRET values (Iad), run in triplicate, were plotted against the concentration of SA (monomer) and a one-site-specific binding model (GraphPad Prism 6.0) was used for curve fitting.
To calculate the dissociation constants (Kd) from simple enhancement or quenching of fluorescence (Figure 7), the change in fluorescence (enhancement or quenching, Iad,400) was normalized and plotted against the concentration of SA (monomer). The experiments were run in triplicate, and a one-site-specific binding model (GraphPad Prism 6.0) was used for curve fitting. For 17, the fluorescence intensity values for the three highest concentrations of the SA monomer (64, 128, and 256 nM) were excluded from the analysis to allow fitting of the one-site-specific binding model.
Determination of Ki Values (Loss of Trp-FRET from Competitive Binding)
The unlabeled ligand was added to a fixed concentration of SA and fluorescent probe 18 (SA = 175 nM, 18 = 25 nM) in PBS (pH 7.4) precomplexed by incubation at room temperature with shaking for 1 h. The concentration of SA was chosen to be close to the Kd value measured for the fluorescent probe, and the concentration of 18 was chosen to be substantially below Kd to assure equilibrium binding. Measurements of raw FRET values (I295, λex = 295 nm, λem = 460 nm) and fluorescence intensity values (I400, λex = 400 nm, λem = 460 nm) were recorded for each sample using a Perkin-Elmer LS55 fluorescence spectrometer. Background subtraction and quenching were factored into each fluorescence measurement to calculate the corrected FRET signal (Iad) as described previously. To calculate IC50 values, the corrected FRET signal (Iad) was plotted against the concentration of the unlabeled ligand. The experiments were run in triplicate, and the log(inhibitor) versus response model (GraphPad Prism 6.0) was used for curve fitting. Inhibitory constants (Ki) were calculated as
ITC
ITC experiments were performed using a MicroCal Auto-Isothermal Titration Calorimeter with protein and ligand solutions prepared in PBS. Titrations were performed at 25 °C and consisted of 25 injections (10 μL) of the ligand (50–250 μM) into SA (1.46 mL, 4–20 μM), with 6 min between injections. The experimental data were fit to a one-site binding model (the Origin software), where ΔH (enthalpy change, kcal/mol), Ka (association constant, M–1), and n (number of binding sites) were variables.
Determination of Kd Values by FP of PB
Fluorescence polarization is very sensitive to fluorescence quenching because of the effects on the lifetime of the fluorophore. This quenching prevented attempts to use FP to independently quantify the affinity of ligands 17 and 18 for SA.3 However, quenching was less significant for the lower affinity ligand 16, and a previously described45 FP method was used to independently analyze this probe. Measurements of fluorescence intensity (I400, λex = 400 nm, λem = 460 nm) and fluorescence polarization (P, λex = 400 nm, λem = 460 nm) were recorded for each sample using a Perkin-Elmer LS55 fluorescence spectrometer. Background subtraction and quenching were factored into each fluorescence measurement to calculate the corrected fraction bound (fa), as described in the Supporting Information. To calculate the dissociation constant (Kd), the corrected fraction bound was plotted against the concentration of SA, and a one-site-specific binding model (GraphPad Prism 6.0) was used for curve fitting (Figure S2).
Acknowledgments
We thank the NIH (P20-GM103638 and P20-GM103418), the G. Harold and Leila Y. Mathers Charitable Foundation, and the KU Cancer Center for financial support. M.M.L. was supported in part by an NIH Dynamic Aspects of Chemical Biology training grant (T32-GM08545).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00356.
Additional experimental methods, data from binding assays and other photophysical measurements, and NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Jelesarov I.; Bosshard H. R. Isothermal titration calorimetry and differential scanning calorimetry as complementary tools to investigate the energetics of biomolecular recognition. J. Mol. Recognit. 1999, 12, 3–18. . [DOI] [PubMed] [Google Scholar]
- Pollard T. D. A guide to simple and informative binding assays. Mol. Biol. Cell 2010, 21, 4061–4067. 10.1091/mbc.E10-08-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson D. M.; Ross J. A. Fluorescence polarization/anisotropy in diagnostics and imaging. Chem. Rev. 2010, 110, 2685–2708. 10.1021/cr900267p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P. G.; Brand L. Resonance energy transfer: Methods and applications. Anal. Biochem. 1994, 218, 1–13. 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Wu Q.; Berezin M. Y. Fluorescence anisotropy (polarization): From drug screening to precision medicine. Expert Opin. Drug Discov. 2015, 10, 1145–1161. 10.1517/17460441.2015.1075001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H.; Sumranjit J.; Kang H. J.; Chung S. J. Discovery of coumarin derivatives as fluorescence acceptors for intrinsic fluorescence resonance energy transfer of proteins. Mol. BioSyst. 2014, 10, 30–33. 10.1039/c3mb70323a. [DOI] [PubMed] [Google Scholar]
- Ghisaidoobe A. B. T.; Chung S. J. Intrinsic tryptophan fluorescence in the detection and analysis of proteins: A focus on Förster resonance energy transfer techniques. Int. J. Mol. Sci. 2014, 15, 22518–22538. 10.3390/ijms151222518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Yang X.; Liu L.; Huang Z.; Pu J.; Long G.; Zhang L.; Liu D.; Xu B.; Liao J.; Liao F. Comparison of Förster-resonance-energy-transfer acceptors for tryptophan and tyrosine residues in native proteins as donors. J. Fluoresc. 2013, 23, 147–157. 10.1007/s10895-012-1128-z. [DOI] [PubMed] [Google Scholar]
- Khazanov N. A.; Carlson H. A. Exploring the Composition of Protein–Ligand Binding Sites on a Large Scale. PLoS Comput. Biol. 2013, 9, e1003321 10.1371/journal.pcbi.1003321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan A. A.; Thorn K. S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- Vivian J. T.; Callis P. R. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 2001, 80, 2093–2109. 10.1016/S0006-3495(01)76183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Barkley M. D. Toward understanding tryptophan fluorescence in proteins. Biochemistry 1998, 37, 9976–9982. 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- Doody M. C.; Gotto A. M.; Smith L. C. 5-(Dimethylamino)naphthalene-1-sulfonic acid, a fluorescent probe of the medium chain fatty acid binding site of serum albumin. Biochemistry 1982, 21, 28–33. 10.1021/bi00530a005. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R.; Gryczynski I.; Cheung H. C.; Wang C. K.; Johnson M. L.; Joshi N. Distance distributions in proteins recovered by using frequency-domain fluorometry. Applications to troponin I and its complex with troponin C. Biochemistry 1988, 27, 9149–9160. 10.1021/bi00426a012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustiananda M.; Liggins J. R.; Cummins P. L.; Gready J. E. Conformation of Prion Protein Repeat Peptides Probed by FRET Measurements and Molecular Dynamics Simulations. Biophys. J. 2004, 86, 2467–2483. 10.1016/S0006-3495(04)74303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink D. W.; Koehler W. R. pH Effects on fluorescence of umbelliferone. Anal. Chem. 1970, 42, 990–993. 10.1021/ac60291a034. [DOI] [Google Scholar]
- Sun W.-C.; Gee K. R.; Haugland R. P. Synthesis of novel fluorinated coumarins: Excellent UV-light excitable fluorescent dyes. Bioorg. Med. Chem. Lett. 1998, 8, 3107–3110. 10.1016/S0960-894X(98)00578-2. [DOI] [PubMed] [Google Scholar]
- Chatterjee A.; Maity B.; Seth D. Photophysics of 7-(diethylamino)coumarin-3-carboxylic acid in cationic micelles: Effect of chain length and head group of the surfactants and urea. RSC Adv. 2014, 4, 34026–34036. 10.1039/C4RA02532F. [DOI] [Google Scholar]
- Darbon H.; Angelides K. J. Structural mapping of the voltage-dependent sodium channel. Distance between the tetrodotoxin and Centruroides Suffusus Suffusus II beta-scorpion toxin receptors. J. Biol. Chem. 1984, 259, 6074–6084. [PubMed] [Google Scholar]
- Liao F.; Xie Y.; Yang X.; Deng P.; Chen Y.; Xie G.; Zhu S.; Liu B.; Yuan H.; Liao J.; Zhao Y.; Yu M. Homogeneous noncompetitive assay of protein via Förster-resonance-energy-transfer with tryptophan residue(s) as intrinsic donor(s) and fluorescent ligand as acceptor. Biosens. Bioelectron. 2009, 25, 112–117. 10.1016/j.bios.2009.06.019. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Shen X.; Chen K.; Jiang H.; Liu D. A New Assay Based on Fluorescence Resonance Energy Transfer to Determine the Binding Affinity of Bcl-xL Inhibitors. Biosci., Biotechnol., Biochem. 2008, 72, 1936–1939. 10.1271/bbb.70735. [DOI] [PubMed] [Google Scholar]
- Xie Y.; Maxson T.; Tor Y. Fluorescent Ribonucleoside as a FRET Acceptor for Tryptophan in Native Proteins. J. Am. Chem. Soc. 2010, 132, 11896–11897. 10.1021/ja105244t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang J.; Shi H.; Li D.; Liao Y.; Lian D.; Xiao Y.; Xue H. Ligand binding and structural properties of segments of GABAA receptor alpha 1 subunit overexpressed in Escherichia coli. J. Biol. Chem. 2000, 275, 18818–18823. 10.1074/jbc.M000193200. [DOI] [PubMed] [Google Scholar]
- Cohen J. D.; Thompson S.; Ting A. Y. Structure-guided engineering of a Pacific Blue fluorophore ligase for specific protein imaging in living cells. Biochemistry 2011, 50, 8221–8225. 10.1021/bi201037r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesl T. N.; Chu W. K.; Stockton A. M.; Amashukeli X.; Grunthaner F.; Mathies R. A. Enhanced Amine and Amino Acid Analysis Using Pacific Blue and the Mars Organic Analyzer Microchip Capillary Electrophoresis System. Anal. Chem. 2009, 81, 2537–2544. 10.1021/ac8023334. [DOI] [PubMed] [Google Scholar]
- Kerkovius J. K.; Menard F. A Practical Synthesis of 6,8-Difluoro-7-hydroxycoumarin Derivatives for Fluorescence Applications. Synthesis 2016, 48, 1622–1629. 10.1055/s-0035-1561603. [DOI] [Google Scholar]
- Green N. M. In Advances in Protein Chemistry; Anfinsen C. B., Frederic J. J. T. E., Avidin M. R., Eds.; Academic Press, 1975; Vol. 29, pp 85–133. [DOI] [PubMed] [Google Scholar]
- Le Trong I.; Wang Z.; Hyre D. E.; Lybrand T. P.; Stayton P. S.; Stenkamp R. E. Streptavidin and its biotin complex at atomic resolution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, D67, 813–821. 10.1107/S0907444911027806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M. L.; Kurzban G. P. Noncooperativity of biotin binding to tetrameric streptavidin. Biochemistry 1995, 34, 11750–11756. 10.1021/bi00037a012. [DOI] [PubMed] [Google Scholar]
- Chilkoti A.; Tan P. H.; Stayton P. S. Site-directed mutagenesis studies of the high-affinity streptavidin-biotin complex: Contributions of tryptophan residues 79, 108, and 120. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 1754–1758. 10.1073/pnas.92.5.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai T.; Kohno M.; Boncompain G.; Sugiyama S.; Saito N.; Fujikake R.; Ueno T.; Komatsu T.; Hanaoka K.; Okabe T.; Urano Y.; Perez F.; Nagano T. Artificial Ligands of Streptavidin (ALiS): Discovery, Characterization, and Application for Reversible Control of Intracellular Protein Transport. J. Am. Chem. Soc. 2015, 137, 10464–10467. 10.1021/jacs.5b05672. [DOI] [PubMed] [Google Scholar]
- Xie Y.; Yang X.; Pu J.; Zhao Y.; Zhang Y.; Xie G.; Zheng J.; Yuan H.; Liao F. Homogeneous competitive assay of ligand affinities based on quenching fluorescence of tyrosine/tryptophan residues in a protein via Forster-resonance-energy-transfer. Spectrochim. Acta, Part A 2010, 77, 869–876. 10.1016/j.saa.2010.08.021. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudelman A.; Marcovici-Mizrahi D.; Nudelman A.; Flint D.; Wittenbach V. Inhibitors of biotin biosynthesis as potential herbicides. Tetrahedron 2004, 60, 1731–1748. 10.1016/j.tet.2003.12.047. [DOI] [Google Scholar]
- Green N. M. Thermodynamics of the binding of biotin and some analogues by avidin. Biochem. J. 1966, 101, 774–780. 10.1042/bj1010774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael M. P.; Rappole C. A.; Kurihara L. K.; Christodoulides J. A.; Qadri S. N.; Byers J. M. Iminobiotin binding induces large fluorescent enhancements in avidin and streptavidin fluorescent conjugates and exhibits diverging pH-dependent binding affinities. J. Fluoresc. 2011, 21, 647–652. 10.1007/s10895-010-0752-8. [DOI] [PubMed] [Google Scholar]
- Melkko S.; Dumelin C. E.; Scheuermann J.; Neri D. On the magnitude of the chelate effect for the recognition of proteins by pharmacophores scaffolded by self-assembling oligonucleotides. Chem. Biol. 2006, 13, 225–231. 10.1016/j.chembiol.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Zhuang Y.-D.; Chiang P.-Y.; Wang C.-W.; Tan K.-T. Environment-sensitive fluorescent turn-on probes targeting hydrophobic ligand-binding domains for selective protein detection. Angew. Chem., Int. Ed. 2013, 52, 8124–8128. 10.1002/anie.201302884. [DOI] [PubMed] [Google Scholar]
- Lee M. J.; Pal K.; Tasaki T.; Roy S.; Jiang Y.; An J. Y.; Banerjee R.; Kwon Y. T. Synthetic heterovalent inhibitors targeting recognition E3 components of the N-end rule pathway. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 100–105. 10.1073/pnas.0708465105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Zhao X.; Chen J.; Chen J.; Kuznetsova L.; Wong S. S.; Ojima I. Mechanism-Based Tumor-Targeting Drug Delivery System. Validation of Efficient Vitamin Receptor-Mediated Endocytosis and Drug Release. Bioconjug. Chem. 2010, 21, 979–987. 10.1021/bc9005656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A. T. R.; Winfield S. A.; Miller J. N. Relative Fluorescence Quantum Yields Using a Computer-Controlled Luminescence Spectrometer. Analyst 1983, 108, 1067–1071. 10.1039/an9830801067. [DOI] [Google Scholar]
- Jones G.; Jackson W. R.; Choi C. Y.; Bergmark W. R. Solvent effects on emission yield and lifetime for coumarin laser dyes. Requirements for a rotatory decay mechanism. J. Phys. Chem. 1985, 89, 294–300. 10.1021/j100248a024. [DOI] [Google Scholar]
- Hink M. A.; Visser N. V.; Borst J. W.; van Hoek A.; Visser A. J. W. G. Practical Use of Corrected Fluorescence Excitation and Emission Spectra of Fluorescent Proteins in Förster Resonance Energy Transfer (FRET) Studies. J. Fluoresc. 2003, 13, 185–188. 10.1023/A:1022947411788. [DOI] [Google Scholar]
- Visser A. J. W. G.; Vysotski E. S.; Lee J.. Critical transfer distance determination between FRET pairs. 2011, http://www.photobiology.info/Experiments/Biolum-Expt.html. [Google Scholar]
- Jing M.; Bowser M. T. Methods for measuring aptamer-protein equilibria: A review. Anal. Chim. Acta 2011, 686, 9–18. 10.1016/j.aca.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.