

Abstract
Multiple sclerosis (MS) is a chronic immune-mediated inflammatory-demyelinating disorder of the central nervous system, with a strong neurodegenerative component. The question whether neurodegeneration in MS is independent or related to neuroinflammation has been long debated, but not yet fully clarified. Furthermore, little is still known on how neuroinflammation and neurodegeneration in MS are related to potential regenerative processes. In this perspective, we briefly discuss main clinical, pathological and experimental evidence on the relationship between neuroinflammation and neurodegeneration in MS, and on their connection with regeneration. We discuss that these processes in MS might represent intercorrelated manifestations of the immune response, especially of the innate immunity.
Keywords: multiple sclerosis, neuroinflammation, neurodegeneration, regeneration, immune response, innate immunity
Introduction
Multiple sclerosis (MS) is a chronic immune-mediated disorder of the central nervous system (CNS) characterized by neuroinflammation and neurodegeneration with demyelination and neuroaxonal loss. The question whether neurodegeneration in MS is a process independent or related to neuroinflammation has been long debated, and it is still not fully resolved.
In this perspective, we briefly present main clinical, pathological and experimental evidence on the relationship between neuroinflammation and neurodegeneration in MS, and on their connection with regeneration. We discuss data that suggest that these processes in MS might represent intercorrelated manifestations of the immune response, and focus on main immune processes in the disease, especially on those of the innate immunity. A better understanding of how neuroinflammation, neurodegeneration, and regeneration are correlated in MS would be extremely valuable in developing more effective and targeted therapeutic strategies.
Neurodegeneration and Neuroinflammation
Evidence from clinico-radiological studies
Most clinical findings support the existence of a strong relationship between inflammation and neurodegeneration in MS (Hutchinson, 2015). For example, early frequent and severe inflammatory exacerbations, as well as delay in starting anti-inflammatory treatments, are known to accelerate either time to disease progression, achievement of severe disability or even time to death.
Conversely, the observation that MS may arise directly with a progressive course or that disease-modifying therapies, which influence prevalently the relapse rate, have little if none efficacy on disability progression has been interpreted as evidence that neuroinflammation and neurodegeneration in MS might be, at least partially, independent from each other (Louapre and Lubetzki, 2015). The relapse rate, however, might not be fully indicative of the level of inflammation in the disease since clinically silent inflammation, as detectable by the presence of gadolinium enhancing lesions, could frequently occur.
Advanced neuroimaging techniques including 1H magnetic resonance (MR) spectroscopy, positron-emission tomography (PET) and quantitative 7 Tesla MRI support the presence of an early cortical damage independent of inflammatory white matter (WM) lesions (Louapre and Lubetzki, 2015). The in vivo study of the role of gray matter (GM) inflammation in neurodegeneration has been hampered so far by the low sensitivity of conventional imaging methods, especially in the cortex. Longitudinal studies are also lacking, making it difficult to establish a causative link between neuroinflammation and neurodegeneration. Recent data using PET imaging demonstrated that GM demyelinating and degenerative pathology is strongly associated with the presence of activated microglia, mostly in the absence of blood-brain barrier (BBB) disruption, and independently of underlying WM inflammation (Herranz et al, 2016).
The presence of local GM inflammation could explain why male MS patients, which show usually a lower inflammatory activity in WM than female patients, tend to exhibit greater GM atrophy with major cognitive impairment and a more frequent progressive MS course (Louapre and Lubetzki, 2015). Gender-MS related differences, however, might be also related to differences in sex hormones and their interactions with the immune system.
Genome-wild association studies in MS demonstrate a prevalent role of the genes involved in inflammatory processes, especially in T-cell mediated immunity (Hutchinson, 2015). A polymorphisms in genes influencing in vivo brain glutamate levels has been found to correlate with neurodegenerative indicators such as decreased N-acetylaspartate level and increased rate of atrophy (Louapre and Lubetzki, 2015). The exact cascade of events, as well as triggering factors, leading to expression of these genes are, however, still unclear.
Evidence from neuropathological studies
Neuropathological studies show that inflammation is present in all MS stages and that putative neurodegenerative lesions are likely driven by inflammation (reviewed in Lassmann, 2007).
In early MS lesions, initial axonal injury, even in absence of demyelination, is associated with both microglia and complement activation (Lassmann, 2007). In later stages, lesions with active demyelination are characterized by massive T-cell infiltration along with microglia and macrophages activation.
Ex vivo pathological-imaging correlations using magnetization-transfer ratio and diffusion-tensor imaging in MS demonstrated the presence of subtle abnormalities in normal-appearing WM (NAWM), close to lesions, which strongly correlate with diffuse microglia activation along with impaired axonal and myelin integrity (Moll et al., 2011).
In primary and secondary progressive MS axonal degeneration and myelin destruction in the NAWM are associated with perivenous and parenchymal T-lymphocytes infiltration and massive microglia activation. Likewise, activated microglia and sparsely infiltrated T- and B-cells are present in cortical active plaques, as well as inflammatory infiltrates of T-, B-lymphocytes and plasma cells are localized in adjacent meninges (Lassmann, 2007). Interestingly, inflammation in progressive MS occurs in the form of compartmentalized immune reaction behind a closed/repaired BBB with a formation of lymph-follicle like structures in the meninges and perivascular spaces (Lassmann, 2007). This determines local cytokines production, chemokines expression and intrathecal immunoglobulin synthesis leading to rapid disease progression and profound brain damage. T-lymphocytes are diffusely infiltrated in the brain parenchyma and concentrated in perivascular cuffs in active lesions whereas B-lymphocytes and plasma cells are accumulated in the connective tissue of perivascular spaces and meninges (Frischer et al., 2009). Plasma cells persist even after extinguishing of T-cell and B-cell inflammation.
The presence of a compartmentalized inflammation provides a plausible explanation for the incongruity between greater brain atrophy and fewer radiological inflammatory lesions in progressive MS supporting a close link between neurodegeneration and neuroinflammation also in this form of MS.
Chronic Immune Activation in MS and other Neurodegenerative Diseases
The CNS “immune privilege” status is determined by different elements including BBB integrity that, together with neurons, glia and the extracellular matrix, form the neurovascular unit regulating immune responses in the CNS (Amor et al., 2014). Cell-contact dependent signals due to neuronal cell adhesion molecule expressed by neurons and glia determine the inhibition of both microglia activation and maturation of antigen-presenting cells. Additionally, neuroimmunoregulatory mediators, including chemokines, neuropeptides, neurotransmitters and neurotrophins produced by neurons, inhibit microglia activation and limit the survival of activated lymphocytes. The impairment of these cell-contact dependent and neuroimmunoregulatory signals due to neuronal damage and loss impoverishes CNS homeostatic protective environment and increases neuroinflammation (Amor et al., 2014). This occurs physiologically in aging due to neuronal loss, genetic mutations, oxidative or metabolic stress with endoplasmic reticulum and mitochondrial dysfunction.
Neurodegeneration seems to be closely associated with neuroinflammation not only in MS, but also in other neurodegenerative disorders including Alzheimer's and Parkinson's diseases, amyotrophic lateral sclerosis, prion disorders, and even in neuropsychiatric and genetic conditions (reviewed in Amor et al., 2014). A common denominator in all these diseases is a chronic activation of the local innate immunity, and in particular of microglia. Microglia are involved in overall brain surveillance and maintenance, including defense against CNS infections and cleaning of cell debris and damaged proteins after stress or tissue damage. Although this represents a primary protective role of the innate immunity, its excessive or prolonged activation may cause tissue damage.
Furthermore, the type of local innate immune response largely determines the extent and nature of any adaptive immune response, and vice versa systemic immune activation influences the local innate immunity. Peripheral infections and/or antecedent insults determine the so-called “primed” environment that increases CNS susceptibility to injury. Experimental studies showed that peripheral inflammation is associated with disease exacerbations in experimental models of either MS, stroke or other neurodegenerative diseases (Amor et al., 2014).
One of the essential functions of innate immunity is to provide the informational input to adaptive immunity, in particular to naïve CD4+ lymphocytes, except for local CNS innate cells that cannot directly initiate adaptive immunity (reviewed in Ransohoff and Brown, 2012). After activation, primed T cells act on resident and recruited innate cells operating together with complement to clear CNS infection or injury. Resident microglia and astrocytes express, in turn, cytokines and chemokines promoting the recruitment of circulating lymphocytes and myeloid cells from the periphery. Neuroinflammation manifests not only with activation of local microglia, astrocytes, oligodendrocytes but also with a recruitment of peripheral innate immune cells such as natural killer, natural killer T cells, mast cells, granulocytes and γδ-T cells. At the same time, MHC class II antigens become up-regulated on microglia and on perivascular microglia/pericytes, facilitating antigen presentation to T cells. The relevance of T cells in neurodegenerative diseases is supported by both their subsets alterations in the periphery and their presence in the CNS.
Chronic immune activation has also been shown to be strongly influenced by the aberrant activity of inflammosomes, multiprotein complexes containing multiple copies of a receptor or sensor of pathogen-derived or damage-derived molecular patterns, which assemble in response to danger signals and regulate the secretion of biologically active IL-1β and IL-18. The influence of inflammasomes on the interrelation between mast cells and myelin-specific T cells in the meninges has been demonstrated recently in an experimental rodent model of early MS (Russi et al, 2016). The crosstalk in the meninges between mast cells and antigen-specific T cells could be critical for immune-mediated disease development.
Regeneration as Generative Result of both Neuroinflammation and Neurodegeneration
The same neuroimmunoregulatory mediators and cells that are involved in neuroinflammation and neurodegeneration, have been shown to provide for CNS repair, growth and development (Amor et al., 2014).
For example, astrocytes induce apoptosis of infiltrating T cells by Fas–FasL interactions, produce interleukin-1 and stimulate influx of regulatory T cells by producing interleukin-27. Moreover, they are the major source of nerve growth factor and glial cell line-derived neurotrophic factor in the CNS. These factors are secreted also by T cells and are required to reduce neurodegeneration and stimulate neuronal regeneration (Amor et al., 2014).
Similarly, different macrophage phenotypes might play opposite roles in both neuroinflammation and regeneration. Acute CNS injury models in vitro have shown that some microglia/macrophages phenotypes (traditionally known as “classically activated M1”) release pro-inflammatory mediators, whereas shift to different phenotypes (traditionally known as “alternatively activated M2”) might be accompanied by phagocytosis and release protective and trophic factors (reviewed in Hu et al., 2015). The latter microglia/macrophages phenotypes have specific functions in CNS repair including neurogenesis, axonal remodeling, angiogenesis, oligodendrogenesis and remyelination. In vivo the M1/M2 dichotomy is more complex than in vitro, including several overlapping functional phenotypes depending on the induction stimuli.
Macrophages/microglia depletion determines accumulation of myelin debris and impairs remyelination (reviewed in Rawji et al., 2016). Remyelination occurs due to oligodendrocyte-precursors differentiation into oligodendrocytes synthesizing new myelin. Depletion of pro-inflammatory macrophages/microglia phenotypes at early post-demyelination time in rat reduces proliferation of oligodendrocyte precursors, whereas a delayed reduction in regulatory macrophages/microglia phenotypes impairs differentiation of oligodendrocyte precursors. Microglia/macrophages release important growth factors and cytochines to stimulate axonal regeneration and oligodendrocyte-precursors maturation. Interestingly, remyelination is impaired after the depletion of tumor necrosis factor-α or interleukin-1β acting directly on oligodendrocyte precursors or indirectly through astrocytes. Additionally, aging macrophages/microglia become dysregulated and this leads to defected cytokine secretion by macrophages and to enhanced pro-inflammatory cytokine production by microglia (Rawji et al., 2016).
Thus, the spontaneous self-repair of damaged CNS tissue is inadequate in neurodegenerative diseases although native neural stem cells (NSCs) persist in the adult mammalian brain (Gonzalez et al, 2016). This is most likely because NSCs are few, restricted to discrete locations and surrounded by a microenvironment not supporting the neuronal differentiation. Therapeutic use of NSC transplantation in neurodegenerative diseases may require the particular conditions with widely distributed gene products and/or replaced cells.
Conclusions
There is ample evidence that neuroinflammation, neurodegeneration and regeneration represent intercorrelated manifestations of the immune response in the healthy brain and in many neurological conditions including MS.
In acute and limited CNS injury, neuroinflammation could circumscribe and contrast neurodegeneration and stimulate regeneration. Conversely, excessive or chronic neuroinflammation leads to increased neurodegeneration that in turn impairs homeostatic protective environment. This further amplifies detrimental neuroinflammation and impoverishes regeneration (Figure 1).
Figure 1.
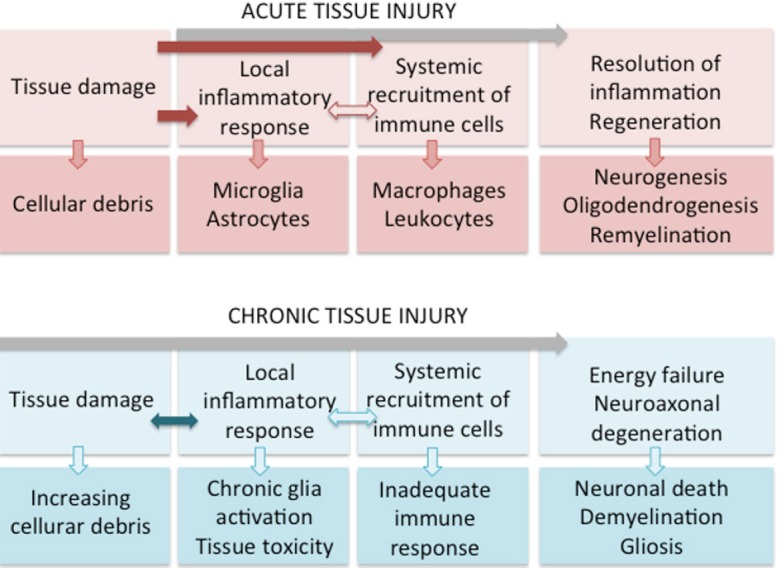
In acute and circumscribed CNS injury, cooperative and mutually potentiating action of local and systemic immune response leads to resolution of neuroinflammation, limitation of neurodegeneration and stimulation of regeneration.
In chronic CNS injury, inadequate local and systemic immune response leads to chronic neuroinflammation and consequent increased neurodegeneration. This further amplifies detrimental neuroinflammation due to both impaired homeostatic protective environment and energy failure interfering with regeneration processes.
In MS, regeneration processes are insufficient compared to the extent of neuroinflammation and neurodegeneration, which co-exist in different degree depending on many factors including tissue localization (GM or WM), lesion formation stage, disease phase and immune system age. Current efforts to combat effectively neurodegeneration and to promote regeneration-remyelination in MS could be targeted to shift the neuroinflammation to its protective-trophic function rather than suppress it altogether.
Footnotes
Conflicts of interest: None declared.
References
- Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D, van Noort JM. Inflammation in neurodegenerative diseases--an update. Immunology. 2014;142:151–166. doi: 10.1111/imm.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Lassmann H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132:1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez R, Hamblin MH, Lee JP. Neural stem cell transplantation and CNS diseases. CNS Neurol Disord Drug Targets. 2016 doi: 10.2174/1871527315666160815164247. doi:10.2174/1871527315666160815164247. [DOI] [PubMed] [Google Scholar]
- Herranz E, Giannì C, Louapre C, Treaba CA, Govindarajan ST, Ouellette R, Loggia ML, Sloane JA, Madigan N, Izquierdo-Garcia D, Ward N, Mangeat G, Granberg T, Klawiter EC, Catana C, Hooker JM, Taylor N, Ionete C, Kinkel RP, Mainero C. The neuroinflammatory component of gray matter pathology in multiple sclerosis. Ann Neurol. 2016 doi: 10.1002/ana.24791. doi:10.1002/ana.24791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, Chen J. Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol. 2015;11:56–64. doi: 10.1038/nrneurol.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson M. Neurodegeneration in multiple sclerosis is a process separate from inflammation: No. Mult Scler. 2015;21:1628–1631. doi: 10.1177/1352458515612244. [DOI] [PubMed] [Google Scholar]
- Lassmann H. Multiple sclerosis: is there neurodegeneration independent from inflammation? J Neurol Sci. 2007;259:3–6. doi: 10.1016/j.jns.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Louapre C, Lubetzki C. Neurodegeneration in multiple sclerosis is a process separate from inflammation: Yes. Mult Scler. 2015;21:1626–1628. doi: 10.1177/1352458515587598. [DOI] [PubMed] [Google Scholar]
- Moll NM, Rietsch AM, Thomas S, Ransohoff AJ, Lee JC, Fox R, Chang A, Ransohoff RM, Fisher E. Multiple sclerosis normal-appearing white matter: pathology-imaging correlations. Ann Neurol. 2011;70:764–773. doi: 10.1002/ana.22521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122:1164–1171. doi: 10.1172/JCI58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawji KS, Mishra MK, Yong VW. Regenerative capacity of macrophages for remyelination. Front Cell Dev Biol. 2016;4:47. doi: 10.3389/fcell.2016.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russi AE, Walker-Caulfield ME, Brown MA. Mast cell inflammasome activity in the meninges regulates EAE disease severity. Clin Immunol. 2016 doi: 10.1016/j.clim.2016.04.009. doi: 10.1016/j.clim.2016.04.009. [DOI] [PubMed] [Google Scholar]