

Peripheral nerve injury causes a partial or total loss of motor and sensory functions as a result of axonal disruption and subsequent axonal disintegration as well as denervation distal from the point of injury. Although peripheral nerves are, in contrast to the central nervous system, able to regenerate and reinnervate, functionality is not always restored completely due to insufficient reinnervation or remyelination, and injury may result in sequelae such as neuropathic pain. The degenerative processes following peripheral nerve injury are generally referred to as Wallerian degeneration (Gaudet et al., 2011).
In rodents, the initial response to injury occurs within 24 hours and is characterized by Schwann cells detaching from their associated axons accompanied by degeneration of the insulating myelin sheaths and a subsequent breakdown of axonal integrity; Schwann cells rapidly dedifferentiate and start proliferating. These dedifferentiated Schwann cells and resident macrophages are among the first cells to recognize the injury and secrete pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and chemokines, e.g., monocyte chemoattractant protein 1 (MCP-1), both of which propagate the recruitment of hematogenous monocytes and macrophages, respectively (Meyer zu Hörste et al., 2007; Gaudet et al., 2011). This well-orchestrated cellular response to injury allows for the timely clearace of cellular and myelin debris in order to enable axon regeneration from the largely unaffected proximal stump. One crucial factor that is known to determine the speed of axonal regrowth is cyclic adenosine monophosphate (cAMP) (Hannila and Filbin, 2008).
We have recently investigated whether modulation of lysophospholipid signaling using the immunomodulatory drug fingolimod (also named FTY720) may propagate nerve regeneration in a mechanical injury model of the peripheral nervous system (Szepanowski et al., 2016). Fingolimod is a first-in-class sphingosine-1-phosphate (S1P) receptor agonist that is thought to exert a “functionally antagonistic” effect on the S1P1 receptor subtype by facilitating its internalization. It thereby prevents the egress of S1P1 expressing activated lymphocytes from lymph nodes (Brinkmann et al., 2010). Additionally, fingolimod has been reported to act as an inhibitor of the lysophospholipase autotaxin, thereby reducing lysophosphatidic acid (LPA) biosynthesis (van Meeteren et al., 2008). Both S1P and LPA are bioactive lysophospholipids that address specific G-protein coupled receptors. S1P and LPA receptors have been recognized to be widely expressed in the nervous system and have been associated with a variety of physiological and pathophysiological processes. Not surprisingly, there have been numerous studies indicating direct effects of fingolimod on cells of the central nervous system, including neuroprotective and remyelinating properties (Groves et al., 2013). To evaluate the regenerative potential of fingolimod and to distinguish its immunosuppressive from potential direct effects on the peripheral nervous system in vivo, sciatic nerve crush injury was performed in wildtype as well as in immunodeficient mice. It was demonstrated that fingolimod treatment improved nerve regeneration by electrophysiological and clinical measures not only in immunocompetent mice, but also independent of its effect on T-lymphocyte sequestration. Interestingly, in combined B- and T-lymphocyte deficient mice, fingolimod treatment failed to cause any improvements by these measures. In order to identify the molecular mechanism underlying this discrepant response to fingolimod treatment, axonal cAMP levels were studied. A significant elevation of axonal cAMP in wildtype and in T-lymphocyte deficient mice was recognizable, whereas combined T- and B-lymphocyte deficient mice displayed overall reduced cAMP. Consistent with these findings, an impairment of regeneration after nerve injury has previously been described for B-lymphocyte deficient mice (Vargas et al., 2010). Vargas and colleagues demonstrated that a timely onset of axonal regrowth is dependent on B lymphocytes producing autoantibodies against myelin debris; the absence of B-lymphocytes resulted in delayed axonal recovery. Myelin debris contains inhibitors of axonal regeneration which are thought to at least partly act through the Nogo-p75 neurotrophin receptor complex, inhibiting axonal cAMP formation in a Gi dependent manner (Hannila and Filbin, 2008). Our finding of significantly reduced cAMP in combined immunodeficient mice, but not in exclusively T-lymphocyte deficient mice, may accentuate the importance of B-lymphocytes for peripheral nerve regeneration, and may provide a plausible explanation for the lack of effectiveness of fingolimod in combined immunodeficient mice.
Another beneficial effect of fingolimod treatment comprised an improvement of myelin thickness in regenerating axons, which surprisingly also occurred in B- and T-lymphocyte deficient mice. Thus, we considered this finding unlikely to be cAMP related, leading us to investigate LPA, which has been linked to demyelination in numerous nerve injury models (Yung et al., 2015). Quantification of LPA from sciatic nerve homogenates via liquid chromatography coupled to tandem mass spectrometry revealed that fingolimod reduces LPA shortly after injury. Although 24 hours post-injury no significant difference in LPA levels between control and fingolimod treated mice was evident anymore, a transient attenuation of LPA signaling may be sufficient to ameliorate tissue damage outcomes and demyelination (Crack et al., 2014). Since we hypothesized the reduction of LPA to be a consequence of fingolimod mediated autotaxin inhibition, mice were treated with the specific autotaxin inhibitor PF-8380 to differentiate between S1P and LPA mediated effects on myelination. The effect of PF-8380 on myelination resembled that of fingolimod, but did not affect axon regeneration, confirming a supportive effect of autotaxin inhibition on myelin integrity.
A previous study investigating the regenerative potential of fingolimod in the peripheral nervous system in vitro proposed a different mode of action (Heinen et al., 2015). Heinen and colleagues suggest that fingolimod may not support axon outgrowth or myelination via direct actions on neurons or Schwann cells, but may induce the secretion of neurotrophic factors from Schwann cells which in turn promote axonal sprouting. The authors report that the cAMP inducible expression of a positive regulator of myelination, Krox-20, was counteracted by fingolimod in forskolin treated Schwann cells. While S1P1 receptor signaling is known to reduce intracellular cAMP levels via inhibition of adenylate cyclase in a Gi dependent manner, the antagonistic effect of fingolimod on S1P1 would be expected to increase cAMP production. Interestingly, it was shown for cell culture experiments involving S1P1 receptor expressing CHO cells that short-term incubation with fingolimod causes persistent S1P signaling from intracellular compartments, leading to sustained inhibition of cAMP formation (Mullershausen et al., 2009). In this context, it has been suggested that the S1P1-Gi-adenylate cyclase system might be internalized as a ternary complex, thereby suppressing enzymatic activity of adenylate cyclase as long as the ligand fingolimod is bound (Jalink and Moleenaar, 2010). In contrast to inhibition of cAMP formation in vitro, the increase in axonal cAMP observed in our recent study may be the result of a long-term treatment regime with fingolimod for more than two weeks, where constantly high concentrations of fingolimod may potentially affect the dynamics of receptor internalization, leading to a spatial segregation of S1P1 and adenylate cyclase by “trapping” de novo synthesized S1P1 in intracellular compartments and allowing for an increased activation of membrane-associated adenylate cyclase during the course of axonal regeneration (Figure 1).
Figure 1.
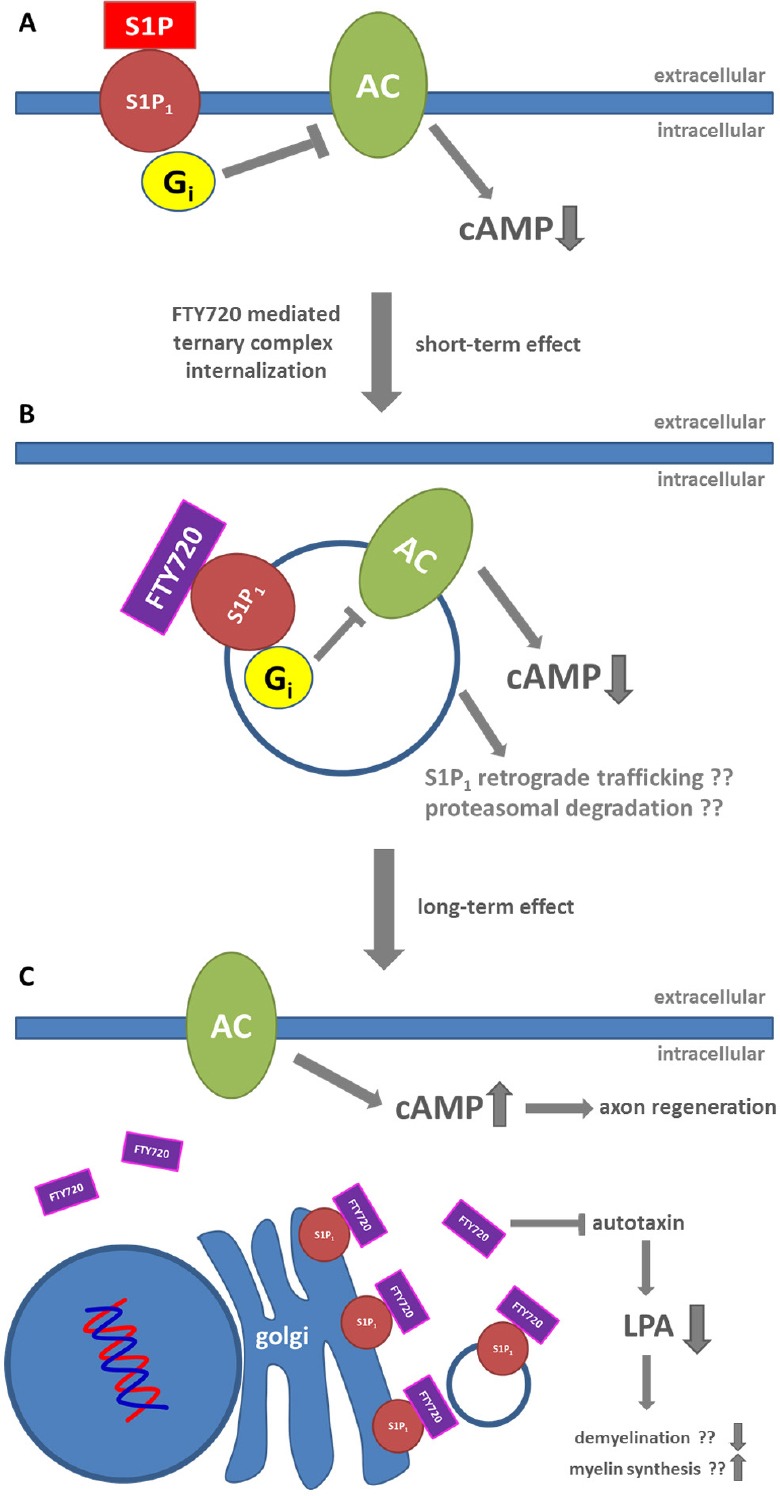
Possible mode of action for fingolimod (FTY720) mediated improvement of nerve regeneration.
In the presence of the natural ligand sphingosine-1-phosphate (S1P), activation of the S1P1 receptor leads to inhibition of adenylate cyclase (AC) through Gi. (A) Binding of phosphorylated FTY720 to S1P1 may lead to internalization of the S1P1-Gi-adenylate cyclase system as ternary complex causing sustained inhibition of cyclic adenosine monophosphate (cAMP) formation. The ternary complex may directly undergo proteasomal degradation or retrograde transport to the Golgi. (B) In the presence of constantly high concentrations of FTY720, de novo synthesized S1P1 may be 'trapped' in intracellular compartments, possibly the Golgi, preventing re-localization of S1P1 to the plasma membrane and thus formation of the presumptive S1P1-Gi-adenylate cyclase complex. This allows for increased generation of cAMP due to a reduction of adenylate cyclase inhibition. Additionally, FTY720 may attenuate demyelination via inhibition of autotaxin catalyzed LPA formation (C).
As such, potentially beneficial effects of fingolimod may be based on an early stimulation of axonal sprouting via neurotrophic factors released by Schwann cells as well as an attenuation of LPA signaling. At later stages, fingolimod may support axon outgrowth via an abrogation of S1P signaling, allowing for an increased cAMP response in the regenerating nerve.
Certainly, there is a need for future studies to further elucidate the molecular mechanisms underlying the presumptive neuroregenerative effects of fingolimod. The current development of novel S1P receptor agonists with greater specificity to S1P receptor subtypes may dramatically expand our understanding of the role of lysophospholipid signaling in physiological and pathophysiological conditions of the nervous system. However, given the emerging body of evidence so far, modulation of lysophospholipid signaling appears not only to be a highly relevant therapeutic target for immunomodulation, but could possibly also represent a promising target for inducing clinically meaningful improvements after primary and secondary nerve damage.
References
- Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9:883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- Crack PJ, Zhang M, Morganti-Kossmann MC, Morris AJ, Wojciak JM, Fleming JK, Karve I, Wright D, Sashindranath M, Goldshmit Y, Conquest A, Daglas M, Johnston LA, Medcalf RL, Sabbadini RA, Pébay A. Anti-lysophosphatidic acid antibodies improve traumatic brain injury outcomes. J Neuroinflammation. 2014;11:37. doi: 10.1186/1742-2094-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet AD, Popovich PG, Ramer MS. Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J Neuroinflammation. 2011;8:110. doi: 10.1186/1742-2094-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves A, Kihara Y, Chun J. Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J Neurol Sci. 2013;328:9–18. doi: 10.1016/j.jns.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannila SS, Filbin MT. The role of cyclic AMP signaling in promoting axonal regeneration after spinal cord injury. Exp Neurol. 2008;209:321–332. doi: 10.1016/j.expneurol.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen A, Beyer F, Tzekova N, Hartung HP, Küry P. Fingolimod induces the transition to a nerve regeneration promoting Schwann cell phenotype. Exp Neurol. 2015;271:25–35. doi: 10.1016/j.expneurol.2015.05.002. [DOI] [PubMed] [Google Scholar]
- Jalink K, Moolenaar WH. G protein-coupled receptors: the inside story. Bioessays. 2010;32:13–16. doi: 10.1002/bies.200900153. [DOI] [PubMed] [Google Scholar]
- Meyer zu Hörste G, Hartung HP, Kieseier BC. From bench to bedside--experimental rationale for immune-specific therapies in the inflamed peripheral nerve. Nat Clin Pract Neurol. 2007;3:198–211. doi: 10.1038/ncpneuro0452. [DOI] [PubMed] [Google Scholar]
- Mullershausen F, Zecri F, Cetin C, Billich A, Guerini D, Seuwen K. Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat Chem Biol. 2009;5:428–434. doi: 10.1038/nchembio.173. [DOI] [PubMed] [Google Scholar]
- Szepanowski F, Derksen A, Steiner I, Meyer Zu Hörste G, Daldrup T, Hartung HP, Kieseier BC. Fingolimod promotes peripheral nerve regeneration via modulation of lysophospholipid signaling. J Neuroinflammation. 2016;13:143. doi: 10.1186/s12974-016-0612-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meeteren LA, Brinkmann V, Saulnier-Blache JS, Lynch KR, Moolenaar WH. Anticancer activity of FTY720: phosphorylated FTY720 inhibits autotaxin, a metastasis-enhancing and angiogenic lysophospholipase D. Cancer Lett. 2008;266:203–208. doi: 10.1016/j.canlet.2008.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas ME, Watanabe J, Singh SJ, Robinson WH, Barres BA. Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc Natl Acad Sci U S A. 2010;107:11993–11998. doi: 10.1073/pnas.1001948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung YC, Stoddard NC, Mirendil H, Chun J. Lysophosphatidic acid signaling in the nervous system. Neuron. 2015;85:669–682. doi: 10.1016/j.neuron.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]