

Abstract
Whether individuals with insulin resistance (IR) but without criteria for diabetes exhibit reduced mitochondrial oxidative capacity is unclear; addressing this question could guide research for new therapeutics. We investigated 248 participants without diabetes from the Baltimore Longitudinal Study of Aging (BLSA) to determine whether impaired mitochondrial capacity is associated with prediabetes, IR, and duration and severity of hyperglycemia exposure. Mitochondrial capacity was assessed as the postexercise phosphocreatine recovery time constant (τPCr) by 31P-magnetic resonance spectroscopy, with higher τPCr values reflecting reduced capacity. Prediabetes was defined using the American Diabetes Association criteria from fasting and 2-h glucose measurements. IR and sensitivity were calculated using HOMA-IR and Matsuda indices. The duration and severity of hyperglycemia exposure were estimated as the number of years from prediabetes onset and the average oral glucose tolerance test (OGTT) 2-h glucose measurement over previous BLSA visits. Covariates included age, sex, body composition, physical activity, and other confounders. Higher likelihood of prediabetes, higher HOMA-IR, and lower Matsuda index were associated with longer τPCr. Among 205 participants with previous OGTT data, greater severity and longer duration of hyperglycemia were independently associated with longer τPC. In conclusion, in individuals without diabetes a more impaired mitochondrial capacity is associated with greater IR and a higher likelihood of prediabetes.
Introduction
Muscle biopsy specimens showed that individuals with type 2 diabetes have lower muscle mitochondrial content and oxidative capacity, mitochondrial proteins, and expression of oxidative genes than control subjects (1), but findings are inconsistent across studies (2,3), especially when accounting for obesity. Not all of the studies using 31P-magnetic resonance spectroscopy (31P-MRS) confirmed that individuals with type 2 diabetes have impaired mitochondrial function (4). Moreover, similar mitochondrial capacity was found in individuals with early-stage type 2 diabetes and prediabetes compared with normoglycemic control subjects (5,6). Thus, whether insulin resistance (IR) and prediabetes are linked to impaired mitochondrial capacity remains unclear. Inconsistent findings among studies may be due to small sample size, use of different diagnostic criteria, and inadequate adjustment for confounders (Supplementary Table 1).
Using data on 31P-MRS collected in 248 participants without diabetes in the Baltimore Longitudinal Study of Aging (BLSA), we tested whether impaired mitochondrial capacity is associated with prediabetes, IR, and the duration and severity of hyperglycemia exposure.
Research Design and Methods
Study Design and Setting
BLSA is a study of human aging established in 1958 by the National Institute on Aging Intramural Research Program (7). BLSA continuously enrolls healthy volunteers ≥20 years of age, followed at intervals of 1–4 years, with more frequent follow-up visits for older persons. The study protocol is reviewed by the National Institute of Environmental Health Sciences Institutional Review Board, and participants consent to participate after receiving an extensive description of the study.
Participants
We studied 248 BLSA participants without diabetes, who were 51–97 years old (mean age 73.5 ± 9.6 years, 46.4% men) with available data from 31P-MRS, oral glucose tolerance test (OGTT) results, and confounders. Information on race (Caucasian vs other), smoking (current smoker, former smoker, or no smoking), and physical activity (questionnaire on different activities transformed in metabolic equivalents of resting oxygen consumption) (8) were self-reported. Body weight in kilograms, and height and waist circumference in centimeters were assessed. BMI was calculated as weight/height (kg/m2). Total body DEXA was performed using the Prodigy Scanner (General Electric) to obtain measures of total body fat mass (TBFM) and trunk fat mass (FM) in kilograms. TBFM was also normalized by body weight and expressed as the percentage of body fatness (i.e., TBFM divided by body weight and multiplied by 100). Serum interleukin-6 (IL-6) and CRP were measured with ELISA (R&D Systems, Minneapolis, MN; and Alpco, Salem, NH, respectively). Plasma leptin level was measured using ELISA (LINCO Research, St. Charles, MO), whereas plasma adiponectin level was measured by radioimmunoassay (LINCO Research).
31P-MRS–Measured Mitochondrial Capacity
31P-MRS measurements of phosphorus-containing metabolites were obtained from the vastus lateralis muscle of the left thigh using a 3-T Achieva MRI scanner (Philips, Best, the Netherlands) and a 10-cm 31P-tuned, flat surface coil (PulseTeq, Surrey, U.K.). Participants performed rapid and intense ballistic knee extensions (9) to maximally recruit quadriceps and rapidly deplete phosphocreatine (PCr) with minimal acidification, which were practiced at least 15 min before starting the actual examination. Exercise duration was controlled to achieve a 33–67% reduction in PCr peak height and never exceeded 42 s, resulting in a postexercise recovery period of 5.8–6.3 min, which was sufficient for reliable fitting of the recovery coefficient. MRI signals were acquired before, during, and after this exercise protocol, with a repetition time of 1.5 s using a pulse-acquire sequence adiabatic radiofrequency excitation over a period of 7.5 min, starting 1 min before exercise to define baseline values, with a time resolution of 6 s (10). Data were analyzed by jMRUI (version 5.0), and metabolites were quantified using a nonlinear least squares algorithm (AMARES), based on the integrated area of the spectral line (11,12). The postexercise PCr recovery time constant (τPCr) was calculated by fitting the time-dependent recovery of PCr to the monoexponential recovery function, as follows:
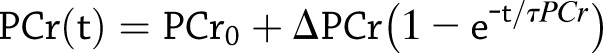 |
where PCr0 represents the amount of PCr immediately after in-magnet exercise, and ΔPCr is the difference in PCr between baseline and postexercise conditions.
Muscle pH was monitored during the protocol based on the chemical shift difference between inorganic phosphate (Pi) and PCr. Subjects showing pH values of <6.8 were excluded from further analysis (13).
Prediabetes Status and IR
Participants were classified as being “normal,” having prediabetes, or having diabetes, according to the American Diabetes Association criteria using fasting plasma glucose (FPG) and/or 2-h post-OGTT glucose (2hG) levels (14). For OGTT, fasting participants consumed 75 g of an oral solution of glucose, and blood samples were collected after fasting and then every 20 min for 2 h. Type 2 diabetes was defined as history of type 2 diabetes and taking hypoglycemic medications; or FPG ≥126 mg/dL and 2hG ≥200 mg/dL at the same visit; or FPG ≥126 mg/dL or 2hG ≥200 mg/dL at consecutive visits. Participants with diabetes were excluded from the current study. Prediabetes was defined as follows: FPG ≥100 mg/dL but <126 mg/dL or 2hG ≥ 140 mg/dL but <200 mg/dL at the same visit; or FPG ≥100 mg/dL but <126 mg/dL or 2hG ≥140 mg/dL but <200 mg/dL during two consecutive visits. IR was estimated by HOMA-IR, calculated as FPG (mg/dL) × fasting insulin (mU/L) divided by 405 (15). Insulin sensitivity was assessed by the Matsuda index (10,000/square root of [(fasting glucose × fasting insulin) × (mean glucose × mean insulin during OGTT)]) (16) and the Reduced Matsuda Index (10,000/square root of [(fasting glucose × fasting insulin) × (glucose at 120 min × insulin at 120 min)]) (17). Duration of hyperglycemia exposure was assessed as the number of years from the onset of prediabetes. Severity of hyperglycemia exposure was estimated for participants with longitudinal data (N = 205) as the average OGTT 2-h glucose level from the time of the first OGTT to mitochondrial capacity assessment.
Statistical Analysis
Variables were summarized as the mean ± SD, median (interquartile range), or number (percentage). The relationships between τPCr (in seconds) and prediabetes, HOMA index, Matsuda index, and reduced Matsuda index were explored using Spearman correlations. Logistic and linear regression models were fitted to test the associations of mitochondrial oxidative capacity with prediabetes, IR/insulin sensitivity, and the severity and duration of hyperglycemia exposure. Covariates included age, sex and percentage of PCr depletion (model I); race, education, smoking status, physical activity level, body weight, body height, and DEXA-measured trunk FM (model II); and levels of plasma IL-6, CRP, leptin, and adiponectin (model III [or the fully adjusted model]). Alternative anthropometric and body composition measures were also tested as confounders (model IV–VII). All analyses were performed by the SAS statistical package, version 9.3 (SAS Institute Inc., Cary, NC) and R version 3.1.2 (R Foundation for Statistical Computing, Vienna, Austria).
Results
The prevalence of prediabetes was 40.3%. The median time from the onset of diabetes was 14 years. The main characteristics of the population according to prediabetes status are presented in Table 1. τPCr was significantly higher in subjects with prediabetes compared with normal subjects and according to HOMA tertiles, and was significantly lower according to tertiles of the Matsuda index and reduced Matsuda index (Fig. 1). Independent of all confounders, τPCr significantly and positively correlated with prediabetes and HOMA, whereas it correlated negatively with the Matsuda index (Supplementary Table 2). Participants with higher τPCr values were more likely to have prediabetes and to be in a higher tertile of IR (odd ratios >1) and were less likely to be a higher tertile of insulin sensitivity (odd ratios <1) than those with lower τPCr values (Table 2). Comparing tertiles of τPCr, both the highest and the medium tertiles were significantly associated with prediabetes and impaired insulin sensitivity compared with the lowest tertiles (Supplementary Table 3).
Table 1.
Characteristics of the sample population according to prediabetes status
| Normal (n = 148) | Prediabetes (n = 100) | P value | |
|---|---|---|---|
| Age (years) | 73.3 ± 10.1 | 73.9 ± 8.9 | 0.570 |
| Male sex | 55 (37.2) | 60 (60) | <0.001 |
| Caucasian race | 110 (74.3) | 69 (69) | 0.388 |
| Education (years) | 18 (16–18) | 17 (16–18) | 0.627 |
| Weight (kg) | 71.5 ± 13.2 | 79.1 ± 13.9 | <0.001 |
| Height (cm) | 165.2 ± 8.2 | 169.2 ± 9.4 | <0.001 |
| BMI (kg/m2) | 26.1 ± 3.8 | 27.5 ± 3.7 | 0.005 |
| TBFM (DEXA-measured), kg | 23.9 ± 8.7 | 27.8 ± 9.3 | <0.001 |
| Body fatness (TBFM/weight*100), % | 33 ± 9 | 35 ± 9 | 0.154 |
| Waist circumference (cm) | 86.6 ± 11.7 | 93.5 ± 11.1 | <0.001 |
| Trunk FM (kg) | 11.05 ± 4.9 | 14.5 ± 5.3 | <0.001 |
| Smokers (former or current) | 55 (37.2) | 47 (47) | 0.148 |
| Physical activity (MET-min/day) | 311 (165.5–490.5) | 243.0 (133.5–445.0) | 0.104 |
| IL-6 (pg/mL) | 3.9 (3.1–5) | 4 (3.3–5.1) | 0.494 |
| CRP (μg/mL) | 0.99 (0.47–2.49) | 0.95 (0.50–2.50) | 0.841 |
| Leptin (ng/mL) | 11.3 (5.2–20.6) | 11.2 (5.3–19.4) | 0.843 |
| Adiponectin (μg/mL) | 13.4 (7.7–21.4) | 12.7 (5.4–18.4) | 0.164 |
| Fasting glucose (mg/dL) | 91.1 ± 8.7 | 102.2 ± 11.6 | <0.001 |
| OGTT 2-h glucose (mg/dL) | 105.7 ± 26.5 | 141.8 ± 39.4 | <0.001 |
| Homa IR units* | 1.31 (0.95–1.76) | 1.91 (1.27–2.96) | <0.001 |
| Matsuda index** | 5.72 (4.07–7.75) | 3.96 (2.59–5.66) | <0.001 |
| Reduced Matsuda index*** | 7.62 (5.09–10.5) | 4.28 (2.77–7.47) | <0.001 |
| Previous BLSA visits with OGTT data (n) | 3 (2–4) | 4 (3–5) | <0.001 |
| Severity of exposure to hyperglycemia (average OGTT 2-h glucose, mg/dL, from previous BLSA visits) | 104.2 ± 19.0 | 134.5 ± 27.5 | <0.001 |
| τPCr (s) | 48.9 (42.4–56.8) | 53.4 (53.4–47.7) | 0.001 |
| %PCr depletion | 34.7 (27.7–45.7) | 35.2 (27.4–45.1) | 0.959 |
Values are reported as the mean ± SD, n (%), or median (interquartile range).
*HOMA index (IR) = [fasting glucose (mg/dL) × fasting insulin (mU/L)]/405.
**Matsuda index (insulin sensitivity) = 10,000/√[(fasting glucose × fasting insulin) × (mean OGTT glucose concentration × mean OGTT insulin concentration).
***Reduced time points − Matsuda index (Reduced Matsuda Index) (insulin sensitivity) = 10,000/√[(fasting glucose × fasting insulin) × (OGTT 2-h glucose × OGTT 2-h insulin).
Figure 1.

Unadjusted median values and range of τPCr (in seconds) according to the presence of prediabetes, IR (HOMA index), and insulin sensitivity (Matsuda index and reduced Matsuda index).
Table 2.
Results from multivariate logistic models testing the association between τPCr (s) and likelihood (odds ratio) of having prediabetes or being in a higher tertile of HOMA index, Matsuda index, and reduced Matsuda index
| Likelihood of prediabetes | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Model I (age, sex, and %PCr depletion adjusted) (N = 248) | Model II* (N = 248) | Model III§ (N = 248) | ||||||||||||||
| OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | |||||||||||
| τPCr (s) | 1.05 (1.03–1.08) | <0.001 | 1.06 (1.02–1.09) | <0.001 | 1.06 (1.03–1.10) | <0.001 | ||||||||||
| Model IV† (N = 247) | Model V# (N = 248) | Model VI** (N = 248) | Model VII‡ (N = 248) | |||||||||||||
| OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | |||||||||
| τPCr (s) | 1.07 (1.03–1.10) | <0.001 | 1.06 (1.03–1.09) | <0.001 | 1.05 (1.02–1.09) | <0.001 | 1.06 (1.03–1.09) | <0.001 | ||||||||
| Higher tertile of HOMA index (IR) | ||||||||||||||||
| Model I (age, sex, and %PCr depletion adjusted) (N = 248) | Model II* (N = 248) | Model III§ (N = 248) | ||||||||||||||
| OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | |||||||||||
| τPCr (s) | 1.04 (1.01–1.06) | 0.002 | 1.04 (1.01–1.07) | 0.002 | 1.04 (1.01–1.07) | 0.003 | ||||||||||
| Model IV† (N = 247) | Model V# (N = 248) | Model VI** (N = 248) | Model VII‡ (N = 248) | |||||||||||||
| OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | |||||||||
| τPCr (s) | 1.05 (1.02–1.08) | 0.001 | 1.04 (1.01–1.07) | 0.003 | 1.03 (1.01–1.06) | 0.015 | 1.05 (1.02–1.08) | 0.001 | ||||||||
| Higher tertile of Matsuda index (insulin sensitivity) | ||||||||||||||||
| Model I (age, sex, and %PCr depletion adjusted) (N = 248) | Model II* (N = 248) | Model III§ (N = 248) | ||||||||||||||
| OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | |||||||||||
| τPCr (s) | 0.96 (0.94–0.98) | 0.001 | 0.96 (0.93–0.99) | 0.004 | 0.96 (0.93–0.99) | 0.003 | ||||||||||
| Model IV† (N = 247) | Model V# (N = 248) | Model VI** (N = 248) | Model VII‡ (N = 248) | |||||||||||||
| OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | |||||||||
| τPCr (s) | 0.95 (0.92–0.98) | 0.001 | 0.96 (0.93–0.98) | 0.003 | 0.97 (0.94–0.99) | 0.011 | 0.95 (0.93–0.98) | 0.001 | ||||||||
| Higher tertile of reduced Matsuda index (insulin sensitivity) | ||||||||||||||||
| Model I (age, sex, and %PCr depletion adjusted) (N = 248) | Model II* (N = 248) | Model III§ (N = 248) | ||||||||||||||
| OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | |||||||||||
| τPCr (s) | 0.95 (0.93–0.97) | <0.001 | 0.95 (0.92–0.98) | <0.001 | 0.95 (0.92–0.98) | <0.001 | ||||||||||
| Model IV† (N = 247) | Model V# (N = 248) | Model VI** (N = 248) | Model VII‡ (N = 248) | |||||||||||||
| OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | |||||||||
| τPCr (s) | 0.94 (0.92–0.97) | <0.001 | 0.95 (0.92–0.98) | <0.001 | 0.96 (0.93–0.98) | 0.001 | 0.95 (0.92–0.97) | <0.001 | ||||||||
OR, odds ratio.
*Age, sex, %PCr depletion, race, education, smoking status, physical activity level, body weight, body height, and DEXA-measured trunk FM.
§Age, sex, %PCr depletion, race, education, smoking status, physical activity level, body weight, body height, DEXA-measured trunk FM, and plasma levels of IL-6, CRP, leptin, and adiponectin.
†Age, sex, %PCr depletion, race, education, smoking status, physical activity level, body height, body weight, waist circumference, and plasma levels of IL-6, CRP, leptin, and adiponectin.
#Age, sex, %PCr depletion, race, education, smoking status, physical activity level, body weight, body height, DEXA-measured TBFM, and plasma levels of IL-6, CRP, leptin, and adiponectin.
**Age, sex, %PCr depletion, race, education, smoking status, physical activity level, body height, percentage of body fatness, and plasma levels of IL-6, CRP, leptin, and adiponectin.
‡Age, sex, %PCr depletion, race, education, smoking status, physical activity level, BMI, and plasma levels of IL-6, CRP, leptin, and adiponectin.
Relationships of τPCr with the Matsuda index and the reduced Matsuda index did not depart from linearity (Supplementary Fig. 1), whereas for HOMA-IR a cubic spline provided the best fit (Supplementary Table 4). Using OGTT-based surrogate indices developed by Abdul-Ghani et al. (18), we found that τPCr was not associated with liver IR, but was inversely and significantly correlated with muscle insulin sensitivity. Adjusting for physical activity and/or trunk FM, the association was attenuated and no longer significant (Supplementary Table 5).
Adjusting for confounders, and greater severity and longer duration of hyperglycemia exposure, even when both were included as predictors in the same model, were independently associated with greater mitochondrial impairment (Table 3).
Table 3.
Results from multivariate regression models testing the association between τPCr and severity (average OGTT 2-h glucose, in mg/dL, from previous BLSA visits) and/or duration (years from the onset of prediabetes) of hyperglycemia exposure (both included as predictors in the same model)
|
τPCr |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Model I (age, sex, and %PCr depletion adjusted) (N = 205) |
Model II* (N = 205) |
Model III§ (N = 205) |
||||||||||
| β (SE) | P value | β (SE) | P value | β (SE) | P value | |||||||
| Severity of hyperglycemia exposure (mg/dL) | 0.05 (0.03) | 0.042 | 0.05 (0.03) | 0.059 | 0.05 (0.03) | 0.055 | ||||||
| Duration of hyperglycemia exposure (years) | 0.16 (0.07) | 0.019 | 0.16 (0.07) | 0.019 | 0.16 (0.07) | 0.023 | ||||||
| Model IV† (N = 204) |
Model V# (N = 205) |
Model VI‡ (N = 205) |
Model VII** (N = 205) |
|||||||||
| β (SE) |
P value |
β (SE) |
P value |
β (SE) |
P value |
β (SE) |
P value |
|||||
| Severity of hyperglycemia exposure (mg/dL) | 0.06 (0.03) | 0.026 | 0.05 (0.03) | 0.051 | 0.05 (0.03) | 0.064 | 0.06 (0.03) | 0.034 | ||||
| Duration of hyperglycemia exposure (years) | 0.17 (0.07) | 0.013 | 0.14 (0.07) | 0.037 | 0.15 (0.07) | 0.028 | 0.16 (0.06) | 0.017 | ||||
Participants with available longitudinal data on OGTT from previous visits were 205 in 248.
*Age, sex, %PCr depletion, race, education, smoking status, physical activity level, body weight, body height, DEXA-measured trunk FM.
§Age, sex, %PCr depletion, race, education, smoking status, physical activity level, body weight, body height, DEXA-measured trunk FM, and plasma levels of IL-6, CRP, leptin, and adiponectin.
†Age, sex, %PCr depletion, race, education, smoking status, physical activity level, body height, body weight, waist circumference, and plasma levels of IL-6, CRP, leptin, and adiponectin.
#Age, sex, %PCr depletion, race, education, smoking status, physical activity level, body weight, body height, DEXA-measured TBFM, and plasma levels of IL-6, CRP, leptin, and adiponectin.
‡Age, sex, %PCr depletion, race, education, smoking status, physical activity level, body height, percentage of body fatness, and plasma levels of IL-6, CRP, leptin, and adiponectin.
**Age, sex, %PCr depletion, race, education, smoking status, physical activity level, BMI, and plasma levels of IL-6, CRP, leptin, and adiponectin.
Discussion
Using data from 248 BLSA participants with diabetes, we examined the relationship between IR and 31P-MRS–measured mitochondrial oxidative capacity. Independent of confounders, a larger τPCr value was significantly associated with a higher likelihood of having prediabetes, more severe HOMA-IR, and lower insulin sensitivity (Matsuda index). The severity and duration of hyperglycemia exposure were independently associated with larger τPCr values.
Time course studies in rodents suggest that mitochondrial impairment follows diet-induced diabetes (3). However, human studies showed mitochondrial impairment in insulin-resistant offspring of patients with types 2 diabetes, suggesting that impaired oxidative capacity may drive IR (19). Weight loss has no effect on mitochondrial capacity (20), but Coen et al. (21) recently found that exercise superimposed on bariatric surgery enhances mitochondrial respiration and improves insulin sensitivity in patients with severe obesity. Similarly, exercise in overweight-to-obese older adults improves insulin sensitivity and increases muscle oxidative capacity (1), perhaps by enhancing mitochondrial biogenesis. Of note, prolonged exercise training can, at least in part, reverse mitochondrial impairment associated with a long-standing diabetic state (6).
Our study suggests that mitochondrial impairment occurs before IR progression to type 2 diabetes. Consistently, in a small group of older adults who did not have diabetes but had IR, a significant reduction in 31P-nuclear magnetic resonance saturation transfer (ST)–measured muscle phosphorylation activity was found compared with young control subjects (22). Our study further supports these findings in a larger and better characterized sample population, accounting for the potential confounding variables of body composition and physical inactivity, and demonstrating the independent association of both the severity and duration of hyperglycemia with reduced mitochondrial capacity. Noteworthy, ST is a technique that allows measurement of the ATP synthesis rate at rest and minimizes the variability of exercise. The postexercise PCr synthesis used in our study expresses the capacity to respond to an energetic challenge and may be more sensitive to even minor metabolic derangements than ST (1).
The mechanism by which mitochondrial impairments could lead to IR is unclear. In impaired mitochondria, the flux of electrons in the electron transport chain is impaired, and spare electrons are more likely to leak in the mitochondrial matrix and produce radical oxygen species. The resulting oxidative stress may further damage mitochondria and affect IR directly or indirectly by triggering a local inflammatory response (4). It also is possible that hyperglycemia plays a key role in mitochondrial impairment. Patients with well-controlled type 2 diabetes have diminished muscle ATP synthesis despite normal glucose transport/phosphorylation (23). In addition, 31P-nuclear magnetic resonance ST–measured mitochondrial activity correlates positively with insulin sensitivity and negatively with insulin secretion, suggesting that reduced insulin signaling might limit mitochondrial oxidative rates (24). Also, mitochondrial activity negatively correlates with hepatic lipid content, supporting a link between altered muscle and liver energy metabolism in promoting IR (24). However, short-term elevation of plasma lipid levels does not affect ATP synthase in human skeletal muscle, suggesting that lipid-induced IR is unlikely to be due to a direct effect of lipid species on mitochondrial function (25).
A major limitation of our study is the cross-sectional design. Hyperinsulinemic-euglycemic clamp would have been more accurate and robust than surrogate indices to evaluate insulin sensitivity, but is time consuming and difficult to perform in large epidemiological studies (18), like the BLSA. Moreover, the severity of hyperglycemia exposure was estimated from OGTT 2-h glucose levels rather than from fasting glucose levels, but the fasting glucose level predominantly reflects liver IR, rather than muscle IR. Also, the severity of hyperglycemia exposure was based on different lengths of follow-up across participants, but we accounted for that in the analysis. Furthermore, we could not investigate race differences in mitochondrial function since BLSA participants are mostly Caucasian. Objective measurements of physical activity by accelerometers would have been more accurate than self-reports, but the accelerometer was recently introduced in the BLSA and was not available for the current sample. Finally, investigating the relationship between mitochondrial function and intramyocellular lipid content would be interesting, but these data are still unavailable in the BLSA.
Despite limitations, our study suggests that reduced mitochondrial oxidative capacity is an integral part of the IR in adult and older persons, and should be considered a possible therapeutic target to reduce the consequences of this condition and prevent progression to type 2 diabetes.
Supplementary Material
Article Information
Funding. This research was supported entirely by the Intramural Research Program of the National Institutes of Health, National Institute on Aging. Data for these analyses were obtained from the Baltimore Longitudinal Study of Aging, a study performed by the National Institute on Aging (03-AG-0325).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. E.F. contributed to the analysis and interpretation of the data, statistical analysis, and drafting of the manuscript. C.W.C. contributed to the acquisition, analysis, and interpretation of the data and to critical revision of the manuscript for important intellectual content. R.G.S. and D.A.R. contributed to the acquisition of the data and critical revision of the manuscript for important intellectual content. K.W.F., D.C., and A.C.Z. contributed to the acquisition of the data. Z.A.M., M.G.-F., M.Z., S.A.S., R.R.K., and J.M.E. contributed to the critical revision of the manuscript for important intellectual content. L.F. contributed to the study concept and design; study supervision; acquisition, analysis, and interpretation of the data; statistical analysis; drafting of the manuscript; and critical revision of the manuscript for important intellectual content. E.F. and L.F. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db16-0754/-/DC1.
References
- 1.Hesselink MK, Schrauwen-Hinderling V, Schrauwen P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat Rev Endocrinol 2016;12:633–645 [DOI] [PubMed] [Google Scholar]
- 2.Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 2011;8:92–103 [DOI] [PubMed] [Google Scholar]
- 3.Montgomery MK, Turner N. Mitochondrial dysfunction and insulin resistance: an update. Endocr Connect 2015;4:R1–R15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schrauwen-Hinderling VB, Kooi ME, Schrauwen P. Mitochondrial function and diabetes: consequences for skeletal and cardiac muscle metabolism. Antioxid Redox Signal 2016;24:39–51 [DOI] [PubMed] [Google Scholar]
- 5.De Feyter HM, van den Broek NM, Praet SF, Nicolay K, van Loon LJ, Prompers JJ. Early or advanced stage type 2 diabetes is not accompanied by in vivo skeletal muscle mitochondrial dysfunction. Eur J Endocrinol 2008;158:643–653 [DOI] [PubMed] [Google Scholar]
- 6.van Tienen FH, Praet SF, de Feyter HM, et al. Physical activity is the key determinant of skeletal muscle mitochondrial function in type 2 diabetes. J Clin Endocrinol Metab 2012;97:3261–3269 [DOI] [PubMed] [Google Scholar]
- 7.Stone JL, Norris AH. Activities and attitudes of participants in the Baltimore Longitudinal Study. J Gerontol 1966;21:575–580 [DOI] [PubMed] [Google Scholar]
- 8.Ainsworth BE, Haskell WL, Leon AS, et al. Compendium of physical activities: classification of energy costs of human physical activities. Med Sci Sports Exerc 1993;25:71–80 [DOI] [PubMed] [Google Scholar]
- 9.Coen PM, Jubrias SA, Distefano G, et al. Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults. J Gerontol A Biol Sci Med Sci 2013;68:447–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi S, Reiter DA, Shardell M, et al. 31P magnetic resonance spectroscopy assessment of muscle bioenergetics as a predictor of gait speed in the Baltimore Longitudinal Study of Aging. J Gerontol A Biol Sci Med Sci 2016;12:1638–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naressi A, Couturier C, Castang I, de Beer R, Graveron-Demilly D. Java-based graphical user interface for MRUI, a software package for quantitation of in vivo/medical magnetic resonance spectroscopy signals. Comput Biol Med 2001;31:269–286 [DOI] [PubMed] [Google Scholar]
- 12.Naressi A, Couturier C, Devos JM, et al. Java-based graphical user interface for the MRUI quantitation package. MAGMA 2001;12:141–152 [DOI] [PubMed] [Google Scholar]
- 13.Smith SA, Montain SJ, Zientara GP, Fielding RA. Use of phosphocreatine kinetics to determine the influence of creatine on muscle mitochondrial respiration: an in vivo 31P-MRS study of oral creatine ingestion. J Appl Physiol 1985;96:2288–2292 [DOI] [PubMed] [Google Scholar]
- 14.American Diabetes Association Standards of medical care in diabetes—2010. Diabetes Care 2010;33(Suppl. 1):S11–S61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412–419 [DOI] [PubMed] [Google Scholar]
- 16.Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 1999;22:1462–1470 [DOI] [PubMed] [Google Scholar]
- 17.DeFronzo RA, Matsuda M. Reduced time points to calculate the composite index. Diabetes Care 2010;33:e93. [DOI] [PubMed] [Google Scholar]
- 18.Abdul-Ghani MA, Matsuda M, Balas B, DeFronzo RA. Muscle and liver insulin resistance indexes derived from the oral glucose tolerance test. Diabetes Care 2007;30:89–94 [DOI] [PubMed] [Google Scholar]
- 19.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 2004;350:664–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toledo FG, Menshikova EV, Azuma K, et al. Mitochondrial capacity in skeletal muscle is not stimulated by weight loss despite increases in insulin action and decreases in intramyocellular lipid content. Diabetes 2008;57:987–994 [DOI] [PubMed] [Google Scholar]
- 21.Coen PM, Menshikova EV, Distefano G, et al. Exercise and weight loss improve muscle mitochondrial respiration, lipid partitioning, and insulin sensitivity after gastric bypass surgery. Diabetes 2015;64:3737–3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petersen KF, Befroy D, Dufour S, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 2003;300:1140–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szendroedi J, Schmid AI, Chmelik M, et al. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med 2007;4:e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szendroedi J, Kaul K, Kloock L, et al. Lower fasting muscle mitochondrial activity relates to hepatic steatosis in humans. Diabetes Care 2014;37:468–474 [DOI] [PubMed] [Google Scholar]
- 25.Brehm A, Krssák M, Schmid AI, Nowotny P, Waldhäusl W, Roden M. Acute elevation of plasma lipids does not affect ATP synthesis in human skeletal muscle. Am J Physiol Endocrinol Metab 2010;299:E33–E38 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.