

Abstract
Cancer cells exhibit multiple epigenetic changes with prominent local DNA hypermethylation and widespread hypomethylation affecting large chromosomal domains. Epigenome studies often disregard the study of repeat elements owing to technical complexity and their undefined role in genome regulation. We have developed NSUMA (Next-generation Sequencing of UnMethylated Alu), a cost-effective approach allowing the unambiguous interrogation of DNA methylation in more than 130,000 individual Alu elements, the most abundant retrotransposon in the human genome. DNA methylation profiles of Alu repeats have been analyzed in colon cancers and normal tissues using NSUMA and whole-genome bisulfite sequencing. Normal cells show a low proportion of unmethylated Alu (1%–4%) that may increase up to 10-fold in cancer cells. In normal cells, unmethylated Alu elements tend to locate in the vicinity of functionally rich regions and display epigenetic features consistent with a direct impact on genome regulation. In cancer cells, Alu repeats are more resistant to hypomethylation than other retroelements. Genome segmentation based on high/low rates of Alu hypomethylation allows the identification of genomic compartments with differential genetic, epigenetic, and transcriptomic features. Alu hypomethylated regions show low transcriptional activity, late DNA replication, and its extent is associated with higher chromosomal instability. Our analysis demonstrates that Alu retroelements contribute to define the epigenetic landscape of normal and cancer cells and provides a unique resource on the epigenetic dynamics of a principal, but largely unexplored, component of the primate genome.
Cancer cells are characterized by the acquisition of new biological properties and the escape from repressive mechanisms. The reprogramming of regulatory circuits arises as a direct consequence of genetic and epigenetic changes. Similarly to genetic aberrations, that may affect a single gene (e.g., a point mutation) or large chromosomal regions (e.g., losses of heterozygosity), cancer cells show epigenetic alterations involving the misregulation of a single gene (e.g., expression silencing by hypermethylation of the promoter CpG island) (Esteller 2007; Jones 2012) or large chromosomal regions (i.e., long-range epigenetic silencing) (Frigola et al. 2006; Coolen et al. 2010; Forn et al. 2013). Although local alterations are excellent pointers for the identification of candidate cancer genes, alterations affecting large chromosomal regions offer limited clues about both the mechanisms underlying the alteration and also the functional impact of the alteration on the disease. A clear example is global DNA hypomethylation in cancer. This is the first epigenetic alteration detected in cancer and probably the most common (Feinberg and Tycko 2004; Wilson et al. 2007; Esteller 2008; Ehrlich 2009; Jones 2012).
DNA methylation mainly occurs in the cytosine of the CpG dinucleotide and is usually associated with chromatin repression. Silencing of repetitive elements, which account for up to 50% of the human genome (International Human Genome Sequencing Consortium 2001), is usually attributed to their heavy methylation (Bird 2002; Goll and Bestor 2005; Bernstein et al. 2007; Suzuki and Bird 2008). The mechanisms responsible for cancer-related DNA hypomethylation are unknown, but multiple studies have unveiled its multiple consequences including gene deregulation, loss of chromatin organization, and genetic instability (Eden et al. 2003; Karpf and Matsui 2005; Rodriguez et al. 2006).
It is commonly accepted that DNA hypomethylation mainly affects repeat elements, but very often this assertion is based on either bulk analyses or the extrapolation of the results obtained from the interrogation of a few surrogate markers. The use of high-throughput genomic technologies to study DNA methylation profiles in cancer cells has demonstrated that DNA hypomethylation preferentially affects large chromatin blocks exhibiting gene expression variability and definite chromatin features (Hansen et al. 2011; Berman et al. 2012; Hon et al. 2012; Timp et al. 2014). Thus, hypomethylation appears to affect both repetitive and unique sequences within these blocks, but it is unknown whether the uneven distribution along the genome of different genetic elements, and especially repeats, determines the hypomethylation profile. Moreover, high resolution DNA methylation maps often have poor or even no coverage of repeat elements. This means that we do not have a precise picture of the epigenomic landscape of repeat elements, even when they are close to functional elements such as genes. Another important factor that underscores the need of characterizing the distribution of hypomethylation in repeats is their heterogeneous scattering along the genome. Short and long interspersed nucleotide elements (SINE and LINE, respectively) account for the two main classes of repeats in the human genome. Alu elements are the most abundant repeat, with more than 1 million copies per haploid genome and spanning ∼10% of the genome sequence (Cordaux and Batzer 2009). Alu repeats tend to accumulate in gene-rich regions (International Human Genome Sequencing Consortium 2001; Chen et al. 2002; Grover et al. 2004) and harbor ∼25% of all CpG dinucleotides in the human genome (Luo et al. 2014; Buj et al. 2016). On the other hand, LINEs, which are depleted in gene-rich regions and span 20% of the human genome, contain ∼12% of the methylated cytosines (Xie et al. 2009; Luo et al. 2014; Buj et al. 2016).
To gain insights into the role of DNA hypomethylation in cancer cells, we have analyzed the DNA methylation profiles in colorectal cancers, paired normal tissues, and colon cancer cell lines. We have focused our analysis on Alu repeats due to their structural and functional features. To improve screening efficiency, we have developed and applied a new technique allowing the unambiguous detection and differential scoring of unmethylated Alu repeats and compared these results with whole-genome bisulfite sequencing (WGBS) data (Hansen et al. 2011).
Results
Detection of unmethylated Alu repeats by NSUMA, technical overview, and coverage
Whole-genome bisulfite sequencing (WGBS) is nowadays the gold standard in genome-scale DNA methylation studies but at a cost (Stirzaker et al. 2014). Indeed, the performance of WGBS is poor when analyzing repeat sequences including Alu and other retrotransposons (Bock et al. 2010). To circumvent, at least in part, these limitations, we have developed the Next-generation Sequencing of UnMethylated Alu (NSUMA) method aimed to selectively enrich for DNA fragments composed by an unmethylated Alu repeat and a flanking nonrepetitive sequence, which allows an unambiguous mapping in the genome. NSUMA method uses genomic DNA digested with the methylation sensitive restriction endonuclease SmaI (cutting unmethylated CCCGGG sites) and the frequent cutter MseI (cutting TTAA). The SmaI site is present in the consensus sequence and a large subset of Alu repeats (Fig. 1A,B). Specific adapters are ligated to the SmaI blunt ends and the MseI sticky ends. Further Alu enrichment is achieved by PCR with primers targeting the consensus Alu sequence and the MseI adapter (Fig. 1A; Supplemental Fig. S1). The NSUMA virtual universe is integrated by 144,108 unique canonical amplicons (Supplemental Data 1), and a high proportion of them (94%) are generated from DNA fragments comprising an Alu repeat and an adjacent unique sequence (Fig. 1B; Supplemental Table S1). The remaining 6% of the NSUMA virtual universe includes amplicons mapping to other repeats, CpG islands, and other unique sequences (Fig. 1B). Additionally, NSUMA generates noncanonical amplicons lacking the consensus SmaI site (Fig. 1A), and as a consequence, their representativeness is unaffected by DNA methylation (Supplemental Table S2). Interestingly, the relative amplification of noncanonical amplicons may be used to determine copy number variation along the chromosome (Supplemental Methods), constituting an integrated alternative to comparative genomic hybridization (CGH).
Figure 1.

(A) Diagram illustrating the principle of Next-generation Sequencing of UnMethylated Alu repeats (NSUMA) technique. Genomic DNA is represented by a blue line, and the Alu repeat is represented by a gray box. DNA is digested with the restriction enzymes SmaI and MseI (impaired and insensitive to DNA methylation, respectively). The digested DNA fragments are ligated to two adapters with ends compatible with each one of the enzyme cuts. Primers complementary to the adapters are used to generate an amplified representation of the fragments. Amplicons generated from DNA fragments containing the SmaI adapter report unmethylated DNA and constitute the NSUMA canonical universe. Amplicons without the SmaI adapter (MseI-MseI) are also sequenced and used for the analysis of copy number variation. (B) Genetic element distribution according to the occupied fraction of the human genome, the content of SmaI sites, and their representation in the canonical NSUMA universe. (C) Relative unmethylation of different genetic elements in each one of the sample types analyzed by NSUMA in regard to the highly hypomethylated cell line DKO. CpG islands show the highest relative representation in all sample types as most of them are unmethylated, whereas Alu repeats show the lowest representation due to their heavy methylation. (D) Number of differentially methylated elements in the comparison between pairs of tissues (adjusted P-value <0.05 and |log2 FC| >1). Full data summary is reported in Supplemental Table S5, and detailed MA plots are shown in Supplemental Figure S13.
NSUMA data processing is summarized in Supplemental Figure S2. The overall representativeness of the SmaI site as a surrogate reporter of the methylation of the entire genomic element was evaluated for Alu repeats (Supplemental Fig. S3) as previously shown for CpG islands (Barrera and Peinado 2012). Alignment of reads to the human genome identified NSUMA amplicons generated from unmethylated SmaI sites (Supplemental Fig. S4). Importantly, the number of reads mapping to a given amplicon is related to the degree of differential DNA methylation when two or more samples are compared (Supplemental Fig. S4B). That is, hypomethylation will be shown as an increase in the number of reads of the specific amplicon; while hypermethylation will be shown as a decreased number of reads in the corresponding amplicon. Technical and biological replicates were performed to evaluate the reproducibility of the technique (Supplemental Fig. S5).
The relative distribution of NSUMA reads illustrates the overall degree of unmethylation of each type of genomic element (Fig. 1C; Supplemental Table S3). Thus, CpG islands covered by NSUMA are highly represented in all the samples (due to their preponderant unmethylated state); repeats are poorly represented in normal samples (in which repeats are heavily methylated), but better represented in tumors and especially in the DNA methyltransferase deficient DKO cell line, due to the overall hypomethylation (Supplemental Table S3).
To perform comparisons, the count of reads mapping to canonical amplicons were normalized by a factor resulting from the analysis of 150 amplicons located in unmethylated CpG islands (Supplemental Fig. S6; Supplemental Table S3). NSUMA achieved a specificity of 95% and sensitivity of 50% in the detection of unmethylated CpG islands (cutoff ≥5 normalized reads) when compared with whole-genome bisulfite sequencing (Lister et al. 2009) using a receiver operating characteristic (ROC) curve analysis (Supplemental Fig. S6). The distributions of amplicon representation in regard to GC content and size (Supplemental Methods) were very similar among samples (Supplemental Figs. S7–S9), indicating that potential PCR biases were very unlikely to have any effect on the assessment of differential methylation.
Rates of unmethylated Alu repeats in normal tissue and cancer samples
Three normal tumor–paired colon tissues as well as the corresponding DNA from whole blood were analyzed by NSUMA. In addition, three biological replicates (independent cell cultures) of the HCT116 cell line and three technical replicates of its DNA methylation deficient variant DKO (Rhee et al. 2002) were performed.
The global unmethylation of each one of the genomic elements considered was estimated by analysis of the relative distribution of normalized reads (nreads) compared with number of nreads in CpG islands, due to their prevalent unmethylation in all samples. Alu repeats displayed the lowest levels of unmethylation, especially in normal tissues (Fig. 1C). A moderate increase in the overall demethylation of Alu repeats was observed in cancer samples and the HCT116 cell line, but other repeats, as LINEs and MIRs, exhibited much higher hypomethylation rates (Supplemental Fig. S10). This result suggests that Alu elements are deeply methylated and are more resistant to hypomethylation in tumor tissues than other genomic elements, including unique sequences (“Rest” group in Fig. 1C; Supplemental Fig. S10). As expected, the severe hypomethylated condition of DKO cells (Sandoval et al. 2011) was evidenced by high representation of all element types in NSUMA (Fig. 1C), but once again, Alu repeats showed the lowest gain (Supplemental Fig. S10A).
To estimate the number of unmethylated elements, a threshold of five or more nreads/amplicon was set based on the comparison of CpG islands analyzed by NSUMA and WGBS (Supplemental Fig. S6). Twenty-five percent of the Alu repeats analyzed by NSUMA were found unmethylated in at least one sample, excluding DKO cells, which showed an additional 39% of unmethylated Alu repeats (Supplemental Fig. S6; Supplemental Table S4). Strikingly, only 225 Alu repeats (<0.3% of informative elements) were found unmethylated in all samples. Eight thousand Alu repeats (6% of the NSUMA universe) appeared unmethylated in at least one normal tissue. The number of unmethylated Alu repeats was lower in blood (range 1.0%–1.6%) than in normal colon tissue (range 1.2%–5.0%) (Supplemental Fig. S10B). This figure was trebled in tumor samples and the colon cancer cell line HCT116 (Supplemental Table S5; Supplemental Fig. S10C).
WGBS data from a previous study of normal and colorectal cancers (Hansen et al. 2011) were used to investigate Alu and LINE repeat methylation. Only elements with a minimum of three informative CpGs for Alu and five for LINE (Supplemental Material and Methods) were considered for this analysis, rendering a total of 117,779 and 111,019 Alu and LINE repeats, respectively. The methylation of each element was calculated as the average fraction of methylation (usually referred to as beta value: 0 to 1, with 0 fully unmethylated, and 1 fully methylated) of all the informative CpGs within the element. Cancer cells showed two- to threefold increase in the proportion of unmethylated Alu repeats compared with normal colon (Supplemental Fig. S11), in line with the figures observed in NSUMA. LINEs exhibited a more pronounced hypomethylation than Alu repeats, especially in carcinomas. A direct comparison of WGBS and NSUMA was not possible as the samples analyzed were different. Moreover, the overlapping coverage of NSUMA and WGBS was very poor (n = 9969), which may be explained by the different constraints imposed by each approach. Besides these limitations, Alu repeats scored by WGBS and NSUMA showed highly concordant DNA methylation states in both normal and tumor samples (Supplemental Fig. S12A). In addition, ROC curve analysis of the NSUMA data using the WGBS data as the gold standard offered a specificity of 94% and a sensitivity of 63% (Supplemental Fig. S12B) when using the same cutoff value previously evaluated for CpG islands (see above).
In summary, Alu repeats appeared as the least unmethylated repeat class, which is suggestive of a stronger pressure to maintain these elements methylated in both normal and malignant cells, and even in DKO cells exhibiting a constitutional deficiency in DNMTs.
Differential DNA methylation of Alu repeats
To compare NSUMA profiles among different samples, the Alu differential methylation ratio (Alu DMR) was calculated as the log2 of the ratio of the normalized reads (nreads) of the two samples. The Alu DMR represents hypermethylation when positive and hypomethylation when negative. Blood and normal colon Alu DMR profiles were very similar in the three patients (Supplemental Fig. S13A). Statistical analysis using the DESeq R package (Anders and Huber 2010) indicated than only 0.5% of Alu repeats displayed differential methylation between these two normal cell types (Fig. 1D; Supplemental Table S5; Supplemental Fig. S13B). Interestingly, LINEs and other repeats, as well as nonrepetitive sequences, showed a higher rate of differences (∼5% of informative elements) with a clear prevalence of hypomethylation in normal colon in regard to blood DNA (Supplemental Table S5).
Indeed, colon tumors exhibited a high number of hypomethylated sites with almost no hypermethylated sites (Fig. 1D; Supplemental Fig. S13). More than 3000 Alu repeats were recurrently hypomethylated in the three tumors analyzed, and about 6,500 were hypomethylated in a least one of the tumors as compared with the paired normal colon mucosa (Supplemental Table S5; Supplemental Data 2). Noteworthily, only 3% of Alu sequences were hypomethylated in tumors, which contrasts with the higher hypomethylation rates observed in LINEs (8%) and MIRs (11%). With the exception of CpG islands, which showed a very low but similar number of hypermethylated and hypomethylated sites, the remaining amplicons located in unique sequences also showed a clear predominance of hypomethylation (6% of the amplicons) (Fig. 1D; Supplemental Table S5).
HCT116 exhibited a high number of hypomethylated Alu repeats (8.7% of informative Alu repeats), but also a high number of hypermethylated sites that affected nearly 12% of the CpG islands and ∼10% of Alu elements (Supplemental Table S5). Exceptionally, DKO displayed extensive demethylation in all types of genomic elements, affecting 65% of the virtual NSUMA coverage (Fig. 1D; Supplemental Figs. S10, S13B). It should be noted that this is probably an underestimation, as the large number of competing amplicons in this cell line is likely to saturate the overall display, precluding a comparable coverage with the rest of the samples. Moreover, the massive demethylation of DKO cells affects the normalization because stably unmethylated CpG islands used as reference (Supplemental Methods) are underrepresented due to signal saturation, which might explain the apparent hypermethylation of some Alu repeats in DKO.
WGBS analysis also illustrated important differences in the hypomethylation of Alu and other repeats in colon cancer (Supplemental Fig. S11A; Supplemental Data 3). In this case, Alu differential methylation (Alu DM) was calculated as the delta of normal-tumor mean beta value within each element. Despite the poor overlap of NSUMA and WGBS, the genomic profiles were similar (Supplemental Fig. S14B). Differential methylation analysis by WGBS in Alu and LINE repeats showed global hypomethylation, although it was more severe in LINEs (Supplemental Fig. S14C). Stratification of elements by the methylation level in normal tissue showed that lowly methylated elements were more resistant to hypomethylation in the tumor (Supplemental Fig. S14D).
Despite that samples analyzed by NSUMA and WGBS were not the same, the differential methylation of the 9969 Alu repeats informative in both studies were comparable (Supplemental Fig. S15A). The differential methylation of a subset of Alu repeats was also confirmed by direct bisulfite sequencing of the same samples and additional normal-tumor pairs and cell lines reinforcing the recurrent nature of their hypomethylation (Supplemental Fig. S15B).
Unmethylated Alu signatures are characteristic of the tissue type
Principal component analysis (PCA) and cluster analysis of the unmethylated Alu repeats revealed specific signatures of each cell type (blood, normal colon, and tumor cells) (Supplemental Fig. S16). Analysis of the remaining genomic elements also classified the samples according to tissue type, except for CpG islands, which grouped samples originating from the same individual rather than by tissue type (Supplemental Fig. S16). As a whole, these data indicate that tissue specific DNA methylation signatures are present in multiple types of sequences, including repeat elements. Differences among individuals were also observed, but larger data sets are needed to confirm the extent and structure of epigenetic diversity.
Structural and functional features of hypomethylated Alu repeats
The features of Alu repeats and the flanking regions were examined for insights into the determinants of hypomethylation in the different types of samples. The NSUMA virtual universe was used as a reference population to analyze feature enrichment to avoid representation biases introduced by the screening methods (Fig. 1D; Supplemental Table S1).
Regarding Alu families, AluY were very well represented in the NSUMA virtual universe (Fig. 1B; Supplemental Table S1) and showed a high resistance to hypomethylation in tumors, whereas the older AluJ family were the most sensitive (Supplemental Fig. S17; Supplemental Table S6). WGBS, with a more balanced representation of all Alu families, showed the same trends as NSUMA, with more frequent hypomethylation in AluJ than AluY (Supplemental Fig. S17; Supplemental Table S6).
Alu elements were classified into different compartments or subsets based on NSUMA coverage and differential representation (Supplemental Methods). The subset of Alu repeats unmethylated in all tissues (UNM All) showed the most striking features: high G+C and CpG content and a lower similarity score (Smith and Waterman 1981), and exceptionally unbalanced CpG content of the flanking regions (one flank rich in CpGs and the other poor) (Fig. 2A; Supplemental Fig. S18; Supplemental Table S7). This result suggests that Alu repeats unmethylated in all tissues (but not the ones differentially methylated) tend to locate at the boundaries of genomic regions with different genetic structure. On the contrary, Alu repeats selectively hypomethylated in tumors contained fewer CpGs and were embedded in regions poor in CpG content when compared with Alu repeats with stable methylation in the tumor (Fig. 2A; Supplemental Table S7). Another striking feature of Alu repeats unmethylated in all tissues was their proximity to transcription start sites (TSS) and CpG islands (Fig. 2B). This trend, although less prominent, was also observed in Alu repeats unmethylated in normal colon, but not in blood. Once again, Alu repeats hypomethylated in tumors showed an opposite behavior: They tended to be far from TSS and CpG islands and closer to LINEs (Fig. 2B).
Figure 2.

Features of genomic compartments based on NSUMA coverage and differential methylation (Supplemental Methods; Supplemental Tables S4, S6). (A) Box plot of the number of CpGs within the Alu (blue boxes) and in the 500-bp flanking regions (yellow boxes). Flanking regions have been arranged by CpG content: low to the left and high to the right. Alu repeats unmethylated in all tissues (UNM All) tend to localize in the boundaries of regions with large differences in CpG content (*). Statistical analyses are shown in Supplemental Table S7. (B) Box plot of the genomic distances between Alu repeats classified in different compartments and TSSs, CpG islands, and LINEs. ANOVA P-values for comparisons between Alu repeat subsets are indicated at the bottom of each graph. (C) Transcription factor binding motif enrichment in different subsets of Alu repeats. Full markers correspond to putative binding sites located inside the Alu sequence, and empty markers indicate motifs located within flanking sequences (500 bp).
Next, we investigated the enrichment of transcription factor binding motifs in the different groups of Alu repeats according to its methylation profile. Compared with the NSUMA virtual universe, the flanking regions (500 bp each side) of Alu elements unmethylated in all tissues showed the highest motif enrichment (six factors) (Fig. 2C). Alu repeats unmethylated in normal colon and blood also showed enrichment of three transcription factors in the flanking regions, one shared by blood and colon (ZFX) and the other two specific of each tissue type. Interestingly, the CEBPA (CCAAT/enhancer-binding protein alpha) binding motif was present inside the Alu sequence in >50% of Alu repeats unmethylated in blood (115 of 206) but absent from those unmethylated in normal colon (0 of 243).
Copy number analysis using noncanonical NSUMA reads
In parallel to canonical amplicons containing a SmaI DNA methylation sensitive target, NSUMA will also competitively amplify DNA fragments flanked by two MseI sites (TTAA), irrespective of the DNA methylation status (Supplemental Fig. S2). Indeed, the amplification dynamics of noncanonical fragments should be similar to the DNA fingerprinting technique arbitrarily primed PCR successfully used to determine copy number variation in cancer (Perucho et al. 1995). Therefore, we analyzed the enrichment of noncanonical reads along the genome in 100-kb windows by applying computational tools developed for comparative genomic hybridization (CGH) (Supplemental Material and Methods). To evaluate the feasibility of this approach, we compared the noncanonical NSUMA copy number analysis with data generated by conventional array CGH (Supplemental Material and Methods) in the three colon tumors and their paired normal tissues as well as the HCT116 and DKO cell lines. Complex copy number variants were found with both techniques in all analyzed tumors (Supplemental Data 4). Noteworthily, the noncanonical NSUMA amplicons reproduced the gains and losses profiles of array CGH with striking accuracy, even when the changes affected regions of <1 Mb (Supplemental Fig. S19).
Two important observations must be underscored from the analysis of canonical and noncanonical amplicons in the NSUMA. First, chromosomal profiles reflecting variation in DNA methylation and copy number are mutually independent (Supplemental Fig. S19). Second, the overall proportion of noncanonical versus canonical reads reflects the global methylation of the sample. For instance, the noncanonical/canonical ratio is ∼10-fold in blood DNA (with heavy global DNA methylation), whereas this ratio is reduced to threefold in the highly demethylated cell line DKO (Supplemental Table S2).
Hypomethylated Alu repeats are clustered in specific chromosomal regions
Plotting of Alu DMR in the UCSC Genome Browser (Supplemental Data 5) revealed a clear clustering of hypomethylated Alu repeats with similar profiles in the three tumors and the HCT116 cell line (Fig. 3A; Supplemental Fig. S19). A circular binary segmentation algorithm (Olshen et al. 2004) was applied to identify hypomethylated genomic compartments (Fig. 3B). The colon tumors and the HCT116 colon cancer cell line showed a bimodal distribution of the segments according to the Alu mean DMR, and a cutoff value was set at the pit (0.8 for tumors and 1.0 for the HCT116 cell line) (Fig. 3C). Segments with an Alu mean DMR below the pit were considered hypomethylated Alu regions (HMAR) (Fig. 3B). The number of HMARs ranged from 28 in tumor 369T to 498 in HCT116 cells and spanned from 5% (138 Mbp in tumor 369T) to 30% (873 Mbp in tumor 557T) of the genome. The shared burden of HMARs (HCT116 and colon tumors) consisted of 325 Mb scattered in 203 regions with a mean size of 1.6 Mb (Supplemental Table S8; Supplemental Data 6).
Figure 3.

(A) Visualization of Chromosome 7 NSUMA Alu DMR for HCT116 and three colon cancer tumors (369T, 544T, and 557T) compared with their corresponding normal tissues (HCT116 was compared against the three normal samples). Values below 0 indicate hypomethylation in the tumor against the normal tissue. Additional tracks show the mean differential methylation of three colon tumors analyzed by WGBS (Hansen et al. 2011) and the density of Alu elements, genes, and CpG islands along the genome. (B) Hypomethylated regions obtained from NSUMA profiles in five colon cancer cell lines, and three primary colon cancers (mTUMOR). Hypomethylated regions obtained from WGBS in three colon cancers considering different repeat types (Alu, LINE, and other repeats). Additional tracks show the hypomethylated block (Colon Cancer HB) (Hansen et al. 2011) and the abundance of genes, CpG islands, and Alu repeats. The inset shows a detailed view of the differential methylation profiles in the region enclosed by a red dotted line. (C) Distribution of hypomethylated regions according to the mean differential methylation of Alu repeats determined by NSUMA and WGBS and LINEs determined by WGBS in colorectal cancers and HCT116 cell line. (D) Distribution of differential methylation in Alu (upper panel) and LINE (lower panel) repeat elements determined by WGBS in regard to the mean differential methylation of the enclosing region: HMAR, no HMAR, and SMAR.
Four additional colon cancer cell lines were also analyzed by NSUMA to confirm the genome compartmentalization based on Alu hypomethylation profiles (Fig. 3B). All the cell lines presented a bimodal distribution of the genomic segments, with HMARs spanning from 14% of the genome in SW480 cells, 30% in HCT116, HT29, and LoVo, and up to 50% in CaCo-2 cells (Supplemental Fig. S20). Notably, a moderate hypomethylation also affected Alu repeats outside HMARs, especially in HT29, CaCo-2, and LoVo cells. This global hypomethylation was not observed in HCT116 cells (Alu mean DMR in segments outside HMARs was 0) and the SW480 cell line, whose genome was largely hypermethylated (Supplemental Fig. S20). Normal colon and whole blood DNA profiles did not show regional differences (Fig. 1D; Supplemental Table S5; Supplemental Fig. S13B).
HMARs were also identified in the three normal-colon carcinoma pairs analyzed by WGBS in a previous study (Hansen et al. 2011). Only Alu elements with at least three informative CpGs in all the samples were included in the analysis (n = 117,779) (Supplemental Data 7). The combined analysis of the three carcinomas (treated as biological replicates) resulted in 192 HMARs (Supplemental Table S9; Supplemental Fig. S20). Although the samples analyzed by NSUMA and WGBS were different and the overlap of Alu repeats analyzed by each technique was very small (<10%, see above), the profiles and the regions identified by both techniques were highly consistent (Fig. 3; Supplemental Fig. S21A). The distribution of Alu mean DM in the tumors analyzed by WGBS was also bimodal and similar to the ones obtained by NSUMA (Fig. 3C). We could also identify 22 chromosomal segments with Alu DM close to 0 (≥ −0.05) in the three tumors and were considered as stable Alu methylation regions (SMAR) (Supplemental Table S9).
Notably, 97% of the HMARs shared by HCT116 and colon cancers analyzed by NSUMA overlapped with the large hypomethylated blocks (HB) obtained by WGBS (Supplemental Tables S10, S11; Hansen et al. 2011). Further analysis of the WGBS data allowed us to compare the differential methylation of other repeat elements inside and outside HMARs. Beyond the parallelism of the hypomethylation profiles among the different types of elements, Alu repeats showed a less pronounced hypomethylation than LINEs and other repeats (Fig. 3B; Supplemental Fig. S21). As expected, Alu DMR (measured by NSUMA) and Alu DM (measured by WGBS) showed consistent behaviors inside and outside HMARs (Supplemental Fig. S21C). LINEs and other repeat elements located outside HMARs also showed hypomethylation, but at lesser extent than inside HMARs (Fig. 3D; Supplemental Fig. S21C). Interestingly, the hypomethylation of Alu elements outside HMAR was very weak and comparable to the impact of hypermethylation, whereas LINE hypomethylation was not restricted to the HMARs and was also apparent in SMARs (Fig. 3D).
Structural, spatial, and functional determinants of Alu hypomethylation
Next, we investigated the features associated with regional Alu hypomethylation. For the sake of simplicity, we applied the genomic segmentation resulting from the overlapping HMARs between HCT116 and the three colon tumors analyzed by NSUMA (Supplemental Table S8).
HMARs were depleted of genes, CpG islands, small nuclear RNAs and, interestingly, of Alu repeats themselves (Fig. 4A). Other repeats as LINEs or MIR and noncoding RNAs showed no differential enrichment (Supplemental Fig. S22A). HMARs identified in the tumors analyzed by WGBS displayed a similar distribution of the different genomic elements (Supplemental Fig. S22B). Segmentation of the genome in bins of 100 kb demonstrated that Alu hypomethylation was restrained to regions with the lowest density of these elements (Fig. 4B). Noteworthily, Alu DNA methylation stable domains (not hypomethylated in any tumor) presented the highest content of genes, Alu repeats, and CpG islands (Supplemental Fig. S22B). Hypomethylated LINE regions (HMLR) showed similar trends, although the differences between the hypomethylated and the unchanged genomic compartments were less accentuated (Supplemental Fig. S22C). LINE density did not appear to affect the degree of hypomethylation (Fig. 4B).
Figure 4.

(A) Density of genomic elements (genes, Alu repeats, CpG islands, and small nuclear RNAs) in genomic segments according to the Alu hypomethylation profiles determined by NSUMA. Overlapping HMAR regions (red line) result from the shared HMARs of three colon tumors and the HCT116 cell line (Supplemental Table S8). Nonoverlapping HMARs (gray line) correspond to hypomethylated regions in tumors or HCT116 but not in both. The rest of the genome (outside HMARs, green line) is neither hypomethylated in tumors nor HCT116 cells. The distribution of other genomic elements is shown in Supplemental Figure S22. (B) Distribution of the Alu differential methylation ratio of HCT116 in regard to Alu and LINE density (elements/100 kb). (C) Distribution of genomic segments according to the Alu mean DMR and the copy number variation in HCT116 and the tumor sample 544T. (D) The upper panel shows the relationship between the extent of the hypomethylated compartment (as percentage of the genome) and the number of regions with chromosomal alterations as detected by array CGH. The lower panel shows the number of chromosome segments with copy number alterations in regard to their location inside or outside HMAR. Tumor samples with a larger hypomethylated compartment show a higher number of alterations in both HMARs and outside HMARs.
A link between global DNA hypomethylation and chromosomal instability has been proposed in tumorigenesis (Eden et al. 2003; Rodriguez et al. 2006). Analysis of our data revealed a correlation between the extent of HMARs and the number of chromosomal losses and gains detected by CGH (Fig. 4C). This correlation was also maintained when chromosomal alterations inside and outside HMARs were considered separately (Fig. 4D), which suggests a global, rather than local, relationship between genetic and epigenetic instabilities.
We also compared the Alu hypomethylation landscapes with histone modification profiles of colonic mucosa produced by the Roadmap Epigenomics Consortium (2015). As shown in Figure 5A, HMARs were enriched in silent chromatin marks (H3K9me3 and H3K27me3) and depleted of active chromatin marks (H3K4me1, H3K4me3, H3K27ac, H3K9ac, and H3K36me3). We could also observe the circumscription of Alu hypomethylation to chromosomal regions attached to the nuclear lamina (Fig. 5B; Guelen et al. 2008), which is consistent with the reported association of long-range hypomethylation and nuclear lamina domains (Berman et al. 2012).
Figure 5.

(A) Relationships in the genomic distribution of colon cancer DNA hypomethylation and histone modification profiles of normal human colonic mucosa. Data have been displayed using the UCSC Genome Browser and represent (from top to bottom): the hypomethylated regions for LINEs analyzed by WGBS in three colorectal carcinomas, for Alu repeats analyzed by NSUMA in three colorectal carcinomas, for Alu repeats analyzed by NSUMA in HCT116 cell line, for Alu repeats analyzed by WGBS in three colorectal carcinomas, and the histone modification profiles for human colonic mucosa produced by the Roadmap Epigenomics Consortium. (B) Distribution of colon cancer hypomethylated regions concurs with nuclear lamina associated domains and late DNA replication. The inset shows a detailed view of genomic elements in the region enclosed by a red dotted line. (C) Alu and LINE differential methylation in colorectal cancer in relation to replication timing in IMR90 cells. Each dot represents a region of 100 kb.
Next, we analyzed the distribution of DNA hypomethylation in relation to DNA replication dynamics (Hansen et al. 2009; Ryba et al. 2010, 2011). A global concordance between hypomethylated domains and late replication was clear (Fig. 5B; Supplemental Fig. S23). Noteworthily, Alu repeats located in early replication domains appeared to be very resistant to tumor hypomethylation, whereas LINEs showed a bimodal distribution (Fig. 5C, left), indicating differential susceptibility. In late replication regions, LINEs exhibited a deeper hypomethylation compared with Alu repeats (Fig. 5C, right).
Transcriptional activity of Alu methylation landscapes
Finally, we explored the impact of DNA methylation and other genomic features on the transcriptional activity of Alu repeats. Two biological replicates of messenger RNA extracted from HCT116 cells were sequenced using paired-end RNA-seq (Supplemental Methods). About 1 million reads representing 0.7% of all unambiguously mapped reads in each replicate overlapped with Alu sequences. Eighteen percent of the 1.1 million Alu repeats were found with at least one read in both replicates. DNA methylation information generated by reduced representation bisulfite sequencing (RRBS) was available for a subset of 9339 Alu repeats (Supplemental Methods; Supplemental Data 8). Parallel analyses were performed with both the RRBS subset and all the Alu repeats, and despite that the genomic fraction covered by RRBS is not representative of the whole genome, similar trends were observed as shown below.
The expression levels of the gene nearest to the Alu appeared as the main determinant of Alu expression (Fig. 6A; Supplemental Fig. S24; Supplemental Table S13). Intragenic location and low methylation were also associated with higher Alu expression although a broad variability was observed (Fig. 6A; Supplemental Fig. S24). Next, we investigated the relationship between the Alu expression levels and DNA methylation status with chromatin functional states annotated using epigenetic signatures (Ernst and Kellis 2010). Highly expressed Alu repeats as well as lowly methylated ones were enriched in active chromatin states, especially in promoters and enhancers, although lowly methylated Alu repeats also showed an increased rate of CTCF insulator states (Fig. 6B; Supplemental Fig. S25).
Figure 6.

Transcriptomic and epigenetic features of Alu elements according to their expression levels in the HCT116 cell line. (A) Distribution of Alu repeats according to the expression levels in regard to DNA methylation, expression of the associated gene, and gene-related location. (B) Enrichment distribution of chromatin functional states in Alu elements according to their expression levels (top) and that of associated gene (middle) and the DNA methylation state. (C) Distribution of Alu repeats according to the expression levels, expression of the associated gene, and DNA methylation in regard to HMAR location. (D) Plotting of 9339 Alu repeats sorted according to their expression (purple dots) and nearest gene expression (black dots). Low DNA methylation state (green bars at the top) and HMAR location (red bars at the bottom) are indicated for each Alu.
Another important determinant of Alu expression was the location inside/outside HMARs. Most Alu repeats located in HMARs were silenced, and the few ones with transcriptional activity (10% versus 44% outside HMAR) were expressed at very low levels (about fourfold lower than outside HMAR) (Fig. 6C; Supplemental Tables S14, S15). Similar differences were observed at the gene expression level, with the highest transcriptional activity outside HMARs (Fig. 6C). Regarding DNA methylation, Alu repeats showed slightly different behaviors inside and outside HMAR: Lower levels of methylation were observed in nonexpressed and lowly expressed Alu sequences inside HMAR, whereas highly expressed Alu elements were more methylated with levels similar to those found outside HMAR (Supplemental Table S14). Combined analysis of Alu expression in regard to DNA methylation, expression of the nearest gene, and HMAR location (Fig. 6D) showed that Alu repeats inside HMAR are strongly repressed independently of the expression levels of the associated gene.
Discussion
Technical considerations
Besides the common methylated state of Alu repeats and the well-established global demethylation in cancer and aging (Esteller 2008; Ehrlich 2009), multiple studies have unveiled physiological and pathological irregularities in the methylation pattern of specific Alu elements (Hellmann-Blumberg et al. 1993; Brohede and Rand 2006; Rodriguez et al. 2008b; Rand and Molloy 2010; Luo et al. 2014). There is a vast repertoire of methodologies to analyze DNA methylation (Esteller 2007; Beck 2010; Gu et al. 2010; Jordà and Peinado 2010; Laird 2010; Stirzaker et al. 2014), and different approaches have been used to make bulk estimates of methylation in repetitive elements (Yang et al. 2004; Weisenberger et al. 2005; Choi et al. 2007; Jintaridth and Mutirangura 2010; Xiang et al. 2010; Wu et al. 2011; Yoshida et al. 2011; Gilson and Horard 2012; Buj et al. 2016). Nevertheless, analysis of repeated sequences remains a challenge in next-generation sequencing approaches (Treangen and Salzberg 2012), and there is still a lack of screening strategies that specifically allow a feasible and consistent identification of DNA methylation states in individualized repeat elements. Moreover, Alu elements pose special difficulties in bisulfite-treated DNA, offering very poor mapability and coverage compared with unique or other repeat sequences (see Supplementary Table S4, in Hansen et al. 2011).
A few methods allow the individualized analysis of DNA methylation in Alu repeats at genome scale by applying massive sequencing to bisulfite and/or enzyme treated DNA (Lister et al. 2009; Xie et al. 2009, 2010; Edwards et al. 2010; Bakshi et al. 2016). These approaches offer interesting alternatives and complementary features to profile the DNA methylation landscape of Alu repeats, although they also present technical and computational constraints precluding a broad applicability. To deal with these limitations, we developed NSUMA, a simple and efficient methodology to uncover unmethylated Alu sequences at the genome scale. NSUMA requires low amounts of biological material, library preparation and sequencing are performed with standard methods and sequencing settings (e.g., no need of long reads, low number of reads to get an acceptable coverage), and simple computational analysis (e.g., conventional mapping, internal normalization, and direct calculation of differential representation among samples).
The main advantages of NSUMA lie in the reduction of complexity of the sample (by biasing the amplification and thus overrepresenting unmethylated Alu repeats) and the unique mapping facilitated by the Alu flanking DNA sequence present in most amplicons. NSUMA represents a >100-fold reduction in the sequencing cost compared with a WGBS offering a similar number of informative Alu elements (approximately 120,000) (Supplemental Table S12). A broader coverage of Alu repeats (up to fourfold) might be achieved by using the methylation sensitive restriction enzyme HpaII (CCGG) instead of SmaI. Nevertheless, the overall efficiency of the approach would be drastically diminished due to the increased presence of non-Alu sequences (up to 68% against just 6% with the current design) (see Table 1, in Buj et al. 2016) and an important raise of multimapping short reads that could not be used for differential methylation assessment. Other approaches, as the use of alternative upstream and downstream PCR primers targeting the Alu consensus SmaI sequence (Rodriguez et al. 2008b), may also expand the coverage up to 180,000 Alu repeats, but the presence of non-Alu elements in the canonical NSUMA would be also significantly increased.
In spite of the strong bias for Alu elements, a small subset of other repeats and unique sequences are also represented in NSUMA, allowing a relative estimate of overall methylation. In particular, we have taken advantage of the presence of CpG islands to normalize the data among samples using the subset with a stable unmethylated state. Another important feature of NSUMA is the coamplification of a large number of MseI-MseI fragments independently of the methylation status. The competitive nature of the reaction results in a copy-number relative representation of these amplicons. The sensitive genome-wide profiles of losses and gains provided by NSUMA are equivalent to those obtained in array-CGH. This information is especially useful when analyzing tumor samples, as chromosomal alterations are a common feature of most cancers and may affect the interpretation of epigenetic changes.
Individual features of unmethylated Alu repeats and functional insights
A few studies have analyzed Alu DNA methylation in different tissues applying different techniques. The rates of unmethylated Alu elements in nontumor tissues range from 1% to 5% (Rodriguez et al. 2008b; Xie et al. 2009, 2010; Molaro et al. 2011), in agreement with our estimations in blood and normal colon. The low number of cases analyzed and the poor overlap between studies preclude direct comparisons and a robust classification of elements based on the methylation variability. On the other side, the finding of 225 elements that were unmethylated in all analyzed samples is highly suggestive of a steady mechanism to maintain this epigenetic state and warrants future studies to reveal the potential function of each one of these elements.
Inter-individual and inter-tissue differences were also observed. Several authors have proposed that Alu elements may act as a large reservoir of functional complexity, constituting alternative mechanisms involved in the assembly and evolution of regulatory networks. Acquired regulatory properties of Alu repeats include the generation of novel transcription factor binding sites, transcriptional regulation of some novel miRNAs, alternative splicing, and gene silencing, among others (Cordaux and Batzer 2009; Keren et al. 2010; Ichiyanagi 2013; Luo et al. 2014).
Nonetheless, and conforming to Sydney Brenner's reasoning about “junk” DNA (Brenner 2009), it is not unreasonable to claim that the massive regulatory potential of more than 1 million Alu elements of the haploid human genome is, and will be, largely latent. Integrative epigenomic analysis has shown the abundance of lowly methylated Alu repeats within active promoters compared to those in weak and poised promoters (Mallona et al. 2016). Moreover, a minority of Alu elements display signs of intrinsic functionality including the presence of activating histone modifications and a DNA unmethylated state (Rodriguez et al. 2008b; Saito et al. 2009; Edwards et al. 2010; Yoshida et al. 2011; Ward et al. 2013; Xie et al. 2013; Su et al. 2014). The presence of diverse transcription factor binding motifs in Alu sequences is common (Ruiz-Narváez and Campos 2008; Cui et al. 2011; Deininger 2011), but motif occupancy appears to be constrained by DNA methylation (Wang et al. 2012; Medvedeva et al. 2014; Domcke et al. 2015; Maurano et al. 2015; Schübeler 2015). Hence, hypomethylation of Alu repeats might enable the binding of transcription and epigenetic factors affecting gene activity and genomic architecture in both normal and cancer cells. Noteworthily, a recent study has provided experimental evidence that reactivation of the MIEN1 gene in prostate cancer is mediated by hypomethylation of an Alu element (Rajendiran et al. 2016).
In line with these postulates, we confirm here that unmethylated Alu repeats show structural and epigenetic traits consistent with functional states (e.g., proximity to promoters and CpG islands, enrichment in active chromatin histone modifications, and TF binding sites). The presence of certain epigenetic features (i.e., enhancer marks) in a specific Alu element are not compelling enough to attribute functional properties per se, and purposeful studies focused in individual repeats are needed. We are currently investigating the contribution of certain Alu repeats exhibiting dynamic DNA methylation to the modulation of genomic structure and expression of the neighboring gene.
Alu-related determinants of global genomic hypomethylation and compartmentalization in cancer
Our data provide a global overview of the methylation dynamics of Alu repeats and delineate the epigenetic landscape of chromatin states and genome compartmentalization of normal and cancer cells (Fig. 7). The hypomethylated domains detected by NSUMA were highly concordant with WGBS results, reinforcing the robustness of the approach. Colorectal cancer and cancers in general display a deregulated common genomic scenario with focal hypermethylation and large hypomethylated blocks (Hansen et al. 2011; Berman et al. 2012; Timp et al. 2014).
Figure 7.
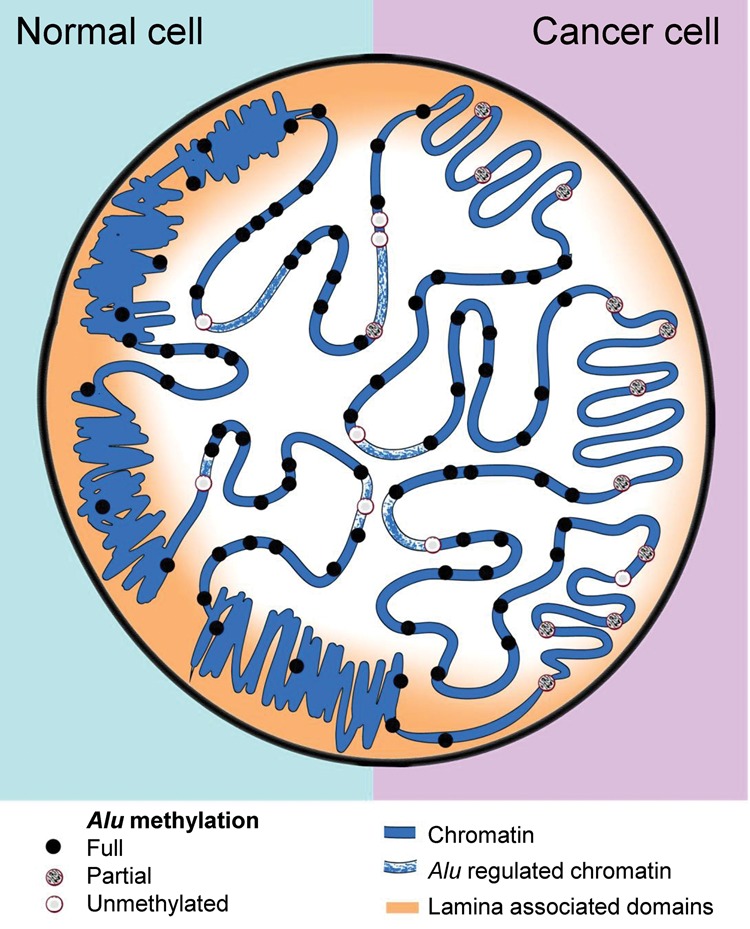
Diagram illustrating the nuclear epigenetic landscape of Alu repeats in normal and cancer cells. Alu-rich regions (dots) display high transcriptional activity and colocalize with active chromatin functional states (central part of the nucleus). In normal cells, most Alu elements remain methylated (black dots), but a few are unmethylated (gray and light gray dots) and show specific structural and functional features. Global DNA hypomethylation in cancer cells affects Alu repeats localized in late replicating lamin-associated domains, but is largely excluded from Alu-rich domains.
Notably, the associations of the hypomethylated regions with lamina-associated domains (Berman et al. 2012) and late replication timing (Fig. 5) indicate a key role of chromatin organization in the maintenance of methylation states. Classically, repeat sequences have been pointed to as main contributors to the burden of DNA hypomethylation in cancer (Ehrlich 2009). Unexpectedly, and in contraposition to other repeat elements as LINEs or MIRs, our study demonstrates the relative low contribution of Alu repeats to global hypomethylation. Indeed, Alu-rich regions retained high methylation levels in cancer cells, whereas hypomethylated blocks showed low Alu density (Fig. 3D). Incidentally, a recent study has shown Alu mediated protection of DNA methylation in placenta, a hypomethylated organ compared to somatic tissues (Chatterjee et al. 2016).
Here, we propose that Alu repeats might act as epigenetic keepers of sensitive regions. Alu repeats may play a role in chromatin interactions and could contribute to the establishment and maintenance of genomic architecture (Gu et al. 2016). Indeed, Rollins et al. reported that Alu repeats are largely excluded from unmethylated domains, but tend to occupy the boundaries of these domains (Rollins et al. 2006; Edwards et al. 2010). The presence of variably methylated Alu elements near promoters might indicate boundary plasticity ensuing changes in chromatin accessibility and transcription factor binding regulation (Domcke et al. 2015; Elliott et al. 2015; Maurano et al. 2015; Schübeler 2015). In this regard, it has been shown that most DNA methylation changes associated with development and cancer do not occur in CpG islands, but rather in sequences up to 2-kb away, which were termed CpG island shores (Doi et al. 2009; Irizarry et al. 2009). A recent study has shown that hypomethylated CpG islands create sloping CpG island shores (Grandi et al. 2015). Interestingly, Alu elements tend to be excluded from promoter regions but are enriched in nearby regions, including CpG island shores. We hypothesize that the epigenetic status of Alu repeats may contribute to set the boundaries of CpG island shores and other regions participating actively in gene regulation.
Besides the large blocks of DNA hypomethylation (Hansen et al. 2011; Berman et al. 2012; Hon et al. 2012; Timp et al. 2014) and hypermethylation (Frigola et al. 2006; Coolen et al. 2010; Forn et al. 2013), other alterations as the activation of gene clusters (Bert et al. 2013) and the increased somatic mutation rates observed in heterochromatic and late replicating regions (Schuster-Böckler and Lehner 2012; Polak et al. 2015) point out that factors regulating high-order chromatin architecture (Dekker et al. 2013; Gómez-Díaz and Corces 2014; Feinberg et al. 2016) might play a principal role in the remodeling of the cancer genome (Rodriguez et al. 2008a; Lizardi et al. 2011; Kemp et al. 2014; Lay et al. 2015; Taberlay et al. 2016). The contribution of Alu repeats to the setting and maintenance of chromatin domains and their regulation in normal and cancer cells is still unknown. Taking into account their unique features and current data, future studies should investigate their role at both the local and global levels.
Epigenetic and functional features of transcribed Alu repeats
Although multiple evidences have pointed out the impact of Alu expression in gene regulation (Cordaux and Batzer 2009; Keren et al. 2010; Ichiyanagi 2013; Luo et al. 2014), most transcriptomic analyses often disregard the presence of Alu and other repeat elements in RNA-seq reads. Our genome-scale survey of Alu transcription indicates that a large number of Alu elements (about 200,000) were present in the transcriptome; nevertheless, the expression levels of most of them were extremely low (Supplemental Table S13), which may partly explain the poor interest they raise. Our preliminary survey exposes global features of Alu transcriptional activity, including its association with the expression levels of the nearest gene, the genomic location, and the epigenetic signatures.
Alu expression contribution to genome regulation involves multiple mechanisms, and different approaches have been used to study it, including the analysis of RNA polymerase III activity, retrotransposition, noncoding RNAs, Alu mediated adenosine to inosine (A-to-I) RNA editing, and exonization among others (Oler et al. 2012; Daniel et al. 2015; Tajnik et al. 2015; Varshney et al. 2015; Klawitter et al. 2016; Lin et al. 2016; Moráles-Hernandez et al. 2016; Nishikura 2016). Therefore, unveiling the specific contribution of each element to genome regulation requires targeted customized studies.
We expect a new generation of important genomic regulators with a direct involvement of Alu repeats and other noncoding elements will be discovered in the coming years. Nevertheless, a note of caution must be sounded about extrapolating individual observations: For a large subset of Alu repeats, expression may just indicate passenger activation related with regional transcriptional activity and without any functional impact.
Final considerations
In addition to the important role of Alu repeats configuring the epigenomic landscape by determining nucleosome positioning (Englander and Howard 1995; Salih et al. 2008) and genome compartmentalization (Rollins et al. 2006; Edwards et al. 2010), a growing number of papers underscore the direct contribution of Alu repeats in gene regulation and as a source of transcriptional plasticity (Jasinska and Krzyzosiak 2004; Häsler and Strub 2006; Cordaux and Batzer 2009; Su et al. 2014). Despite this, most studies addressed to explore the regulatory complexity of cell processes devote little or no attention at all to these elements. This is explained by pragmatic and technical constraints: Just a fraction of these elements are likely to have a specific functional role, and their repeat nature impairs their individualized analysis. The feasibility of identifying repeat elements as unique loci and characterizing their epigenetic signatures offers an opportunity to illuminate the specific contribution of these elements to genomic organization and the dynamic regulation of cell programs. The identification of the unmethylated Alu repeats by NSUMA provides a cost-effective resource of candidate regulatory elements and sets the starting line in a quest for the mechanisms and functional consequences of epigenetic diversity.
Methods
Samples
Three colorectal tumors and their paired nonadjacent normal colonic mucosa as well as the corresponding whole blood DNA were used in this analysis. Samples were obtained from the Hospital Germans Trias i Pujol following a protocol approved by the Local Ethics Committee (Ref. CEIC EO-11-134). Human colon cancer cell lines were obtained and cultured as described in Supplemental Material.
Next-generation sequencing of unmethylated Alu repeats (NSUMA)
Briefly, 1 μg of genomic DNA was double digested with the methylation-sensitive restriction endonuclease SmaI (CCC/GGG) and MseI (T/TAA). The fragments were ligated to blunt (SmaI-ends) and sticky (MseI-ends) adapters, PCR amplified using primers specific of each adapter, and with additional nucleotides at the 3′ end to enrich for Alu elements (Fig. 1A; Supplemental Fig. S1). Sequencing libraries were generated using standard procedures and ran on an Illumina Genome Analyzer IIx or a HiSeq 2000 (Supplemental Methods).
RNA sequencing
Poly(A)+ messenger RNA was obtained from two independent cultures of the HCT116 cell line and sequenced separately using paired-end RNA-seq using standard Illumina protocols (Supplemental Methods).
Data analysis summary
An overview of data processing is shown in Supplemental Figure S2, and a more detailed description is provided in Supplemental Methods. Briefly, only reads mapping unambiguously to the human reference genome (NCBI build 37/hg19) were considered. This reference genome was chosen based on the rich availability of functionally relevant data (used in our study) compared with the NCBI GRCh38 build at the time of the analysis. Moreover, our analyses consisted of direct comparisons between samples of amplicon representation, which are unaffected by the specific genome assembly. Therefore, using the NCBI GRCh38 genome assembly instead of GRCh37 would not significantly affect our conclusions. Each read was assigned to a canonical amplicon as determined in a virtual NSUMA universe based on hg19 sequence (regions flanked by the restriction sites plus primer extended nucleotides). Differential methylation in every canonical amplicon was determined based in the number of reads mapping in each amplicon after normalization. DESeq R package (Anders and Huber 2010) was used for differential representation analysis. Regional differential methylation and copy number variation were determined using the DNAcopy R package (Venkatraman and Olshen 2007). RNA-seq data was mapped using TopHat2 (Kim et al. 2013) and summarized using featureCounts from the subread package.
Data access
All genomic sequencing data generated for this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number NCBI GSE72751.
Competing interest statement
M.A.P. is cofounder and equity holder of Aniling, a biotech company developing technologies focused on the improvement of DNA sequencing methods to integrate genetic and epigenetic information.
Supplementary Material
Acknowledgments
We thank Mar Muñoz, Anna Ferrer, Heinz Himmelbauer, and Lauro Sumoy for technical advice and support; Kasper D. Hansen and Rafael Irizarry for data sharing; and the Biobank IGTP-HUGTiP for providing the samples. A.D.-V. was supported in part by a contract PTC2011-1091 from Ministerio de Economía y Competitividad. V.B. was supported by a PFIS fellowship from Instituto de Salud Carlos III. This work was supported by grants from FEDER, the Ministerio de Economía y Competitividad (CSD2006/49, SAF2011/23638, and SAF2015-64521-R to M.A.P.), the Instituto de Salud Carlos III (FIS PI14/00308 to M.J.), Fundación Merck Serono (to M.J.), and Fundació Olga Torres (to M.J.).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.207522.116.
References
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakshi A, Herke S, Batzer MA, Kim J. 2016. DNA methylation variation of human-specific Alu repeats. Epigenetics 11: 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera V, Peinado MA. 2012. Evaluation of single CpG sites as proxies of CpG island methylation states at the genome scale. Nucleic Acids Res 40: 11490–11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck S. 2010. Taking the measure of the methylome. Nat Biotechnol 28: 1026–1028. [DOI] [PubMed] [Google Scholar]
- Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CP, van Dijk CM, Tollenaar RA, et al. 2012. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina–associated domains. Nat Genet 44: 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Meissner A, Lander ES. 2007. The mammalian epigenome. Cell 128: 669–681. [DOI] [PubMed] [Google Scholar]
- Bert SA, Robinson MD, Strbenac D, Statham AL, Song JZ, Hulf T, Sutherland RL, Coolen MW, Stirzaker C, Clark SJ. 2013. Regional activation of the cancer genome by long-range epigenetic remodeling. Cancer Cell 23: 9–22. [DOI] [PubMed] [Google Scholar]
- Bird A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21. [DOI] [PubMed] [Google Scholar]
- Bock C, Tomazou EM, Brinkman AB, Müller F, Simmer F, Gu H, Jäger N, Gnirke A, Stunnenberg HG, Meissner A. 2010. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 28: 1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. 2009. Interview with Sydney Brenner by Soraya de Chadarevian. Stud Hist Philos Biol Biomed Sci 40: 65–71. [DOI] [PubMed] [Google Scholar]
- Brohede J, Rand KN. 2006. Evolutionary evidence suggests that CpG island-associated Alus are frequently unmethylated in human germline. Hum Genet 119: 457–458. [DOI] [PubMed] [Google Scholar]
- Buj R, Mallona I, Díez-Villanueva A, Barrera V, Mauricio D, Puig-Domingo M, Reverter JL, Matias-Guiu X, Azuara D, Ramírez JL, et al. 2016. Quantification of unmethylated Alu (QUAlu): a tool to assess global hypomethylation in routine clinical samples. Oncotarget 7: 10536–10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Macaulay EC, Rodger EJ, Stockwell PA, Parry MF, Roberts HE, Slatter TL, Hung NA, Devenish CJ, Morison IM. 2016. Placental hypomethylation is more pronounced in genomic loci devoid of retroelements. G3 (Bethesda) 6: 1911–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Gentles AJ, Jurka J, Karlin S. 2002. Genes, pseudogenes, and Alu sequence organization across human chromosomes 21 and 22. Proc Natl Acad Sci 99: 2930–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi IS, Estecio MR, Nagano Y, Kim do H, White JA, Yao JC, Issa JP, Rashid A. 2007. Hypomethylation of LINE-1 and Alu in well-differentiated neuroendocrine tumors (pancreatic endocrine tumors and carcinoid tumors). Mod Pathol 20: 802–810. [DOI] [PubMed] [Google Scholar]
- Coolen MW, Stirzaker C, Song JZ, Statham AL, Kassir Z, Moreno CS, Young AN, Varma V, Speed TP, Cowley M, et al. 2010. Consolidation of the cancer genome into domains of repressive chromatin by long-range epigenetic silencing (LRES) reduces transcriptional plasticity. Nat Cell Biol 12: 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA. 2009. The impact of retrotransposons on human genome evolution. Nat Rev Genet 10: 691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui F, Sirotin MV, Zhurkin VB. 2011. Impact of Alu repeats on the evolution of human p53 binding sites. Biol Direct 6: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel C, Behm M, Öhman M. 2015. The role of Alu elements in the cis-regulation of RNA processing. Cell Mol Life Sci 72: 4063–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deininger P. 2011. Alu elements: Know the SINEs. Genome Biol 12: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Marti-Renom MA, Mirny LA. 2013. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet 14: 390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al. 2009. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet 41: 1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domcke S, Bardet AF, Adrian Ginno P, Hartl D, Burger L, Schübeler D. 2015. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 528: 575–579. [DOI] [PubMed] [Google Scholar]
- Eden A, Gaudet F, Waghmare A, Jaenisch R. 2003. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300: 455. [DOI] [PubMed] [Google Scholar]
- Edwards JR, O'Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, Chanrion B, Fu Y, Su T, Hibshoosh H, et al. 2010. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res 20: 972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M. 2009. DNA hypomethylation in cancer cells. Epigenomics 1: 239–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott G, Hong C, Xing X, Zhou X, Li D, Coarfa C, Bell RJ, Maire CL, Ligon KL, Sigaroudinia M, et al. 2015. Intermediate DNA methylation is a conserved signature of genome regulation. Nat Commun 6: 6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander EW, Howard BH. 1995. Nucleosome positioning by human Alu elements in chromatin. J Biol Chem 270: 10091–10096. [DOI] [PubMed] [Google Scholar]
- Ernst J, Kellis M. 2010. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol 28: 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. 2007. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 8: 286–298. [DOI] [PubMed] [Google Scholar]
- Esteller M. 2008. Epigenetics in cancer. N Engl J Med 358: 1148–1159. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Tycko B. 2004. The history of cancer epigenetics. Nat Rev Cancer 4: 143–153. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Koldobskiy MA, Gondor A. 2016. Disease mechanisms: epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 17: 284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forn M, Muñoz M, Tauriello DV, Merlos-Suárez A, Rodilla V, Bigas A, Batlle E, Jordà M, Peinado MA. 2013. Long range epigenetic silencing is a trans-species mechanism that results in cancer specific deregulation by overriding the chromatin domains of normal cells. Mol Oncol 7: 1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigola J, Song J, Stirzaker C, Hinshelwood RA, Peinado MA, Clark S. 2006. Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat Genet 38: 540–549. [DOI] [PubMed] [Google Scholar]
- Gilson E, Horard B. 2012. Comprehensive DNA methylation profiling of human repetitive DNA elements using an MeDIP-on-RepArray assay. Methods Mol Biol 859: 267–291. [DOI] [PubMed] [Google Scholar]
- Goll MG, Bestor TH. 2005. Eukaryotic cytosine methyltransferases. Annu Rev Biochem 74: 481–514. [DOI] [PubMed] [Google Scholar]
- Gómez-Díaz E, Corces VG. 2014. Architectural proteins: regulators of 3D genome organization in cell fate. Trends Cell Biol 24: 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi FC, Rosser JM, Newkirk SJ, Yin J, Jiang X, Xing Z, Whitmore L, Bashir S, Ivics Z, Izsvák Z, et al. 2015. Retrotransposition creates sloping shores: a graded influence of hypomethylated CpG islands on flanking CpG sites. Genome Res 25: 1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover D, Mukerji M, Bhatnagar P, Kannan K, Brahmachari SK. 2004. Alu repeat analysis in the complete human genome: trends and variations with respect to genomic composition. Bioinformatics 20: 813–817. [DOI] [PubMed] [Google Scholar]
- Gu H, Bock C, Mikkelsen TS, Jäger N, Smith ZD, Tomazou E, Gnirke A, Lander ES, Meissner A. 2010. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat Methods 7: 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Jin K, Crabbe MJ, Zhang Y, Liu X, Huang Y, Hua M, Nan P, Zhang Z, Zhong Y. 2016. Enrichment analysis of Alu elements with different spatial chromatin proximity in the human genome. Protein Cell 7: 250–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. 2008. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453: 948–951. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. 2009. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci 107: 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, et al. 2011. Increased methylation variation in epigenetic domains across cancer types. Nat Genet 43: 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häsler J, Strub K. 2006. Alu elements as regulators of gene expression. Nucleic Acids Res 34: 5491–5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann-Blumberg U, Hintz MF, Gatewood JM, Schmid CW. 1993. Developmental differences in methylation of human Alu repeats. Mol Cell Biol 13: 4523–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE, et al. 2012. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res 22: 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyanagi K. 2013. Epigenetic regulation of transcription and possible functions of mammalian short interspersed elements, SINEs. Genes Genet Syst 88: 19–29. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. 2009. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41: 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasinska A, Krzyzosiak WJ. 2004. Repetitive sequences that shape the human transcriptome. FEBS Lett 567: 136–141. [DOI] [PubMed] [Google Scholar]
- Jintaridth P, Mutirangura A. 2010. Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol Genomics 41: 194–200. [DOI] [PubMed] [Google Scholar]
- Jones PA. 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492. [DOI] [PubMed] [Google Scholar]
- Jordà M, Peinado MA. 2010. Methods for DNA methylation analysis and applications in colon cancer. Mutat Res 693: 84–93. [DOI] [PubMed] [Google Scholar]
- Karpf AR, Matsui S. 2005. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res 65: 8635–8639. [DOI] [PubMed] [Google Scholar]
- Kemp CJ, Moore JM, Moser R, Bernard B, Teater M, Smith LE, Rabaia NA, Gurley KE, Guinney J, Busch SE, et al. 2014. CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell Rep 7: 1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H, Lev-Maor G, Ast G. 2010. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 11: 345–355. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klawitter S, Fuchs NV, Upton KR, Muñoz-Lopez M, Shukla R, Wang J, Garcia-Cañadas M, Lopez-Ruiz C, Gerhardt DJ, Sebe A, et al. 2016. Reprogramming triggers endogenous L1 and Alu retrotransposition in human induced pluripotent stem cells. Nat Commun 7: 10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird PW. 2010. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet 11: 191–203. [DOI] [PubMed] [Google Scholar]
- Lay FD, Liu Y, Kelly TK, Witt H, Farnham PJ, Jones PA, Berman BP. 2015. The role of DNA methylation in directing the functional organization of the cancer epigenome. Genome Res 25: 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Jiang P, Park JW, Wang J, Lu ZX, Lam MP, Ping P, Xing Y. 2016. The contribution of Alu exons to the human proteome. Genome Biol 17: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizardi PM, Forloni M, Wajapeyee N. 2011. Genome-wide approaches for cancer gene discovery. Trends Biotechnol 29: 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Lu X, Xie H. 2014. Dynamic Alu methylation during normal development, aging, and tumorigenesis. Biomed Res Int 2014: 784706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallona I, Jordà M, Peinado MA. 2016. A knowledgebase of the human Alu repetitive elements. J Biomed Inform 60: 77–83. [DOI] [PubMed] [Google Scholar]
- Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, Stamatoyannopoulos JA. 2015. Role of DNA methylation in modulating transcription factor occupancy. Cell Rep 12: 1184–1195. [DOI] [PubMed] [Google Scholar]
- Medvedeva YA, Khamis AM, Kulakovskiy IV, Ba-Alawi W, Bhuyan MS, Kawaji H, Lassmann T, Harbers M, Forrest AR, Bajic VB. 2014. Effects of cytosine methylation on transcription factor binding sites. BMC Genomics 15: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, Smith AD. 2011. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 146: 1029–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moráles-Hernandez A, González-Rico FJ, Román AC, Rico-Leo E, Alvarez-Barrientos A, Sánchez L, Macia Á, Heras SR, García-Pérez JL, Merino JM, et al. 2016. Alu retrotransposons promote differentiation of human carcinoma cells through the aryl hydrocarbon receptor. Nucleic Acids Res 44: 4665–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. 2016. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17: 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oler AJ, Traina-Dorge S, Derbes RS, Canella D, Cairns BR, Roy-Engel AM. 2012. Alu expression in human cell lines and their retrotranspositional potential. Mob DNA 3: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. 2004. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5: 557–572. [DOI] [PubMed] [Google Scholar]
- Perucho M, Welsh J, Peinado MA, Ionov Y, McClelland M. 1995. Fingerprinting of DNA and RNA by arbitrarily primed polymerase chain reaction: applications in cancer research. Methods Enzymol 254: 275–290. [DOI] [PubMed] [Google Scholar]
- Polak P, Karlić R, Koren A, Thurman R, Sandstrom R, Lawrence MS, Reynolds A, Rynes E, Vlahoviček K, Stamatoyannopoulos JA, et al. 2015. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 518: 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendiran S, Gibbs LD, Van Treuren T, Klinkebiel DL, Vishwanatha JK. 2016. MIEN1 is tightly regulated by SINE Alu methylation in its promoter. Oncotarget. 10.18632/oncotarget.11675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand KN, Molloy PL. 2010. Sensitive measurement of unmethylated repeat DNA sequences by end-specific PCR. Biotechniques 49: xiii–xvii. [DOI] [PubMed] [Google Scholar]
- Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, et al. 2002. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416: 552–556. [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Frigola J, Vendrell E, Risques RA, Fraga MF, Morales C, Moreno V, Esteller M, Capellá G, Ribas M, et al. 2006. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res 66: 8462–9468. [DOI] [PubMed] [Google Scholar]
- Rodriguez J, Muñoz M, Vives L, Frangou CG, Groudine M, Peinado MA. 2008a. Bivalent domains enforce transcriptional memory of DNA methylated genes in cancer cells. Proc Natl Acad Sci 105: 19809–19814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Vives L, Jordà M, Morales C, Muñoz M, Vendrell E, Peinado MA. 2008b. Genome-wide tracking of unmethylated DNA Alu repeats in normal and cancer cells. Nucleic Acids Res 36: 770–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins RA, Haghighi F, Edwards JR, Das R, Zhang MQ, Ju J, Bestor TH. 2006. Large-scale structure of genomic methylation patterns. Genome Res 16: 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Narváez EA, Campos H. 2008. Evolutionary rate heterogeneity of Alu repeats upstream of the APOA5 gene: Do they regulate APOA5 expression? J Hum Genet 53: 247–253. [DOI] [PubMed] [Google Scholar]
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. 2010. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res 20: 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T, Hiratani I, Sasaki T, Battaglia D, Kulik M, Zhang J, Dalton S, Gilbert DM. 2011. Replication timing: a fingerprint for cell identity and pluripotency. PLoS Comput Biol 7: e1002225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Suzuki H, Tsugawa H, Nakagawa I, Matsuzaki J, Kanai Y, Hibi T. 2009. Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of Mcl-1 in human gastric cancer cells. Oncogene 28: 2738–2744. [DOI] [PubMed] [Google Scholar]
- Salih F, Salih B, Kogan S, Trifonov EN. 2008. Epigenetic nucleosomes: Alu sequences and CG as nucleosome positioning element. J Biomol Struct Dyn 26: 9–16. [DOI] [PubMed] [Google Scholar]
- Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. 2011. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 6: 692–702. [DOI] [PubMed] [Google Scholar]
- Schübeler D. 2015. Function and information content of DNA methylation. Nature 517: 321–326. [DOI] [PubMed] [Google Scholar]
- Schuster-Böckler B, Lehner B. 2012. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 488: 504–507. [DOI] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. 1981. Identification of common molecular subsequences. J Mol Biol 147: 195–197. [DOI] [PubMed] [Google Scholar]
- Stirzaker C, Taberlay PC, Statham AL, Clark SJ. 2014. Mining cancer methylomes: prospects and challenges. Trends Genet 30: 75–84. [DOI] [PubMed] [Google Scholar]
- Su M, Han D, Boyd-Kirkup J, Yu X, Han JD. 2014. Evolution of Alu elements toward enhancers. Cell Rep 7: 376–385. [DOI] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465–476. [DOI] [PubMed] [Google Scholar]
- Taberlay PC, Achinger-Kawecka J, Lun AT, Buske FA, Sabir K, Gould CM, Zotenko E, Bert SA, Giles KA, Bauer DC, et al. 2016. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res 26: 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajnik M, Vigilante A, Braun S, Hänel H, Luscombe NM, Ule J, Zarnack K, König J. 2015. Intergenic Alu exonisation facilitates the evolution of tissue-specific transcript ends. Nucleic Acids Res 43: 10492–10505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timp W, Bravo HC, McDonald OG, Goggins M, Umbricht C, Zeiger M, Feinberg AP, Irizarry RA. 2014. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med 6: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Salzberg SL. 2012. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 13: 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney D, Vavrova-Anderson J, Oler AJ, Cowling VH, Cairns BR, White RJ. 2015. SINE transcription by RNA polymerase III is suppressed by histone methylation but not by DNA methylation. Nat Commun 6: 6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman ES, Olshen AB. 2007. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 23: 657–663. [DOI] [PubMed] [Google Scholar]
- Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, Lee K, Canfield T, Weaver M, Sandstrom R, et al. 2012. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res 22: 1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward MC, Wilson MD, Barbosa-Morais NL, Schmidt D, Stark R, Pan Q, Schwalie PC, Menon S, Lukk M, Watt S, et al. 2013. Latent regulatory potential of human-specific repetitive elements. Mol Cell 49: 262–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberger DJ, Campan M, Long TI, Kim M, Woods C, Fiala E, Ehrlich M, Laird PW. 2005. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res 33: 6823–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AS, Power BE, Molloy PL. 2007. DNA hypomethylation and human diseases. Biochim Biophys Acta 1775: 138–162. [DOI] [PubMed] [Google Scholar]
- Wu HC, Delgado-Cruzata L, Flom JD, Kappil M, Ferris JS, Liao Y, Santella RM, Terry MB. 2011. Global methylation profiles in DNA from different blood cell types. Epigenetics 6: 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang S, Liu Z, Zhang B, Zhou J, Zhu BD, Ji J, Deng D. 2010. Methylation status of individual CpG sites within Alu elements in the human genome and Alu hypomethylation in gastric carcinomas. BMC Cancer 10: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Wang M, Bonaldo Mde F, Smith C, Rajaram V, Goldman S, Tomita T, Soares MB. 2009. High-throughput sequence-based epigenomic analysis of Alu repeats in human cerebellum. Nucleic Acids Res 37: 4331–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Wang M, Bonaldo MD, Rajaram V, Stellpflug W, Smith C, Arndt K, Goldman S, Tomita T, Soares MB. 2010. Epigenomic analysis of Alu repeats in human ependymomas. Proc Natl Acad Sci 107: 6952–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Hong C, Zhang B, Lowdon RF, Xing X, Li D, Zhou X, Lee HJ, Maire CL, Ligon KL, et al. 2013. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nat Genet 45: 836–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. 2004. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res 32: e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Yamashita S, Takamura-Enya T, Niwa T, Ando T, Enomoto S, Maekita T, Nakazawa K, Tatematsu M, Ichinose M, et al. 2011. Alu and Satα hypomethylation in Helicobacter pylori-infected gastric mucosae. Int J Cancer 128: 33–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.