

Abstract
Improvement of variant calling in next-generation sequence data requires a comprehensive, genome-wide catalog of high-confidence variants called in a set of genomes for use as a benchmark. We generated deep, whole-genome sequence data of 17 individuals in a three-generation pedigree and called variants in each genome using a range of currently available algorithms. We used haplotype transmission information to create a phased “Platinum” variant catalog of 4.7 million single-nucleotide variants (SNVs) plus 0.7 million small (1–50 bp) insertions and deletions (indels) that are consistent with the pattern of inheritance in the parents and 11 children of this pedigree. Platinum genotypes are highly concordant with the current catalog of the National Institute of Standards and Technology for both SNVs (>99.99%) and indels (99.92%) and add a validated truth catalog that has 26% more SNVs and 45% more indels. Analysis of 334,652 SNVs that were consistent between informatics pipelines yet inconsistent with haplotype transmission (“nonplatinum”) revealed that the majority of these variants are de novo and cell-line mutations or reside within previously unidentified duplications and deletions. The reference materials from this study are a resource for objective assessment of the accuracy of variant calls throughout genomes.
Recent disruptive changes in sequencing technology (Bentley et al. 2008; Drmanac et al. 2010) have led to a massive growth in the use of DNA sequencing in research and clinical applications (The 1000 Genomes Project Consortium 2010; The International Cancer Genome Consortium 2010; Erikson et al. 2016). Accurate calling of genetic variants in sequence data is essential as sequencing moves into new settings such as clinical laboratories (Gullapalli et al. 2012; Goldfeder et al. 2016). It is anticipated that genomic sequence information will improve the precision of clinical diagnosis as part of the new initiatives in precision medicine (Ashley 2015; Marx 2015). The field of next-generation sequencing (NGS) is evolving rapidly: Continual improvements in technology and informatics underline the need for effective ways to measure the quality of sequence data and variant calls, so that it is possible to perform objective comparisons of different methods. Robust benchmarking enables us to better understand the accuracy of sequence data, to identify underlying causes of error, and to quantify the improvements obtained from algorithmic developments.
It is important to assess aspects of variant calling accuracy such as the fraction of true variants detected (recall) and the fraction of the variants called that are true (precision). One approach is to test variant calls made by an NGS method using an orthogonal technology (e.g., array-based genotyping or Sanger sequencing) and then to measure the degree of concordance between results (Ajay et al. 2011; The 1000 Genomes Project Consortium 2012; Pirooznia et al. 2014). This approach can provide a measure of precision of a variant caller, but not recall, as recall estimates require knowledge of what is missed. Additionally, the resulting measure of precision is typically based on a few hundred variants and is then extrapolated to the entire variant call set. Limitations in this approach to validation include cost and incompleteness due to failed or erroneous results from the orthogonal technology. A second approach is to compare technical and/or informatic replicates of a data set (Lam et al. 2012; O'Rawe et al. 2013; Zook et al. 2014). It is assumed that a variant call is correct if it is seen in multiple analyses or data sets. Although this approach allows rapid comparison of large variant call sets, estimates of precision and recall of one variant call set can only be expressed relative to a second set; it is not possible to know which variants are true in either set. Additionally, calls found in two data sets may be categorized as correct even where they are in fact systematic errors in both sets. A significant limitation of this approach is that some of the variants called by just one method may be correct and may provide valuable insights on how to improve variant calling, but these variants are excluded from further consideration by this approach. A third approach is to sequence parent–parent–child trios and test for Mendelian consistency (Boland et al. 2013; Patel et al. 2014). Although this approach can detect a subset of errors, it falls short of identifying genotyping errors that do not violate inheritance in a trio (Supplemental Tables S3, S4).
In the present study, we generated a genome-wide catalog of 5.4 million phased “platinum” variants. We included variant calls from six different informatics pipelines (Conrad et al. 2011; Garrison and Marth 2012; Iqbal et al. 2012; Saunders et al. 2012; Raczy et al. 2013; Rimmer et al. 2014) and two different sequencing technologies (Bentley et al. 2008; Drmanac et al. 2010). We used an inheritance-based validation based on a family of two parents and 11 children to resolve conflicts between different call sets and to include high confidence variants called by a single informatics pipeline. Compared to previous studies, the genetic inheritance provided by this large family serves as a validation of all of the variants within this study and provides an unbiased assessment of different technologies and software pipelines. We also examined features of the SNVs that failed to segregate consistently with the inheritance and conclude that the majority of these conflicts are caused by SNVs that colocalize with copy number variants (CNVs). The data from this study are fully open source so that groups can reanalyze and provide feedback to the catalog as needed. As the technologies improve, this community resource will continue to evolve by adding more validated variants, including CNVs and structural variants (SVs) identified from new sequencing technologies and analysis methods.
Results
Phasing the pedigree
To obtain phased variants across a large pedigree, we sequenced each of the four grandparents, two parents, and 11 children of CEPH pedigree 1463 (Fig. 1; Dausset et al. 1990) on an Illumina HiSeq2000 to an average depth of 50× using 2 × 100 bp reads and PCR-free sample preparation. We determined the genome-wide transmission of the parental haplotypes to each of the 11 children in the pedigree and identified 731 inheritance vectors between parents and children within the autosomes, plus 16 distinct inheritance vectors on Chromosome X (Methods). In agreement with previous studies (Kong et al. 2010), we observed a higher number of crossovers in maternal versus paternal autosomal haplotypes: 58% (415/709, 1.35 cM/Mb) compared to 42% (302/709, 0.98 cM/Mb) (Supplemental Fig. S1).
Figure 1.
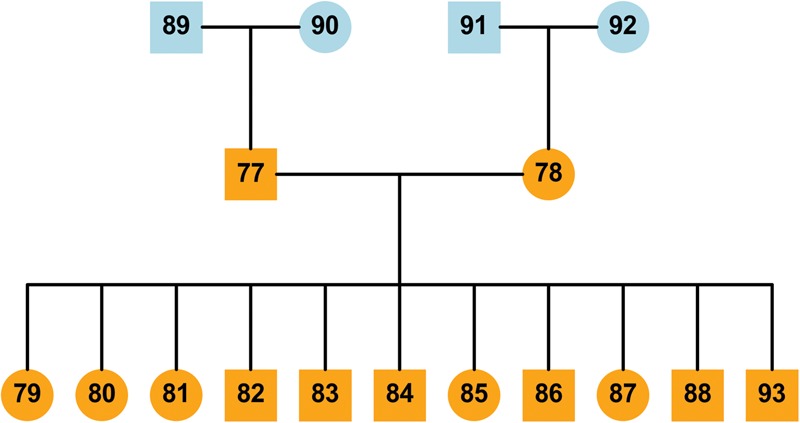
Pedigree of the family sequenced for this study (CEPH pedigree 1463). The Coriell ID for each sample is defined by adding the prefix NA128 to each numbered individual: e.g., 77 = NA12877. Samples filled with dark orange are used in this analysis but the founder generations (blue) were also sequenced and used as further validation of the haplotypes generated in this study. The trio, 91-92-78, was also sequenced during Phase I of the 1000 Genomes Project (The 1000 Genomes Project Consortium 2010).
Sequencing and phasing a larger pedigree increases the ability to detect errors and assess the accuracy of more of the variants compared to a trio analysis. Theoretically, by sequencing the parents and 11 children, both parental haplotypes will be represented in at least one of the children for 99.8% of the genome, and single errors in variant calls can be detected in all of these regions in any member of the pedigree for which sequence data are available (Supplemental Table S6). To this effect, combining the genotype calls with the haplotype inheritance vectors enables identification of genotype errors as well as other factors that lead to Mendelian inheritance inconsistency, such as when variants cosegregate with deletions and duplications.
Platinum variants
To generate a comprehensive catalog of pedigree-validated variants, we used six analysis pipelines (Supplemental Table S1) to call SNVs and indels. For each individual call set, we identified variants in which the genotypes are present in all 13 individuals and are also consistent with the transmission of the parental haplotypes (Supplemental Material, section 1). Next we merged each of the six call sets into one catalog, using the following inclusion criteria: (1) The genotypes and alleles were concordant between call sets (154,681 variants removed by this rule), and (2) for monoallelic variants (i.e., homozygous for the alternate allele in every sample), the variant was included in call sets using at least two different sequence aligners (103,603 variants removed by this rule). Note that we included all variants that met the above criteria regardless of whether or not they passed the default quality filters of each analysis pipeline. Instead, we relied on the sensitivity of the genetic inheritance to detect genotyping errors and maximize the chance of including true variants that might otherwise be removed by suboptimal filtering.
It is still possible that misalignments may lead to false genotypes that are consistent with the inheritance or a single variant may be represented as two different, nonoverlapping variants when merging the call sets. To identify errors such as these, we required that some of the reads supporting each variant call also supported the flanking sequence using a k-mer approach. In brief, we declared each variant valid only if the supporting reads contained sequence confirming each allele, and the flanking sequence in a 51-bp window centered on the variant (Supplemental Figs. S2, S3). We performed this check for every variant in the catalog and removed 132,528 SNVs and 64,917 indels in which the genotype calls were consistent with inheritance but failed this additional flanking-sequence test (Supplemental Table S7).
In total, we identified 5,426,236 phased variants (4,729,676 SNVs; 693,623 indels; and 2937 complex variants with overlapping SNVs and indels) that segregate consistently with haplotype inheritance and also pass the flanking-sequence test; including 159,167 pedigree-consistent variants that were identified from a single sequencing pipeline and 1042 SNVs that were unique to the Complete Genomics (CGI) data (Drmanac et al. 2010). These variants have features consistent with high-quality sequencing data. For example, the ratio of transitions to transversions in the pedigree-validated SNVs is 2.11 (all) and 2.42 (coding), and the het:hom ratios of SNVs and indels in both the parents of this family are in agreement with both previous studies and theoretical predictions (Supplemental Table S9). In addition to the 2937 complex variants, the set also includes 59,310 multiallelic variants (2728 SNVs and 56,582 indels). The indel loci include short tandem repeats such as homopolymer tracts and di- and trinucleotide repeats (Supplemental Material, section 1.5), which are known to be less stable than SNVs; hence, we expect a significant number of the indels to be multiallelic, as observed here.
To identify invariant positions of high confidence, we collated all genomic positions that were called as homozygous reference in every sample by at least two pipelines. As for the monoallelic variants (described above), we required that the different pipelines included at least two different sequence aligners. Additionally, we filtered out all positions where any pipeline made a variant call in any sample to exclude missed variants and regions of high error rates. Based on these rules, we identified 2,737,246,156 positions that are homozygous reference across the pedigree. These positions can be used to calculate false positive rates when assessing variant calling pipelines.
Using founder haplotypes to validate platinum variants
We utilized the founders to provide an additional assessment of the phasing and variant quality by testing whether the predicted haplotype variants are observed in the appropriate founders (i.e., individuals 89, 90, 91, or 92) (Fig. 1). All the platinum variants identified by this study are phased, and one of the haplotypes will be present in each founder. To quantify the presence of the variant in the founders without relying on the variant callers, we tested whether the k-mers described above were observed in the appropriate founders. This analysis supported 99.5% of the 5,426,236 variants in all of the founders, leaving 28,371 lower-confidence variants (Supplemental Table S8). Variants with a low normalized k-mer value are more likely to fail this test due to either lower average depth or higher error rate. For example, ∼97.3% of the platinum variants have at least 5 k-mers supporting each allele (Supplemental Table S7) compared with just 16.4% (4,652) of the variants that failed the founder test. After excluding the 23,719 variants with fewer than 5 k-mers supporting each allele, many of the remaining variants exhibit signs of clustering around biological mutational events in a grandparental haplotype. We presume, therefore, that they arose either in early development or during generation or culture of the cell line: Specifically, 2066 (46.7%) occur in 135 clusters of between five and 260 spatially adjacent variants in the same founder (Fig. 2; Supplemental Fig. S6).
Figure 2.
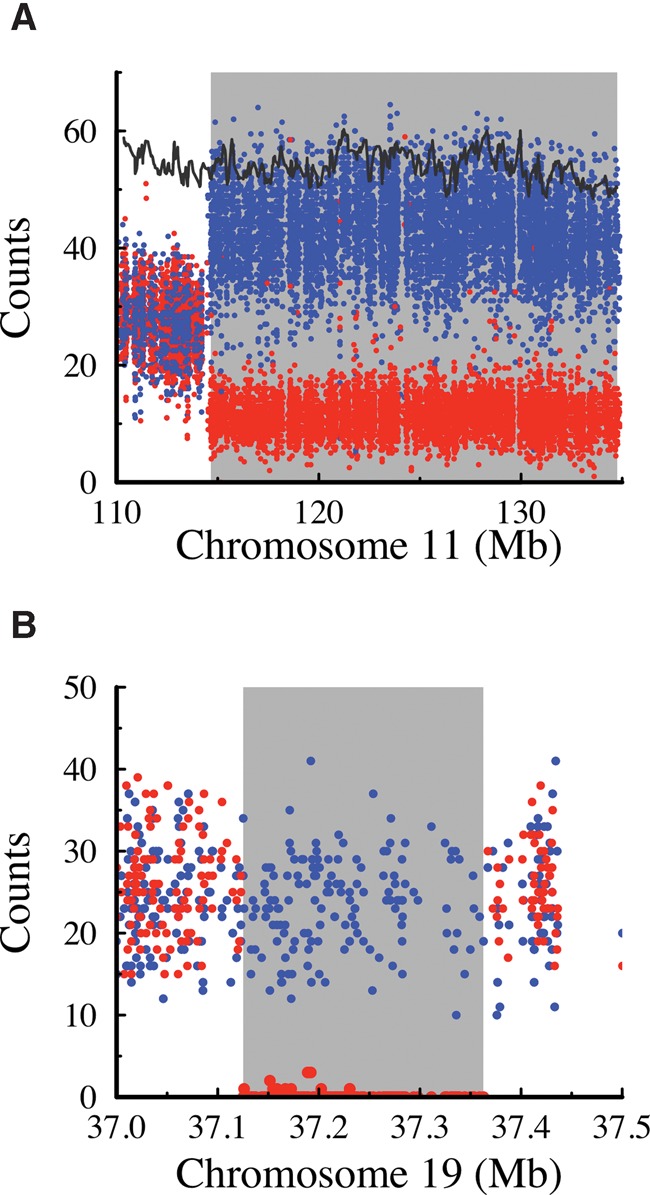
Structural abnormalities account for most of the inconsistencies in detection of the platinum haplotypes in the founders. (A) A 20-Mb structural abnormality identified by 177 variants in nine clusters that failed the founder haplotype validation test in NA12889. Based on the marked skew in frequency of the SNV alleles within the rearrangement (4:1) compared to the proximal flanking region (1:1), this event is likely a mosaic on the distal end of Chromosome 11, specific to NA12889. Each point represents the allele count at a SNV location expected to be heterozygous in NA12889 based on seeing the nontransmitted allele at least six times. Red points show the allele counts (average n = 11 within the mosaic) of the haplotype transmitted to NA12877, and blue points show the allele counts (average n = 43 in the mosaic) of the nontransmitted haplotype. The black line shows the average total depth in windows of 100 SNVs, highlighting that this mosaic is not associated with a change in copy number. (B) Allele counts for SNV positions in a possible cell-line somatic deletion in NA12891 identified by a cluster of 174 k-mer failures. Points are colored the same as in A. Within this deletion, there are virtually no reads corresponding to the transmitted haplotype, whereas a relatively constant read depth was observed across the region for the nontransmitted haplotype.
It should be noted that these biological mutational events are limited to the founders and thus do not affect the pedigree consistency or our assessment that these variants are accurately called in the main pedigree. Overall, for the variants in which at least one of the founders did not have the appropriate k-mer, 99.2% show strong supporting evidence for the variant, either because the k-mer was seen as predicted in another founder (72.2%) and/or the k-mer was seen multiple (5+) times in the pedigree (99.3%). Because of this, we conclude that most of the failures in the founders are caused by complex genetic events in the grandparents and do not affect the concordance of transmission. These variants are flagged but not removed from the Platinum catalog.
Nonplatinum variants
The nonplatinum variants that failed our validation process revealed important biological insights. We examined a total of 334,652 high-quality SNV calls, in which at least two pipelines provided the same answer for all genotype calls in the parents and all children, but the genotypes were inconsistent with haplotype inheritance. This analysis excluded all nonplatinum SNV calls that might be of low quality, e.g., in which there were missing data, or discrepancies between two callers, or variants called by only one caller. We classified the high-quality failed SNVs into four categories. Category 1 SNVs (191,087) are heterozygous in all thirteen individuals. Most (91%) of the SNVs in this category are clustered at specific locations in the genome and also deviate significantly (P < 0.01) from Hardy Weinberg Equilibrium (HWE) in a population of European ancestry, having an excess of heterozygous genotypes (Supplemental Fig. S7). Together these observations indicate that most of these variants overlap real duplications or higher-order CNVs and are not false positive variants (for an example of a 25-kb duplication, see Fig. 3A). Category 2 SNVs (3861) are consistent with the occurrence of underlying hemizygous deletions in the family. Most (83%) of the SNVs are clustered (Fig. 3B) and have low read depth in samples predicted to be hemizygous for the deletion (25.6× compared to the 50.3× diploid genome average) (Supplemental Fig. S8). Many (81%) are also significantly out of HWE in the European cohort. Category 3 SNVs (49,800) are positions with a single heterozygous call in the pedigree. These singletons are not clustered; they may be isolated false positive calls or somatic mutations (Supplemental Fig. S9). Of these, 48.8% (24,299) were called identically in two independent data sets generated by different sequencing chemistries and analysis pipelines, suggesting that they are likely to be mostly true somatic mutations, arising either in the individual or during culture of the cell line, plus an expected roughly 50–100 de novo germline mutations in each of the children. The remaining 51.2% (25,501) are just identified in one sequencing technology, and there is no evidence to indicate how many are errors versus low frequency somatic mutations. Category 4 SNVs (89,904) are the remaining positions that are not pedigree consistent. Compared to the previous categories, these may be accounted for by a variety of reasons. Although some SNVs are genotyping errors, most (79%) of the common SNVs are significantly out of HWE and approximately half (54%) of all the SNVs are clustered (Supplemental Fig. S10), which is consistent with the occurrence of both somatic deletions and germline deletions or duplications (but does not meet the criteria for inclusion in Categories 1–3). An example of a complex event is shown in Figure 3C, in which somatic instability in different individuals has occurred in 22q11.2, a region in the genome that is known to be highly mutable (Mikhail et al. 2014). Additionally, there are approximately 589 SNVs in 322 clusters that failed to match the inheritance vectors, that is consistent with a double crossover or gene conversion (Supplemental Table S14).
Figure 3.

CNVs in this pedigree identified from nonplatinum variants. (A) Duplication on Chromosome 1 containing 242 Category 1 nonplatinum SNVs in a region of elevated read depth. Colored lines show the depths for the parents (NA12877 in purple and NA12878 in orange), and the gray lines show the depths for each of the children. Points along the bottom highlight the platinum variants (blue) and the nonplatinum variants (red). Note that there are 16 platinum variants in this duplication, because the presence of duplicated sequence can still produce genotypes that are consistent with those predicted by a diploid model (Supplemental Table S15). (B) Deletion on Chromosome 4 identified by 176 Category 2 nonplatinum SNVs that were consistent with the presence of a large hemizygous deletion. In addition to the large deletion, the depth supports the presence of several segmental duplications both overlapping and flanking the deletion. Lines and points are colored as in A. (C) Cell line or somatic deletions in multiple members of the pedigree on Chromosome 22 identified by 926 Category 4 nonplatinum SNVs. Lines are colored as in A, except for the addition of a black line that shows the depth for NA12893 (child). Although the other children (gray lines) do not appear to be deleted in any part of this region, the depths are highly variable, which may indicate somatic instability and mosaicism. The variability of this region within different cell line passages is evident when we compared these sequence data for NA12878 against corresponding data from the 1000 Genomes Project (1000 Genomes Project Consortium 2010) and NIST (Zook et al. 2014) (Supplemental Fig. S11).
Comparing our nonplatinum SNVs with a set of common, population-level CNVs (Sudmant et al. 2015a) and CNV calls from this pedigree (Roller et al. 2016) shows a significant excess of Category 1 and Category 4 SNVs overlapping duplications and significantly more Category 2 SNVs overlapping deletions (Supplemental Table S13). Overall, we conclude that the majority (∼75%) of nonplatinum SNVs in this analysis colocate with an unidentified germline or somatic variant; 67.3% were accounted for in 23,442 clusters (indicative of CNVs). Improvements in CNV calling and modifying the inheritance analysis to utilize the copy number information will lead to more accurate characterization of these events. Although many of these are likely to be true variant locations, assessing algorithm performance should exclude these sites until improved CNV calling is available and variants pass the necessary criteria for inclusion in the Platinum variant catalog (Discussion).
Using the Platinum variant catalog for benchmarking
We used the Platinum variant catalog to benchmark the performance of four commonly used informatics pipelines on sample NA12878, using WGS at 30×, 40× or 50× average aligned read depths (Fig. 4; Supplemental Tables S17–S19). We performed joint calling with the parental data sets for those callers that have a multisample analysis mode. For precision, we utilized the homozygous reference positions identified as high confidence. For SNVs, FreeBayes has the highest recall at the cost of lower precision compared to the other algorithms. The results for Strelka (Saunders et al. 2012) and GATK3 (DePristo et al. 2011) are similar, with GATK3 showing slightly higher recall and lower precision than Strelka. For indels, Strelka and GATK3 have very similar recall and precision, whereas Platypus has the highest precision values at a cost of lower recall. We used default filters for these comparisons, but recall can be improved for any algorithm with a resulting loss in precision by altering filter thresholds. The receiver operator characteristic (ROC) curves for GATK3 and Strelka are shown as an example in Figure 4B.
Figure 4.
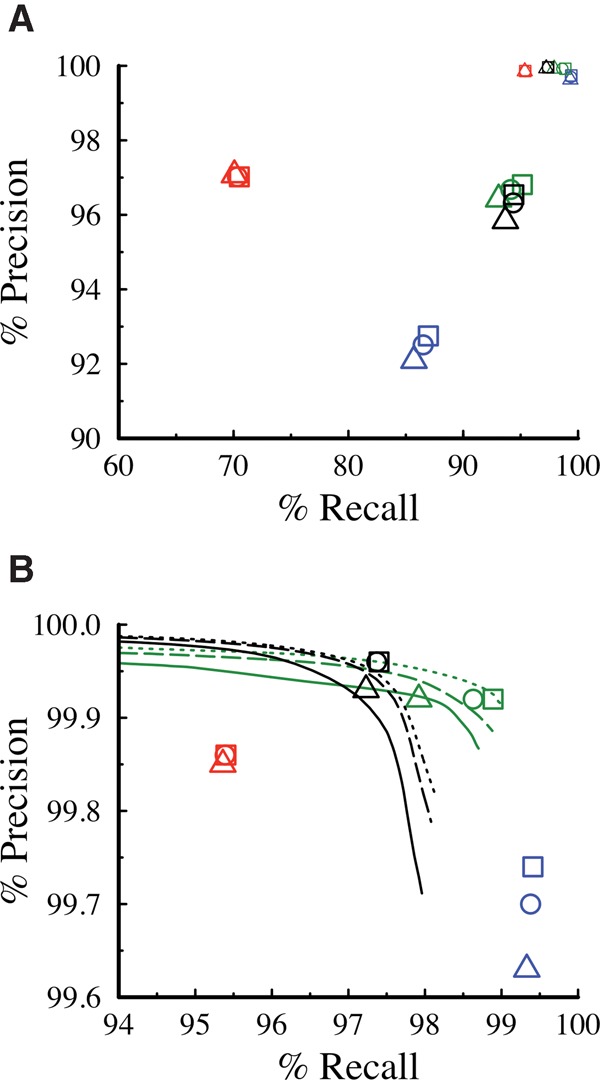
Precision versus recall in NA12878 evaluated against the Platinum catalog data set. Triangles, circles, and squares, respectively, represent the results from 30×, 40×, and 50× sequencing depth for Platypus (red), FreeBayes (blue), GATK3 (green), and Strelka (black). Excluding Strelka, all callers are run in joint calling mode incorporating the parents. (A) Indels (large symbols) and SNV (small symbols) results plotted on the same axis. (B) Expansion of SNV results, also showing ROC curves for GATK3 and Strelka that reflect the trade-off of recall versus precision that is obtained by altering specific variable parameters when using the algorithms.
Comparison to other studies
The NIST Genome-in-a-Bottle (Zook et al. 2014) and the 1000 Genomes (1KG) (The 1000 Genomes Project Consortium 2015) both included data from NA12878, and we compared variant calls from these studies with our Platinum variant catalog. All three studies generated high-confidence, genome-wide variant sets and then used different approaches to filter the calls and assess quality. The goal of the NIST study was to generate a high-confidence set of variants as a resource for benchmarking SNV and indel calls and is therefore directly aligned with the aim of the present study. The goal of the 1000 Genomes Project was to generate a comprehensive catalog of human genetic variation across a diverse set of individuals from multiple populations across the globe.
The NIST study was a replicate-based analysis of a single sample (NA12878) using 14 separate sequencing experiments (total depth = 831×) (Zook et al. 2014). Variants were excluded from the final set by arbitration based on inconsistent genotype calls between pipelines and/or occurrence in repetitive/duplicated sequence or structural variants. In contrast to our platinum genome calls, NIST used neither pedigree information nor flanking-sequence analysis to identify errors, although visual inspection was used for some variants. We found that the genotype concordance was very high for the SNVs (>99.99%) and indels (99.92%) called in both data sets (Table 1). The Platinum data set also contained 800,564 SNVs and 223,780 indels not included in the NIST set. Additionally we identified 912,018 SNVs and 261,647 indels that are more difficult (based on the observation that they were not called consistently by all of the pipelines), including 347,186 (38.2%) SNVs and 142,964 (54.6%) indels not included in the NIST set (Supplemental Table S23). Identifying these “difficult” variants is particularly important because they highlight areas where the variant calling methods can be improved. Given that these additional variants passed our inheritance and flanking-sequence tests, these can be considered true positives. Most of these variants (91.3% of the SNVs and 63.5% of the indels) were also found independently in the 1KG data set (see below). Conversely, the NIST data set contained 62,946 SNVs and 60,057 indels absent from the Platinum data set. The majority of these were observed in this study but were then filtered out (Supplemental Table S22).
Table 1.
Comparison of platinum genotype calls in NA12878 with NIST and 1KG
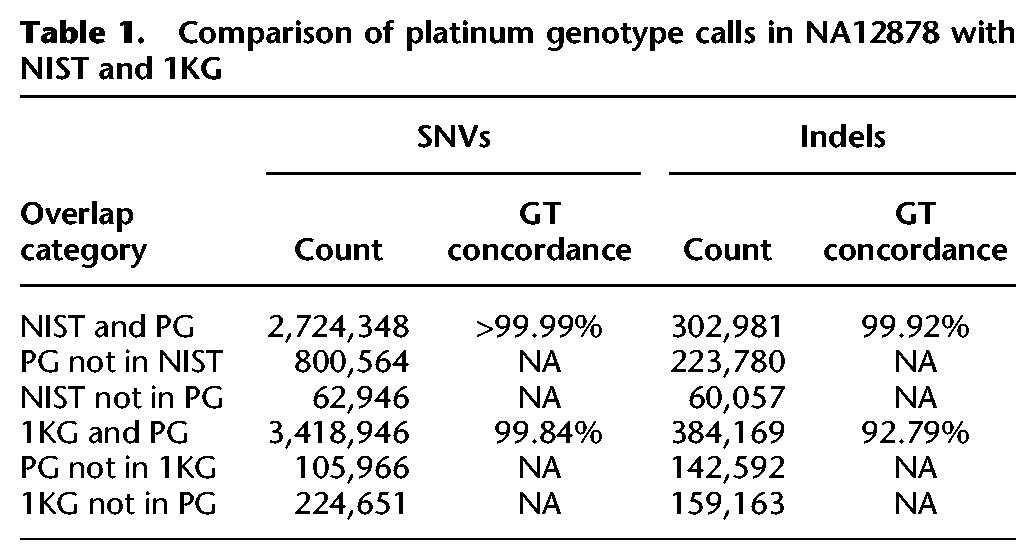
The 1KG data set was generated from 2504 individuals (including NA12878) using 7.4× average depth WGS data combined with 65.7× exome sequence data and array-based genotype data (The 1000 Genomes Project Consortium 2015). Haplotype-based imputation was used for error correction and to impute missing data. This cohort-based analysis was calibrated to maintain a population-level false discovery rate (FDR) of <5% for most variants, including SNVs and small indels. Genotyping concordance for the 3.4M SNVs shared between the two data sets is high (>99.8%), but lower (92.8%) for the indels. The Platinum data set contains 105,966 SNVs and 142,592 indels that are absent from the 1KG data for NA12878. When we examined the entire 1KG data set, we found 28% of these SNVs and 30% of the indels were in fact found in other individuals of the 1KG cohort. The rest were not found in any of the 1KG individuals and these variants may have low population frequency (<0.1%). Conversely, the 1KG calls for NA12878 contain 224,651 SNVs and 159,163 indels absent from the Platinum catalog, and most of these were observed in our study but were filtered out (Supplemental Table S22).
Estimating the extent of platinum genome coverage
To estimate the fraction of the total reference genome that has high-confidence base calls in this study, we combined our Platinum variant catalog with all high-confidence invariant positions (i.e., explicitly called homozygous reference throughout the pedigree). We concluded that 96.72% of the known reference genome (hg19: autosomes and Chromosome X) is covered on the basis of these combined criteria (Table 2), although coverage of the autosomes (97.0%) is significantly better than Chromosome X (92.5%) (Supplemental Table S11). Platinum coverage in genes, and particularly exons, is higher than the genome average, as expected on the assumption that highly repetitive motifs, which make up most of the nonplatinum regions of the reference, are depleted in transcribed and translated sequences compared to the genome average.
Table 2.
Platinum coverage of the genome, entire genes, and exons and in each major category of repeat sequence, based on UCSC (hg19)
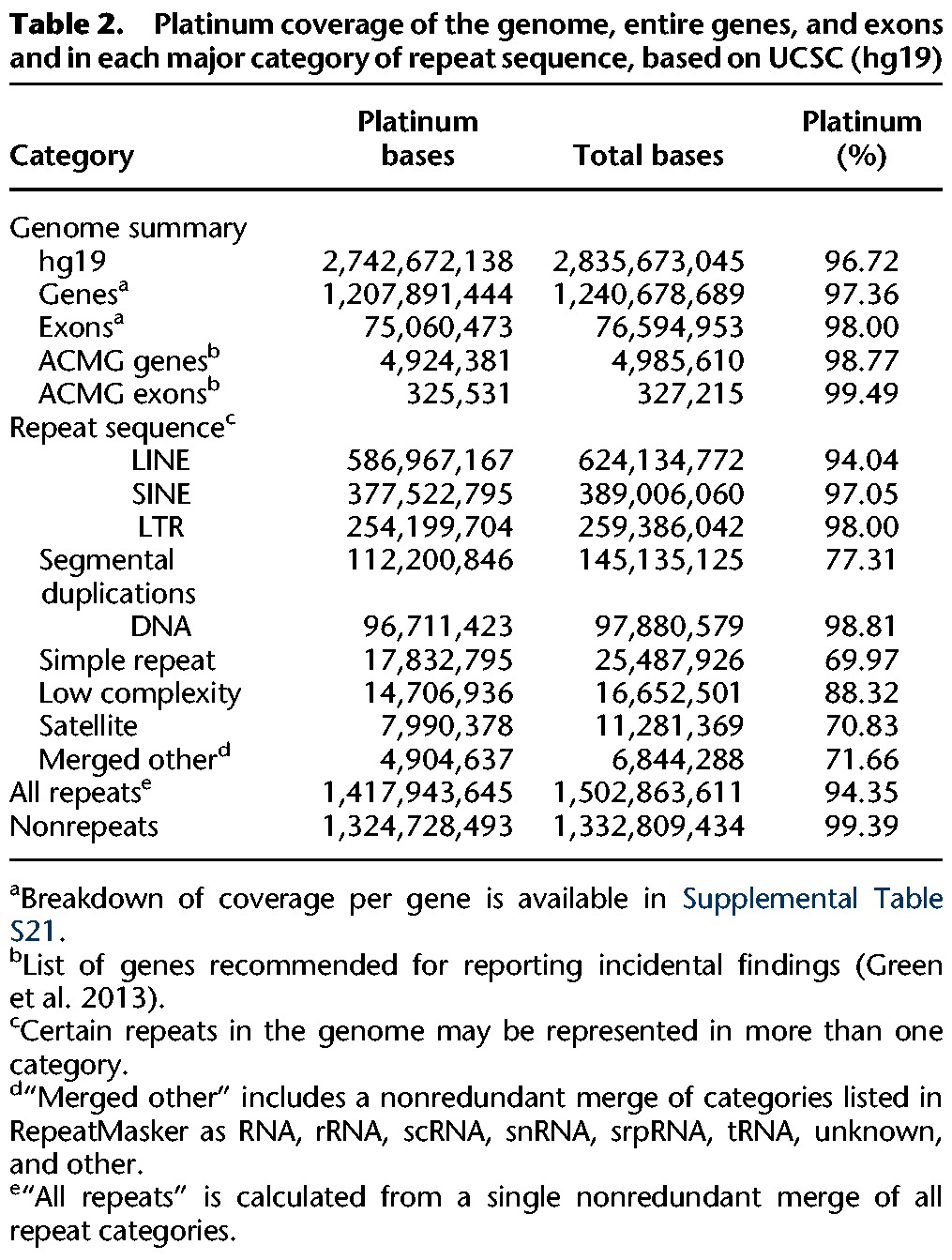
By this analysis, 93,000,907 reference bases of hg19 (∼3.3%) are classified as nonplatinum. However 84,919,966 of these bases lie within segmental duplications or one of the categories of known repeats listed in RepeatMasker, which leaves just 8,080,941 nonplatinum bases in the reference that are not accounted for by one of the annotation classes incorporated here. Further work is required to address this remaining 8.1 Mb along with the full characterization of undetected CNVs (see above). This result indicates that we can locate and identify the vast majority of nonplatinum bases, that most of them are known repeats, and that they can all be flagged for inclusion or exclusion as dictated by the needs of individual studies in the future.
Discussion
We combined data from two different sequencing technologies and six different informatics pipelines to identify a ground truth catalog of 5.4M phased SNVs and indels in this pedigree. A definitive feature of this study is that every platinum variant has been validated using inheritance patterns defined by phased haplotypes in a large pedigree. We avoided the user-defined filters of the mapping and variant calling algorithms, thus maximizing utility of the input data and minimizing potential biases. The platinum variant set is not intended to be an exhaustive list of variants. Instead, it is a comprehensive genome-wide set of phased small variants that has been validated to high confidence. We discovered that many nonplatinum SNVs and small indels in this analysis are likely real, but their inheritance patterns were confounded by colocalized CNVs. For example, the present study highlighted 23,442 locations that are likely to be confounded by CNVs. Next steps will include revision of the assumption of autosomal diploidy so as to include models of multivariate positions. Future work will also need to correct and incorporate the true variants that fail the flanking-sequence analysis; many of these are due to issues with merging and normalizing variant calls from multiple analysis pipelines. A future study might benefit from the use of nonimmortalized samples, not confounded by cell culture somatic mutation artifacts. Future studies should also include pedigrees from other ethnic origins.
Compared to other studies, utilizing the genetic inheritance allows us to create a more comprehensive catalog of platinum variants that reflects both high accuracy and completeness. A comprehensive set of highly accurate SNVs and indels in both the easy and difficult parts of the genome is vital for software developers because these are the areas where the current methods most need training data to improve their methods. Additionally, because every one of the variants in this catalog is phased, this data set provides a resource to accurately assess emerging technologies designed to provide phasing information (Kitzman et al. 2011; Suk et al. 2011; Zheng et al. 2016).
We demonstrated the use of the platinum variants to make an accurate comparison of different analysis pipelines. This exercise can be repeated using any pipeline to track progress in development and to measure improvements in SNV and indel calling. As tools improve, the same path forward will allow expansion of truth data, with the overriding principle that validation by genetic inheritance enables consideration of variants without being dependent on the strengths and weaknesses of each caller. In particular, 2% of the exons are not within our platinum regions, and thus these represent locations where the sequencing workflows fall short. These regions should be prioritized for improvement in future development efforts. In addition, this same method can be used to rapidly assess new genome builds once the inheritance vectors are available, and some of the nonplatinum variants may be caused by reference issues that will be solved in future genome builds (platinum calls for GRCh38 are also available with the hg19 calls described in this study).
Ultimately a Platinum variant catalog should contain comprehensive, genome-wide sets of all types of variant calls: SNVs, insertions, deletions, large structural rearrangements, and CNVs. In this study, a single pedigree provided a very large number of SNVs and indels. Inclusion of CNVs and SVs will become possible as methods for detection improve. However, compared to the small variants, the haplotypes in a single pedigree contain relatively few large structural events. A population-based approach, using aggregated whole-genome data, is a more appropriate strategy to identify comprehensive genome-wide sets of CNVs and SVs that can be used to improve large variant callers (Sudmant et al. 2015b). The more common large variants will be identified relatively early on in an aggregate genome study and can then be validated by inheritance in this and other pedigrees. An initial set of common platinum CNVs and SVs will be invaluable to aid algorithm improvement. The rare CNVs and SVs will be discovered over time, as individual genomes are accumulated on a large scale in population-based studies.
Methods
Sequence data and variant calls
Two sets of sequence data from the various members of CEPH pedigree (1463) were generated or used in this study. The raw sequence data were aligned to the hg19 reference, and SNVs and indels were called using a variety of sequence data and variant calling pipelines (Supplemental Table S1).
Calculating inheritance vectors
Inheritance vectors describing the transmission of the parental haplotypes were calculated for the entire genome using the GATK3 SNV calls (DePristo et al. 2011) combined with the linkage software Merlin (Abecasis et al. 2002). Because genotype errors or other deviations from Mendelian inheritance (e.g., hemizygous deletions) will be problematic for the phasing, we only used the SNVs that passed the quality filters in all 13 samples and showed no Mendelian conflicts. Additionally, some parts of the genome such as the regions around centromeres are prone to mapping errors that may lead to an excess of heterozygous calls that will confound the phasing algorithm. To account for this, we developed an automated approach to merge large regions that show the same familial inheritance but are separated by smaller blocks exhibiting multiple double crossovers. After removing these unlikely crossover events, we were left with 747 distinct inheritance regions for the autosomes and Chromosome X. Because we filtered out many SNVs when calculating the initial inheritance vectors, there may be significant gaps where a crossover event occurs. We used the similarity between siblings to narrow these gaps so that the majority of SNVs are included (Supplemental Table S2). Our final set of defined inheritance blocks cover 2.95 Gb (∼97% of the genome).
Parsimony analysis
Assuming any variant is biallelic and each sample is diploid, there are a limited number (24−1 = 15 for the autosomes and 23−1 = 7 for X) of possible phased genotype combinations in the parents, excluding sites that are homozygous reference in every individual. Because the children are not independent of the parents, there are also only 15 (7 for X) possible genotype combinations in the entire pedigree within each of the regions defined by the 747 inheritance regions described above. For each of these regions, we calculated the possible genotype combinations across the family defined by the inheritance vectors. For each variant position, we compared the observed genotypes against each of the possible genotype combinations based on the known haplotype transmission. If exactly one of the 15 predefined genotype combinations agrees with the observed genotype calls, then the site is defined as accurate and by definition is also phased. See the Supplemental Section 1 for a full description of the method and rules used to identify and merge the confident variant calls.
Data access
Sequence data, merged variant calls, and transmission vectors for the two-generation pedigree analyzed here, as well as merged variant calls and transmission vectors for GRCh38, have been submitted to the Database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap) under accession number phs001224.v1.p1. In addition, the variant calls and sequence data for just NA12877 and NA12878 have been submitted to http://www.platinumgenomes.org and the sequence data for NA12877, NA12878, NA12889, NA12890, NA12891, and NA12892 have been submitted to the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena) under accession number ERP001960.
Supplementary Material
Acknowledgments
We thank Andrew Gross, Mark Ross, and Ryan Taft for helpful discussions and comments.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.210500.116.
Freely available online through the Genome Research Open Access option.
References
- 1000 Genomes Project Consortium. 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. 2002. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30: 97–101. [DOI] [PubMed] [Google Scholar]
- Ajay SS, Parker SC, Abaan HO, Fajardo KV, Margulies EH. 2011. Accurate and comprehensive sequencing of personal genomes. Genome Res 21: 1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley EA. 2015. The precision medicine initiative: a new national effort. JAMA 313: 2119–2120. [DOI] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland JF, Chung CC, Roberson D, Mitchell J, Zhang X, Im KM, He J, Chanock SJ, Yeager M, Dean M. 2013. The new sequencer on the block: comparison of Life Technology's Proton sequencer to an Illumina HiSeq for whole-exome sequencing. Hum Genet 132: 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 2011. Variation in genome-wide mutation rates within and between human families. Nat Genet 43: 712–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dausset J, Cann H, Cohen D, Lathrop M, Lalouel JM, White R. 1990. Centre d'Etude du Polymorphisme Humain (CEPH): collaborative genetic mapping of the human genome. Genomics 6: 575–577. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81. [DOI] [PubMed] [Google Scholar]
- Erikson GA, Bodian DL, Rueda M, Molparia B, Scott ER, Scott-Van Zeeland AA, Topol SE, Wineinger NE, Niederhuber JE, Topol EJ, et al. 2016. Whole-genome sequencing of a healthy aging cohort. Cell 165: 1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G. 2012. Haplotype-based variant detection from short-read sequencing. arXiv 1207.3907. [Google Scholar]
- Goldfeder RL, Priest JR, Zook JM, Grove ME, Waggott D, Wheeler MT, Salit M, Ashley EA. 2016. Medical implications of technical accuracy in genome sequencing. Genome Med 8: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O'Daniel JM, Ormond KE, et al. 2013. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 15: 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullapalli RR, Desai KV, Santana-Santos L, Kant JA, Becich MJ. 2012. Next generation sequencing in clinical medicine: challenges and lessons for pathology and biomedical informatics. J Pathol Inform 3: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Cancer Genome Consortium. 2010. International network of cancer genome projects. Nature 464: 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G. 2012. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat Genet 44: 226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzman JO, Mackenzie AP, Adey A, Hiatt JB, Patwardhan RP, Sudmant PH, Ng SB, Alkan C, Qiu R, Eichler EE, et al. 2011. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol 29: 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, Walters GB, Jonasdottir A, Gylfason A, Kristinsson KT, et al. 2010. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467: 1099–1103. [DOI] [PubMed] [Google Scholar]
- Lam HY, Clark MJ, Chen R, Chen R, Natsoulis G, O'Huallachain M, Dewey FE, Habegger L, Ashley EA, Gerstein MB, et al. 2012. Performance comparison of whole-genome sequencing platforms. Nat Biotechnol 30: 78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx V. 2015. The DNA of a nation. Nature 524: 503–505. [DOI] [PubMed] [Google Scholar]
- Mikhail FM, Burnside RD, Rush B, Ibrahim J, Godshalk R, Rutledge SL, Robin NH, Descartes MD, Carroll AJ. 2014. The recurrent distal 22q11.2 microdeletions are often de novo and do not represent a single clinical entity: a proposed categorization system. Genet Med 16: 92–100. [DOI] [PubMed] [Google Scholar]
- O'Rawe J, Jiang T, Sun G, Wu Y, Wang W, Hu J, Bodily P, Tian L, Hakonarson H, Johnson WE, et al. 2013. Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med 5: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel ZH, Kottyan LC, Lazaro S, Williams MS, Ledbetter DH, Tromp H, Rupert A, Kohram M, Wagner M, Husami A, et al. 2014. The struggle to find reliable results in exome sequencing data: filtering out Mendelian errors. Front Genet 5: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirooznia M, Kramer M, Parla J, Goes FS, Potash JB, McCombie WR, Zandi PP. 2014. Validation and assessment of variant calling pipelines for next-generation sequencing. Hum Genomics 8: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raczy C, Petrovski R, Saunders CT, Chorny I, Kruglyak S, Margulies EH, Chuang HY, Källberg M, Kumar SA, Liao A, et al. 2013. Isaac: ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics 29: 2041–2043. [DOI] [PubMed] [Google Scholar]
- Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SR, WGS500 Consortium, Wilkie AO, McVean G, Lunter G. 2014. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet 46: 912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roller E, Ivakhno S, Lee S, Royce T, Tanner S. 2016. Canvas: versatile and scalable detection of copy number variants. Bioinformatics 32: 2375–2377. [DOI] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. 2012. Strelka: accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics 28: 1811–1817. [DOI] [PubMed] [Google Scholar]
- Sudmant PH, Mallick S, Nelson BJ, Hormozdiari F, Krumm N, Huddleston J, Coe BP, Baker C, Nordenfelt S, Bamshad M, et al. 2015a. Global diversity, population stratification, and selection of human copy-number variation. Science 349: aab3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, et al. 2015b. An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suk EK, McEwen GK, Duitama J, Nowick K, Schulz S, Palczewski S, Schreiber S, Holloway DT, McLaughlin S, Peckham H, et al. 2011. A comprehensively molecular haplotype-resolved genome of a European individual. Genome Res 21: 1672–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Lau BT, Schnall-Levin M, Jarosz M, Bell JM, Hindson CM, Kyriazopoulou-Panagiotopoulou S, Masquelier DA, Merrill L, Terry JM, et al. 2016. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat Biotechnol 34: 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, Salit M. 2014. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol 32: 246–251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.