

Abstract
Given the central role of genetic factors in the pathogenesis of common neurodegenerative disorders, it is critical that mechanistic studies in human tissue are interpreted in a genetically enlightened context. To address this, we performed exome sequencing and copy number variant analysis on 1511 frozen human brains with a diagnosis of Alzheimer's disease (AD, n = 289), frontotemporal dementia/amyotrophic lateral sclerosis (FTD/ALS, n = 252), Creutzfeldt-Jakob disease (CJD, n = 239), Parkinson's disease (PD, n = 39), dementia with Lewy bodies (DLB, n = 58), other neurodegenerative, vascular, or neurogenetic disorders (n = 266), and controls with no significant neuropathology (n = 368). Genomic DNA was extracted from brain tissue in all cases before exome sequencing (Illumina Nextera 62 Mb capture) with variants called by FreeBayes; copy number variant (CNV) analysis (Illumina HumanOmniExpress-12 BeadChip); C9orf72 repeat expansion detection; and APOE genotyping. Established or likely pathogenic heterozygous, compound heterozygous, or homozygous variants, together with the C9orf72 hexanucleotide repeat expansions and a copy number gain of APP, were found in 61 brains. In addition to known risk alleles in 349 brains (23.9% of 1461 undergoing exome sequencing), we saw an association between rare variants in GRN and DLB. Rare CNVs were found in <1.5% of brains, including copy number gains of PRPH that were overrepresented in AD. Clinical, pathological, and genetic data are available, enabling the retrieval of specific frozen brains through the UK Medical Research Council Brain Banks Network. This allows direct access to pathological and control human brain tissue based on an individual's genetic architecture, thus enabling the functional validation of known genetic risk factors and potentially pathogenic alleles identified in future studies.
The past 20 years have witnessed major advances in our understanding of the genetic landscape of common neurodegenerative disorders including Alzheimer's disease (AD), Parkinson's disease (PD), motor neuron disease (MND)/amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and prion disorders such as Creutzfeldt-Jakob Disease (CJD). Highly penetrant alleles causing familial forms of these diseases have now been identified in approximately 50 genes (Supplemental Methods; for review, see Mead 2006; Chen et al. 2013; Guerreiro and Hardy 2014; Lin and Farrer 2014), and both genome-wide association and candidate gene studies in sporadic cases have detected numerous genetic risk alleles (for review, see Ramanan and Saykin 2013).
The complex genetic architecture of neurodegenerative diseases presents several challenges. First, the implications of a novel genetic variant are particularly difficult to define (MacArthur et al. 2014). The clinical phenotype often does not correspond to the anticipated neuropathology, and specific disease alleles are associated with a wide phenotypic spectrum (Caslake et al. 2008; Grau-Rivera et al. 2015). This makes it extremely difficult to establish whether a novel genetic variant is responsible for the disorder—an issue that is gaining importance, given the widespread use of clinical exome and genome sequencing.
Second, our understanding of the functional consequences of specific mutations is far from clear. This is partly because the effects of genetic variants can vary between tissue and cell type (Dimas et al. 2009) and also because several neurodegenerative diseases are modulated by age-related comorbidity. Both factors are extremely challenging to model in vitro and in animals.
The analysis of postmortem human brain tissue circumvents some of these difficulties, but understanding the genetic background is of critical importance. This knowledge enables the stratification of mechanistic studies and the dissection of disease pathways downstream from specific genetic lesions. It also avoids the inadvertent enrichment of a study group for specific genetic disorders, thus preventing results being inappropriately generalized across a broad disease category.
To address these issues, we performed exome sequencing and array-based gene dosage analysis of samples from 1511 frozen brains from a brain tissue resource: the UK Medical Research Council Brain Bank. Our analysis provides a genetic compendium for this accessible tissue resource.
Results
Demographics
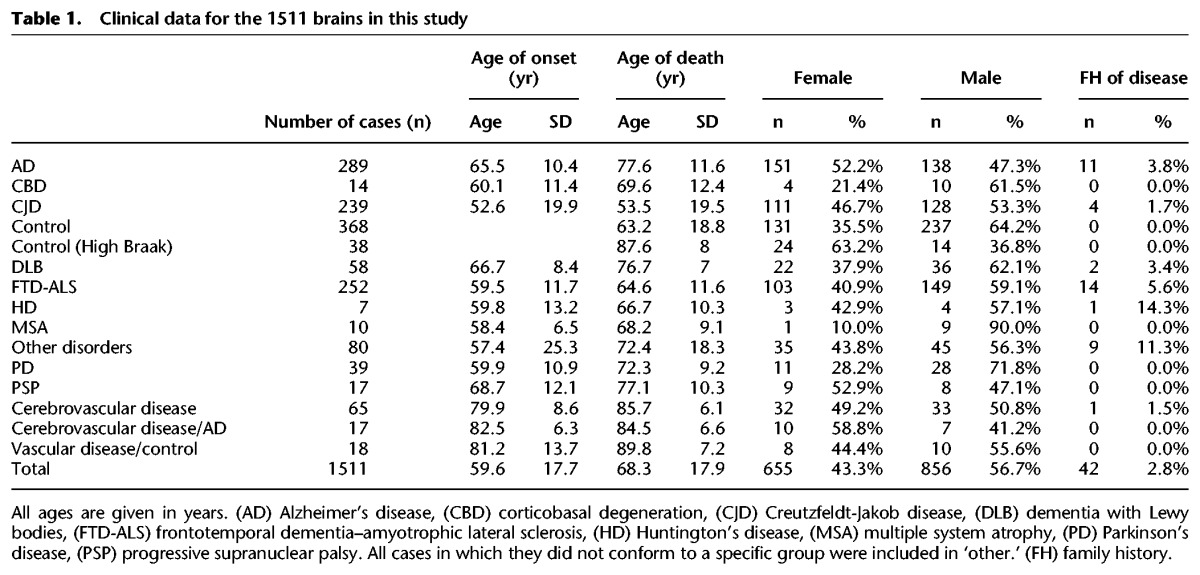
The mean age at death across all cases was 68.3 (sd = 17.9) and ranged from 3 to 103 (Table 1; Supplemental Table S1). Only 2.8% of cases had a family history of disease, as defined by a recorded first- or second-degree relative with the same or a related neurodegenerative disease (Table 1). The CJD cohort comprised sporadic (n = 126), variant (n = 62), and other subtypes, as shown Supplemental Fig S1. All demographic data are available online (https://ega-archive.org/studies/EGAS00001001599).
Table 1.
Clinical data for the 1511 brains in this study
Exome sequencing
The mean depth of sequencing of the whole exome was 51.9 (sd = 12.9), and 49.1 (sd = 13.8) in genes associated with neurodegenerative disease (Supplemental Tables S2, S3). Following quality control (QC), 1241 brains had data available using all four genetic approaches (exome sequencing, array single nucleotide variant [SNV] genotyping, C9orf72 PCR, and APOE genotyping) (Fig. 1; Supplemental Fig. S2).
Figure 1.

Summary of the molecular genetic approach and the data passing quality control (QC) in 1511 postmortem brains. Algorithm showing the genotyping strategy and headline positive results.
Single heterozygous SNVs, small insertions, or deletions in genes known to cause common neurodegenerative diseases
We initially identified rare heterozygous variants present in genes known to cause autosomal dominant forms of common neurodegenerative diseases (Supplemental Methods; Supplemental Table S2). In total, 313 variants were seen in 817 cases (Fig. 2), with 261 (83.4%) absent from the 1000 Genomes Project (The 1000 Genomes Project Consortium 2012), 212 (67.7%) absent from ESP6500 databases (Exome Variant Server 2016), and 149 (47.6%) absent from the ExAC database (Lek et al. 2016). Sixty-two percent of variants (n = 194) were initially determined to be of uncertain significance given the lack of any prior available data, and 39 variants in 57 of the 1461 cases (3.9%) undergoing exome sequencing were considered to be either pathogenic or likely pathogenic according to the American College of Medical Genetics criteria for variant pathogenicity (Fig. 2; Richards et al. 2015).
Figure 2.

Exome sequencing in 1461 postmortem brains. Assessment of heterozygous variants in genes known to cause familial forms of neurodegenerative disease. All variants were initially assessed against the American College of Medical Genetics (ACMG) criteria and all evidence relating to pathogenicity was recorded according to the guidelines of MacArthur et al. (2014). Comparing variants to clinical and neuropathological data in their respective cases enabled a significant refinement of likely pathogenicity and, in particular, an increase in the number of variants likely to be benign, allowing the reclassification of previous variants considered pathogenic.
Following comparison and correlation of variants with the case diagnoses in our data set, 28 pathogenic and likely pathogenic heterozygous variants were confirmed in 38 cases (3.46% of 1099 cases of disease) (Fig. 3; Supplemental Table Clinical data; Supplemental Table Variant data). Importantly, cross-correlating our genomic data with clinical and pathological data enabled the reclassification of 149 of the 194 variants that initially were classified as being of uncertain significance. The initial classification of the 194 variants as being of uncertain significance was because, prior to our study, they had never been reported in human reference databases, nor had they been described in either clinical cases of disease, nor studied in vitro or in vivo. However, by detecting 149 of these in control subjects or in cases with a discordant phenotype, we were able to conclude that they were likely to be benign (Supplemental Table Clinical data; Supplemental Table Variant data). Forty-five variants in 52 cases remained classified as being of uncertain significance after initial assessment (3.6% of 1461 cases undergoing exome sequencing), with these variants present in phenotypically appropriate cases for the genotype or in young control subjects. Over 50% of these variants are entirely novel (n = 23), having never been previously detected in human subjects not in any other cases in our data set, nor the three on-line databases accessed in this study (n = 69,660).
Figure 3.

Frequency of known pathogenic mutations and risk alleles in 1511 postmortem brains. (A) Number of brains with mutations in known familial neurodegenerative disease genes, and (B) the nature of pathogenic and likely pathogenic variants in each disease category. (C) Number of brains with a known genetic risk factor for neurodegenerative disease, and (D) known genetic risk factors in each subgroup (APOE*E4 alleles excluded but including rare coding variants). Variants which were significantly associated with each disease cohort are highlighted by an asterisk (P < 0.05, Fisher's exact test). All variant associations with disease can be seen in Supplemental Tables 7 and 8.
Homozygous or compound heterozygous SNVs, insertions, or deletions in genes known to cause common neurodegenerative diseases
Homozygous or compound heterozygous variants known to cause disease were seen in only four cases and were considered to be pathogenic (Fig. 1; Supplemental Table Clinical data; Supplemental Table Variant data).
Single heterozygous SNVs, small insertions, or deletions in genes known to cause rare neurodegenerative diseases
In 10 cases with rare neurological disorders, pathogenic variants were detected in three brains: Case 24, adult-onset ganglosidosis (HEXA; p.R499C); Case 29, Kuf's disease (DNAJC5; p.L115R); and Case 44; chorea-acanthocytosis (VPS13A; p.L1841*/p.W2347fs*36) (Fig. 1; Supplemental Table S4; Supplemental Table Clinical data; Supplemental Table Variant data).
C9orf72 screening
The C9orf72 hexanucleotide repeat expansion was present in 15/1482 brains cases (1% of the total disease cohort, and 6.14% of all FTD or ALS cases (n = 244) which had C9orf72 analysis). Of note, the ‘FTD-ALS’ cohort combined all isolated FTD cases, isolated MND cases, and FTD-ALS cases into one cohort due to the genetic overlap between these disorders (Supplemental Methods; Supplemental Table S5). These findings were confirmed by Southern blot.
Identity By Descent (IBD) analysis
IBD using the SNV genotypes revealed nine pairs of individuals with cryptic 1st–4th degree ancestry (Supplemental Table S6; Supplemental Fig. S3). This led to a molecular diagnosis of spinocerebellar ataxia (SCA) because one of two first-degree relatives within the cohort was diagnosed with SCA7 in a parallel study, leading to the diagnosis in the remaining relative.
Copy number variation
Rare copy number variants (CNVs) spanning the coding region of any of the 49 common neurodegenerative disease genes included in our study (see Supplemental Methods for criteria and Supplemental Table S2 for gene IDs) were seen in 1.46% of cases (n = 19). Only one CNV occurred in a gene known to convey pathogenicity through copy number variation (Case 61: Alzheimer's disease with an APP triplication) (Supplemental Table S7) and was considered to be pathogenic. Copy number gains were also seen in APOE (n = 1), DAO (n = 1), SCARB2 (n = 1), and SPG11 (n = 1), and copy number loss in LRRK2 (n = 1) and PARK2 (n = 4). Nine cases showed a copy number gain in PRPH comprising three different CNVs, with a mean length of 13.8 kb (SD = 0.9 kb) (Fig 4; Supplemental Table S7). Six of the nine cases had AD, resulting in PRPH copy number gains having a significant association with AD vs. all controls with CNV data (n = 383, P = 0.015, Fisher's exact test), and when compared to all forms of neurodegenerative disease (n = 1060, P = 0.002). The three other brains with PRPH copy number gains showed: pathologically atypical tauopathy with AD-like features; ALS; and a control case with an age at death two standard deviations below the mean of the confirmed AD cases with PRPH copy number gains.
Figure 4.
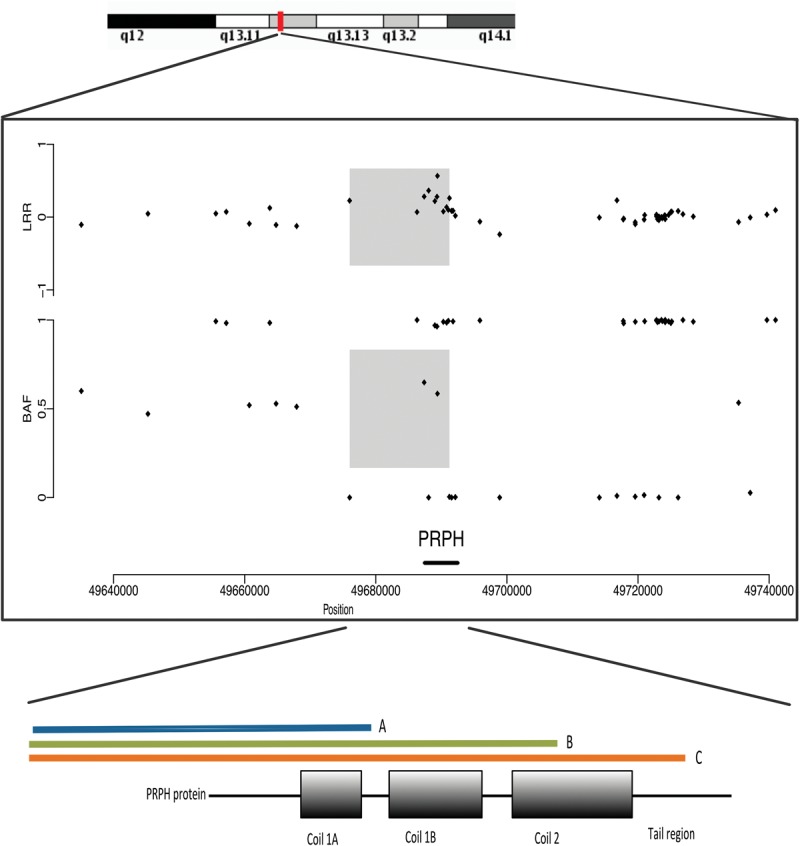
Copy number gain in PRPH. (Upper) q13 region of Chromosome 12 is shown. (Middle) Individual points show the Log R Ratio (LRR) and beta allele frequency (BAF) of SNV genotyping SNPs between base position 49640000 and 49740000 on Chromosome 12 (GRCh37 build). Positive LRR values in the gray shaded region are consistent with a copy number gain in that region (>3 SDs above the mean of the cohort) (see Supplemental Methods). The PRPH gene within that genomic region is identified on the x-axis. (Lower) 5′ position of the final SNP in the copy number region in the three different CNVs identified and their relevant position in the PRPH protein (orange, green, blue), highlighting that three different CNV end positions were identified in patients within the study within the PRPH gene.
Risk factor SNVs
Alleles previously described as conferring a risk of AD, DLB, FTD-ALS, or PD were identified (Supplemental Table S8), with 38 cases possessing established risk factors in TREM2, GBA, LRRK2 (Supplemental Table S8). We also compared the total burden of rare coding variants in genes known to act as risk factors for disease for each case cohort compared to aged (>55 yr) neuropathologically normal controls. We confirmed associations between AD and variants in TREM2 (cases; n = 20 [6.9%], controls n = 8 [3.3%]; P = 0.017), DLB with GBA (cases; n = 8 [13.8%], controls: n = 10 [4.1%]; P = 0.0075), and TREM2 (cases; n = 7 [12.1%], controls: n = 8 [3.3%]; P = 0.0084), and FTD-ALS with SQSTM1 (cases; n = 29 [13.2%], controls: n = 16 [6.6%]; P = 0.0076) (Supplemental Table S8). We also identified a putative novel risk factor for disease: the TREM2 p.R62H variant with DLB (P = 0.0024, OR;3.2 [95% CI 1.7–27]) which was seen in 7/58 cases (12.1%) and 5/244 controls (2.05%). We also found supporting evidence that the SQSTM1 p.P392L variant is a risk factor for FTD-ALS (P = 0.05). APOE*E3/4 alleles and E4/4 alleles were strongly associated with AD (P < 0.0001) and DLB (P < 0.001) (Supplemental Table S9). In total, 349 cases or controls had at least one protein-altering variant in a gene previously associated with an appropriate neurodegenerative disease (Fig. 2; Supplemental Tables S8, S9).
Genotype-pathology and genotype-clinical correlations
There was no association between the presence of variants in any risk-factor gene (TREM2, GBA, LRRK2, SQSTM1, GRN) and any neuropathological assessment score or any clinical parameter (age of onset, death, or disease duration). When substratifying by disease, TREM2 variants in AD cases were associated with a shorter disease duration (Kaplan-Meier Mantel-Cox, P = 0.027) (Supplemental Fig. S4), but there were no other associations between any risk-factor variants and any clinical or pathological feature.
Novel risk-factor associations
To determine whether variants in any established risk-factor gene conferred risk for alternative forms of neurodegenerative disease we performed burden testing of all risk-factor genes across all large cohorts (AD, FTD-ALS, CJD, DLB) compared to an extended version of all controls (n = 380). Cases possessing highly penetrant pathogenic variants (Fig. 1; Supplemental Table S10), were removed from each group before testing. Variants in GRN were associated with DLB (P = 0.005), with eight variants seen in 12.1% of patients and only 3.2% of controls, though no single allele was significantly associated with disease (Supplemental Tables S10, S11).
Discussion
Using a multimodal sequencing approach, we have identified highly penetrant alleles causing disease in 61 individuals (heterozygous alleles n = 40, homozygous or compound heterozygous alleles n = 5, C9orf72 hexanucleotide repeat expansion n = 15, and pathogenic CNV n = 1) at postmortem with clinical and pathological evidence of neurodegenerative disease and a further 349 individuals with established protein-coding heterozygous genetic risk factors for neurodegeneration (Fig. 1). This integrated genomic and pathological data set provides a unique resource to genetically stratify human brain tissue research and will ensure that future work using this tissue resource is not confounded by hitherto unknown genetic factors. The genetic data is available to all researchers, enabling the rapid identification and provision of brain tissue containing genetic variants which emerge as likely risk factors for neurodegeneration in future years but are currently not known to be important.
Using current guidelines (Richards et al. 2015), 62% (n = 194) of rare variants (MAF < 0.5%) were initially classified as being of uncertain significance. Just under half of these variants were not present in international reference databases (n = 95, 48.9%) and with no previous clinical or functional in vitro modeling to support or refute pathogenicity (Supplemental Tables Clinical data and Variant data; MacArthur et al. 2014). However, analysis of our clinical and pathological data showed that 149 of these 194 variants (76.8%) were found in control brain donors or in patients with incongruous clinical phenotypes, enabling the reclassification of 149 variants as likely to be benign. These findings illustrate the importance of studying postmortem tissue in late-onset disorders where the clinical phenotype alone may be unreliable.
In addition to the reclassification of uncertain variants as benign, our data also shows the converse: that specific alleles previously dismissed as benign, may be pathogenic. For example, the PSEN2 p.D439A variant was first described in patients with AD but subsequently found in asymptomatic controls. This led some to conclude that PSEN2 p.D439A was not pathogenic (Sassi et al. 2014). In this study, we saw PSEN2 p.D439A in both an AD and a control brain. However, the control brain had moderate Alzheimer-type pathology (Braak stage 3/4), supporting possible pathogenicity with age-related penetrance. Our findings therefore show that the presence of a variant in a clinically defined ‘healthy control’ is not reliable, and it is unwise to reject a putative pathogenic variant as benign without a comprehensive assessment of the neuropathology in an ostensibly healthy control subject. For neurodegenerative diseases, this means a postmortem examination.
Related to this, another unique and valuable feature of the resource is the inclusion of young clinically unaffected controls (n = 135 under age 55). Genotyped control brains provide an opportunity to explore the preclinical effects of neurodegenerative risk alleles, before significant cell loss and secondary pathological change. In this study, this was only seen once, in a young control harboring a known pathogenic mutation (compound heterozygous PARK2 mutations). Larger cohorts of control brain tissue are likely to yield more examples and thus provide valuable insight into the preclinical pathology of late-onset neurological disorders, potentially revealing early targets for therapeutic intervention.
We also found exceptionally rare genotypes of disease, including a homozygous p.R217* OPTN mutation and a novel p.G216R missense variant in HNRNPA1, both causing ostensibly sporadic cases of motor neuron disease (Fig. 5). Other than the original case series (Maruyama et al. 2010), this is one of only a handful of known pathogenic homozygous OPTN cases, and HNRNPA1 variants have failed to be detected in several large clinical cohorts (Seelen et al. 2014). Identification of brain tissue from these rare forms of disease provides an invaluable opportunity to further define the molecular pathogenesis of these disorders and establish genotype-phenotype relationships. For example, although both mutations were associated with a TDP-43 proteinopathy (Fig. 5), the neuropathological pattern was atypical in the homozygous p.R217* OPTN patient, demonstrating dominant subcortical and glial proteinopathy.
Figure 5.

Neuropathology in the brain donors found to harbor an OPTN (A,B) and HNRNPA1 mutation (C). (A,B) Severe degeneration with spongiosis of the primary motor cortex (haematoxylin and eosin, and atypical, predominantly subcortical white matter TDP-43 proteinopathy, A) in the patient with the OPTN mutation. In contrast, in C, the HNRNPA1 mutation was associated with a lower motor neuron phenotype with classical skein-like cytoplasmic mislocalization of TDP-43 (C, hypoglossal nucleus). (A, 200×; B,C, 400×.) (Cx) Cortex, (SCxWm) subcortical white matter.
Analyzing the coding region variants across the entire cohort enabled a compilation of a comprehensive database of genotype-phenotype correlations and facilitated the detection of rare phenotypes and presymptomatic cases of disease. This is perhaps best highlighted by the homozygous SOD1 p.D91A variant, which classically presents with distal lower motor-neuron features and progresses slowly (Andersen et al. 1996). Here, we detected the same homozygous variants in a patient with atypical dementia and some clinical features overlapping with multiple system atrophy (MSA). In addition, we found a 35-yr-old clinical control male with a previously identified pathogenic compound heterozygous mutation in PARK2. Subsequent repeat neuropathological assessment showed no evidence of neuronal loss within the substantia nigra. Given his age at postmortem, it is not clear whether this individual was presymptomatic, or whether the alleles have a reduced clinical penetrance in this case. Larger cohorts of neuropathologically stratified cases are likely to assist in determining pathogenicity in such cases.
We also made number of novel observations. Firstly, p.R62H TREM2 variant appears to be a risk factor for DLB (P = 0.0024, OR;3.2 [95% CI 1.7–27]). This variant was previously reported in association with AD (Jin et al. 2014), and although these findings need replication, these observations further emphasize the overlapping genetic etiology of DLB and AD and endorse the view that TREM2 increases the risk of developing several neurodegenerative diseases (Rayaprolu et al. 2013). Second, we show the first association of GRN variants with DLB. Again, this will require replication but suggests a novel genetic overlap between DLB and FTD. Thus, a combination of genetic risk factors contributes to the prevalence of DLB, which affects ∼5% of those aged over 80 yr but rarely affected more than one individual in a family (Meeus et al. 2012). Finally, we provide the first evidence of an association between a 13.8-kb copy number gain within PRPH and AD, found in six patients with typical AD pathology (Fig. 4). Although point mutations have previously been found in association with ALS/MND (Gros-Louis et al. 2004), the recent finding that peripherin may regulate amyloid metabolism (Muresan et al. 2014) provides a potential mechanism linking amyloid deposition and the first possible overlapping genetic mechanism between ALS/MND and AD.
There are several explanations for the absence of a relevant family history in most (73.8%) of the 61 likely genetically determined cases we identified (n = 45), including early death in previous generations, incomplete penetrance, de novo mutation, and false paternity. Although we cannot resolve this issue for individual cases, our findings do highlight the importance of considering highly penetrant single gene defects in patients with an ostensibly sporadic neurodegenerative disease.
Although a number of known genetic causes of neurodegeneration were not included in our analysis, including large insertions, deletions, and large-scale inversions, these are only likely to be relevant for a small minority of cases. In addition, being an analysis of a legacy collection, newer neuropathological assessment criteria were not available in brains collected some time ago. However, the fine-detailed genetic characterization of specific alleles and contemporary histopathological assessments will be added in the near future. This genetic compendium of 1511 brains includes 40 brains with highly penetrant SNVs in 13 known disease genes, five cases with homozygous or heterozygous variants in four genes, together with pathological expansions of C9orf72 in 15 cases, and a copy number gain of APP in one case, resulting in a total of 61 cases of genetically determined disease. The compendium also includes 349 cases and aged controls with established risk protein coding alleles for neurodegenerative disease (Fig. 1). The whole data set will facilitate variant interpretation and provides a framework for the genetic stratification of future human postmortem analysis studies.
Methods
Clinical and pathological data
Ethical approval for the genetic analysis of postmortem brain tissue was obtained from the ethical review board of each participating center. One thousand five hundred eleven brains were selected from four centers within the MRC Brain Bank Network to include a representative range of neurodegenerative disorders and controls who died from unrelated causes (Table 1; Supplemental Table S1). Demographic data (age of disease onset and death, disease duration, and family history of disease), together with the antemortem clinical diagnosis and postmortem neuropathological diagnosis were recorded for all cases. Quantitative neuropathological scores and stages (e.g., Braak neurofibrillary tangle stage [Braak et al. 2006]) were also supplied for each case as appropriate (see Supplemental Methods). For cases conforming to diagnostic criteria for Creutzfeld-Jakob disease (Budka et al. 1995), the clinical subgroup (sporadic, familial, variant, or iatrogenic) was also recorded.
Antemortem clinical and postmortem pathological data were reviewed for all 1511 cases to ensure the antemortem history was consistent with the neuropathological diagnosis for all disorders (Supplemental Methods). Given the known neuropathological and genetic overlap, cases with either frontotemporal dementia, motor neuron disease /amyotrophic lateral sclerosis, were included in a combined FTD-ALS subgroup. When antemortem symptoms were either absent or unavailable, the case was categorized as ‘other,’ alongside rare neurological diagnoses. A full description of the cohort stratification process together with the specific diagnoses of rare cases can be found in Supplemental Table S1 and the Supplemental Methods.
Molecular genetic and bioinformatic analysis
DNA extraction
DNA was extracted from either the cerebellum, cerebral cortex, or basal ganglia as described in the Supplemental Methods.
Exome sequencing and analysis
Genomic DNA was fragmented, exome enriched, and sequenced (Nextera Rapid Exome Capture 62 Mb and HiSeq 2000, 100-bp paired-end reads). Bioinformatic analysis was performed using an in-house pipeline including alignment (human reference genome hg19, UCSC) using Burrows-Wheeler Aligner (BWA) (Langmead et al. 2009). Variant calling was performed using FreeBayes (Garrison and Marth 2012). Subsequent analysis was restricted to on-target homozygous, heterozygous, and compound heterozygous variants with a minimum read depth of 10 in any case or control, and base quality score of 20 within the cohort. Further analysis was performed on frameshift, in-frame indel, or start/stop codon change, missense variants, and splice site loss variants with a minor allele frequency < 0.5% or < 3% (for familial forms of disease or risk-factor alleles, respectively) in the 1000 Genomes Project Database (The 1000 Genomes Project Consortium 2012), European American cases from the NHLBI ESP exomes database (Exome Variant Server 2016), and ExAC server (Lek et al. 2016), using Qiagen Ingenuity Variant Analysis software. Variants in genes known to cause familial forms of neurodegenerative disease (Supplemental Methods; Supplemental Table S2) with the appropriate inheritance pattern were assessed in all cases according to both the 2015 American College of Medical Genetics (ACMG) (Richards et al. 2015) and the MacArthur Criteria (MacArthur et al. 2014), irrespective of phenotype or neuropathological diagnosis (Fig. 2). The ACMG criteria remain the world's leading criteria for the interpretation of sequencing variants in a clinical context, and the MacArthur criteria are a second stringent criteria that propose that researchers summarize and present a spectrum of evidence in order to truly attribute pathogenicity of identified alleles in sequencing studies. The same criteria were also applied to a specific assessment of a small number of cases with rare neurological diseases, undertaken independently (adult onset gangliosidosis, cerebello-olivary atrophy, chorea-acanthocytosis, Huntington-like disease, Kuf's disease, infantile onset mitochondrial disease, neurodegeneration with brain iron accumulation, and primary familial basal ganglia calcification) (Supplemental Methods; Supplemental Table S4).
Associations at the gene level
Initially, known pathogenic and likely pathogenic causes of disease were removed. Known risk-factor associations at the gene level were then tested through case burden testing using Sequence Kernel Association Test (SKAT-O) (Wu et al. 2011) against a group of control cases aged 55 or over with no features of neurodegenerative disease (Supplemental Methods) (n = 244) within Ingenuity Variant Analysis. Subsequent allele-level associations were assessed through χ2 testing with a Mantel-Haenszel correction when a count of zero occurred in either group. Subsequent analyses for novel associations between disease cohorts and controls were performed against an expanded cohort of controls across the whole age range (n = 380 including n = 244 aged controls, plus n = 136 young controls and vascular controls) (see Supplemental Methods).
Array genotyping and analysis
Array genotyping was performed on all samples using the Illumina HumanOmniExpress-12 BeadChip array (Illumina) and subsequently utilized to perform Identity by Descent to determine related individuals within the data set. Single nucleotide variant genotyping data was also used to determine copy number variants within genes known to cause familial forms of disease or act as known risk factors (Supplemental Methods). Rare CNVs were defined as those present in <1% of cases in references databases.
Additional allele-specific genotyping
Specific genotyping to determine the APOE genotype was also performed independently because of poor exome and SNV array coverage (Supplemental Methods). Screening for the C9orf72 hexanucleotide repeat expansion was also performed as described (Renton et al. 2011). Positive results were confirmed by Southern blot (Beck et al. 2013), allowing an estimate of the repeat size.
Data access
Clinical, pathological, and genetic data from this study have been submitted to the European Genome-phenome Archive (EGA, https://www.ebi.ac.uk/ega/home) under accession number EGAS00001001599. VCF files and associated and annotated metadata (clinical and neuropathological diagnosis, age of disease onset, and age of death) are available for download through this archive. All requests for data should be made to the Data Access Committee as identified through the portal. Access to brain tissue is available through http://www.mrc.ac.uk/research/facilities/brain-banks/.
Supplementary Material
Acknowledgments
This work was funded by the UK Medical Research Council (13044). M.J.K. is a Wellcome Trust Clinical Research Training Fellow. P.F.C. is a Wellcome Trust Senior Fellow in Clinical Science (101876/Z/13/Z) and a UK NIHR Senior Investigator, who receives support from the Medical Research Council Mitochondrial Biology Unit (MC_UP_1501/2), the Wellcome Trust Centre for Mitochondrial Research (096919Z/11/Z), the Medical Research Council (UK) Centre for Translational Muscle Disease (G0601943), EU FP7 TIRCON, and the National Institute for Health Research (NIHR) Biomedical Research Centre based at Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health. We thank Thahira Rahman and Mikyung Jang for assistance with the initial database management. We are grateful to Dr. Mark Head, Ms. Helen Yull, and Ms. Suzanne Lowrie for tissue sampling, DNA extraction, and management of the CJD samples used in this study.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.210609.116.
Freely available online through the Genome Research Open Access option.
References
- The 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PM, Forsgren L, Binzer M, Nilsson P, Ala-Hurula V, Keranen ML, Bergmark L, Saarinen A, Haltia T, Tarvainen I, et al. 1996. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain 119(Pt 4): 1153–1172. [DOI] [PubMed] [Google Scholar]
- Beck J, Poulter M, Hensman D, Rohrer JD, Mahoney CJ, Adamson G, Campbell T, Uphill J, Borg A, Fratta P, et al. 2013. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet 92: 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. 2006. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F, Haltia M, Hauw JJ, Ironside JW, Jellinger K, et al. 1995. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol 5: 459–466. [DOI] [PubMed] [Google Scholar]
- Caslake R, Moore JN, Gordon JC, Harris CE, Counsell C. 2008. Changes in diagnosis with follow-up in an incident cohort of patients with parkinsonism. J Neurol Neurosurg Psychiatry 79: 1202–1207. [DOI] [PubMed] [Google Scholar]
- Chen S, Sayana P, Zhang X, Le W. 2013. Genetics of amyotrophic lateral sclerosis: an update. Mol Neurodegener 8: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, et al. 2009. Common regulatory variation impacts gene expression in a cell type–dependent manner. Science 325: 1246–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exome Variant Server. 2016. NHLBI GO Exome Sequencing Project (ESP). Vol. 2016, Seattle, WA: http://evs.gs.washington.edu/EVS/. [Google Scholar]
- Garrison EM, Marth G. 2012. Haplotype-based variant detection from shortread sequencing. arXiv: 1207.3907v1202. [Google Scholar]
- Grau-Rivera O, Gelpi E, Nos C, Gaig C, Ferrer I, Saiz A, Llado A, Molinuevo JL, Graus F, Sanchez-Valle R, et al. 2015. Clinicopathological correlations and concomitant pathologies in rapidly progressive dementia: a brain bank series. Neurodegener Dis 15: 350–360. [DOI] [PubMed] [Google Scholar]
- Gros-Louis F, Lariviere R, Gowing G, Laurent S, Camu W, Bouchard JP, Meininger V, Rouleau GA, Julien JP. 2004. A frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosis. J Biol Chem 279: 45951–45956. [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Hardy J. 2014. Genetics of Alzheimer's disease. Neurotherapeutics 11: 732–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D, Norton JB, Hsu S, Harari O, Cai Y, et al. 2014. Coding variants in TREM2 increase risk for Alzheimer's disease. Hum Mol Genet 23: 5838–5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MK, Farrer MJ. 2014. Genetics and genomics of Parkinson's disease. Genome Med 6: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, et al. 2014. Guidelines for investigating causality of sequence variants in human disease. Nature 508: 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, et al. 2010. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465: 223–226. [DOI] [PubMed] [Google Scholar]
- Mead S. 2006. Prion disease genetics. Eur J Hum Genet 14: 273–281. [DOI] [PubMed] [Google Scholar]
- Meeus B, Theuns J, Van Broeckhoven C. 2012. The genetics of dementia with Lewy bodies: What are we missing? Arch Neurol 69: 1113–1118. [DOI] [PubMed] [Google Scholar]
- Muresan V, Villegas C, Ladescu Muresan Z. 2014. Functional interaction between amyloid-β precursor protein and peripherin neurofilaments: a shared pathway leading to Alzheimer's disease and amyotrophic lateral sclerosis? Neurodegener Dis 13: 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan VK, Saykin AJ. 2013. Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer's disease, Parkinson's disease, and related disorders. Am J Neurodegener Dis 2: 145–175. [PMC free article] [PubMed] [Google Scholar]
- Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, Hatanpaa KJ, Lomen-Hoerth C, Kertesz A, Bigio EH, et al. 2013. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener 8: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassi C, Guerreiro R, Gibbs R, Ding J, Lupton MK, Troakes C, Al-Sarraj S, Niblock M, Gallo JM, Adnan J, et al. 2014. Investigating the role of rare coding variability in Mendelian dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) in late-onset Alzheimer's disease. Neurobiol Aging 35: 2881 e2881–2881 e2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelen M, Visser AE, Overste DJ, Kim HJ, Palud A, Wong TH, van Swieten JC, Scheltens P, Voermans NC, Baas F, et al. 2014. No mutations in hnRNPA1 and hnRNPA2B1 in Dutch patients with amyotrophic lateral sclerosis, frontotemporal dementia, and inclusion body myopathy. Neurobiol Aging 35: 1956 e1959–1956 e1911. [DOI] [PubMed] [Google Scholar]
- Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. 2011. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet 89: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.