

Abstract
Parahydrogen-induced polarization (PHIP) is an NMR hyperpolarization technique that increases nuclear spin polarization by orders of magnitude, and it is particularly well-suited to study hydrogenation reactions. However, the use of high-field NMR spectroscopy is not always possible, especially in the context of potential industrial-scale reactor applications. On the other hand, the direct low-field NMR detection of reaction products with enhanced nuclear spin polarization is challenging due to near complete signal cancellation from nascent parahydrogen protons. We show that hydrogenation products prepared by PHIP can be irradiated with weak (on the order of spin–spin couplings of a few hertz) alternating magnetic field (called Spin-Lock Induced Crossing or SLIC) and consequently efficiently detected at low magnetic field (e.g., 0.05 T used here) using examples of several types of organic molecules containing a vinyl moiety. The detected hyperpolarized signals from several reaction products at tens of millimolar concentrations were enhanced by 10000-fold, producing NMR signals an order of magnitude greater than the background signal from protonated solvents.
Introduction
Nuclear magnetic resonance (NMR) spectroscopy is a powerful analytical tool used for a broad range of applications.1−3 One of the main limitations of NMR is low detection sensitivity dictated by the weak interaction energy of nuclear spins with the static magnetic field B0.4−6 Hence, methods for increasing the sensitivity of NMR detection are welcome because they decrease the detection limit and acquisition time. Nuclear spin hyperpolarization techniques such as dissolution dynamic nuclear polarization (d-DNP),6 spin exchange optical pumping (SEOP) of noble gases,7,8 and parahydrogen-induced polarization (PHIP)5,9−11 can temporarily increase nuclear spin polarization (P) by several orders of magnitude (>10000 at high magnetic fields6 and hundreds of millions at low fields12) and thus have become very popular in the past decade.13,14 Motivated by biomedical applications, d-DNP and SEOP have been introduced into the clinical research realm to probe metabolism, function, response to treatment, etc.13,15−17
Recent PHIP innovations have demonstrated relatively inexpensive chemistries for production of contrast agents18,19 and use of aqueous media20 and heterogeneous catalysts,10,21 making PHIP a promising means for generating new classes of hyperpolarized (HP) molecular contrast agents for in vivo applications.22 PHIP offers a number of advantages compared to d-DNP and SEOP, that is, (i) very fast (<1 min) hyperpolarization production speed, (ii) low cost, and (iii) straightforward scalability.8 In addition, PHIP naturally employs hydrogenation reactions and therefore can find promising applications beyond biomedicine. For example, it could be a useful modality for in situ detection and imaging of industrial-scale hydrogenation and hydrogen-involving reactions,23,24 which represent a significant fraction of all industrial chemical processes.25
In principle, while the PHIP hyperpolarization technique is inexpensive and high-throughput, high-resolution NMR spectroscopic detection is most often conducted at high fields using expensive superconducting magnets, which additionally have significant limitations of small sample size. Therefore, despite the low-cost nature of the PHIP hyperpolarization process, the high-field detection renders the entire analysis process generally expensive and limited to small samples—counter to the goals of industrial-scale applications.26 Alternatively, cheap low-field magnets can be efficiently used for PHIP signal detection27−31 because detection sensitivity for HP states has a very weak (B01/4) magnetic field dependence.27,32 Moreover, low-field detection offers other advantages: (i) reduced B0 susceptibility gradients, and (ii) the possibility of construction of relatively low-cost large homogeneous magnets that, in principle, can encompass large chemical reactors.24,31−34
However, the direct NMR detection of PHIP hyperpolarization in low magnetic fields is fundamentally challenging. Indeed, even if the magnetic equivalence of the parahydrogen (p-H2) singlet state is broken during the hydrogenation reaction, two p-H2-nascent spins will still reside in the non-observable pseudo-singlet state35,36 at low magnetic field (Figure 1a). In practice, this results in the collapse of the NMR lines because the difference in the chemical shift of the two nascent protons is too small with respect to the spin–spin coupling (JHH) and also with respect to the magnetic field homogeneity. As a result, the direct detection of PHIP products suffers from massive (more than 2 orders of magnitude) signal cancellation.37−39
Figure 1.
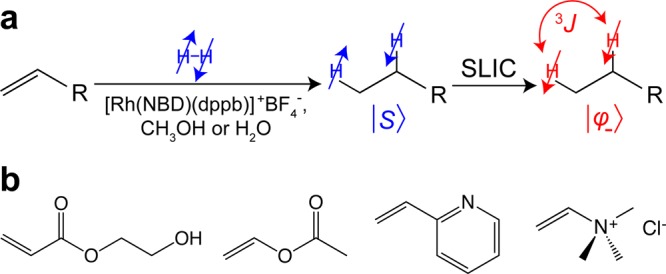
(a) Molecular diagram of unsaturated precursor hydrogenation by p-H2, leading to the hydrogenated product with p-H2-nascent protons residing in the pseudo-singlet state;35 then, pseudo-singlet state is converted to observable magnetization by using spin-lock induced crossing (SLIC) sequence. (b) Chemical structures of investigated organic molecules containing vinyl moiety: 2-hydroxyethyl acrylate (HEA), vinyl acetate (VA), 2-vinylpyridine (VPy), and (vinyl)trimethylammonium chloride (VTMA).
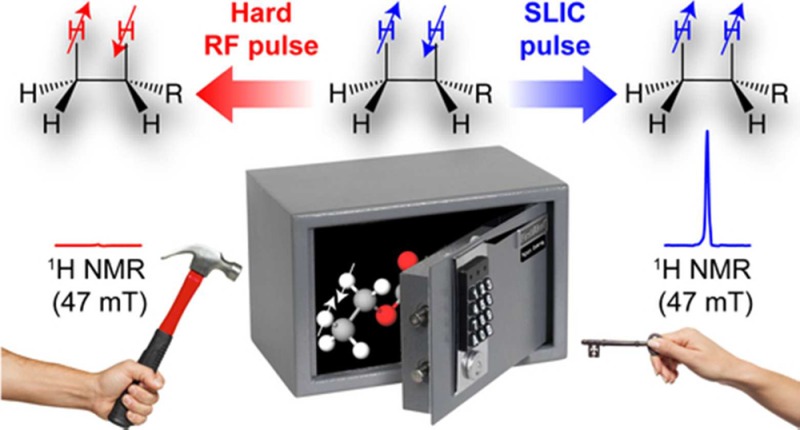
Here, we show that application of spin-lock induced crossing (SLIC)40 allows for direct proton readout of HP products at low magnetic field (e.g., 47.5 mT used here). The SLIC sequence is a simple low-power radio frequency (RF) pulse with B1 strength on the order of JHH. Specifically, more than 10000-fold NMR signal enhancement enabled direct 1H low-field detection of 80 mM solutions of 2-hydroxyethyl propionate, ethyl acetate, 2-ethylpyridine, and (ethyl)trimethylammonium chloride hyperpolarized via PHIP (Figure 1). The presented methodology of PHIP-enhanced milli-Tesla NMR with SLIC sensing (vs conventional NMR where a hard RF pulse is applied for signal detection) can be used for fast screening of potential HP contrast agents by PHIP and potentially without expensive high-field NMR instruments and isotopic labeling. Moreover, the presented methodology may be potentially conveniently applied to the visualization of industrial-scale processes in situ.
Methods
Preparation of Catalyst/Precursor Solutions
Methanol (80 mL) was placed in four square bottles (431430, Corning Life Sciences, NY, USA) and degassed by the repetitive (three times) sequence: argon flushing, closing the cap, and vortexing the solution. Rhodium catalyst [(bicyclo[2.2.1]hepta-2,5-diene)[1,4-bis(diphenylphosphino)butane]rhodium(I) tetrafluoroborate, 0.40 mmol, 0.150 g, 5.00 mM final concentration] was placed in each bottle. Vinyl acetate (6.40 mmol, 0.551 g, 80.0 mM final concentration), 2-hydroxyethyl acrylate (6.40 mmol, 0.742 g, 80.0 mM final concentration), trimethyl(vinyl)ammonium chloride (6.40 mmol, 1.06 g, 80.0 mM final concentration), and 2-vinylpyridine (6.40 mmol, 0.672 g, 80.0 mM final concentration) were added to individual bottles. Water-soluble rhodium catalyst was prepared as described earlier.20 Vinyl acetate (8.00 mmol, 0.688 g, 80.0 mM final concentration) and 2-hydroxyethyl acrylate (8.00 mmol, 0.928 g, 80.0 mM final concentration) were dissolved in the aqueous solution of rhodium catalyst (100 mL, 2.60 mM) each. Trimethyl(vinyl)ammonium chloride (4.00 mmol, 0.664 g, 80.0 mM final concentration) and 2-vinylpyridine (4.00 mmol, 0.420 g, 80.0 mM final concentration) were dissolved in aqueous rhodium catalyst solution (50 mL, 5.30 mM). Each bottle containing the catalyst/precursor solutions was connected to an automated PHIP polarizer41 for further experiments.
Preparation of Parahydrogen Gas
For parahydrogen (p-H2) preparation, normal hydrogen was passed through a spiral copper tube packed with FeO(OH) (Sigma-Aldrich, P/N 371254, 30–50 mesh) and immersed into a liquid N2 Dewar. This procedure produces a stream of hydrogen enriched with ca. 50% para-isomer.10
PHIP Polarizer and Hydrogenation Reaction
A fully automated parahydrogen-based polarizer was employed for hydrogenation.42 The prepared stock solutions containing catalyst and precursor molecule were connected to the heated injection loop of the polarizer. Hydrogenation of the unsaturated compounds (Figure 1b) was conducted in a chemical reactor (∼56 mL volume) of the polarizer at an ∼7.8 atm p-H2 pressure by injecting the warmed solution from the injection loop and spraying it into the atmosphere of hot p-H2 gas using the back pressure (∼17 atm) of N2 gas (Figure 2a). Reactor temperature was held within the range of 55–60 °C. After a variable reaction time, tR (Figure 2b), 2–2.5 mL of the solution was ejected from the polarizer into a ∼50 mL detection chamber located inside the RF probe within a 47.5 mT magnet. The magnet was located ∼0.5 m away from the polarizer, allowing very short (<1 s) ejection time of the reacted solution from the reactor to the detection chamber (Corning 50 mL PP centrifuge tube). A TTL microcontroller of the PHIP polarizer was used to switch solenoid valves that control gas and chemical delivery to the high-pressure reactor and ejection to the detection chamber. The detection chamber was cleaned after each NMR signal acquisition before a new portion of the solution from the polarizer was ejected.
Figure 2.
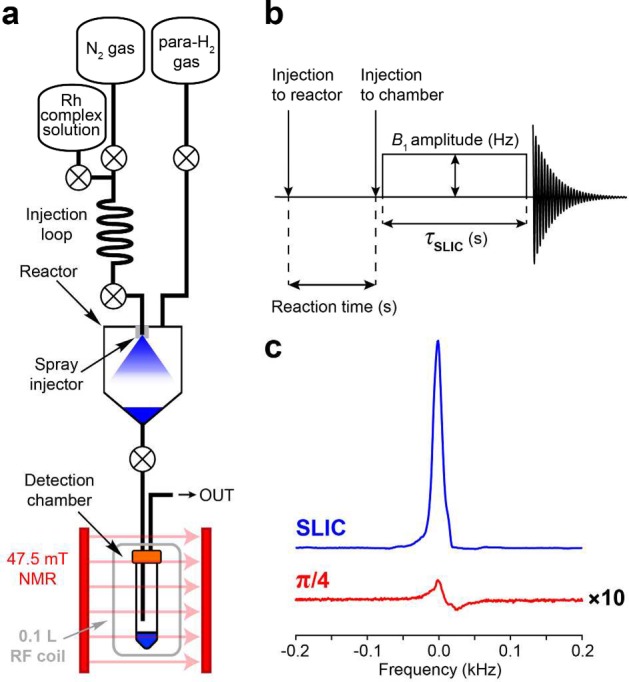
(a) Schematic diagram of experimental setup. The reactor was kept at 50–60 °C. (b) Sequence of events: injection of Rh complex solution (in CH3OH or in H2O) into reactor filled with ∼7.8 atm of p-H2, variable reaction time, injection into the detection chamber located inside a 47.5 mT magnet, subsequent application of SLIC pulse and signal acquisition. (c) 1H NMR signal of HEP in methanol obtained after application of SLIC pulse (blue) and NMR signal obtained after application of a hard π/4 RF pulse (red); note the scaling by a factor of 10.
Low-Field (2 MHz) NMR Detection
A commercially available MR Kea2 spectrometer (Magritek, Wellington, New Zealand) was used for NMR detection as described by Waddell et al.43 The magnet (2 MHz Magritek core analyzer, Halbach array, radial field direction) had a homogeneity of 20 ppm over 4 cm diameter of spherical volume. The detection chamber was placed in the home-built 1H RF coil44 located in the magnet (Figure S1). Radio frequency calibration using a 5 mM aqueous solution of CuSO4 in a 2.8 mL spherical phantom yielded a 90° 1H excitation pulse width of 177 μs at 0.22 W.
SLIC RF Pulse Sequence
Spin order of the p-H2 singlet state was converted to observable magnetization using the SLIC sequence developed by DeVience et al.40 In order to generate low-power (∼1 μW) SLIC pulses, additional attenuators (Bird Technologies, 10 W, A series, male/female N connector, 30 and 20 dB) were inserted between the output of the Tomco RF amplifier (P/N BT00250-AlphaS-Dual, Tomco Technologies, Stepney, Australia) and TR switch of the spectrometer (Figure S1). The SLIC pulse amplitude was calibrated by measuring the TR switch voltage output on an oscilloscope (Tektronix, TDS 3034C) and comparing it to the measurements for the π/2 RF pulse calibrated by nutation experiment. Acquisition of the 1H NMR signal started immediately after injection of the reactor content to the detection chamber followed by a SLIC pulse (Figure 2b). Optimization of SLIC parameters (B1 amplitude and duration, τSLIC) was performed for the 2-hydroxyethyl propionate (the product of 2-hydroxyethyl acrylate hydrogenation), and then parameters found were used for the detection of all other substrates under study.
Results
We carried out hydrogenation of several molecules containing a vinyl moiety (Figure 1): 2-hydroxyethyl acrylate (HEA), vinyl acetate (VA), 2-vinylpyridine (VPy), and (vinyl)trimethylammonium chloride (VTMA), with p-H2, using a home-built automated PHIP polarizer (Figure 2a). The compounds were chosen based on their importance in the context of potential PHIP applications. For example, ethyl acetate can be potentially employed to trace the metabolism of brain damage and cancer; 2-ethylpyridine (EPy) has shown the potential for 15N pH mapping/imaging;45 (ethyl)trimethylammonium is structurally similar to choline, which is a key metabolic signature in many cancers;46−48 2-hydroxyethyl propionate can be used for in vivo angiography, and it is also a typical test molecule for PHIP studies.49,50 Hydrogenation of the vinyl motif (−CH=CH2) by p-H2 for the molecules studied leads to ethyl group (−CH2–CH3) formation, where two hydrogens come from the same p-H2 molecule, and these nascent parahydrogen nuclei are incorporated into two chemically inequivalent positions. Due to the identical structure of the intentionally chosen hydrogenated motif for all substrates investigated (Figure 1b), their NMR parameters and J coupling patterns are relatively similar (Table S1).
Use of the automated home-built low-field PHIP polarizer made possible the fast conversion of 80 mM unsaturated substrates into hydrogenation reaction products. Reactions were carried out by fast (<4 s) injection of 3–5 mL of the catalyst/precursor solution into an atmosphere of p-H2. Then reaction solutions were quickly (<1 s) pushed to the detection chamber located in the bore of a 47.5 mT magnet; the SLIC RF pulse was applied immediately, and it was followed by 1H NMR signal acquisition (Figure 2a,b). The intensity of the NMR signal obtained after SLIC was at least 2 orders of magnitude greater than the signal intensity obtained after application of a hard π/4 pulse as typically employed in high-field PHIP experiments11 (Figure 2c).
We note that the hydrogenation reactions were carried out in nondeuterated solvents, such as methanol and water. The NMR signal resulting from ∼80 mM of hydrogenated material after SLIC was 10–30 times greater than the NMR signal originating from the solvent (Figure 3a,b). Besides providing a direct comparison with the HP signal, utilization of nondeuterated solvents here advantageously allowed calibration of experimental parameters (e.g., RF pulses, B1 frequency offset, and adjustments to account for minor magnetic field drift typical for low-field scanners based on permanent magnets and in the absence of a deuterium spin-lock apparatus). The signal enhancement of HP resonances was evaluated by computing the ratio of HP signal to the signal originating from the thermally polarized solvent (since the amount of material injected into the detection chamber each time may vary). Maximal apparent polarization percentage (PAPP) of ∼0.23% was found for 2-hydroxyethyl propionate (HEP) when hydrogenation reaction of HEA was performed in methanol. Lower values were found for ethyl acetate and when water was used as a solvent. Hydrogenation of VPy and VTMA was also detected by SLIC, but their NMR signals were significantly lower (see Discussion).
Figure 3.
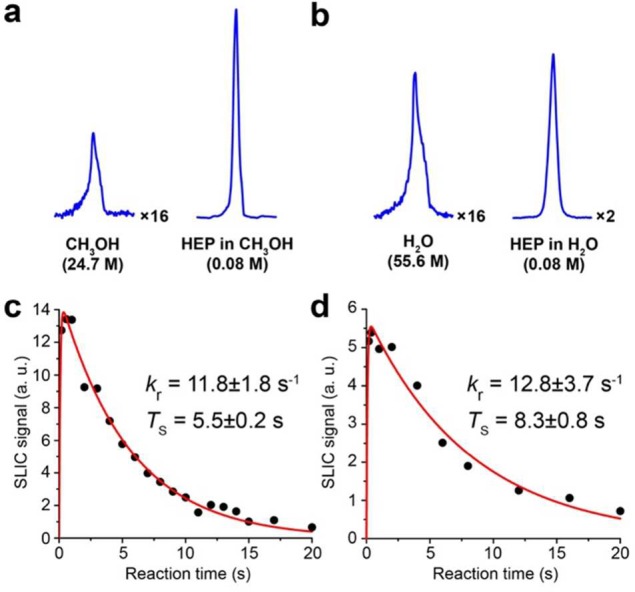
1H NMR spectroscopy of HEP at 47.5 mT. (a) Left: 1H NMR signal of reaction mixture injected into the detection chamber at equilibrium nuclear spin polarization; the NMR signal originates primarily from methanol solvent. Right: 1H HP NMR signal obtained after SLIC pulse applied to the reaction mixture (∼0.08 M of 2-hydroxyetyl propionate (HEP) in methanol). (b) Left: 1H NMR signal of reaction mixture injected into the detection chamber at equilibrium nuclear spin polarization; the NMR signal originates primarily from water solvent. Right: 1H HP NMR signal obtained after SLIC pulse applied to the reaction mixture (∼0.08 M of HEP in water). (c) Dependence of the SLIC signal (normalized to the thermal signal of the solvent) on the reaction time for HEP in methanol. (d) Dependence of the SLIC signal (normalized to the thermal signal of the solvent) on the reaction time for HEP in water.
Varying the reaction time allowed the build-up and decay of the hyperpolarized signal to be detected (Figure 3c,d). We found that the signal decayed with the time constant TS ranging between 5 and 15 s depending on the studied molecule and the solvent. The TS values obtained correlate well with prior results in the literature. For example, TS of 6.4 ± 1.2 s was measured for EA at Earth’s magnetic field in ALTADENA conditions,18 while the present study yielded TS of EA to be 7.2 ± 0.5 s in methanol (see Table 1 for the TS values for all studied substrates in methanol and in water). One may also estimate the effective hydrogenation reaction kinetic constant kr by fitting the experimental data with suitable analytical expression (eq S1) describing the build-up and decay of a hyperpolarized signal (Figure 3c,d and Supporting Information).
Table 1. Kinetic and Relaxation Parameters Extracted from Fitting of the Build-up and Decay Curves for HEP, EA, ETMA, and EPy (Catalyst Concentration was 5 mM).
| methanol | HEP | EA | ETMA | EPy |
|---|---|---|---|---|
| kr (s–1) | 11.8 ± 1.8 | 11.5 ± 2.3 | 14.1 ± 2.5 | 2.8 ± 0.5 |
| TS (s) | 5.5 ± 0.2 | 7.2 ± 0.5 | 17.1 ± 1.2 |
| water | HEP | EA | ETMA | EPy |
|---|---|---|---|---|
| kr (s–1) | 12.8 ± 3.9 | 14.4 ± 5.9 | 21.9 ± 4.2 | |
| TS (s) | 8.3 ± 0.8 | 4.3 ± 0.5 | 13.2 ± 1.4 |
In principle, the use of an automated PHIP polarizer with a high-pressure injection reactor is not mandatory; that is, high-pressure NMR tubes with p-H2 bubbling can be employed (similar to the recent studies of 13C-VA hydrogenation and hyperpolarization with signal amplification by reversible exchange).18,51 Nevertheless, the use of an automated PHIP polarizer benefited the present study because a series of experiments could be performed routinely and identically, which allowed us to quickly find optimal conditions for performing a singlet-to-magnetization transformation, that is, optimal RF pulse amplitude, frequency offset, and duration of the SLIC pulse (Figure 4).
Figure 4.
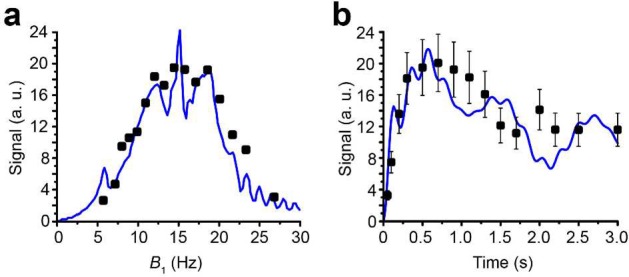
(a) Dependence of the SLIC signal of HEP in methanol (normalized to the thermal signal of the solvent) vs B1 amplitude at the SLIC duration (τSLIC) of 0.6 s; black squares correspond to experimental results, and blue line corresponds to simulation assuming ±5 Hz RF pulse offset (i.e., simulating B0 inhomogeneity). (b) Dependence of the SLIC signal vs SLIC duration (time) at B1 amplitude of 15 Hz; black squares correspond to experimental results, and blue line corresponds to simulation.
Discussion
The true singlet state of two spins (i.e., the state with a total spin of 0) is not NMR detectable because the singlet state has no magnetic moment.52 The best example is the nuclear spin singlet state of p-H2 that produces no NMR signal. Another way to explain the absence of the observable NMR signal is the realization that the transitions between exchange antisymmetric singlet state and exchange symmetric triplet states are forbidden. However, once the magnetic equivalence of the two H atoms is broken (e.g., by introducing two hydrogen atoms from the same p-H2 molecule into a nonsymmetric molecular environment), the spin order of the singlet state can be manifested as a nearly 100% nuclear spin polarization.53 Hydrogenation reactions can be employed in a way that both hydrogen atoms from the same p-H2 molecule are transferred to the product as a pair (pairwise addition), resulting in a canonical PHIP effect.11,53,54 However, the singlet state is considered to be truly broken (i.e., resulting in two well-resolved resonances in the NMR spectrum) only if the chemical shift difference between p-H2-nascent protons (δHA and δHB) is greater than the spin–spin coupling constant JHA–HB between them (corresponding to the condition of a weak coupling regime).55 Otherwise—in a strong coupling regime (sometimes referred to as inverse weak coupling regime56), that is, (δHA – δHB) < JHA-HB—the two nascent protons reside in a pseudo-singlet state even after the act of pairwise addition of p-H2.35 For example, for the case where (δHA – δHB) is 3 ppm and J coupling is 7 Hz, the strong coupling regime occurs for magnetic fields below 0.055 T. The ethyl moiety (−CH2–CH3) is a chemical motif found in a wide range of organic molecules which has a typical J coupling constant of ∼7 Hz between methylene and methyl groups, and the corresponding chemical shift difference (δMETHYLENE – δMETHYL) ranges from 0.5 to 3 ppm (Table S1). Since hydrogenation of vinyl or acrylate groups produces ethyl or propionate groups respectively (note that both cases are considered as an isolated five spin system −CH2−CH3), all four molecules studied form strongly coupled spin systems if hydrogenation by p-H2 is carried out in the fields below 0.055 T.
Although the pseudo-singlet state is not directly detectable by NMR in the strong coupling regime,38,56−58 the spin order from the pseudo-singlet spin state can be transformed into the observable magnetization using the SLIC sequence introduced by DeVience and co-workers.40,59 They demonstrated that low-power continuous wave decoupling (with alternating magnetic field B1 amplitude on the order of JHA–HB and with frequency set at (δHA + δHB)/2) enables coherent transfer of population between the singlet state |S⟩ = (|αβ⟩ – |βα⟩)/√2 and the state |φ–⟩ = (|αα⟩ – |αβ⟩ – |βα⟩ + |ββ⟩)/2. The latter term corresponds to magnetization aligned along the −x axis in a rotating frame, and it is readily observable by NMR.52 The optimum duration of SLIC RF irradiation depends on the combination of NMR resonance frequency, JHA–HB, and (δHA – δHB). Advantageously, this transformation does not require any other RF pulses, making it relatively straightforward to implement from the hardware perspective.
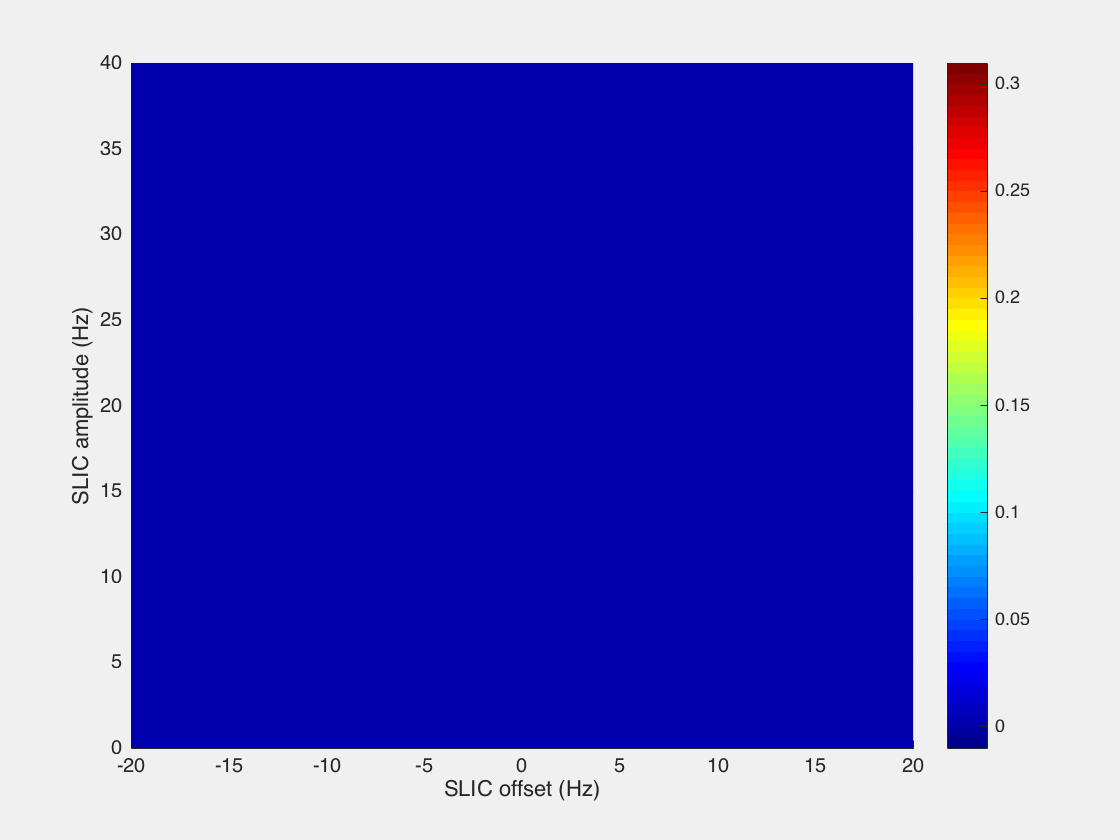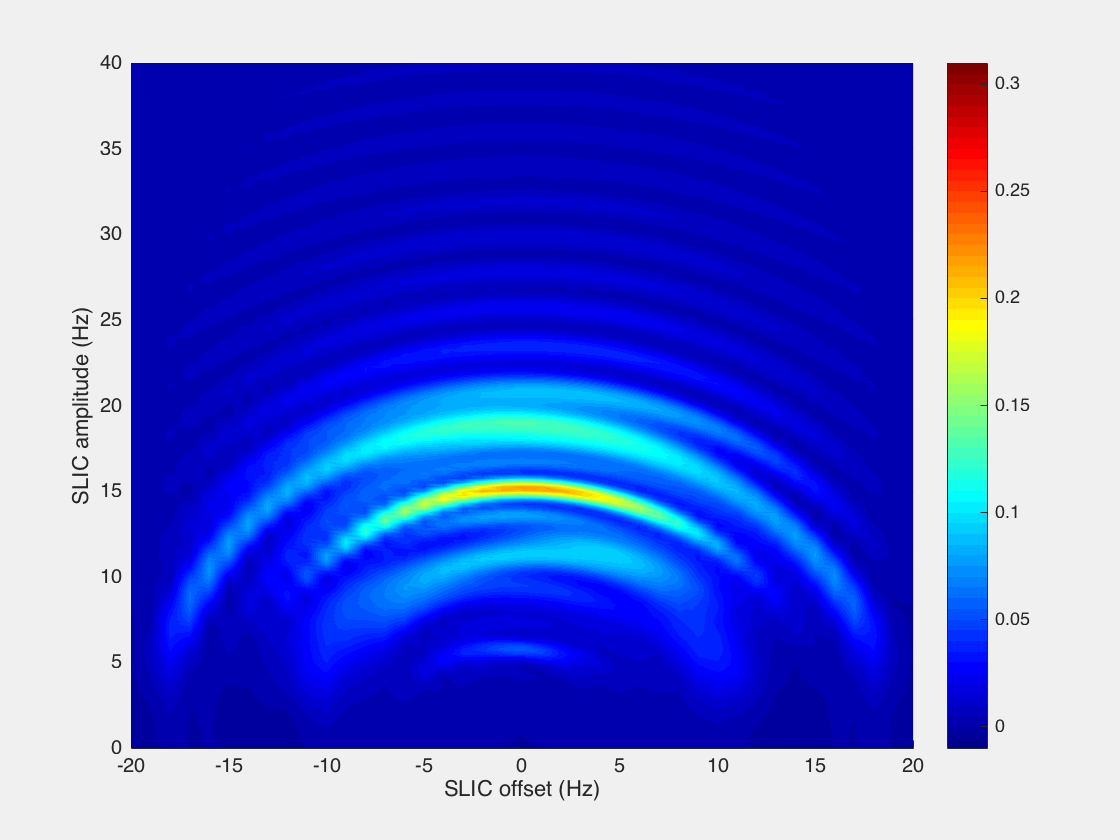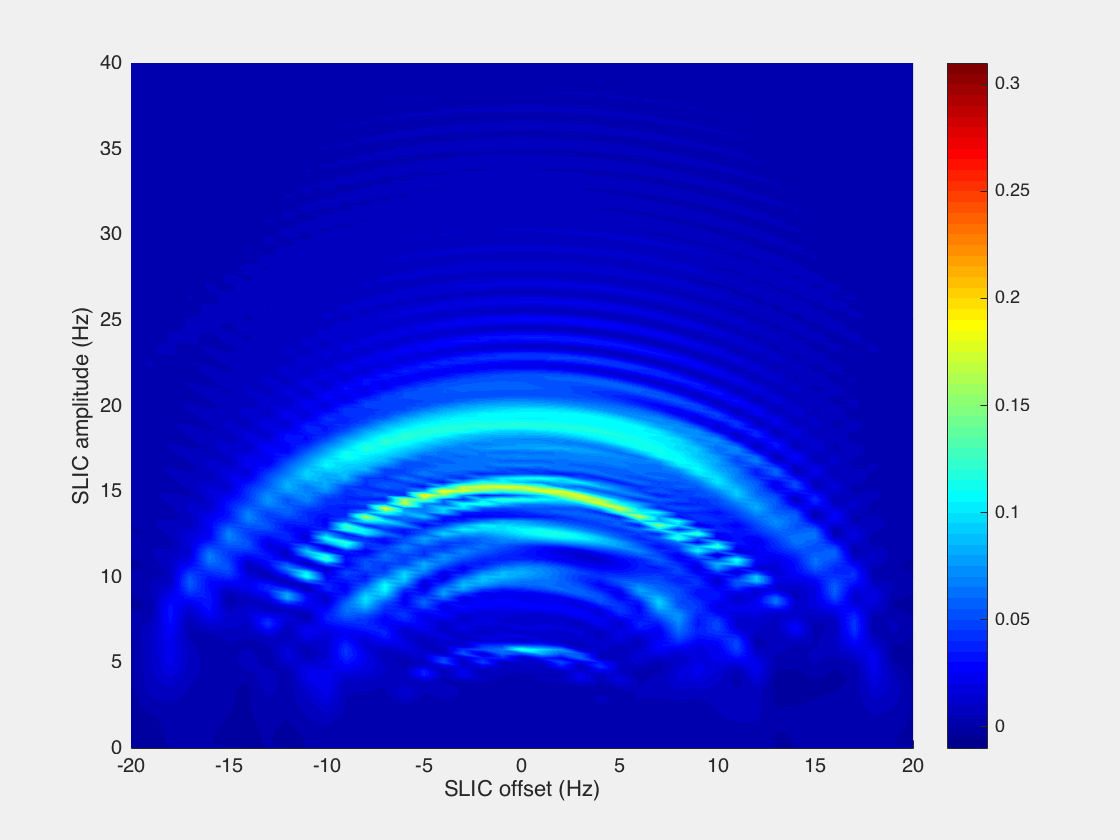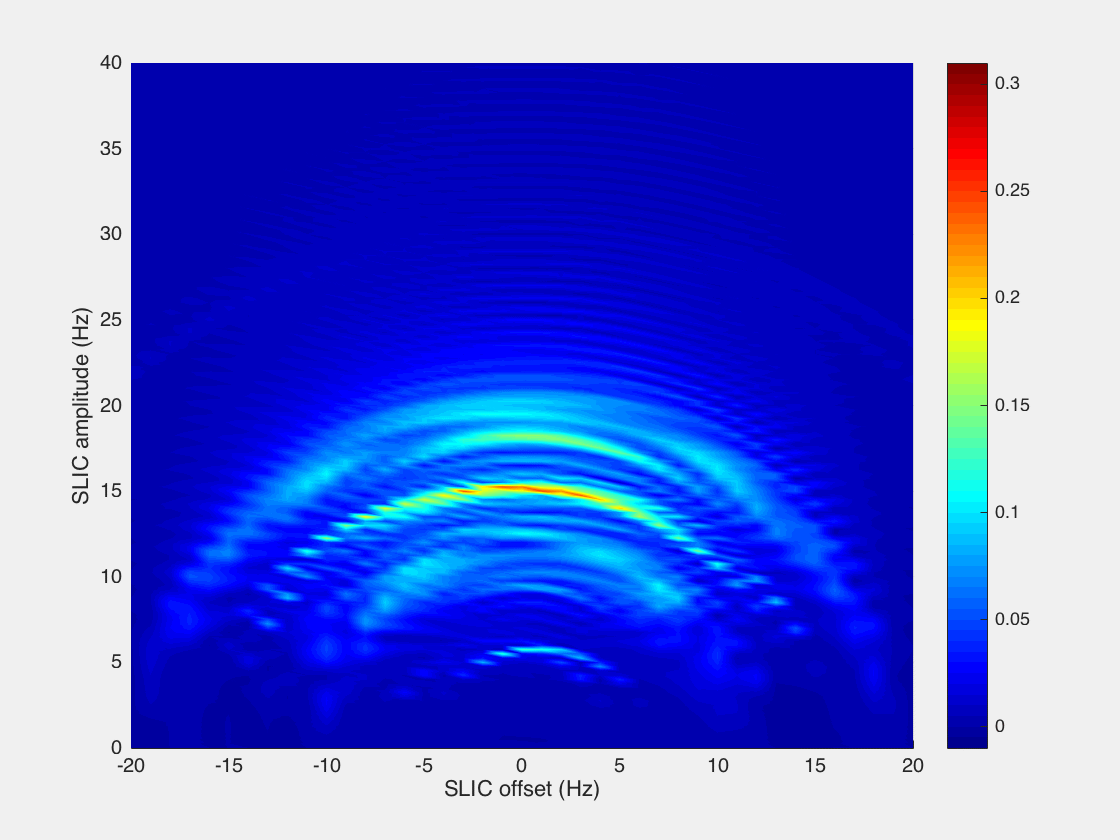
However, we found that while the analytical model presented by DeVience et al. works well for the simple case of a two-spin system, it cannot properly describe the observed patterns obtained in the experiment with the five-spin systems of ethyl groups studied here. For example, our results show that instead of a relatively narrow maximum at B1 = JHA–HB as predicted by the simple theory, there is a broad maximum at B1 ∼ 2JHA–HB, and the optimal SLIC pulse duration is about √2/Δυ (where Δυ is the chemical shift difference (δMETHYLENE – δMETHYL) expressed in Hz), that is, two times longer than that predicted for the two-spin model40 (Figure 4). Here, detailed spin dynamics simulations were carried out (see Supporting Information), resulting in the graphs (Figure 5) where SLIC signal is plotted versus SLIC B1 amplitude, SLIC B1 offset, and SLIC duration, where NMR signal maxima form concentric waves with radii of approximately 3JHA–HB/2, 2JHA–HB, and 5JHA–HB/2. The pattern of the map also changes with SLIC duration, although there is a clear indication of an optimal pulse duration (τSLIC) yielding the global maximum of the produced signal. We note that the spin dynamics for five-spin systems is much more complex than that for two-spin systems but yet relatively easily predictable using the density matrix formalism.
Figure 5.

Simulation of 1H NMR signal dependence in −CH2CH3 system on SLIC parameters: B1 amplitude (Hz), SLIC pulse offset (Hz) (position of zero offset corresponds to the center frequency between CH3 and CH2 resonances), and SLIC pulse duration (τSLIC). Relaxation effects were not included in the simulation. See Supporting Information for animated gif files and the corresponding signal dependences for two-spin system.
Although low-field PHIP hyperpolarizers have been used previously to prepare HP molecules via pairwise p-H2 addition,43,49 prior attempts to perform direct 1H NMR signal detection of nascent HP protons resulted in a very weak antiphase NMR signal.60 Without SLIC or other singlet-to-magnetization pulse sequences,61 direct proton detection at low fields is unlikely to yield high signal-to-noise ratio to study reaction conversion and pairwise selectivity. Building on our previous experience with SLIC detection of HP propane gas prepared via heterogeneous PHIP,38 SLIC proton detection of HP liquid was employed in a low magnetic field of 47.5 mT, and the 1H NMR signal obtained was approximately 2 orders of magnitude greater than the 1H NMR signal obtained using conventional hard (i.e., short duration and high amplitude) RF pulses (Figure 2c). To the best of our knowledge, the fact that the action of hard RF pulses on a spin system leads to NMR signal significantly lower than that obtained by low-frequency irradiation is somewhat unique in the field of NMR spectroscopy. Moreover, one can entertain an analogy with a safe, which is hard to open using brute force (e.g., a hammer), but a tiny key with appropriate symmetry can easily crack the lock. It should also be emphasized that unlike the vast majority of hyperpolarization techniques, where the preparation of singlet states requires additional preparation steps, direct creation of pseudo-singlet states is an inherent and unique feature of the PHIP technique.
By varying the “reaction” time (i.e., the time period that the reaction solution remains in the reactor following the injection), we measured singlet state lifetime (TS) (Figure 3c,d). It was found that the NMR signal decays with a time constant of about 5–15 s depending on the solvent nature (i.e., methanol vs water) (Table 1 and Supporting Information). These values indicate that, despite the spin systems being in pseudo-singlet states, their lifetimes were not significantly longer compared to T1; that is, they were not several fold greater. This is, however, not surprising because the existence of long-lived spin states requires specific symmetry properties, which may not be present in the systems studied here.62−64 One should note, however, that such examples can occur, for example, in previously reported long-lived HP propane states.38,65 Future studies are certainly warranted to identify other examples of long-lived HP spin states that could find use in biomedical and material science applications.66
The efficiency of a singlet-to-magnetization conversion by SLIC may be analyzed using the boundary transformation methodology presented by Levitt.67 In the case of the two-spin system, it is possible to “extract” nearly 100% of the singlet spin order and transform it into observable magnetization (∼91% when using the SLIC pulse sequence). At the same time for a five-spin system, such as −CH2–CH3, it is fundamentally possible to transform up to only 55% of spin order (Supporting Information). Our calculations for SLIC show ∼27% transformation efficiency (Figure 5), indicating that there could be a more efficient RF pulse sequence alternative to the SLIC implementation employed here.68,69 This inefficiency partially explains the relatively low apparent polarization level of ∼0.23% obtained for HEP and even lower values for other studied molecules. Other factors that likely had a significant negative impact on the efficiency of SLIC spin transformation include spin relaxation processes and B0 magnetic field inhomogeneity. Indeed, our calculations show that the efficiency of spin order transfer depends dramatically on the B1 RF frequency offset (Figure 5). Static B0 magnetic field drifts and imperfections across the sample can therefore cause significant shifts away from the optimal transfer conditions, thus leaving a potentially large fraction of the population hidden in the “dark” unobservable nuclear pseudo-singlet state. The use of more homogeneous B0 and B1 fields can likely significantly improve SLIC efficiency and consequently PAPP in the future. Strong dependence of singlet-to-magnetization transformation on the magnetic field inhomogeneity is a substantial limitation of the presented SLIC-based low-field detection method.
Simple analysis also determines the limits of the B0 magnetic field strength, which should be optimal for parahydrogen-based SLIC sensing presented here. First of all, the magnetic field should not be too high (the weakly coupled regime); otherwise, the singlet spin state is no longer an eigenstate of the nuclear spin Hamiltonian. The strong coupling condition can provide a quick estimate of the upper limit of the low magnetic field range, B0 < 2πJHA–HB/γΔυ, where γ is the proton gyromagnetic ratio. At the same time, very low magnetic fields can result in prohibitively long SLIC pulse duration (since, generally, τSLIC ∼ 1/Δυ), resulting in significant relaxation losses and/or decoherences during an excessively long SLIC pulse. Thus, the magnetic field of 0.0475 T employed here lies in the “SLIC-safe range” for the studied spin systems; however, the optimal field should be calculated for a particular spin system under study.
We used 1H and 13C high-field (400 MHz) NMR spectroscopy to determine reaction conversion levels by taking aliquots of stock solutions before and after the reaction. One can see that measured conversion values were relatively high for all studied molecules (Figure 6). Lower levels for conversion of VPy and VTMA (25–40%) compared to nearly 100% conversion for HEA and VA can be explained by the presence of nitrogen in the former molecules. In case of VPy, the electron-donating N site can potentially compete with the double bond for a binding event to the Rh center, thereby lowering the probability of forming the active catalytic species and, consequently, decreasing reaction yields. For VTMA, the nitrogen atom possesses positive charge, which can lead to repulsion of the molecule from the positively charged cationic Rh center due to electrostatic interactions of charged ions. The determined conversion can be compared with the intensity of SLIC signal for the same experiments (Figure 6). The SLIC signal from reaction products does not linearly correlate with the conversion and decreases in the following order of substrates used in hydrogenation: HEA > VA > VTMA > VPy.
Figure 6.
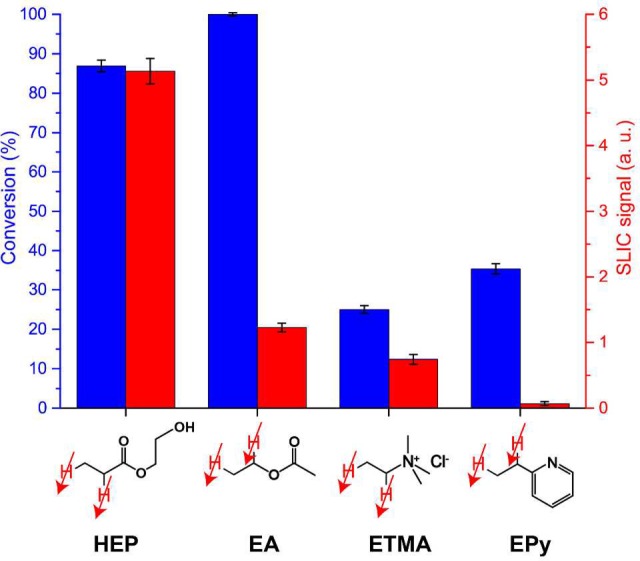
Hydrogenation reaction conversion and SLIC signal (normalized to the signal of the solvent) for substrates in water. Conversion was measured using high-resolution high-field 1H 400 MHz NMR of aliquots before and after the reaction.
One should not find surprising that the different substrates show a different SLIC signal. This is a result of different chemical dynamics and pairwise addition behavior in hydrogenation reactions. It is known that the interplay between the substrate and the catalyst is very important, and variations of the substrate or catalyst structure that seem insignificant at the first glance may have drastic consequences on a pairwise addition performance.70 A good recent example supporting this statement is the homogeneous batch-mode hydrogenation of propylene in methanol using two Rh-based catalysts: [Rh(COD)(dppb)]BF4 and [Rh(NBD)(dppb)]BF4 (where COD = 1,5-cyclooctadiene, NBD = norbornadiene, dppb = 1,4-bis(diphenylphosphino)butane).71 Despite the fact that their structure is very similar and differ only in the structure of the ligand, PHIP effects observed for propane are 3–8 times higher when the latter catalyst is used. While discrepancy between the conversion and the SLIC signal can be treated as a disadvantage for a general applicability of the presented method for low-field monitoring of hydrogenation reactions, this observation is advantageous for probing the pairwise nature of p-H2 addition (i.e., reaction selectivity) for HP NMR and MRI. This means that large libraries of compounds can be screened to identify promising candidates for PHIP HP contrast agents and for optimization of PHIP processes and hyperpolarization equipment. Moreover, since such selectivity probing does not require chemical shift dispersion (which is generally lacking at low magnetic fields), the low-field NMR modality presented here can be used to monitor the production of HP products in larger, more complex reactors operating with high pressures and temperatures; for example, the reactor used here already operated at >17 atm of gas pressure and >55 °C. We foresee that low-field NMR (and MRI) of large-scale industrial hydrogenation processes—hydrogenation of vegetable oils, hydrodesulfurization of petroleum, and other large-scale applications of hydrogenation in the industry—can become a useful spectroscopic and imaging tool.
Conclusions
In conclusion, we have shown that low-field NMR and low-amplitude RF irradiation termed spin-lock induced crossing can be used to detect the signal originating from HP molecules produced via hydrogenation reactions with p-H2. Signal enhancement of more than 10000 allowed the build-up and decay of HP reaction products upon hydrogenation of several organic molecules (2-hydroxyethyl acrylate, vinyl acetate, 2-vinylpyridine, (vinyl)trimethylammonium chloride) to be detected. Moreover, since the signal from ≤80 mM HP reaction products was significantly greater than the signal of thermally polarized solvents, direct proton detection was demonstrated in protonated solvents such as methanol and water, which can provide a significant potential application for molecular sensing of industrial-scale processes in the presence of large concentrations of background species. While it was shown that chemical conversion and SLIC signal are not directly correlated, this finding can be very useful for quick analysis of selectivity of hydrogen addition in catalysis and for production of HP contrast agents by PHIP technique; that is, despite the efficient overall hydrogenation, some compounds may exhibit a lower degree of p-H2 pairwise addition. Moreover, the presented method allows for a quality assurance of the HP state of the molecules before performing experiments with more expensive isotopically enriched (e.g., 13C) compounds29,72 using polarization transfer schemes. Lastly, we showed that spin dynamics during the SLIC pulse for five-spin systems (e.g., molecules such as presented here, containing CH3–CH2– moiety) is much more complex than SLIC for two-spin systems. However, it is possible to adequately predict optimal detection parameters, such as B1 amplitude, offset, and SLIC time. Low-field NMR signals are generally far less sensitive to susceptibility-induced magnetic field gradients (because these gradients scale linearly with B0 strength), which is a useful property for studies of heterogeneous reactions (e.g., liquid/gas, liquid/solid, gas/solid) frequently practiced in industrial hydrogenation processes. Combined with greater penetration depth, which also scales inversely with B0, low-field SLIC sensing may potentially provide a complementary analytical technology for analysis of hydrogenation reactions on a large scale.
Acknowledgments
This work was supported by NIH 1R21EB018014, 1R21EB020323, and 1F32EB021840, NSF CHE-1416268, and CHE-1416432, DOD CDMRP W81XWH-12-1-0159/BC112431 and W81XWH-15-1-0271, ExxonMobil Research and Engineering Company. The Russian team thanks the Russian Science Foundation (Grant 14-35-00020) for support of the hydrogenation experiments. K.V.K. thanks President's of Russian Federation Funding (Grant # MK-4498.2016.3) for catalysts testing.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcc.6b07555.
The authors declare no competing financial interest.
Supplementary Material
{kind=link}
{kind=link}
References
- Ernst R. Nuclear Magnetic Resonance Fourier Transform Spectroscopy (Nobel Lecture). Angew. Chem., Int. Ed. Engl. 1992, 31, 805–823. 10.1002/anie.199208053. [DOI] [PubMed] [Google Scholar]
- Zalesskiy S.; Danieli E.; Bluemich B.; Ananikov V. Miniaturization of NMR Systems: Desktop Spectrometers, Microcoil Spectroscopy, and ″NMR on a Chip″ for Chemistry, Biochemistry, and Industry. Chem. Rev. 2014, 114, 5641–5694. 10.1021/cr400063g. [DOI] [PubMed] [Google Scholar]
- Gladden L. Magnetic Resonance in Reaction Engineering: Beyond Spectroscopy. Curr. Opin. Chem. Eng. 2013, 2, 331–337. 10.1016/j.coche.2013.05.005. [DOI] [Google Scholar]
- Ardenkjaer-Larsen J.; Boebinger G.; Comment A.; Duckett S.; Edison A.; Engelke F.; Griesinger C.; Griffin R.; Hilty C.; Maeda H.; et al. Facing and Overcoming Sensitivity Challenges in Biomolecular NMR Spectroscopy. Angew. Chem., Int. Ed. 2015, 54, 9162–9185. 10.1002/anie.201410653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R.; Adams R.; Duckett S.; Mewis R.; Williamson D.; Green G. The Theory and Practice of Hyperpolarization in Magnetic Resonance Using Parahydrogen. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 67, 1–48. 10.1016/j.pnmrs.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Ardenkjaer-Larsen J.; Fridlund B.; Gram A.; Hansson G.; Hansson L.; Lerche M.; Servin R.; Thaning M.; Golman K. Increase in Signal-to-noise Ratio of > 10,000 Times in Liquid-state NMR. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 10158–10163. 10.1073/pnas.1733835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson B. Nuclear Magnetic Resonance of Laser-polarized Noble Gases in Molecules, Materials, and Organisms. J. Magn. Reson. 2002, 155, 157–216. 10.1006/jmre.2001.2341. [DOI] [PubMed] [Google Scholar]
- Nikolaou P.; Goodson B.; Chekmenev E. NMR Hyperpolarization Techniques for Biomedicine. Chem. - Eur. J. 2015, 21, 3156–3166. 10.1002/chem.201405253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natterer J.; Bargon J. Parahydrogen Induced Polarization. Prog. Nucl. Magn. Reson. Spectrosc. 1997, 31, 293–315. 10.1016/S0079-6565(97)00007-1. [DOI] [Google Scholar]
- Kovtunov K.; Zhivonitko V.; Skovpin I.; Barskiy D.; Koptyug I. Parahydrogen-Induced Polarization in Heterogeneous Catalytic Processes. Top. Curr. Chem. 2012, 338, 123–180. 10.1007/128_2012_371. [DOI] [PubMed] [Google Scholar]
- Bowers C.; Weitekamp D. Parahydrogen and Synthesis Allow Drammatically Enhanced Nuclear Alignment. J. Am. Chem. Soc. 1987, 109, 5541–5542. 10.1021/ja00252a049. [DOI] [Google Scholar]
- Nikolaou P.; Coffey A.; Barlow M.; Rosen M.; Goodson B.; Chekmenev E. Temperature-Ramped Xe-129 Spin-Exchange Optical Pumping. Anal. Chem. 2014, 86, 8206–8212. 10.1021/ac501537w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurhanewicz J.; Vigneron D.; Brindle K.; Chekmenev E.; Comment A.; Cunningham C.; DeBerardinis R.; Green G.; Leach M.; Rajan S.; et al. Analysis of Cancer Metabolism by Imaging Hyperpolarized Nuclei: Prospects for Translation to Clinical Research. Neoplasia 2011, 13, 81–97. 10.1593/neo.101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardenkjaer-Larsen J. On the Present and Future of Dissolution-DNP. J. Magn. Reson. 2016, 264, 3–12. 10.1016/j.jmr.2016.01.015. [DOI] [PubMed] [Google Scholar]
- Harzstark A.; Weinberg V.; Grycz K.; Hurd R.; Ardenkjaer-Larsen J.; Murray J.; Chen A.; Ferrone M.; Park I.; Reed G. A First-in-human Phase I Imaging Study Using Hyperpolarized (1)C-13 Pyruvate (H-Py) in Patients (Pts) With Localized Prostate Cancer (I-PCa). J. Clin. Oncol. 2012, 30, 4660. [Google Scholar]
- Nelson S.; Kurhanewicz J.; Vigneron D.; Larson P.; Harzstark A.; Ferrone M.; van Criekinge M.; Chang J.; Bok R.; Park I.; et al. Metabolic Imaging of Patients with Prostate Cancer Using Hyperpolarized [1-C-13]Pyruvate. Sci. Transl. Med. 2013, 5, 198ra108. 10.1126/scitranslmed.3006070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugler J.; Altes T. Hyperpolarized 129Xe MRI of the Human Lung. J. Magn. Reson. Imaging 2013, 37, 313–331. 10.1002/jmri.23844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchepin R.; Barskiy D.; Coffey A.; Manzanera Esteve I.; Chekmenev E. Efficient Synthesis of Molecular Precursors for Para-Hydrogen-Induced Polarization of Ethyl Acetate-1–13C and Beyond. Angew. Chem., Int. Ed. 2016, 55, 6071–6074. 10.1002/anie.201600521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineri F.; Boi T.; Aime S. Parahydrogen-Induced Polarization of C-13 Carboxylate Resonance in Acetate and Pyruvate. Nat. Commun. 2015, 6, 5858. 10.1038/ncomms6858. [DOI] [PubMed] [Google Scholar]
- Shchepin R.; Coffey A.; Waddell K.; Chekmenev E. Parahydrogen Induced Polarization of 1-C-13-Phospholactate-d(2) for Biomedical Imaging with > 30,000,000-fold NMR Signal Enhancement in Water. Anal. Chem. 2014, 86, 5601–5605. 10.1021/ac500952z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glöggler S.; Grunfeld A. M.; Ertas Y. N.; McCormick J.; Wagner S.; Schleker P. P. M.; Bouchard L. S. A Nanoparticle Catalyst for Heterogeneous Phase Para-Hydrogen-Induced Polarization in Water. Angew. Chem., Int. Ed. 2015, 54, 2452–2456. 10.1002/anie.201409027. [DOI] [PubMed] [Google Scholar]
- Bhattacharya P.; Harris K.; Lin A.; Mansson M.; Norton V. A.; Perman W.; Weitekamp D. P.; Ross B. D. Ultra-fast Three Dimensional Imaging of Hyperpolarized 13C In Vivo. MAGMA 2005, 18, 245–256. 10.1007/s10334-005-0007-x. [DOI] [PubMed] [Google Scholar]
- Lysova A.; Koptyug I. Magnetic Resonance Imaging Methods for In Situ Studies in Heterogeneous Catalysis. Chem. Soc. Rev. 2010, 39, 4585–4601. 10.1039/b919540h. [DOI] [PubMed] [Google Scholar]
- Barskiy D.; Kovtunov K.; Koptyug I.; He P.; Groome K.; Best Q.; Shi F.; Goodson B.; Shchepin R.; Truong M.; et al. In Situ and Ex Situ Low-Field NMR Spectroscopy and MRI Endowed by SABRE Hyperpolarization. ChemPhysChem 2014, 15, 4100–4107. 10.1002/cphc.201402607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R.; Menon R. K. An Overview of Industrial Uses of Hydrogen. Int. J. Hydrogen Energy 1998, 23, 593–598. 10.1016/S0360-3199(97)00112-2. [DOI] [Google Scholar]
- Blumich B.; Casanova F.; Appelt S. NMR at Low Magnetic Fields. Chem. Phys. Lett. 2009, 477, 231–240. 10.1016/j.cplett.2009.06.096. [DOI] [Google Scholar]
- Gong Q.; Gordji-Nejad A.; Blumich B.; Appelt S. Trace Analysis by Low-Field NMR: Breaking the Sensitivity Limit. Anal. Chem. 2010, 82, 7078–7082. 10.1021/ac101738f. [DOI] [PubMed] [Google Scholar]
- Gloggler S.; Blumich B.; Appelt S.; Heise H.; Matthews S. NMR Spectroscopy for Chemical Analysis at Low Magnetic Fields. Top. Curr. Chem. 2011, 335, 1–22. 10.1007/128_2011_304. [DOI] [PubMed] [Google Scholar]
- Bhattacharya P.; Chekmenev E.; Reynolds W.; Wagner S.; Zacharias N.; Chan H.; Bunger R.; Ross B. Parahydrogen-Induced Polarization (PHIP) Hyperpolarized MR Receptor Imaging In Vivo: a Pilot Study of C-13 Imaging of Atheroma in Mice. NMR Biomed. 2011, 24, 1023–1028. 10.1002/nbm.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovener J.; Schwaderlapp N.; Lickert T.; Duckett S.; Mewis R.; Highton L.; Kenny S.; Green G.; Leibfritz D.; Korvink J. A Hyperpolarized Equilibrium for Magnetic Resonance. Nat. Commun. 2013, 4, 2946. 10.1038/ncomms3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suefke M.; Liebisch A.; Blumich B.; Appelt S. External High-quality-factor Resonator Tunes Up Nuclear Magnetic Resonance. Nat. Phys. 2015, 11, 767–771. 10.1038/nphys3382. [DOI] [Google Scholar]
- Coffey A.; Truong M.; Chekmenev E. Low-field MRI Can Be More Sensitive Than High-field MRI. J. Magn. Reson. 2013, 237, 169–174. 10.1016/j.jmr.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovener J.; Schwaderlapp N.; Borowiak R.; Lickert T.; Duckett S.; Mewis R.; Adams R.; Burns M.; Highton L.; Green G.; et al. Toward Biocompatible Nuclear Hyperpolarization Using Signal Amplification by Reversible Exchange: Quantitative In Situ Spectroscopy and High-Field Imaging. Anal. Chem. 2014, 86, 1767–1774. 10.1021/ac403653q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruset I.; Tsai L.; Mair R.; Patz S.; Hrovat M.; Rosen M.; Muradian I.; Ng J.; Topulos G.; Butler J.; et al. A System for Open-access 3He Human Lung Imaging at Very Low Field. Concepts Magn. Reson., Part B 2006, 29, 210–221. 10.1002/cmr.b.20075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singlet and triplet spin states are eigenstates of the nuclear spin Hamiltonian for a two-spin system in the condition of a strong coupling regime (sometimes referred to as an inverse weak coupling regime). However, for multispin systems (e.g., a five-spin system studied here) versus two-spin systems, this “singlet–triplet” terminology is applicable only in qualitative terms, but it is still quite illustrative: indeed, at the magnetic field of 0.0475 T, the methyl and methylene proton spins of the studied molecular systems are strongly coupled and essentially represent a system of five almost magnetically equivalent spins. This is why we use the term “pseudo-singlet state” to describe the nuclear state of the of the −CH2CH3 system populated after molecular addition of p-H2 to the unsaturated precursor.
- Olaru A. M.; Roy S. S.; Lloyd L. S.; Coombes S.; Green G. G. R.; Duckett S. B. Creating a Hyperpolarised Pseudo Singlet State Through Polarisation Transfer From Parahydrogen Under SABRE. Chem. Commun. 2016, 52, 7842–7845. 10.1039/C6CC02020H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczyk T.; Gutmann T.; Dillenberger S.; Abdulhussaein S.; Frydel J.; Breitzke H.; Bommerich U.; Trantzschel T.; Bernarding J.; Magusin P. C.; et al. Time Domain Parahydrogen Induced Polarization. Solid State Nucl. Magn. Reson. 2012, 43, 14–21. 10.1016/j.ssnmr.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Kovtunov K.; Truong M.; Barskiy D.; Koptyug I.; Coffey A.; Waddell K.; Chekmenev E. Long-Lived Spin States for Low-field Hyperpolarized Gas MRI. Chem. - Eur. J. 2014, 20, 14629–32. 10.1002/chem.201405063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prina I.; Buljubasich L.; Acosta R. Parahydrogen Discriminated PHIP at Low Magnetic Fields. J. Magn. Reson. 2015, 251, 1–7. 10.1016/j.jmr.2014.11.010. [DOI] [PubMed] [Google Scholar]
- DeVience S.; Walsworth R.; Rosen M. Preparation of Nuclear Spin Singlet States Using Spin-Lock Induced Crossing. Phys. Rev. Lett. 2013, 111, 173002. 10.1103/PhysRevLett.111.173002. [DOI] [PubMed] [Google Scholar]
- Coffey A.; Kovtunov K.; Barskiy D.; Koptyug I.; Shchepin R.; Waddell K.; He P.; Groome K.; Best Q.; Shi F.; et al. High-Resolution Low-Field Molecular Magnetic Resonance Imaging of Hyperpolarized Liquids. Anal. Chem. 2014, 86, 9042–9049. 10.1021/ac501638p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey A.; Shchepin R.; Truong M.; Wilkens K.; Pham W.; Chekmenev E. An Open-Source Automated Parahydrogen Hyperpolarizer for Molecular Imaging Using 13C Metabolic Contrast Agents. Anal. Chem. 2016, 88, 8279–8288. 10.1021/acs.analchem.6b02130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell K.; Coffey A.; Chekmenev E. In Situ Detection of PHIP at 48 mT: Demonstration Using a Centrally Controlled Polarizer. J. Am. Chem. Soc. 2011, 133, 97–101. 10.1021/ja108529m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey A.; Shchepin R.; Wilkens K.; Waddell K.; Chekmenev E. A Large Volume Double Channel H-1-X RF Probe for Hyperpolarized Magnetic Resonance at 0.0475 T. J. Magn. Reson. 2012, 220, 94–101. 10.1016/j.jmr.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W.; Lumata L.; Chen W.; Zhang S.; Kovacs Z.; Sherry A. D.; Khemtong C. Hyperpolarized 15N-pyridine Derivatives as pH-sensitive MRI Agents. Sci. Rep. 2014, 5, 9104–9104. 10.1038/srep09104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurhanewicz J.; Vigneron D. B.; Hricak H.; Narayan P.; Carroll P.; Nelson S. J. Three-dimensional H-1 MR Spectroscopic Imaging of the In Situ Human Prostate With High (0.24–0.7-cm3) Spatial Resolution. Radiology 1996, 198, 795–805. 10.1148/radiology.198.3.8628874. [DOI] [PubMed] [Google Scholar]
- Gillies R. J.; Morse D. L. In vivo magnetic resonance spectroscopy in cancer. Annu. Rev. Biomed. Eng. 2005, 7, 287–326. 10.1146/annurev.bioeng.7.060804.100411. [DOI] [PubMed] [Google Scholar]
- Eliyahu G.; Kreizman T.; Degani H. Phosphocholine as a Biomarker of Breast Cancer: Molecular and Biochemical Studies. Int. J. Cancer 2007, 120, 1721–1730. 10.1002/ijc.22293. [DOI] [PubMed] [Google Scholar]
- Goldman M.; Johannesson H.; Axelsson O.; Karlsson M. Design and Implementation of C-13 Hyperpolarization from Parahydrogen, for New MRI Contrast Agents. C. R. Chim. 2006, 9, 357–363. 10.1016/j.crci.2005.05.010. [DOI] [Google Scholar]
- Golman K.; Axelsson O.; Johannesson H.; Mansson S.; Olofsson C.; Petersson J. Parahydrogen-Induced Polarization in Imaging: Subsecond C-13 Angiography. Magn. Reson. Med. 2001, 46, 1–5. 10.1002/mrm.1152. [DOI] [PubMed] [Google Scholar]
- Truong M.; Shi F.; He P.; Yuan B.; Plunkett K.; Coffey A.; Shchepin R.; Barskiy D.; Kovtunov K.; Koptyug I.; et al. Irreversible Catalyst Activation Enables Hyperpolarization and Water Solubility for NMR Signal Amplification by Reversible Exchange. J. Phys. Chem. B 2014, 118, 13882–13889. 10.1021/jp510825b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M.Spin Dynamics: Basics of Nuclear Magnetic Resonance; John Wiley & Sons, 2001. [Google Scholar]
- Bowers C.; Weitekamp D. Transformation of Symmetrization Order to Nuclear Spin Magnetization by Chemical Reaction and Nuclear Magnetic Resonance. Phys. Rev. Lett. 1986, 57, 2645–2648. 10.1103/PhysRevLett.57.2645. [DOI] [PubMed] [Google Scholar]
- Eisenschmid T.; Kirss R.; Deutsch P.; Hommeltoft S.; Eisenberg R.; Bargon J.; Lawler R.; Balch A. Parahydrogen Induced Polarization in Hydrogenation Reactions. J. Am. Chem. Soc. 1987, 109, 8089–8091. 10.1021/ja00260a026. [DOI] [Google Scholar]
- Appelt S.; Hasing F.; Sieling U.; Gordji-Nejad A.; Gloggler S.; Blumich B. Paths from Weak to Strong Coupling in NMR. Phys. Rev. A: At., Mol., Opt. Phys. 2010, 81, 023420. 10.1103/PhysRevA.81.023420. [DOI] [Google Scholar]
- Colell J.; Turschmann P.; Gloggler S.; Schleker P.; Theis T.; Ledbetter M.; Budker D.; Pines A.; Blumich B.; Appelt S. Fundamental Aspects of Parahydrogen Enhanced Low-Field Nuclear Magnetic Resonance. Phys. Rev. Lett. 2013, 110, 137602. 10.1103/PhysRevLett.110.137602. [DOI] [PubMed] [Google Scholar]
- Turschmann P.; Colell J.; Theis T.; Blumich B.; Appelt S. Analysis of Parahydrogen Polarized Spin System in Low Magnetic Fields. Phys. Chem. Chem. Phys. 2014, 16, 15411–15421. 10.1039/c4cp01807a. [DOI] [PubMed] [Google Scholar]
- Bernarding J.; Buntkowsky G.; Macholl S.; Hartwig S.; Burghoff M.; Trahms L. J-coupling Nuclear Magnetic Resonance Spectroscopy of Liquids in nT Fields. J. Am. Chem. Soc. 2006, 128, 714–715. 10.1021/ja055273e. [DOI] [PubMed] [Google Scholar]
- DeVience S.; Walsworth R.; Rosen M. Probing Scalar Coupling Differences via Long-Lived Singlet States. J. Magn. Reson. 2016, 262, 42–49. 10.1016/j.jmr.2015.12.003. [DOI] [PubMed] [Google Scholar]
- Kovtunov K.; Barskiy D.; Shchepin R.; Coffey A.; Waddell K.; Koptyug I.; Chekmenev E. Demonstration of Heterogeneous Parahydrogen Induced Polarization Using Hyperpolarized Agent Migration from Dissolved Rh(I) Complex to Gas Phase. Anal. Chem. 2014, 86, 6192–6. 10.1021/ac5013859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayler M.; Levitt M. Singlet Nuclear Magnetic Resonance of Nearly-Equivalent Spins. Phys. Chem. Chem. Phys. 2011, 13, 5556–60. 10.1039/c0cp02293d. [DOI] [PubMed] [Google Scholar]
- Levitt M. Singlet Nuclear Magnetic Resonance. Annu. Rev. Phys. Chem. 2012, 63, 89–105. 10.1146/annurev-physchem-032511-143724. [DOI] [PubMed] [Google Scholar]
- Vinogradov E.; Grant A. K. Long-Lived States in Solution NMR: Selection Rules for Intramolecular Dipolar Relaxation in Low Magnetic Fields. J. Magn. Reson. 2007, 188, 176–182. 10.1016/j.jmr.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Stevanato G.; Roy S. S.; Hill-Cousins J.; Kuprov I.; Brown L. J.; Brown R. C.; Pileio G.; Levitt M. H. Long-Lived Nuclear Spin States Far from Magnetic Equivalence. Phys. Chem. Chem. Phys. 2015, 17, 5913–5922. 10.1039/C4CP05704J. [DOI] [PubMed] [Google Scholar]
- Kovtunov K.; Truong M.; Barskiy D.; Salnikov O.; Bukhtiyarov V.; Coffey A.; Waddell K.; Koptyug I.; Chekmenev E. Propane-d(6) Heterogeneously Hyperpolarized by Parahydrogen. J. Phys. Chem. C 2014, 118, 28234–28243. 10.1021/jp508719n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Duan X.; Soon P. C.; Sychrovský V.; Canary J. W.; Jerschow A. Limits in Proton Nuclear Singlet-State Lifetimes Measured with Para-Hydrogen-Induced Polarization. ChemPhysChem 2016, 17, 2967. 10.1002/cphc.201600663. [DOI] [PubMed] [Google Scholar]
- Levitt M. Symmetry Constraints on Spin Dynamics: Application to Hyperpolarized NMR. J. Magn. Reson. 2016, 262, 91–99. 10.1016/j.jmr.2015.08.021. [DOI] [PubMed] [Google Scholar]
- Theis T.; Feng Y.; Wu T.-l.; Warren W. S. Composite and Shaped Pulses for Efficient and Robust Pumping of Disconnected Eigenstates in Magnetic Resonance. J. Chem. Phys. 2014, 140, 014201. 10.1063/1.4851337. [DOI] [PubMed] [Google Scholar]
- Pravdivtsev A. N.; Kiryutin A. S.; Yurkovskaya A. V.; Vieth H.-M.; Ivanov K. L.. Robust Conversion of Singlet Spin Order in Coupled Spin-1/2 Pairs by Adiabatically Switched RF-fields. arXiv:1607.00539 2016. [DOI] [PubMed] [Google Scholar]
- Reineri F.; Aime S.; Gobetto R.; Nervi C. Role of the Reaction Intermediates in Determining PHIP (Parahydrogen Induced Polarization) Effect in the Hydrogenation of Acetylene Dicarboxylic Acid With the Complex [Rh(dppb)](+) (dppb: 1,4-bis(diphenylphosphino)butane. J. Chem. Phys. 2014, 140, 094307. 10.1063/1.4867269. [DOI] [PubMed] [Google Scholar]
- Salnikov O. G.; Barskiy D. A.; Coffey A. M.; Kovtunov K. V.; Koptyug I. V.; Chekmenev E. Y. Efficient Batch-Mode Parahydrogen-Induced Polarization of Propane. ChemPhysChem 2016, 10.1002/cphc.201600564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya P.; Chekmenev E.; Perman W.; Harris K.; Lin A.; Norton V.; Tan C.; Ross B.; Weitekamp D. Towards Hyperpolarized C-13-succinate Imaging of Brain Cancer. J. Magn. Reson. 2007, 186, 150–155. 10.1016/j.jmr.2007.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.