

Abstract
Purpose
We previously have reported that chondrocyte-derived extracellular matrix (CDECM) suppresses the growth of pterygium in athymic nude mice. The aim of this study is to demonstrate the effect of CDECM on the pterygium epithelial cells and molecular signaling pathways in human primary pterygium epithelial cells (hPECs).
Methods
Human conjunctival epithelial cells (hConECs) were used for identification of the effect of CDECM on normal conjunctiva. The effects of CDECM on proliferation were measured with the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfenyl)-2H-tetrazolium (MTS) assay. Cell migration was evaluated according to the scratch wound closure assay and the Transwell invasion assay. Pterygium-related angiogenesis, inflammation, and extracellular matrix remodeling were analyzed with immunoblot and enzyme-linked immunosorbent assay (ELISA). The level of oxidative stress was detected with 2′,7′-dichlorofluorescein diacetate (DCFH-DA). Protein kinase signaling was also analyzed with immunoblot.
Results
CDECM did not show cytotoxicity until 1 mg/ml in the hConECs and hPECs. Cell migration and invasion were markedly reduced by treatment of 1 mg/ml CDECM in the hPECs to 34% of the control, but not in the hConECs. CDECM significantly downregulated matrix metallopeptidase 9 (MMP-9) and fibronectin and upregulated tissue inhibitor of metalloprotease 1 (TIMP-1) and -2 in the hPECs. Angiogenic factors, such as vascular endothelial growth factor (VEGF), antivascular cellular adhesion molecule 1 (VCAM-1), and cluster of differentiation 31 (CD31), and proinflammatory factors, including tumor necrosis factor-α (TNF-α), cyclooxygenase-2 (Cox2), interleukin 6 (IL-6), and prostaglandin E2 (PGE2), were dramatically reduced by CDECM in the hPECs. Furthermore, CDECM significantly inhibited the generation of intracellular reactive oxygen species and the expression of NADPH oxidase subunits, Nox2 and p47phox. CDECM induced nuclear factor erythroid-2 related factor 2 (Nrf2) mediated-antioxidant enzyme heme oxygenase-1 (HO-1). CDECM also suppressed nuclear factor-kappa B (NF-κB) activation and the phosphorylation of p38 mitogen-activated protein kinase (MAPK), protein kinase C alpha (PKCα), and PKCθ.
Conclusions
CDECM was markedly effective in pathogenesis of hPECs. CDECM-suppressed migration of hPECs resulted from the inhibition of NF-κB activation and the improvement of Nrf2 induction by blocking the p38 MAPK and PKC signaling pathways.
Introduction
Pterygium which may be caused by chronic ultraviolet (UV)-B irradiation is an invasive and proliferative disease in humans [1,2]. Pterygium takes the formation of triangular strap-like fibrovascular tissue that lies over the epibulbar surface of the conjunctiva, with the bottom of the triangle on the nasal conjunctiva and pointing to the cornea [3,4]. In advanced cases, pterygium extends to the optical center of the cornea and causes disruption of vision. The principal treatment for pterygium is surgical removal. This approach can have high success rates, but there can be complications and recurrences requiring repeat surgery. Conjunctival autografts, amniotic membrane transplantation, and treatment with radiation or chemotherapeutic agents, usually mitomycin C, are often employed in attempts to reduce recurrence [5,6]. Unfortunately, side effects have been reported for these treatments, including the development of cataract and glaucoma and the increased risk of infection. Therefore, new effective therapeutic methods for treating pterygium are still required [7-9].
Pterygia are characterized by the hyperplastic and centripetally directed growth of altered limbal epithelial cells accompanied by dissolution of Bowman’s layer and epithelial–mesenchymal transition. Recent studies also have shown activated fibroblastic stroma with inflammation, neovascularization, and matrix remodeling, mediated through the concerted actions of cytokines, growth factors, and matrix metalloproteinases (MMPs) in pterygial tissue [10-12]. This histopathological evidence indicates that inhibition of fibroblastic growth through suppression of inflammation, neovascularization, and matrix remodeling may be a concern for reducing pterygial tissue.
Chondrocytes are affected directly by vessel invasion, which may reduce the matrix synthesis of cells, cause apoptosis, and subsequently interfere with the maturation of cells in the new tissue in vivo [13,14]. Choi et al. reported that chondrocyte-derived extracellular matrix (CDECM) constructs showed less vessel invasion on the surface and inside the constructs than polyglycolic acid constructs [15]. CDECM inhibits the adhesion, proliferation, and tube formation of human umbilical vein endothelial cells and suppresses the formation of vessel-like structures and the markers of angiogenesis, including vascular endothelial growth factor (VEGF), in nude mice [16]. These studies indicate that CDECM mitigates migration, angiogenesis, and neovascularization. Furthermore, we have previously demonstrated that CDECM suppresses pterygial lesion growth in human primary pterygial cell-induced pterygium in the eyes of athymic nude mice [17], suggesting that inhibitory effects of CDECM on migration, angiogenesis, and neovascularization may modulate pterygial lesion growth. We also have reported that CDECM suppresses corneal neovascularization and opacification by inhibition of the translocation of nuclear factor-kappa B (NF-κB) in corneal alkaline burns [18]; however, no studies concerning the mechanism by which CDECM suppresses pterygium have been conducted. In this study, we identified the effects of CDECM on pathogenesis and underlying mechanisms, including angiogenesis, inflammation, extracellular matrix remodeling, and oxidative stress in human pterygium epithelial cells (hPECs).
Methods
Preparation of CDECM
A CDECM powder was prepared as described previously [19]. Briefly, primary chondrocytes from porcine knee joints were cultured in monolayers at high density (3×106 cells/ml) for 3 weeks to form a cell–ECM composite membrane. The membrane was then treated with DNase I and sodium dodecyl sulfate (SDS) to remove the cell components and was finally fabricated to a thin film 30–60 µm thick. To produce powder forms of CDECM, the film was frozen at –70 °C, freeze-dried, and comminuted to a particulate form using a cryogenic sample crusher (JFC-300, JAI, Tokyo, Japan). All products were sterilized with ethylene oxygen gas for 24 h at 27 °C before use.
Culture of primary human pterygium epithelial cells and conjunctival epithelial cells
The hPECs were isolated and cultured from specimens during surgical removal, as previously described [17]. The experiments followed the Declaration of Helsinki and the ARVO statement for the use of human subjects and the protocols were approved by the institutional review board (IRB) of Busan Paik Hospital, Inje University College of Medicine, Busan, Korea (IRB No. 12,128). Ten male and seven female pterygium patients (age 47 to 65) without any other clinical problem provided written informed consent after having received a comprehensive explanation of the study procedures. A total of 16 fresh pterygium specimens from 16 donors were placed in six-well culture plates that contained 500 μl of Dulbecco’s PBS (DPBS-2.7 mM KCl, 1.5 mM KH2PO4, 137.9 mM NaCl, 8 mM Na2HPO4-7H2O, pH 7.4; Gibco, Carlsbad, CA). The epithelium was separated from the underlying stroma and subsequently cut into several 1–2 mm2 pieces. The epithelial tissue was cultured on surfaces coated with collagen (rat tail collagen type I; Sigma, St. Louis, MO) for 3 days in Dulbecco’s modified Eagle’s medium/F12 (DMEM/F12; Gibco) supplemented with 10% bovine calf serum (BCS; Gibco), 0.5% dimethyl sulfoxide (DMSO; Sigma), and 1% antibiotic/antimycotic (Gibco), during which time the cells migrated from the explant. Then, the explant was removed, and the medium was replaced with keratinocyte-serum free medium (Gibco) with 5% BCS and 1% antibiotic/antimycotic to further promote epithelial cell growth. When the cells were 60–80% confluent, they were trypsinized with 0.25% trypsin-EDTA (Gibco). The cells were then placed in DMEM/F12 medium supplemented with 5% fetal bovine serum (FBS; Hyclone, Logan, UT), 0.5% DMSO, and 1% antibiotic/antimycotic. The medium was renewed every 2 to 3 days and maintained in an incubator at 37 °C in an atmosphere of 5% CO2.
Human conjunctival epithelial cells (hConECs) were obtained from Innoprot (P10870, Bizkaia, Spain). The cells were incubated with corneal epithelial cell complete medium supplemented with 10% FBS and 1% penicillin/streptomycin in poly-L-lysin–coated cell culture plate at 37 °C in an atmosphere of 5% CO2, according to the manufacturer’s instructions. The medium was changed every 48 h.
Cell viability assay
Cell viability was measured with a CellTiter 96 AQueous One Solution colorimetric cell proliferation assay kit (Promega, Madison, WI), according to the manufacturer’s instructions. Briefly, hPECs (1×104 cells/well) and hConECs (1×104 cells/well) were dispensed in 96-well plates and supplemented with various concentrations of CDECM (0.1–10 mg/ml). After incubation for 24 h, 20 μl of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfenyl)-2H-tetrazolium (MTS) solution was added to each well. The cells were further incubated for 1 h at 37 °C in 5% CO2, and the absorbance was measured at 490 nm using a microplate reader (Molecular Devices, Sunnyvale, CA).
Cell migration assay
Cell migration behavior was measured by using the iBidi Culture-Insert (iBidi, Martinsried, Germany), according to the manufacturer’s instructions. Briefly, the hPECs and hConECs were inoculated into the Culture-Insert of the six-well plate. After incubation overnight until confluence, the Culture-Inserts were gently removed. The cells were washed once with serum-free medium and then incubated for 24 h in each medium containing 1% serum with or without 1 mg/ml CDECM. After incubation, cell migration into the blank surface was monitored with a microscope (DM2500, Leica Microsystems, Wetzlar, Germany) at various time points. Migration was quantified by measuring the distance between the wound edge of the migrating cells and the start point from three independent experiments.
Transwell invasion assay
The cell invasion was assessed in a 24-Transwell cell culture chamber using a polycarbonate membrane with 8-μM pores (Corning, Lowell, MA). The lower surface of the membrane was precoated with 0.2 mg/ml collagen-type I for the hPECs and poly-L-lysine for the hConECs. The cells were suspended in medium supplemented with 0.2% BSA (Sigma) at a concentration of 1×106 cells/ml. The lower compartment contained 0.5 ml of complete medium with or without 1 mg/ml CDECM before use. Cells were incubated at 37 °C in 5% CO2 for 24 h. After incubation, the non-migrated cells on the upper surface were removed gently, and the filters were fixed in methanol and stained with hematoxylin and eosin (H&E) solution. The stained cells were observed under a microscope.
Immunoblot analysis
The cells were incubated with or without 1 mg/ml CDECM for 24 h. After incubation, the cells were washed twice using ice-cold PBS and harvested by scraping in 1 ml of ice-cold PBS. Whole cell lysates were extracted with the Pro-prep protein extraction solution (Intron Biotechnology, Gyeonggi-do, Korea). The nuclear protein extracts obtained from the cells were prepared using the NucBuster Protein Extraction Kit (Novagen, Darmstadt, Germany). The protein concentration was determined with a commercial protein assay kit (Bio-Rad, Hercules, CA). BSA was used as a standard. Equal protein concentrations were subjected to 7.5–12.5% SDS–polyacrylamide gel electrophoresis (PAGE) and transferred to a polyvinylidene fluoride membrane (GE Healthcare, Piscataway, NJ). After blocking with PBS containing 5% nonfat dry milk for 1 h at room temperature, each membrane was incubated with a specific primary antibody overnight at 4 °C: anti-cluster of differentiation (CD31, 1:1,000), anti-MMP-9 (1:1,000, all from Bioss Antibodies, Woburn, MA), anti-VEGF (Abbiotec, San Diego, CA), anti-heme oxygenase-1 (HO-1,1:1,000), anti-Nox2 (1:1,000, Millipore, Temecula, CA), anti-cyclooxygenase-2 (Cox2, 1:1,000), anti-tumor necrosis factor-α (TNF-α, 1:1,000), anti-extracellular signal–regulated kinase (ERK; 1:2,000), anti-stress-activated protein kinase/c-Jun N terminal kinase (SAPK/JNK; 1:2,000), anti-p38 mitogen-activated protein kinase (p38 MAPK; 1:2,000), anti-Akt (1:2,000), anti-phospho-ERK (1:1,000), anti-phospho-SAPK/JNK (1:1,000), anti-phospho-p38 MAPK (1:1,000), anti-phospho-Akt (Thr308; 1:1,000), anti-nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκBα, 1:1,000), anti-phospho-IκBα (Ser32/36; 1:1,000, all from Cell Signaling Technology, Beverly, MA), anti-protein kinase C θ (Thr538; 1:1,000, Invitrogen, Carlsbad, CA), anti-PKCα (1:2,000), anti-phospho-PKCα (Ser657; 1:1,000), anti-vascular cellular adhesion molecule (VCAM)-1 (1:1,000), anti-tissue inhibitor of metalloprotease (TIMP)-1 (1:1,000), anti-TIMP-2 (1:1,000), anti-nuclear factor erythroid-2 related factor 2 (Nrf2; 1:1,000), anti-p47phox (1:1,000), anti-NF-κB p65 (1:1,000), and anti-β-actin (1:5,000, all from Santa Cruz Biotechnology, Santa Cruz, CA). After washing twice with PBS containing 0.1% Tween-20 (PBST), each membrane was immunoblotted with horseradish peroxidase-conjugated secondary immunoglobulin G (IgG) antibodies (1:5,000, Santa Cruz Biotechnology) for 1 h at room temperature. The immune complex was detected using an enhanced chemiluminescence (ECL) detection kit (GE Healthcare). Densitometric analysis of the data was performed using the Fusion FX image acquisition system (Vilber Lourmat, Torcy, France).
Enzyme-linked immunosorbent assay
The hConECs (1×106) and hPECs (1×105) were incubated with or without 1 mg/ml CDECM for 24 h. The culture supernatants were subsequently isolated, and the amounts of interleukin (IL)-1β, IL-6, and prostaglandin E2 (PGE2) production were measured using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. The cytokine concentrations were determined using a microplate reader at 450 nm.
Reactive oxygen species generation
Intracellular reactive oxygen species (ROS) levels were assessed using a ROS-sensitive fluorescent dye, 2′,7′-dichlorofluorescein diacetate (DCFH-DA, Sigma), as previously described [20]. The hConECs (1×106) and hPECs (1×105) were incubated with or without 1 mg/ml CDECM for 24 h. In the last 30 min of treatment, 10 μM DCFH-DA was added to the incubated cells. The cells were protected from light. Each group was washed three times using PBS. The stained cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Carlsbad, CA) and viewed under an automatic fluorescence in situ hybridization (FISH) imager (BX51, Olympus, Tokyo, Japan).
Statistical analysis
All experiments were performed at least three times by conducting each assay. Data comparisons of MTS assay, cell migration, cell invasion, and immunoblot between the control group and the CDECM group were analyzed with a Student t test using SPSS for Windows (Chicago, IL). Statistical significance is indicated as *p<0.05, **p<0.01, and ***p<0.001.
Results
Effect of CDECM on the cell viability of hConECs and hPECs
To evaluate the effect of CDECM on cell proliferation in hConECs and hPECs, viable cells were estimated with the MTS assay. We used two pterygial tissues collected from two donors for the MTS assay. CDECM did not have a discernable cytotoxic effect at the concentration of 1 mg/ml in the hConECs and hPECs (Figure 1). However, cell viability was significantly suppressed to 85.9±5.1% at 5 mg/ml (p<0.01, n = 5) and 73.8±6.9% at 10 mg/ml (p<0.01, n = 5) in the hConECs and 72.8±1.3% at 5 mg/ml (p<0.001, n = 5) and 59.4±2.3% at 10 mg/ml (p<0.001, n = 5) in the hPECs.
Figure 1.
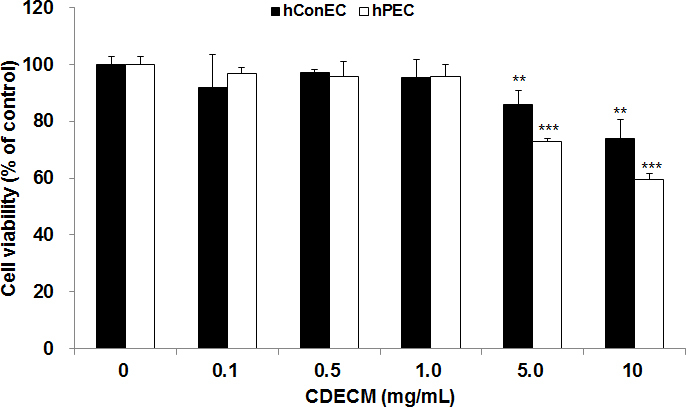
Effect of CDECM on cell viability in hConECs and hPECs. Cell viability was evaluated with the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfenyl)-2H-tetrazolium (MTS) assay. Human conjunctival epithelial cells (hConECs) and human pterygium epithelial cells (hPECs) were incubated with different concentrations of chondrocyte-derived extracellular matrix (CDECM) for 24 h. Values are mean ± standard deviation (SD; n = 5). Statistical significance is indicated as **p<0.01 and ***p<0.001.
Inhibitory effect of CDECM on the migration of hPECs
The effects of CDECM on hConEC and hPEC migration were evaluated with the scratch wound closure assay and the Transwell invasion assay. We used four pterygial tissues collected from four donors for this test. In the scratch wound closure assay, the CDECM markedly inhibited migration of the hPECs to 55.7±12.9% of control at 24 h (p<0.001, n = 4, Figure 2A,B), but not in the hConECs. In the Transwell invasion assay, CDECM also significantly suppressed the movement of the hPECs but not that of the hConECs. As shown in Figure 2C,D, fewer hPECs passed through the membrane in the CDECM treatment (24.7±2.5 cells/area, p<0.001, n = 4) than in the control group (69.3±4.9 cells/area, n = 4).
Figure 2.
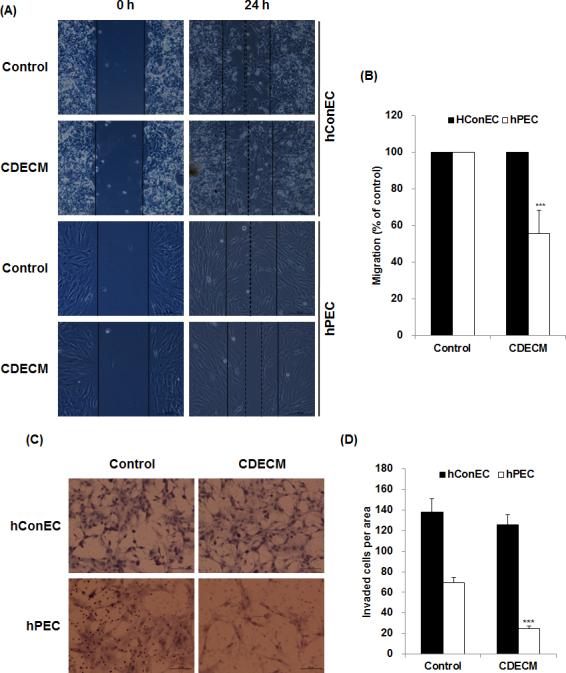
Effect of CDECM on the migration of hConECs and hPECs. A: The effects of chondrocyte-derived extracellular matrix (CDECM; 1 mg/ml) on human conjunctival epithelial cell (hConEC) and human pterygium epithelial cell (hPEC) migration were evaluated through a scratch wound closure assay. Scale bar = 200 μm. B: The graph shows the relative migration rate normalized to the control. C: The Transwell invasion assay tests the influence of CDECM on the invasion ability of hPECs. Scale bar = 200 μm. D: The graph shows invaded cells per area. Data are expressed as the means ± standard deviation (SD; n = 4). Statistical significance is indicated as ***p<0.001.
Inhibitory effect of CDECM on the extracellular matrix remodeling in hPECs
We performed immunoblot analysis to identify the effects of CDECM on extracellular matrix remodeling in hConECs and hPECs. We used three pterygial tissues collected from three donors for this test. The cell lysates were immunostained with extracellular remodeling–related markers, including MMP-9, TIMP-1, and TIMP-2. As shown in Figure 3, the hPECs induced overexpression of MMP-9, compared with the hConECs (7.4±5.5%, n = 5). However, the overexpression of MMP-9 was significantly decreased to 51.3±10.4% (p<0.01, n = 5) by treatment with CDECM, compared with the untreated hPECs. The tissue inhibitor MMPs, TIMP-1 and TIMP-2, were induced by CDECM treatment to 859.8±60.7% (p<0.01, n = 5) and 700.6±49.1% (p<0.01, n = 5), respectively, compared with the untreated hPECs. Moreover, fibronectin, which is one of the extracellular matrix components, increased in the hPECs, compared with the hConECs (33.3±6.8%, n = 5). CDECM significantly decreased fibronectin expression to 66.7±8.4% (p<0.05, n = 5), compared with the untreated hPECs. CDECM did not show statistically significant changes in the hConECs.
Figure 3.
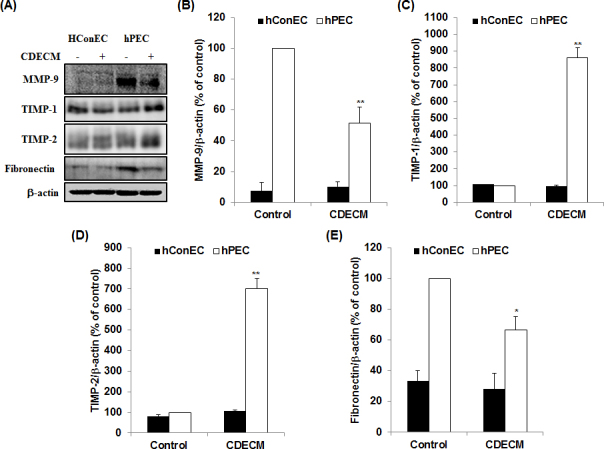
Effect of CDECM on the remodeling of the extracellular matrix in hConECs and hPECs. The samples were immunostained for matrix metallopeptidase 9 (MMP-9), tissue inhibitor of metalloprotease 1 (TIMP-1), TIMP-2, and fibronectin. A: The membranes were photographed with the Fusion FX image acquisition system. The graphs show the densitometry quantification of MMP-9 (B), TIMP-1 (C), TIMP-2 (D), and fibronectin (E), compared with the untreated human pterygium epithelial cells (hPECs). β-actin was used as the control. Values are mean ± standard deviation (SD; n = 5). Statistical significance is indicated as *p<0.05 and **p<0.01.
Inhibitory effect of CDECM on angiogenesis and inflammation in hPECs
The proinflammatory markers, including TNF-α, VCAM-1, and Cox2, were not detected in the hConECs, compared with the hPECs. We used three pterygial tissues collected from three donors for this test. The hPECs showed higher expression levels of the proinflammatory proteins, whereas this overexpression was significantly decreased by CDECM (Figure 4A). The level of expression of TNF-α, VCAM-1, and Cox2 was significantly decreased to 32.8±11.1% (p<0.01, n = 5), 38.6±15.0% (p<0.01, n = 5), and 8.5±2.8% (p<0.001, n = 5) compared with the untreated hPECs, respectively (Figure 4B–D). In addition, we evaluated the effect of CDECM on the expression of the proangiogenic markers. As expected, when the cells were incubated with CDECM, the protein expression of VEGF and CD31 was decreased to 30.7±20.8% (p<0.01, n = 5) and 51.2±28.4% (p<0.05, n = 5), respectively, compared with the untreated hPECs. Furthermore, the ELISA results for the amounts of the proinflammatory cytokines (Figure 4G–I) showed the inhibitory effect of CDECM on IL-6 and PGE2 production in hPECs. The production of IL-6 was 153.16±9.22 pg/ml in the hPECs without CDECM and 114.76±0.71 pg/ml in the hPECs with CDECM (p<0.001, n = 3). The amount of PGE2 was markedly decreased to 8.85±0.18 pg/ml compared with the control (13.57±0.38 pg/ml, p<0.001, n = 3). However, the levels of IL-1β did not show statistically significant changes.
Figure 4.
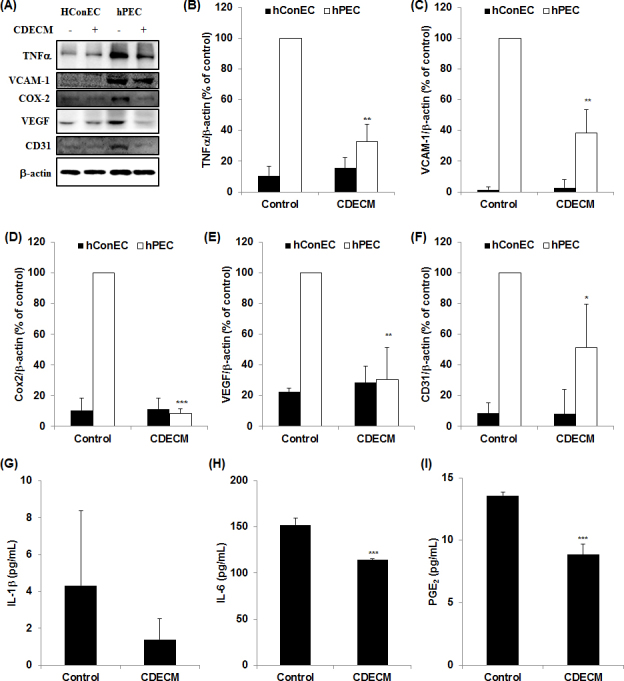
Effect of CDECM on angiogenesis and inflammation in hConECs and hPECs. A: The cells were immunostained for tumor necrosis factor-α (TNF-α), antivascular cellular adhesion molecule 1 (VCAM-1), cyclooxygenase-2 (Cox2), vascular endothelial growth factor (VEGF), and cluster of differentiation 31 (CD31). Images of the membranes were photographed with the Fusion FX image acquisition system. The graphs show the densitometry quantification of TNF-α (B), VCAM-1 (C), Cox2 (D), VEGF (E), and CD31 (F), compared with the untreated human pterygium epithelial cells (hPECs). β-actin was used as the control. Values are mean ± standard deviation (SD; n = 5). Statistical significance is indicated as *p<0.05, **p<0.01, and ***p<0.001 (B). The levels of interleukin 1 beta (IL-1β) (G), IL-6 (H), and prostaglandin E2 (PGE2) (I) production were measured with enzyme-linked immunosorbent assay (ELISA). Data are expressed as the means ± SD (n = 3). Statistical significance is indicated as ***p<0.001.
Inhibition of oxidative stress in hPECs
Figure 5A depicts the levels of intracellular ROS generation detected with the oxidative stress-sensitive fluorescent dye, DCFH-DA. For this test, we used two pterygial tissue samples collected from two donors. The hPECs absorbed high levels of the fluorescent dye for ROS compared with the hConECs. However, the fluorescence was weak in the CDECM-treated hPECs compared with the untreated cells. In addition, we evaluated whether CDECM suppressed nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which is an enzyme-producing ROS. As shown in Figure 5B, the hPECs expressed high levels of the NADPH oxidase subunits, including Nox2 and p47phox, compared with the hConECs. However, these were markedly decreased by CDECM to 33.2±20.5% (p<0.01, n = 5 from three donors) and 66.5±15.0% (p<0.01, n = 5 from three donors) of control, respectively (Figure 5C,D). Furthermore, CDECM induced the upregulation of the antioxidant enzyme HO-1, as well as the expression of nucleus Nrf2 (Figure 5E). The levels of expression of HO-1 and nucleus Nrf2 were slightly increased in the CDECM-treated hConECs to 153.6±14.1% (p<0.05, n = 5) and 150.2±14.1 (p<0.05, n = 5), respectively, compared with the untreated hConECs (Figure 5F,G). In the hPECs, the expression of HO-1 and nucleus Nrf2 protein was significantly increased by the CDECM treatment to 220.2±23.4% (p<0.01, n = 5 from three donors) and 393.4±100.6% (p<0.05, n = 5 from three donors), respectively, compared with the untreated hPECs (Figure 5F,G).
Figure 5.
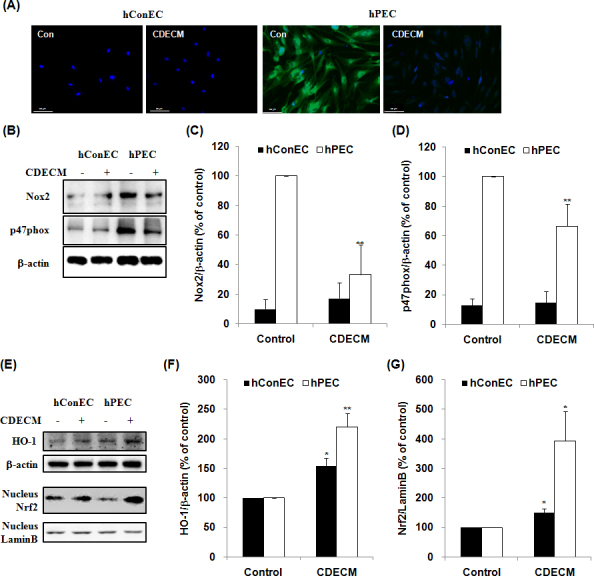
Effect of CDECM on oxidative stress in hConECs and hPECs. A: Intracellular reactive oxygen species (ROS) were detected using 2′,7′-dichlorofluorescein diacetate (DCFH-DA), a ROS-sensitive fluorescent dye. The stained cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and viewed under an automatic fluorescence in situ hybridization (FISH) imager. Scale bar = 50 μm. The cell lysates were immunostained for Nox2 and p47phox (B). The graphs show the densitometry quantification of Nox2 (C) and p47phox (D). β-actin was used as the control. Values are mean ± standard deviation (SD; n = 5). Statistical significance is indicated as **p<0.01. E: The cell lysates were immunostained for heme oxygenase-1 (HO-1) and nuclear factor erythroid-2 related factor 2 (Nrf2). The graphs show the densitometry quantification of HO-1 (F) and Nrf2 (G). β-actin and Lamin B were used as the control for the whole cell fraction and the nucleus fraction, respectively. Values are mean ± SD (n = 5). Statistical significance is indicated as *p<0.05 and **p<0.01.
Inhibitory effect of CDECM on NF-κB activation in hPECs
We evaluated whether CDECM decreased NF-κB activation in the pterygium. The hPECs showed statistically significantly induced IκBα degradation and phosphorylation in the cytoplasm, as well as the nuclear translocation of NF-κBp65 (Figure 6A). In the CDECM-treated hPECs, the IκBα protein was increased to 143.3±7.7% (p<0.01, n = 5 from three donors), and the pIκBα protein was decreased to 44.4±11.3% (p<0.01, n = 5 from three donors) compared with the untreated hPECs, indicating that IκBα protein degradation and phosphorylation in hPECs were suppressed by the CDECM treatment. Furthermore, the expression of the NF-κB p65 protein in the nucleus was also suppressed in the CDECM treatment group (31.8±9.2%, p<0.001, n = 6 from four donors). There were no statistically significant changes in the expression of these proteins in hConECs by the CDECM treatment.
Figure 6.
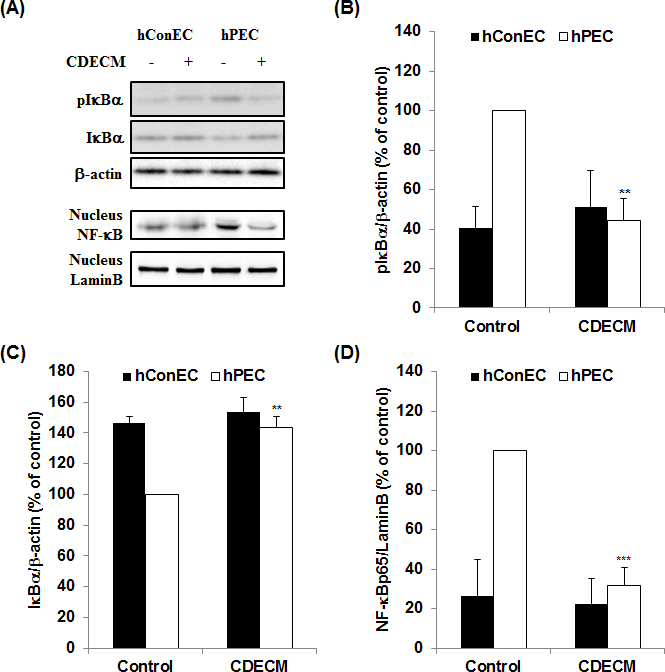
Effect of CDECM on the NF-κB activation in hConECs and hPECs. A: The cell lysates were immunostained for nuclear factor-kappa B p65 (NF-κB p65), nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκBα), and phospho-IκBα (Ser32/36). The membranes were photographed with the Fusion FX image acquisition system. The graphs show the densitometry quantification of pIκBα (B, n = 5), IκBα (C, n = 5), and NF-κB p65 (D, n = 6). β-actin and Lamin B were used as the controls for the whole cell fraction and the nucleus fraction, respectively. Values are mean ± standard deviation (SD). Statistical significance is indicated as **p<0.01 and ***p<0.001.
The effect of CDECM on intracellular signaling in hConECs and hPECs
To examine which upstream signaling pathways are associated with NF-κB activation in hPECs, we tested the activation of intracellular signaling, such as MAPKs, PKC, and Akt. The phosphorylation levels of ERK and SAPK/JNK in hPECs were similar compared to those in the hConECs (Figure 7). However, the phosphorylation of p38 MAPK, PKCα (Ser657), and PKCθ (Thr538) increased in the hPECs, compared with the hConECs (Figure 7). The phosphorylation of p38 MAPK, but not ERK and JNK, was significantly decreased by the CDECM treatment (Figure 7A). The phosphorylation of PKCα (Ser657) and PKCθ (Thr 538) was also markedly decreased in the CDECM treatment group (Figure 7B). The phosphorylation of Akt (Thr308) was also increased in the hPECs, compared with the hConECs (Figure 7C). However, the phosphorylation level of Akt (Thr308) was not statistically significantly changed by the CDECM treatment (Figure 7C).
Figure 7.
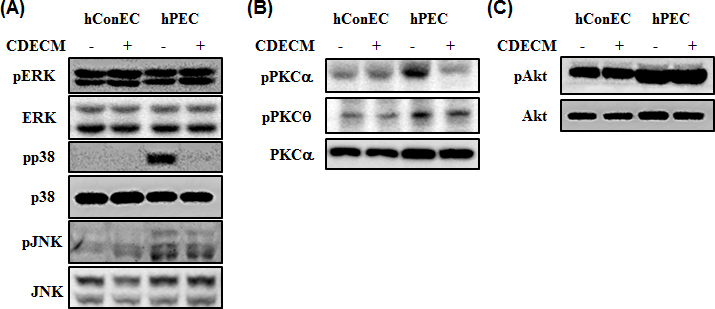
Mechanism by which CDECM suppresses the growth of hConECs and hPECs. The cell lysates were immunostained for mitogen-activated protein kinases (MAPKs) (A), protein kinase C (PKC) (B), and Akt (C). The membranes were photographed with the Fusion FX image acquisition system.
Discussion
Extracellular matrix remodeling is a prominent feature in pterygium, and this remodeling includes elastosis and the breakdown of Bowman’s layer. Some of these changes are attributed to the actions of MMPs, which contribute to the local invasive nature of this disease [2]. In 2010, Tsai et al. reported that MMP-9 may participate in pterygium formation, and TIMPs contribute to inhibiting invasion by pterygium [21]. Furthermore, Girolamo et al. reported that MMPs and TIMPs may contribute to the inflammation, tissue remodeling, and angiogenesis that characterizes pterygium [22]. In the present study, we evaluated whether CDECM inhibited cell migration and invasion by regulating MMPs and TIMPs in hPECs. As shown in Figure 1, the invasion and migration ability of the hPECs was inhibited by the CDECM treatment. In addition, CDECM significantly decreased the expression of MMP-9, whereas CDECM induced the expression of TIMP-1 and TIMP-2 in hPECs (Figure 3). Fibronectin, a major component of extracellular matrix, which induces migration of corneal epithelium [23], was highly expressed in the hPECs, but the CDECM treatment reduced the protein expression level of fibronectin (Figure 3). With our previous report about the suppressive effect of CDECM on pterygial lesion growth in a nude mouse model of pterygium [17], our present results indicate that CDECM contributes to the inhibition of pterygium growth by the reduction of cell invasion and migration and extracellular matrix remodeling through downregulation of MMP-9 and fibronectin.
Recently, several proinflammatory cytokines and growth factors have been shown to play key roles in inflammatory, fibrogenic, and angiogenic processes, all of which are commonly observed in pterygium [24]. In particular, IL-1, IL-6, IL-8, and TNF-α are increased in pterygium, contributing to inflammatory mediators and MMPs and potentially to pterygium pathogenesis [2]. Cox2 is also overexpressed in pterygium, yielding a further increase in prostaglandin products and the inflammatory cascade [25]. Pterygium samples show elevated levels of cell signaling and adhesion molecules, including VCAM-1 and ICAM-1 [26,27]. Therefore, we investigated the effect of CDECM on proinflammatory cytokines and growth factors in hPECs. As shown in Figure 4, hPECs showed high levels of proangiogenic markers, including VCAM-1, VEGF, and CD31, whereas expression of those proteins was significantly decreased by CDECM. In addition, CDECM markedly inhibited the expression of proinflammatory factors, such as TNF-α, Cox2, IL-6, and PGE2. Our data provide evidence that CDECM suppresses the inflammatory and angiogenic processes of hPECs by inhibiting the expression of cytokines and growth factors. Ultraviolet (UV)-B light has been considered a major cause of pterygium [2]. UV-B exposure causes oxidative stress, leading to the upregulation of many potential mediators of pterygium growth [27]. ROS are harmful to cells because they injure cellular NAD, proteins, and lipids in a process called oxidative stress [28]. The NADPH oxidases of the Nox family are important enzymatic sources of ROS. For activation of NADPH oxidases, Nox2 interacts with p22phox in membrane via supported by p47phox, which acts as an adaptor protein [29]. In the present study, we evaluated the effect of CDECM on ROS generation followed by the modulation of NADPH oxidase in pterygium pathogenesis. We found that hPECs showed elevated ROS production, which was dramatically inhibited by treatment with CDECM (Figure 5A). In addition, CDECM suppressed the expression of NADPH oxidase subunits, including Nox2 and p47phox (Figure 5B). These results suggest that CDECM may act as an antioxidant in pterygium. Therefore, we examined whether CDECM has an antioxidant effect. HO-1 has been shown to exert anti-inflammatory, antiapoptotic, and antioxidant actions [30]. The promoter of the HO-1 gene contains consensus binding sites for numerous transcription factors, including Nrf2, 'activator protein (AP)-1, AP-2, NF-κB, Signal transducer and activator of transcription-x (STATx), and hepatocyte nuclear factor (HNF)-1 [31]. One of the most crucial appears to be Nrf2, which induces the expression of genes that encode phase II detoxifying enzymes and antioxidant proteins [32]. As shown in Figure 5E, CDECM induced the upregulation of the antioxidant enzyme HO-1, as well as expression of its transcriptional factor, Nrf2. Interestingly, there were no effects on cell invasion and migration ability and protein expression in hConECs by CDECM treatment, except HO-1 and Nrf2 protein expression. CDECM induced Nrf2 translocation into the nucleus and HO-1 protein expression in the hConECs (Figure 5). Nrf2-mediated HO-1 induction by bioactive materials is already well-known as a strategy for increasing cellular antioxidant capacity [33,34]. Although the role of antioxidants and iron chelation therapy in the management of pterygium is unknown, CDECM is expected to act as a scavenger of ROS based on the antioxidant capacity of CDECM in hConECs and hPECs.
NF-κB is a ubiquitous transcription factor that, through target genes, regulates key processes, such as inflammation, apoptosis, stress response, wound healing, angiogenesis, and lymphangiogenesis [35]. The pathogenesis of pterygium is unknown, although sun exposure and free radical production have been implicated in the formation of pterygium. Because NF-κB is involved in oxidative stress signaling, it is unsurprising that NF-κB may contribute to pterygium pathology [35,36]. It is well-known that the p65, the most commonly studied form of NF-κB, interacts with IκB in the cell cytoplasm. Upon activation by various types of pathological or physiologic stimulation, IκB is phosphorylated and degraded. p65 then translocates into the cellular nucleus and regulates the transcription of the NF-κB target genes [35]. Therefore, we investigated the role of CDECM in NF-κB activation in hPECs. As shown in Figure 6, the hPECs showed marked phosphorylation and degradation of IκBα in the cytoplasm, as well as nuclear translocation of NF-κBp65. However, CDECM treatment slightly decreased the nuclear translocation of NF-κBp65 and markedly increased the level of cytosolic IκBα. Siak et al. reported that phosphorylation of IκBα is increased in pterygium tissues relative to control conjunctival tissues, and this may start a chain of events leading to the formation or progression of inflammation [37]. Lan et al. demonstrated that inflammation and angiogenesis can be consequences of the production of cytokines, chemokines, growth factors, and proteolytic enzymes in the pathology of pterygium [36]. Consistent with the previous observations, our findings support the role of activated NF-κB in pterygium. Furthermore, we observed that CDECM downregulated NF-κB signaling, leading to the inhibition of pterygium pathogenesis.
In ocular disease, NF-κB activation may be triggered by MAPK, leading to cytokine transcription, and it is also downstream of the phosphoinositide 3-kinase (PI3K)/Akt pathway [38,39]. Therefore, we evaluated specific upstream pathways associated with NF-κB activation. The phosphorylation of p38 MAPK, PKCα (Ser657), and PKCθ (Thr 538) was elevated in hPECs and was significantly decreased by CDECM treatment (Figure 7A,B). However, ERK and SAPK/JNK were not changed (Figure 7A). hPECs expressed Akt at normal levels, but the phosphorylation of Akt (Thr308) was slightly increased by the CDECM treatment (Figure 7C). These results are consistent with our previous finding that CDECM inhibited corneal neovascularization and inflammation by blocking NF-κB activation via regulation of the PKC signaling pathway in the cornea from alkali-burned rabbits [19].
In conclusion, in this study, we identified the pathogenic effects of CDECM, including angiogenesis, inflammation, extracellular matrix remodeling, and oxidative stress, in hPECs, as well as the underlying signaling pathways. The CDECM markedly inhibited cell migration and invasion, as well as suppressing inflammation and remodeling of the extracellular matrix, by influencing the expression of TNF-α, IL-6, and PGE2, and MMP and fibronectin in hPECs. CDECM significantly inhibited the expression of NADPH oxidase and generation of ROS. The CDECM induced the Nrf2 mediated-antioxidant enzyme HO-1 and reduced NF-κB activation. Furthermore, CDECM decreased the phosphorylation of p38 MAPK, PKCα (Ser657), and PKCθ (Thr538) in the hPECs. Our findings suggest that CDECM suppresses pterygium pathogenesis through the inhibition of NF-κB activation and the upregulation of Nrf2 by blocking the p38 MAPK and PKC signaling pathways (Figure 8).
Figure 8.

Role of CDECM in regulating the pathogenesis of hPECs.
Acknowledgments
This study was supported by a grant from the Korea Healthcare Technology R&D Project, Ministry of Health and Welfare Affairs, Republic of Korea (grant #: HI12C0005) to JWY. JWY is a director in the Ocular Neovascular Disease Research Center of Inje University Busan Paik Hospital.
References
- 1.Coroneo M. Pterygium as an early indicator of ultraviolet insolation: a hypothesis. Br J Ophthalmol. 1993;77:734. doi: 10.1136/bjo.77.11.734. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=8280691&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Girolamo N, Chui J, Coroneo MT, Wakefield D. Pathogenesis of pterygia: role of cytokines, growth factors, and matrix metalloproteinases. Prog Retin Eye Res. 2004;23:195–228. doi: 10.1016/j.preteyeres.2004.02.002. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15094131&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 3.Livezeanu C, Craitoiu MM, Manescu R, Mocanu C, Craitoiu S. Angiogenesis in the pathogenesis of pterygium. Rom J Morphol Embryol. 2011;52:837–44. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21892527&dopt=Abstract [PubMed] [Google Scholar]
- 4.Ray J, Stetler-Stevenson W. The role of matrix metalloproteases and their inhibitors in tumour invasion, metastasis and angiogenesis. Eur Respir J. 1994;7:2062–72. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=7533104&dopt=Abstract [PubMed] [Google Scholar]
- 5.Ang LP, Chua JL, Tan DT. Current concepts and techniques in pterygium treatment. Curr Opin Ophthalmol. 2007;18:308–13. doi: 10.1097/ICU.0b013e3281a7ecbb. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17568207&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 6.Jaworski CJ, Aryankalayil-John M, Campos MM, Fariss RN, Rowsey J, Agarwalla N, Reid TW, Dushku N, Cox CA, Carper D, Wistow G. Expression analysis of human pterygium shows a predominance of conjunctival and limbal markers and genes associated with cell migration. Mol Vis. 2009;15:2421–34. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19956562&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta D, Illingworth C. Treatments for corneal neovascularization: a review. Cornea. 2011;30:927–38. doi: 10.1097/ICO.0b013e318201405a. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21389854&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 8.Roodhooft JM. Leading causes of blindness worldwide. Bull Soc Belge Ophtalmol. 2002;283:19–25. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=12058483&dopt=Abstract [PubMed] [Google Scholar]
- 9.Roth H. The Etiology of Ocular Irritation in Soft Lens Wearers: Distribution in a Large Clinical Sample. Eye Contact Lens. 1978;4:38–47. [Google Scholar]
- 10.Chui J, Coroneo MT, Tat LT, Crouch R, Wakefield D, Di Girolamo N. Ophthalmic pterygium: a stem cell disorder with premalignant features. Am J Pathol. 2011;178:817–27. doi: 10.1016/j.ajpath.2010.10.037. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21281814&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chui J, Di Girolamo N, Wakefield D, Coroneo MT. The pathogenesis of pterygium: current concepts and their therapeutic implications. Ocul Surf. 2008;6:24–43. doi: 10.1016/s1542-0124(12)70103-9. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18264653&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 12.Di Girolamo N, Wakefield D, Coroneo MT. UVB-mediated induction of cytokines and growth factors in pterygium epithelial cells involves cell surface receptors and intracellular signaling. Invest Ophthalmol Vis Sci. 2006;47:2430–7. doi: 10.1167/iovs.05-1130. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16723453&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 13.Hooiveld M, Roosendaal G, Wenting M, van den Berg M, Bijlsma J, Lafeber F. Short-term exposure of cartilage to blood results in chondrocyte apoptosis. Am J Pathol. 2003;162:943–51. doi: 10.1016/S0002-9440(10)63889-8. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=12598327&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lima EG, Tan AR, Tai T, Bian L, Stoker AM, Ateshian GA, Cook JL, Hung CT. Differences in interleukin-1 response between engineered and native cartilage. Tissue Eng Part A. 2008;14:1721–30. doi: 10.1089/ten.tea.2007.0347. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18611148&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 15.Choi KH, Choi BH, Park SR, Kim BJ, Min BH. The chondrogenic differentiation of mesenchymal stem cells on an extracellular matrix scaffold derived from porcine chondrocytes. Biomaterials. 2010;31:5355–65. doi: 10.1016/j.biomaterials.2010.03.053. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20394983&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 16.Choi BH, Choi KH, Lee HS, Song BR, Park SR, Yang JW, Min BH. Inhibition of blood vessel formation by a chondrocyte-derived extracellular matrix. Biomaterials. 2014;35:5711–20. doi: 10.1016/j.biomaterials.2014.03.083. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24768193&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 17.Lee HS, Lee JH, Yang JW. Effect of porcine chondrocyte-derived extracellular matrix on the pterygium in mouse model. Graefes Arch Clin Exp Ophthalmol. 2014;252:609–18. doi: 10.1007/s00417-014-2592-8. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24562465&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 18.Lee HS, Lee JH, Kim CE, Yang JW. Anti-neovascular effect of chondrocyte-derived extracellular matrix on corneal alkaline burns in rabbits. Graefes Arch Clin Exp Ophthalmol. 2014;252:951–61. doi: 10.1007/s00417-014-2633-3. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24789464&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 19.Li TZ, Jin CZ, Choi BH, Kim MS, Kim YJ, Park SR, Yoon JH, Min BH. Using cartilage extracellular matrix (CECM) membrane to enhance the reparability of the bone marrow stimulation technique for articular cartilage defect in canine model. Adv Funct Mater. 2012;22:4292–300. [Google Scholar]
- 20.Bouloumie A1 Marumo T, Lafontan M, Busse R. Leptin induces oxidative stress in human endothelial cells. FASEB J. 1999;13:1231–8. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10385613&dopt=Abstract [PubMed] [Google Scholar]
- 21.Tsai YY, Chiang CC, Yeh KT, Lee H, Cheng YW. Effect of TIMP-1 and MMP in pterygium invasion. Invest Ophthalmol Vis Sci. 2010;51:3462–7. doi: 10.1167/iovs.09-4921. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20207965&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 22.Di Girolamo N, McCluskey P, Lloyd A, Coroneo MT, Wakefield D. Expression of MMPs and TIMPs in human pterygia and cultured pterygium epithelial cells. Invest Ophthalmol Vis Sci. 2000;41:671–9. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10711680&dopt=Abstract [PubMed] [Google Scholar]
- 23.Kimura K, Hattori A, Usui Y, Kitazawa K, Naganuma M, Kawamoto K, Teranishi S, Nomizu M, Nishida T. Stimulation of corneal epithelial migration by a synthetic peptide (PHSRN) corresponding to the second cell-binding site of fibronectin. Invest Ophthalmol Vis Sci. 2007;48:1110–8. doi: 10.1167/iovs.06-0704. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17325153&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 24.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, Fauci AS. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci USA. 1986;83:4167–71. doi: 10.1073/pnas.83.12.4167. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=2424019&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiang CC, Cheng YW, Lin CL, Lee H, Tsai FJ, Tseng SH, Tsai YY. Cyclooxygenase 2 expression in pterygium. Mol Vis. 2007;13:635–8. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17515883&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 26.Beden U, Irkec M, Orhan D, Orhan M. The roles of T-lymphocyte subpopulations (CD4 and CD8), intercellular adhesion molecule-1 (ICAM-1), HLA-DR receptor, and mast cells in etiopathogenesis of pterygium. Ocul Immunol Inflamm. 2003;11:115–22. doi: 10.1076/ocii.11.2.115.15913. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=14533030&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 27.Bradley JC, Yang W, Bradley RH, Reid TW, Schwab IR. The science of pterygia. Br J Ophthalmol. 2010;94:815–20. doi: 10.1136/bjo.2008.151852. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19515643&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge J. Oxidative stress and antioxidant protection: some special cases. Oxford University Press. 2015. [Google Scholar]
- 29.Brandes RP, Weissmann N, Schroder K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic Biol Med. 2014;76:208–26. doi: 10.1016/j.freeradbiomed.2014.07.046. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25157786&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 30.Dulak J, Loboda A, Jozkowicz A. Effect of heme oxygenase-1 on vascular function and disease. Curr Opin Lipidol. 2008;19:505–12. doi: 10.1097/MOL.0b013e32830d81e9. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18769232&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 31.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16601269&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 32.Alam J, Stewart D, Touchard C, Boinapally S, Choi AMK, Cook JL. Nrf2, a Cap'n'Collar Transcription Factor, Regulates Induction of the Heme Oxygenase-1 Gene. J Biol Chem. 1999;274:26071–8. doi: 10.1074/jbc.274.37.26071. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10473555&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 33.Jun YJ, Lee M, Shin T, Yoon N, Kim JH, Kim HR. Eckol Enhances Heme Oxygenase-1 Expression through Activation of Nrf2/JNK Pathway in HepG2 Cells. Molecules. 2014;19:15638–52. doi: 10.3390/molecules191015638. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25268719&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee MS, Lee B, Park KE, Utsuki T, Shin T, Oh CW, Kim HR. Dieckol enhances the expression of antioxidant and detoxifying enzymes by the activation of Nrf2-MAPK signalling pathway in HepG2 cells. Food Chem. 2015;174:538–46. doi: 10.1016/j.foodchem.2014.11.090. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25529716&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 35.Srivastava SK, Ramana KV. Focus on molecules: nuclear factor-kappaB. Exp Eye Res. 2009;88:2–3. doi: 10.1016/j.exer.2008.03.012. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18472097&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lan W, Petznick A, Heryati S, Rifada M, Tong L. Nuclear Factor-kappaB: central regulator in ocular surface inflammation and diseases. Ocul Surf. 2012;10:137–48. doi: 10.1016/j.jtos.2012.04.001. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22814642&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 37.Siak JJ, Ng SL, Seet LF, Beuerman RW, Tong L. The nuclear-factor kappaB pathway is activated in pterygium. Invest Ophthalmol Vis Sci. 2011;52:230–6. doi: 10.1167/iovs.10-5735. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20811049&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 38.Rajaiya J, Xiao J, Rajala RV, Chodosh J. Human adenovirus type 19 infection of corneal cells induces p38 MAPK-dependent interleukin-8 expression. Virol J. 2008;5:17. doi: 10.1186/1743-422X-5-17. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18221537&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajala MS, Rajala RV, Astley RA, Butt AL, Chodosh J. Corneal cell survival in adenovirus type 19 infection requires phosphoinositide 3-kinase/Akt activation. J Virol. 2005;79:12332–41. doi: 10.1128/JVI.79.19.12332-12341.2005. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16160160&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]