

Abstract
Purpose
Studies of neuronal development in the retina often examine the stages of proliferation, differentiation, and synaptic development, albeit independently. Our goal was to determine if a known neurotoxicant insult to a population of retinal progenitor cells (RPCs) would affect their eventual differentiation and synaptic development. To that end, we used our previously published human equivalent murine model of low-level gestational lead exposure (GLE). Children and animals with GLE exhibit increased scotopic electroretinogram a- and b-waves. Adult mice with GLE exhibit an increased number of late-born RPCs, a prolonged period of RPC proliferation, and an increased number of late-born rod photoreceptors and rod and cone bipolar cells (BCs), with no change in the number of late-born Müller glial cells or early-born neurons. The specific aims of this study were to determine whether increased and prolonged RPC proliferation alters the spatiotemporal differentiation and synaptic development of rods and BCs in early postnatal GLE retinas compared to control retinas.
Methods
C57BL/6N mouse pups were exposed to lead acetate via drinking water throughout gestation and until postnatal day 10, which is equivalent to the human gestation period for retinal neurogenesis. RT-qPCR, immunohistochemical analysis, and western blots of well-characterized, cell-specific genes and proteins were performed at embryonic and early postnatal ages to assess rod and cone photoreceptor differentiation, rod and BC differentiation and synaptic development, and Müller glial cell differentiation.
Results
Real-time quantitative PCR (RT-qPCR) with the rod-specific transcription factors Nrl, Nr2e3, and Crx and the rod-specific functional gene Rho, along with central retinal confocal studies with anti-recoverin and anti-rhodopsin antibodies, revealed a two-day delay in the differentiation of rod photoreceptors in GLE retinas. Rhodopsin immunoblots supported this conclusion. No changes in glutamine synthetase gene or protein expression, a marker for late-born Müller glial cells, were observed in the developing retinas. In the retinas from the GLE mice, anti-PKCα, -Chx10 (Vsx2) and -secretagogin antibodies revealed a two- to three-day delay in the differentiation of rod and cone BCs, whereas the expression of the proneural and BC genes Otx2 and Chx10, respectively, increased. In addition, confocal studies of proteins associated with functional synapses (e.g., vesicular glutamate transporter 1 [VGluT1], plasma membrane calcium ATPase [PMCA], transient receptor potential channel M1 [TRPM1], and synaptic vesicle glycoprotein 2B [SV2B]) revealed a two-day delay in the formation of the outer and inner plexiform layers of the GLE retinas. Moreover, several markers revealed that the initiation of the differentiation and intensity of the labeling of early-born cells in the retinal ganglion cell and inner plexiform layers were not different in the control retinas.
Conclusions
Our combined gene, confocal, and immunoblot findings revealed that the onset of rod and BC differentiation and their subsequent synaptic development is delayed by two to three days in GLE retinas. These results suggest that perturbations during the early proliferative stages of late-born RPCs fated to be rods and BCs ultimately alter the coordinated time-dependent progression of rod and BC differentiation and synaptic development. These GLE effects were selective for late-born neurons. Although the molecular mechanisms are unknown, alterations in soluble neurotrophic factors and/or their receptors are likely to play a role. Since neurodevelopmental delays and altered synaptic connectivity are associated with neuropsychiatric and behavioral disorders as well as cognitive deficits, future work is needed to determine if similar effects occur in the brains of GLE mice and whether children with GLE experience similar delays in retinal and brain neuronal differentiation and synaptic development.
Introduction
The adult retina consists of six different types of neurons (i.e., rod and cone photoreceptors and horizontal, bipolar, amacrine, and ganglion cells) and one glial cell (i.e., Müller glia). These seven cell types develop in a specific histogenic order that is conserved across species [1,2]. In mice, early-born neurons (i.e., cone photoreceptors and horizontal, amacrine, and ganglion cells) exhibit peak neurogenesis before birth, while late-born neurons (i.e., rod photoreceptors and bipolar cells [BCs]) and Müller glia peak after birth [3]. Despite the significant time span it takes for retinal cells to develop (i.e., embryonic day [E] = 10.5; postnatal day [PN] = 10 in mice), all these cells originate from one common pool of multipotent retinal progenitor cells (RPCs) [4,5]. To ensure the production of the correct number of cell types, the developing retina must maintain a constant regulation of mitosis, proliferation, and differentiation throughout development [6].
In the later stages of retinal development, differentiated retinal neurons undergo a regulated sequence of synaptic development in the outer and inner plexiform layers (OPLs and IPLs). For example, the developmental sequence of synapses in the OPL, as determined by vesicular glutamate transporter 1 (VGluT1) protein expression, is as follows: 1) cone pedicles, 2) horizontal synapses, 3) rod spherules, and 4) BC innervation [7]. In the IPL, the OFF pathway develops before the ON pathway [7]. Much of what is known about functional synaptic development in the retina is in the context of mediated activity-dependent mechanisms [8-10].
Other than the coordinated regulation that occurs within the individual stages of the proliferation, differentiation, and functional development of neurons in the young retina, little is known about the interdependence of each phase within populations of RPCs. Many studies have examined the effects of various genes (i.e., cyclin-dependent kinases, transcription factors, cell signaling proteins, etc.) [11-14], soluble neurotrophic factors [15], and/or neurotransmitters [16] on cell proliferation, the cell cycle, and/or cell fate decisions. The goal of the current study was to determine how the increased and prolonged proliferation of RPCs would affect the subsequent structural and functional maturation of the mouse retina.
Gestational lead exposure (GLE) in children, monkeys, and rodents results in dose-dependent supernormal scotopic electroretinograms [17-20]. Consistent with these results, we observed a dose-dependent increase in the number of RPCs, prolongation of RPC proliferation, and selective increase in the number of rods and BCs in adult mice [21] and Long-Evans hooded rats [19]. This is in marked contrast to postnatal or adult lead exposure, at similar blood lead levels, which produces rod-selective apoptosis [22,23], photoreceptor synaptic degeneration [24], and hippocampal granule cell apoptosis [25,26], synaptic dysfunction, and degeneration [27].
We hypothesized that the increased number of RPCs accelerates the development of rods and BCs and increases the abundance of the functional synaptic markers of these cell types as they differentiate and mature. We used a murine model of GLE. Unexpectedly, we found a two- to three-day delay in the initial expression of proteins associated with rod and BC differentiation, synaptic development and maturation.
Methods
Murine model of GLE
All experimental and animal care procedures complied with the National Institutes of Health (NIH) Public Health Service Policy on Humane Care and Use of Laboratory Animals (NIH 2002), were approved by the Institutional Animal Care and Use Committee of the University of Houston, and adhered to the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Research. Data was collected from litters bred at our facility and maintained on a 14:10 light:dark cycle (100 lux: maximum cage luminance) with food and water available ad libitum [21,28]. Wild-type C57BL/6N mice (Harlan Sprague-Dawley, Indianapolis, IN) were used for all the experimental data shown herein. We observed the same phenotypic effects of GLE in C57BL/6J mice [29] and Long-Evans hooded rats [19]. Despite the rd8 mutation [30,31], we did not observe dysplasia, irregularities in retinal layer thickness, or the displacement of photoreceptors at any experimental time point in this or in our previous studies [21,24,29,32]. Briefly, naïve females were given either tap (control) or 55 parts per million (ppm) lead acetate containing drinking water (GLE group) for two weeks before mating, during pregnancy, and until PN10, after which the lead was replaced with tap water. For the E16.5 and E18.5 retinas, the dams were mated overnight and checked for vaginal plugs in the morning, and the pups were used for timed-pregnant experiments. Mice were sacrificed by decapitation between 1,000 and 1,200 h. The Institutional Animal Care and Use Committee of the University of Houston approved decapitation without anesthetic for all ages so we have not included information of this type. As published, the control and GLE groups had peak blood lead concentrations at PN0 and/or PN10 of 0.75 ± 0.06 and 22.11 ± 1.05 μg/dl, respectively. In the E14, the blood lead concentration of the dams was similar to that of the PN0 pups; at PN30, the blood lead concentration in the GLE mice was not different from the control mice [28]. There were no statistical differences between the control and GLE groups for any dam measure, litter measure, or bodyweight in the males or females [28]. No age- or sex-dependent differences were found in the analysis; therefore, all the data were combined for presentation herein.
RNA isolation
Real-time quantitative PCR (RT-qPCR) studies were conducted using retinas from E16.5, E18.5, PN2, PN6, and PN10 control and GLE mice. E16.5 and E18.5, entire litters were used for control and GLE mice (N=4-5 litters). For the postnatal ages, four to five mice from the control and GLE litters were used. All the supplies and equipment as well as the surgical and laboratory procedures followed RNase-free conditions essentially as described [29]. Briefly, retinas were dissected and immediately placed into TRIzol (Thermo Fisher Scientific, Waltham, MA) and vortexed; chloroform was added and the retinas were vortexed again, then centrifuged. Next, 2-propanol was added, and the retinas were vortexed and centrifuged. The pellet was saved at −80 °C until the RNA was isolated.
RT-qPCR
Total RNA was synthesized into cDNA as described [33]. Briefly, 1 µg of total RNA was used for first-strand cDNA synthesis and added to oligo dT and random hexamer primers according to Bio-Rad specifications. All RT-qPCR experiments were run on the Bio-Rad iCycler platform (Bio-Rad Laboratories, Hercules, CA). The RT-qPCR primers were designed and validated within the following parameters: GC content = 50%–60%, melt temperature = 55–65 °C, and no secondary structures, primer dimers, or homodimers. A primer quality analysis was conducted using IDT DNA’s OligoAnalyzer. All the primers designed were intron spanning, and they were selected from the Roche Applied Science Universal Probe Library and Assay Design Center database (https://lifescience.roche.com/shop/products/universal-probelibrary-system-assay-design). The primers were tested for alternative sites of homology with NCBI’s BLAST and the UCSC Genome Browser. The primer sequences are shown in Table 1. The primers were validated by a melt-curve analysis for a single peak of fluorescence.
Table 1. RT-qPCR primer list.
| Gene | Accession number | Forward primer (5’-3’) | Reverse primer (3’-5’) | Product size (bp) |
|---|---|---|---|---|
|
Actb |
NM_007393 |
AGAGAGGTATCCTGACCCTGAAGT |
CACGCAGCTCATTGTAGAAGGTGT |
105 |
|
Chx10 |
NM_007701 |
TGTTTCCTCCAGTGACCGAA |
ACATCTGGGTAGTGGGCTTCAT |
144 |
|
Crx |
NM_007770 |
CACGTGAGGAGGTTGCTCTT |
TCGCCCTACGATTCTTGAAC |
75 |
|
Glul |
NM_008131 |
AGGACTGCGCTGCAAGAC |
CCATCAAAGTTCCACTCAGGT |
78 |
|
Nr2e3 |
NM_013708 |
GCTAAGCCAGCATAGCAAGG |
GGAGCAATTTCCCAAACCTC |
62 |
|
Nrl |
NM_008736 |
TTCTGGTTCTGACAGTGACTACG |
GGACTGAGCAGAGAGAGGTGTT |
75 |
|
Otx2 |
NM_144841 |
TTTGCGCCTCCAAACAACCT |
ATGCAGCAAGTCCATACCCGAA |
102 |
| Rho | NM_145383 | TGCAAGCCGATGAGCAACTT | AACGCCATGATCCAGGTGAA | 77 |
All the RT-qPCR experiments were performed in triplicate using SYBR green (Bio-Rad Laboratories, Hercules, CA) with four to five independent biologic samples per age per treatment. β-Actin was used as the internal control. The PCR mixture consisted of 12.7 µl of Bio-Rad supermix, 1 µl of cDNA template, and 1.5 µl of gene specific forward and reverse primers, all combined in a 0.5 ml PCR tube on a 96-well plate. A no template control, water control, and air [empty well] control were run for each plate. The PCR cycle ran at 95 °C for 3 min, followed by 40 cycles of 95 °C for 30 s and 60 °C for 30 s. A melt-curve analysis was run at the end of each plate to ensure the proper performance of the primer pairs starting at 60 °C increased by 0.5 °C per min to 95 °C. The threshold for the Ct values was manually adjusted for each plate to coincide with entrance into the exponential growth phase of the PCR. The Ct values were then exported to Microsoft Excel for further analysis. The level of gene expression relative to the controls was determined using the ∆∆Ct method [34]: ∆∆Ct = ∆Ct of the gene of interest - ∆Ct of β-actin. 2∆∆Ct was used to compare the fold change in the gene expression between the GLE and control retinas.
Tissue processing and immunohistochemistry
All tissue processing and immunohistochemistry techniques were performed as described [19,21,33]. Briefly, developing or adult mice were decapitated, and their eyes were quickly enucleated and placed in ice-cold phosphate-buffered saline (PBS) (144 mM NaCl, 7.7 mM Na2HPO4, 2.7 mM NaH2PO4-H2O, 305 mOsm, pH 7.40), in which the corneas were gently slit. The eyes were immersion fixed at room temperature, buffered with 4% paraformaldehyde (Ladd Research, Williston VT) for 30 min, washed in PBS 3X for 10 min, and cryoprotected in 30% w/v sucrose for 48 h. The anterior segments were removed, and the eyes were embedded in Tissue-Tek optimum cutting temperature (OCT) Compound (Electron Microscopy Services, Fort Washington, PA), flash frozen in liquid nitrogen, and stored at −80 °C. Three nonadjacent fixed and frozen transverse sections (10 μm) were taken from the vertical meridians of four to nine independent biological samples per age per treatment. The retinal sections were mounted on Superfrost Plus glass slides (Thermo Fisher Scientific, San Francisco, CA) and stored at −20 °C.
All sections were taken from the central retina 200–400 μm from the optic nerve. The slides were thawed slowly, and the sections were post-fixed with 4% paraformaldehyde. They were washed and briefly immersed in a 1% sodium-borohydride (Sigma-Aldrich, St. Louis, MO) solution to reduce double bonds, to allow for proper epitope access for antibodies, and to decrease auto-fluorescence. The sections were washed and incubated for 2 h with a blocking buffer containing 10% normal goat serum (Jackson ImmunoResearch Labs Inc., West Grove, PA) and 0.3% Triton-X100 in PBS. Primary antibodies were applied for 2 days at 4 °C, after which the slides were washed with PBS and blocked for 30 min; then, secondary antibodies were applied. Table 2 lists the well-characterized and commercially available primary antibodies that were used. For the double-labeling experiments, primary antibodies from different host animals were applied simultaneously. Dilutions of Alexa 488, 555, and 647 (1:400) conjugated secondary antibodies were made in a blocking buffer and applied to the slides, which were left to incubate in the dark at room temperature for 1 h. The slides were washed with PBS, double-distilled H2O, dried, and mounted with Vectashield Mounting Medium (Vector Laboratories Inc., Burlinghame, CA) and a No. 1 coverslip; then, they were stored at 4 °C. For the DAPI-labeling experiments, the sections were incubated with DAPI for 1 h in the dark after the secondary incubation. All antibodies were titrated through a broad range of working dilutions (with three orders of magnitude) to determine the optimal working dilutions. Immunolabeling specificity was confirmed by processing the retinal sections in the absence of a primary antibody and/or with blocking antibodies. Outer nuclear layer (ONL) thickness was measured from slides using a calibrated Filar micrometer eyepiece at a total magnification of 1000x (Reichert Scientific Instruments, Buffalo, NY) [21]. The ONL thickness from three nonadjacent coded retinal sections from control and GLE mice were analyzed (N=4-5 litters).
Table 2. Cell-specific primary antibodies used for immunohistochemistry.
| Primary Antigen | Imunogen | Source | Catalog. No. | Target | Host | Dilution |
|---|---|---|---|---|---|---|
| CHX10 |
Recombinant amino acids 1-131 N-terminal of human protein |
Exalpha Biologicals |
X1180P |
RPCs in development
Bipolar cells in adult |
Sheep |
1:200 |
| CcnD1 |
Recombinant fusion protein from mouse |
Santa Cruz |
SC-450 |
Proliferative RPCs |
Mouse |
1:200 |
| PCNA |
Purified PCNA |
Chemicon |
MA4076 |
Proliferative RPCs |
Mouse |
1:100 |
| PKCα |
Protein kinase C alpha |
AbD Serotec |
MCA1572 |
Rod bipolars |
mouse |
1:100 |
| PMCA |
Purified human erythrocyte ATPas |
Affinity Bioreagents |
MA3-914 |
OPL, IPL some amacrine and bipolar cells |
Mouse |
1:500 |
| Recoverin |
KLH-conjugated linear peptide corresponding to a sequence from the EF-hand 4 domain of human recoverin |
Chemicon |
AB5585 |
Photoreceptors |
Rabbit |
1:1500 |
| Rhodopsin |
Bovine rhodopsin C-terminus, last 9 amino acids |
Chemicon |
MAB5356 |
Rod photoreceptors |
Mouse |
1:5000 |
| Secretagogin |
Amino acids 186-276 C-Terminus of SCGN of human origin |
Santa Cruz |
SC-135395 |
Cone bipolars |
Rabbit |
1:1000 |
| SV2B |
amino acids 1-126 mapping at the rat N-terminust |
Gift from Roger Janz |
|
Photoreceptor and bipolar cell ribbon synapses |
Rabbit |
1:1000 |
| TRPM1 |
Synthetic peptide of transient potential cation, subfamily M, member 1 variant delta |
Osenses |
OST00037W |
Dendrites of ON bipolar cells |
Sheep |
1:300 |
| VGluT1 | Synthetic peptide from rat VGLUT1 protein | Chemicon | AB5905 | Synaptic layers | Guinea Pig | 1:1000 |
Western blotting
Immunoblotting for rhodopsin was done as described [21,29]. The blots were probed with anti-rhodopsin and -glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies (Table 2), followed by incubation with an HRP-conjugated secondary antibody. The blots were visualized using enhanced chemiluminescence (ECL) Plus (Thermo Fisher Scientific). Densitometry measurements were obtained from non-saturated blots using NIH-Image J. Four mice from different litters per age per treatment group were used.
Statistical analyses
For all data, animals from the same litter represented only one data point. The data are presented as the mean ± Standard Error of the Mean (SEM) for four to nine animals per treatment group. Data were analyzed using a two-way ANOVA (treatment by age) followed by a post-hoc analysis using the Fisher’s Least Significant Difference Test or the Student t test when only two means were compared. Values of p < 0.05 were considered significantly different from the controls and were noted in the figures by asterisks where appropriate. In the text, values of p < 0.05 were noted as significantly different from the controls. IHC images were compiled and presented using Adobe Photoshop CS (Adobe Systems Inc., Mountain View, CA). Graphs were generated using KaleidaGraph (Synergy Software, Reading, PA).
Results
GLE delays the expression of genes and proteins associated with the differentiation of rod photoreceptors
Figure 1 shows the relative gene expression of the rod-specific transcription factors Nrl (Figure 1A), Nr2e3 (Figure 1B), Crx (Figure 1C) and the rod-specific functional gene Rho (Figure 1D) from E16.5 to PN10. The RT-qPCR data were normalized with β-actin so as not to reflect the overall increased number of cells associated with the GLE model [21]. In both the control and GLE retinas, the developmental gene expression pattern was similar for all four genes (i.e., a continuous increase from E18.5 to PN10). In the GLE retinas, all four of the rod-associated genes decreased significantly at PN2. In addition, Nrl expression decreased significantly at PN6. These data reveal an overall decrease in rod-specific gene expression in the GLE retinas at PN2, suggesting a possible delay in development.
Figure 1.

GLE decreased relative expression of Nrl, Nr2e3, Crx, and Rho genes. In the control and GLE retinas, the developmental patterns of (A) Nrl, (B) Nr2e3, (C) Crx, and (D) Rho gene expression were similar, and their relative gene expression significantly increased from E16.5 to PN10. A: In the GLE retinas, Nrl expression significantly decreased at PN2 and PN6 relative to the age-matched controls. B–D: In the GLE retinas, Nr2e3, Crx, and Rho expression significantly decreased at PN2 relative to the age-matched controls. The mean ± SEM values represent the triplicate samples from four to five animals per treatment group per age. Values with an asterisk indicate p < 0.05 compared to the controls. GLE = Gestational lead exposure; E = embryonic; PN = postnatal; SEM = standard error of the mean.
To determine the functional development of rod photoreceptors, we analyzed the spatiotemporal appearance of the photoreceptor specific proteins rhodopsin and recoverin by immunohistochemistry. Rhodopsin is a rod-specific photopigment that initiates the phototransduction cascade in the presence of light [35,36]. Recoverin, a calcium-sensing protein present in all photoreceptors, regulates phototransduction by deactivating rhodopsin kinase [37,38]. Rhodopsin and recoverin protein expression were determined in the control and GLE retinas at E16.5, E18.5, PN1, PN3, PN5, PN7, PN10, and PN60 (Figure 2 and Figure 3). Recoverin-immunoreactive (IR) cells represent cone photoreceptors, while rods were double labeled with recoverin and rhodopsin. In the control retinas, rhodopsin-IR cell somas were first observed in the developing ONL at PN1 (Figure 2C). This is consistent with the published data for rats [39]. Rhodopsin expression increased throughout development, consistent with the increased differentiation of the RPCs into rods [40]. By PN10 in control retinas (Figure 3C), rhodopsin-IR was seen in the outer segments of rod photoreceptors. In the GLE retinas, rhodopsin-IR cells were observed first in the ventricular zone at PN3, two days after controls (Figure 2P; Table 3). Rhodopsin expression in the GLE retinas increased throughout development, and the ONLs of the GLE retinas were consistently thicker than the age-matched controls beginning at PN5 (compare Figure 3A-M). Moreover, at PN10, rhodopsin-IR in the outer segments was not seen in the GLE retinas (Figure 3O) as it was in the controls (Figure 3C). In contrast, the onset of and developmental increase in recoverin-IR (using an antibody at a low concentration of 1:1500 and with a short fixation time to selectively label photoreceptors [41]) was similar in both the control and GLE retinas. Recoverin-IR cells were first observed at E18.5 in the control and GLE mice (Figure 2B,N), consistent with the published data in C57BL/6 mice [42]. Although a few double-labeled recoverin-IR and rhodopsin-IR photoreceptors were seen in the PN1 control retinas (Figure 2K), these were not observed until PN3 in the GLE retinas (Figure 2X). We were unable to detect M or S cone opsin-IR cells at the embryonic and early postnatal ages tested. This is in harmony with what is known about M and S cone opsin expression, which does not occur until PN5 [43]. Since the double-labeled rhodopsin- and recoverin-IR rods did not appear until PN1 and PN3 in the control and GLE retinas, respectively, and since recoverin-IR cells are observed in both the control and GLE retinas at E18.5, we take this to possibly suggest that the cones differentiate at similar time points. Our Affymetrix data (Fox et al, unpublished) from E16.5 to PN10 for the control and GLE mice show that the gene expression of Opn1sw (S cones) and Opnmw1 (M cones) were not significantly different in the control and GLE retinas at any age. Together, these data indicate that the rods were functionally delayed by approximately two days in the GLE retinas.
Figure 2.

GLE delayed rhodopsin-IR, but not recoverin-IR, in developing retinas (E16.5-PN3). The developing retinas (E16.5-PN3) from (A–L) the control and (M–X) GLE mice were double labeled with antibodies against rhodopsin (red: A–D and M–P) and recoverin (green: E–H and Q–T), and colabeling was examined in the merged images (yellow: I–L and U–X). A–D: In the control retinas, a faint amount of rhodopsin-IR was first observed in the ISs at E18.5. At PN1 and PN3, rhodopsin-IR increased in the ISs and in the ONL. E–H: In the control retinas, recoverin-IR was first observed in cone ISs at E16.5. From E18.5 to PN3, recoverin-IR increased in the ISs and ONL. I–L: The colabeling of recoverin and rhodopsin-IR was first detected at PN1. M–P: In the GLE retinas, a few faint rhodopsin-IR rod ISs were detected at E18.5. At PN1 and PN3, rhodopsin-IR increased in the rod ISs and ONL. Thus, there was a two-day delay in the appearance of rhodopsin-IR in GLE retinas (Table 3). Q–T: In the GLE retinas, like the control retinas, recoverin-IR was first seen at E16.5. From E18.5 to PN3, recoverin-IR increased in the ISs and ONL. U–X: In the GLE retinas, the colabeling of recoverin and rhodopsin was first detected at PN3 as opposed to at PN1 in the control retinas. Scale bar = 40 μm. GLE = Gestational lead exposure; IR = immunoreactivity; E = embryonic; PN = postnatal; IS = inner segment; ONL = outer nuclear layer.
Figure 3.

GLE delayed rhodopsin-IR, but not recoverin-IR, in developing retinas (PN5-PN60). The developing retinas at PN5-PN60 from (A–L) the control and (M–X) GLE mice were double labeled with antibodies against rhodopsin (red: A–D and M–P) and recoverin (green: E–H and Q–T), and colabeling was examined in merged images (yellow: I–L and U–X). In the control retinas at (A, E, and I) PN5 and (B, F, and J) PN7, the ISs, ONL, and OPL were colabeled with rhodopsin and recoverin, which increased at PN7. C, G, and K: At PN10, the OS expressed rhodopsin-IR, and extensive colabeling with recoverin occurred in the ISs, ONL, and OPL. D, H, and L: In young adult control retinas (PN60), the OSs were intensely rhodopsin-IR, and there was extensive labeling in the ISs, ONL, and OPL. In the distal OPL, the smaller rod spherules [24] were colabeled (yellow pixels), whereas the larger cone pedicles in the proximal OPL [24] were only recoverin-IR (green pixels). In the GLE retinas at (M, Q, and U) PN5 and (N, R, and V) PN7, the ISs, ONL, and OPL were rhodopsin-IR and recoverin-IR, and an increased amount of colabeling was seen in all the layers. Relative to the age-matched controls, the ONL thickness increased. O, S, and W: At PN10, OSs were rhodopsin-IR, and extensive colabeling with recoverin occurred in the ISs, ONL, and OPL. Relative to the age-matched controls, the ONL thickness increased. P, T, and X: In the PN60 GLE retinas, the OSs, ONL, and OPL were intensely rhodopsin-IR and almost completely colabeled with recoverin. Relative to the age-matched controls, the ONL and OPL thickness increased, and the number of rod spherules increased as described [21]. Scale bar = 40 μm. GLE = Gestational lead exposure; IR = immunoreactivity; PN = postnatal; IS = inner segment; OS = outer segment; ONL = outer nuclear layer; OPL = outer plexiform layer.
Table 3. Age of first appearance of immunoreactivty in rod and bipolar cell compartments in controls and GLE mice.
| Primary antigen | Cell or compartment source |
Age of first appearance |
|
|---|---|---|---|
| Control | GLE | ||
| Recoverin |
Cone inner segments & somas |
E18.5 |
E18.5 |
| Rhodopsin |
Rod inner segments & somas |
PN1 |
PN3 |
| Chx10 |
Inner nuclear layer |
PN3 |
PN5 |
| Secretagogin |
INL (cone bipolar cells) |
PN5 |
PN7 |
| PKCα |
OPL and IPL |
PN3 |
PN3 (reduced) |
| VGluT1 |
OPL |
PN3 |
PN5 |
| VGluT1 |
IPL-a |
PN7 |
PN10 |
| VGluT1 |
IPL-b |
PN10 |
after PN10 |
| PMCA |
ISs |
PN3 |
PN5 |
| PMCA |
OPL |
PN5 |
PN5-PN7 |
| SV2B |
OPL |
PN3 |
PN5 |
| TRPM1 |
ISs |
PN5 |
PN5-PN7 |
| TRPM1 | OPL | PN7 | PN7 (reduced) |
To more precisely determine the timing of the delay in the appearance of rhodopsin-IR in the GLE retinas, we conducted western blot experiments using PN1, PN2, and PN3 control and GLE retinas. Figure 4 shows that rhodopsin was detectable in whole homogenized control retinas at PN1 and PN2, consistent with published data [39]. Rhodopsin was detected at PN1 and PN2 in the GLE retinas but significantly reduced by ≥ 20%. By PN3, the rhodopsin concentration was ≥ 10% higher in GLE than in the control retinas. The rhodopsin concentration increased approximately 40% between PN1 and PN2, and between PN2 and PN3 in the control and GLE mice. Together, these immunohistochemistry and immunoblot data indicate that there is a two-day delay in relative rhodopsin expression in GLE mice retinas.
Figure 4.
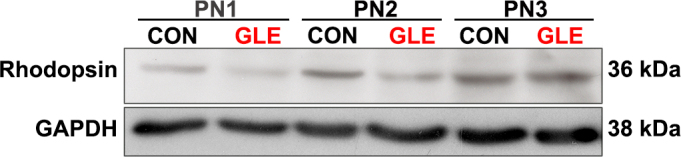
GLE decreased rhodopsin protein content in developing retinas. At PN3, the rhodopsin content significantly increased by 10% in the GLE retinas relative to the age-matched controls. GAPDH was used as the protein loading control. GLE = Gestational lead exposure; PN = postnatal; GAPDH = glyceraldehyde 3-phosphate dehydrogenase.
GLE does not alter the gene or protein expression of glutamine synthetase (GS) in Müller glial cells during development
Figure 5 reveals that the developmental profile of GS protein expression was similar in the control (Figure 5A-E) and GLE (Figure 5F-J) retinas throughout development. Figure 5K shows that GS gene expression was similar and low from E16.5 to PN6, then significantly increased by PN10 in the control and GLE retinas. These GS results are consistent with the birth-dating and adult retinal immunohistochemistry data for GSs, which shows that GLE did not alter the initiation or pattern of gliogenesis or the number of Müller glial cells [21].
Figure 5.

GLE did not alter GS protein or gene expression in developing retinas. The developing and adult retinas from (A–E) control and (F–J) GLE mice were labeled with an antibody against GS. GS exhibited an age-dependent increase in expression from PN3 to PN60. A and F: At PN 3, only the proximal retina was labeled. B and G: By PN5, the entire retina was immunolabeled. C, D, H, and I: PN7 and PN10 images show that the GS-IR labeling pattern became more organized as the retina developed. E and J: The intensity of GS-IR Müller glial cells was not different in PN60 control and GLE retinas, as previously described [21]. The number of cyclin D3-IR Müller glial cells in the PN60 control and GLE retinas was not different [21]. Scale bar = 40 μm. K: In the control and GLE retinas, GS gene expression (Glul) was low from E16.5 to PN10 and then increased at PN10. There were no significant differences at any age. The mean ± SEM values represent the triplicate samples from four to five animals per treatment group per age. GLE = Gestational lead exposure; GS = glutamine synthetase; PN = postnatal; E = embryonic; SEM = standard error of the mean.
GLE increases proneural and BC gene expression but delays the expression of proteins associated with BC differentiation
Figure 6 shows the relative gene expression of the rod and bipolar proneural genes Otx2 (Figure 6A) and Chx10 (Vsx2; Figure 6B) in developing retinas. In the control retinas, Otx2 expression increased continually from E18.5 to PN6 and peaked at PN6, when most late-born retinal neurons were differentiated [44]. This is consistent with in situ hybridization data for similar ages [45]. The pattern of Otx2 expression in GLE retinas followed that in the control retinas; however, gene expression in the GLE retinas increased significantly at PN2 and PN6. In the developing control retinas, Chx10 expression declined from E16.5 to PN2 and increased from PN2 to PN10. In contrast, in the GLE retinas, a decline in Chx10 expression was never observed; rather, the pattern of expression continually increased, and PN2 and PN6 expression levels were significantly increased relative to those in the controls. Thus, the Otx2 and Chx10 expression patterns in PN2 to PN6 GLE retinas were consistent with the increased number of rods and BCs observed in the GLE retinas [21].
Figure 6.

GLE increased gene expression of Otx2 and Chx10 (Vsx2) in developing retinas. In the control and GLE retinas, (A) Otx2 and (B) Chx10 gene expression significantly increased from E16.5 to PN10. A: For Otx2, the pattern of expression was similar in the control and GLE retinas, peaking at PN6, when most late-born retinal neurons were differentiated [29]. In the GLE retinas, Otx2 expression significantly increased at PN2 and PN6 relative to the age-matched controls. B: For Chx10, the pattern of gene expression in the control and GLE retinas differed. In the GLE retinas, Chx10 expression significantly increased at PN2 and PN6 relative to the age-matched controls. The mean ± SEM values represent the triplicate samples from four to five animals per treatment group per age. The values with an asterisk indicate p < 0.05 compared to the control retinas. GLE = Gestational lead exposure; E = embryonic; PN= postnatal; SEM = standard error of the mean.
To examine the differentiation of rod BCs, we analyzed the spatiotemporal protein expression of Chx10 and PKCα in PN3, PN5, PN7, PN10, and PN60 control and GLE retinas (Figure 7). We also colabeled Chx10 with CCND1 at PN1 and PN5 in the control retinas to illustrate the difference between RPC-associated and BC-associated Chx10 (Figure 8). Chx10 is a homeobox domain transcription factor that is initially expressed in RPCs during the earliest stages of neuroretinal development [46]. Chx10 expression persists in RPCs until the early postnatal ages, after which its expression permanently localizes to rod and cone BCs [46,47]. Immunohistochemically, this switch from neuronal- to bipolar-associated Chx10-IR can be determined by the appearance of the Chx10 antibody label in BC somas. In the PN3 control retinas, we observed Chx10-IR in the inner segments (ISs), inner nuclear layer (INL), and ganglion cell layer (GCL) (Figure 7A). In the INL, cytoplasmic and nuclear labeling was seen in RPCs and immature BCs, respectively. Furthermore, the strongly labeled Chx10-IR cells localized only to the center of the INL. The appearance of Chx10-IR BCs at PN3 correlates with the peak of BC differentiation [21,48]. In the PN5 controls, Chx10-IR was restricted to BCs and the GCL, and the diffuse label associated with RPCs was no longer present (Figure 7B). Figure 8 illustrates the difference between RPC-associated and BC-associated Chx10 in the control retinas. At PN1, Chx10-IR and CCND1-IR colabeled (Figure 8C), whereas at PN5, they did not (Figure 8F). In the PN7 and PN10 controls (Figure 7C,D, respectively), the appearance of Chx10-IR BCs was similar to that at PN60 (Figure 7E [21]). In the PN3 GLE retinas, Chx10 labeled the ISs, INL, and GCL (Figure 7K). However, the strongly observed Chx10-IR BCs in the controls were not present in the GLE retinas, and Chx10-IR in the INL was restricted to proliferative cells. However, the Chx10-IR in the ‘inner plexiform layer (IPL) and GCL of the control and GLE retinas was similar at all ages (Figure 7A-E,K-O), confirming that the effect of GLE was selective for late-born neurons. In the PN5 GLE retinas, immature BCs were strongly IR for Chx10 as in the age-matched controls; however, the cells appeared as spindles and were elongated (Figure 7L; Table 3). By PN7, the appearance of Chx10-IR BCs in the GLE retinas was similar to that in the control retinas (Figure 7 M). At PN10 and PN60, an increased number of Chx10-IR BCs consistent with the GLE phenotype was observed (Figure 7N,O, respectively) [21].
Figure 7.

GLE delayed the differentiation of Chx10-IR and PKCα-IR BCs. The developing retinas from (A–J) the control and (K–T) GLE mice were double labeled with antibodies against Chx10 (green: A–E and K–O) and PKCα (red: F–J and P–T) A: In the PN3 controls, Chx10 labeled both the proliferative and the newly differentiated BCs in the INL, as denoted by diffuse and intense labelin,g respectively, as well as cells in the GCL. B: At PN5 and (C) PN7, Chx10 labeled differentiated BCs in the INL as well as cells in the GCL. For confirmation, Figure 8 shows Chx10 and CCND1, a cell cycle marker, labeling at PN1 and PN5. At PN7, a well-defined OPL was visible. D and E: The patterns at PN10 and PN60 was similar to that at PN7. K: In contrast, in the PN3 GLE retinas, Chx10 labeled only proliferative cells, and no differentiated Chx10-IR BCs were present. L: At PN5, Chx10-IR BCs were present, although they were spindle-shaped compared to the age-matched controls. M: At PN7, the patterns were similar in the control and GLE retinas. N and O: At PN10 and PN60, there were significantly more Chx10-IR cells in the GLE retinas, as described [21]. F: In the PN3 control retinas, PKCα-IR was in ISs, the IPL, and the GCL. G: At PN5, PKCα-IR increased in the ISs, IPL, and GCL and appeared in the OPL. H: At PN7, PKCα-IR was selectively and intensely expressed in the rod BCs, from their dendrites in the OPL and to their axon terminals in the most proximal INL. I: At PN10, the rod BCs continued to develop, as evidenced by the increased extent of the PKCα-IR. J: At PN60, PKCα-IR labeled the complete mature rod BC. P: In the PN3 GLE retinas, PKCα-IR was less intense and more diffuse than in the age-matched controls, as seen in the ISs, IPL, and GCL. Q and R: At PN5 and PN7, the GLE retinas were similar to the controls. However, the OPL was less developed, and fewer PKCα-IR rod BCs were present. S: At PN10, the BCs in the GLE increased in their number and thickness. T: In PN60, the BC number and thickness increased in the GLE retinas relative to the age-matched controls, as described [21]. Thus, GLE delayed the appearance and localization of Chx10 and PKCα (Table 3). Scale bar = 40 μm. GLE= Gestational lead exposure; IR = immunoreactive; BC = bipolar cell; PN = postnatal; INL = inner nuclear layer; GCL = ganglion cell layer; CCND1 = cyclin D1; OPL = outer plexiform layer; IPL = inner plexiform layer.
Figure 8.

Chx10 colocalized with CCND1 before BC differentiation. The developing (A–C) PN1 and (D–F) PN3 control retinas double labeled with antibodies against Chx10 (green: A and D) and CCND1 (red: B and E), and colabeling was examined in the merged images (yellow: C and F). A: In the PN1 controls, Chx10-IR was diffusely located throughout the retina in the cytosolic compartment. B: CCND1, a cell cycle protein, was localized in the nucleus of the proliferating cells in the VZ. C: Chx10-IR colocalized with CCND1-IR in the VZ. D: In the PN5 control retinas, Chx10-IR was localized in the somas in the INL and the cytosol of cells in the ganglion cell layer. The shape of the Chx10-IR in the INL cells changed from spindly to compact and round. E: CCND1 localized to spindly proliferating cells in the INL. F: Chx10 did not colabel with CCND1 in the INL. Scale bar = 40 μm. CCND1 = cyclin D1; BC = bipolar cell; PN = postnatal; VZ = ventricular zone; INL = inner nuclear layer.
Figure 7 also shows the spatiotemporal profile of PKCα in relation to Chx10-IR. PKCα, a calcium-activated serine/threonine protein kinase, selectively labels the soma, dendrites, axons, and synaptic terminals of adult rod BCs in mouse retinas [48]. In the PN3 control retinas, PKCα minimally labeled the INL and strongly labeled the developing ISs, IPL, and GCL (Figure 7F). In the PN5 control retinas, PKCα-IR increased in the ISs, IPL, and GCL and appeared in the OPL (Figure 7G). By PN7, PKCα-IR was restricted to rod BC somas, dendrites, axons, and bipolar cell terminals (Figure 7H). For comparison to the GLE retinas, the adult phenotype of control PKCα-IR BCs is shown at PN60 (Figure 7J). In the PN3 GLE retinas, PKCα-IR was less intense and more diffuse than in the age-matched controls (Figure 7P). The GLE retinas were similar to the controls at PN5 and PN7 (Figures 7Q,R, respectively), although the OPL was less developed and fewer PKCα-IR rod BCs were present. Similar to Chx10-IR, PKCα-IR in the IPL and GCL of the PN3 and PN5 control and GLE retinas was similar, again showing that the effect of GLE was selective for late-born neurons. At PN10 and PN60 in the GLE retinas (Figure 7S,T, respectively), there was an increased number of PKCα-IR rod BCs relative to the controls (Figure 7I,J, respectively).
To examine the differentiation of the cone BCs, we analyzed the spatiotemporal protein expression of secretagogin (SCGN) in the control and GLE retinas (Figure 9). SCGN is an EF-hand calcium-binding protein that localizes to subpopulations of ON and OFF cone BCs, but not rod BCs, in mammalian retinas [49]. Figure 9 shows the spatiotemporal SCGN-IR for PN5, PN7, PN10, and PN60 retinas in control (Figure 9A-L) and GLE mice (Figure 9M-X) in relation to PKCα-IR, the adult rod BC marker [48]. In the control mice, SCGN-IR cone BCs were first observed in the INL at PN5 (Figure 9A). These SCGN-IR cells were elongated, and their projections extended distally to the IS and proximally toward the GCL. By PN7, most of the SCGN-IR was restricted to the somas of the INL, dendritic processes of the OPL, and axon terminals in IPL sublamina a (IPL-a) and b (IPL-b; Figure 9B). SCGN labeled more intensely in IPL-a (the OFF layer) than in IPL-b (the ON layer). In the PN10 controls, the pattern of SCGN-IR in the INL changed from moderately labeled linear projections to intensely labeled soma. Furthermore, the dendrites and axon terminals of these SCGN-IR cone BCs exhibited punctate and well-defined labeling in the OPL and IPL, respectively (Figure 9C). In the PN10 controls, the appearance of SCGN-IR somas, dendrites, and axon terminals closely resembled the appearance seen at PN60 (Figure 9C,D, respectively). These results are consistent with the images [49]. The control retinas double labeled for PKCα and SCGN showed no colocalization at any age (Figure 9I-L).
Figure 9.

GLE delayed the differentiation of SCGN-IR cone BCs. The developing retinas from (A–L) the control and (M–X) GLE mice were double labeled with antibodies against SCGN (red: A–D and M–P) and PKCα (green: E–H and Q–T), and colabeling was examined in the merged images (yellow: I–L and U–X). A-D: In the PN5 controls, the SCGN labeled cone BC somas in the INL, their dendritic processes in the OPL, and processes in the GCL. At PN7, SCGN-IR increased and localized to immature cone BC somas in the INL, dendritic processes in the OPL, and axonal terminals in the OFF layer (IPL-a) and the ON layer (IPL-b). SCGN-IR was more intense in IPL-a than in IPL-b. At PN10, SCGN strongly labeled cone BC somas located throughout the INL, dendrites, and axon terminals. By PN60, SCGN-IR cone BC somas were in the distal half of the INL, and the OPL and IPL reached their adult pattern and thickness. E–H: The pattern of PKCα-IR in the developing controls was similar to that shown in Figure 7. I–L: Although SCGN and PKCα were in close approximation in the proximal IPL and especially in the OPL, they did not colabel. M–P: In the PN5 GLE retinas, there were no SCGN-IR cells or processes. At PN7, SCGN-IR dramatically increased and was localized to immature cone BC somas in the INL, the OPL, IPL-a, and IPL-b, the former exhibiting more intense labeling. Relative to the age-matched controls, the INL was thicker and the IPL was less organized. At PN10, SCGN moderately labeled cone BC somas located throughout the INL, dendrites, and axon terminals. Relative to the age-matched controls, the INL and IPL were less developed and organized (Table 3). By PN60, SCGN-IR cone BC somas were in the distal half of the INL, and the OPL and IPL reached their adult pattern and thickness. Relative to the age-matched controls, the OPL, INL, and IPL were significantly thicker. Q–R: The pattern of PKCα-IR in the developing GLE retinas was similar to that shown in Figure 7. U–X: Although SCGN and PKCα were in close approximation in the proximal IPL, and especially in the OPL, they did not colabel. Scale bar = 40 μm. GLE =Gestational lead exposure; SCGN = secretagogin; IR = immunoreactive; BC = bipolar cell; PN = postnatal; INL = inner nuclear layer; OPL = outer plexiform layer; IPL = inner plexiform layer; PKCα = protein kinase c alpha.
In the GLE retinas, SCGN expression was delayed at PN5 (Figure 9M; Table 3). By PN7, however, there was an increase in the number of SCGN-IR cone BCs when compared to the age-matched controls (Figure 9N). Interestingly, the SCGN-IR pattern and intensity in PN10 GLE retinas (Figure 9O) appeared more similar to the PN7 than the PN60 retinas, unlike that seen in the controls. That is, the punctate dendritic and axon terminals observed in the PN10 control retinas were not present in the age-matched GLE retinas. Together, these data indicate that BC differentiation and synaptic development are delayed two to three days in GLE retinas.
GLE delays the onset of functional rod synapses in the OPL
Since the initial expression of rod-specific genes (Figure 1) and proteins (Figure 2 and Figure 4) was delayed, we examined the expression of proteins associated with the functional synapse in the OPL of rod photoreceptors to determine if the entire rod was similarly and coordinately affected. Figure 10A-J shows the spatiotemporal expression of VGluT1 in the control and GLE retinas at ages PN3, PN5, PN7, PN10, and PN60. In the control retinas, sporadic VGluT1-IR puncta were first observed below the ventricular zone at PN3 (Figure 10A). At PN5, VGluT1 labeling in the control retinas localized to the developing OPL (Figure 10B). In the control retinas, VGluT1-IR increased in the OPL at both PN7 and PN10 as the rods developed and matured (Figure 10C,D, respectively) toward the adult phenotype (Figure 10E). In the GLE retinas, VGLUT-IR puncta were first observed in the OPL at PN3 (Figure 10F), similar to the controls. However, in the PN5 GLE retinas, the extent of VGluT1-IR in the OPL decreased (Figure 10G; Table 3). In the PN7 GLE retinas, VGluT1-IR in the OPL decreased; although it was detected in the PN7 retinas, it still decreased (Figure 10H; Table 3). By PN10, VGluT1-IR in the OPL of GLE was greater than that in the controls (Figure 10I; Figure 10D). In the GLE retinas at PN60, VGluT1-IR in the OPL and IPL (Figure 10J) was greater than in the controls, as reported [21]. Thus, GLE caused a two to three day in the appearance and development of the age-matched extent and intensity of VGluT1-IR in the OPL.
Figure 10.

GLE delayed the synaptic expression of VGluT1 and PMCA in developing plexiform layers. The developing retinas from (A–E) the control and (F–J) GLE mice were labeled with an antibody against VGluT1. A: In the PN3 controls, sporadic VGluT1- IR was seen in the OPL. B–C: In the PN5 and PN7 retinas, the VGluT1-IR amount and intensity increased in the OPL and was seen in IPL-a (OFF layer). D: At PN10, the VGluT1-IR in the OPL laminated into discrete puncta in the distal (rod spherules) and clumps in the proximal (cone pedicles) layers as well as into IPL-a and IPL-b (ON layer). E: At PN60, the adult pattern of the VGluT1 occurred in the OPL and IPL. F–G: In the PN3 and PN5 GLE retinas, the sporadic VGluT1-IR occurred in the OPL. In contrast to the controls, the IPL-a was not have the VGluT1-IR at PN5. H: In the PN7 retinas, a minimal amount of VGluT1-IR was seen in the OPL and IPL-a. I: At PN10, the VGluT1-IR in the OPL increased in the GLE retinas and was similar to the controls. Although the VGluT1-IR was present in IPL-a, only a minimal amount occurred in IPL-b. Thus, the GLE caused a two- to three-day delay in the appearance and development of the age-matched extent and intensity of VGluT1-IR in the OPL and IPL (Table 3). J: At PN60, the amount and intensity of VGluT1 in the OPL and IPL of the GLE retinas increased relative to the age-matched controls such that the layers were thicker. The developing retinas from (K–O) the control and (P–T) GLE mice were labeled with an antibody against pan-PMCA. K: In the PN3 controls, PMCA-IR was seen in ISs and throughout the IPL and GCL. L–M: In the PN5 and PN7 retinas, PMCA-IR was visible in the ISs and intensely labeled the OPL, IPL, and GCL. N: At PN10, PMCA-IR was more localized. Intensely PMCA-IR occurred in the OPL and IPL, and labeling was visible in the distal and proximal INL. O: At PN60, PMCA-IR laminated in the OPL such that the distal layer (rod spherules) was intensely labeled, whereas the proximal OPL (cone pedicles) was only lightly labeled. Somas occurred throughout the INL in the PMCA-IR somas were observed throughout the INL. Intense PMCA-IR was observed in both the IPL-a and IPL-b. P: In the PN3 GLE retinas, PMCA-IR occurred in the IPL and GCL but not in ISs. Q: In the PN5 GLE retinas, PMCA-IR occurred in the ISs but was less intense in the OPL, IPL, and GCL compared to the age-matched controls. R–S: At PN7 and PN10, the pattern of PMCA-IR was similar in the control and GLE retinas. Thus, GLE caused a two-day delay in the appearance and development of the age-matched extent and intensity of PMCA-IR in the ISs and OPL (Table 3). T: At PN60, the pattern of PMCA-IR was similar in the control and GLE retinas, except that the distal OPL and entire IPL were thicker and had more intense PMCA-IR in the GLE retinas. Scale bars = 40 μm. GLE = Gestational lead exposure; VGluT1 = vesicular glutamate transporter 1; PMCA = plasma membrane calcium ATPase; PN = postnatal; IR = immunoreactivity; OPL = outer plexiform layer; IPL = inner plexiform layer; GCL = ganglion cell layer; INL = inner nuclear layer.
Neurons use four different plasma membrane calcium ATPase (PMCA) isoforms to return to or maintain the basal levels of intracellular calcium after the stimulation of voltage-operated calcium channels and the subsequent release of neurotransmitters [33,50]. In the OPL of adult mammalian retinas, a commercially available pan-PMCA antibody (Table 2) is able to recognize all four isoforms and label both rod spherules and the apical portion of cone pedicles; however, intense and widespread PMCA-IR is associated with rod spherules [33]. Figure 10K-T shows the labeling profile of PMCA-IR at ages PN3, PN5, PN7, PN10, and PN60 in the control and GLE retinas. In the PN3 control retina (Figure 10K), but not the GLE retina (Figure 10P), the PMCA-IR was in the ISs, supporting the delay in rhodopsin appearance. The PMCA-IR in the IPL and GCL of PN3 was similar in the control and GLE retinas, confirming that the effect of GLE was selective for late-born neurons. At PN5, the controls exhibited strong PMCA-IR, which was first observed in the OPL (Figure 10L). In the PN5 GLE retinas, the extent and intensity of PMCA–IR in the OPL decreased (Figure 10Q; Table 3). At PN7 and PN10, the pattern and intensity of PMCA-IR appeared similar in the control (Figure 10M,N) and GLE retinas (Figure 10R,S). However, at PN60, the increased number of rod synapses in GLE retinas was evidenced by a higher number of PMCA-IR spherules (Figure 10T) relative to the controls (Figure 10O).
To further examine the development of the proteins associated with glutamatergic synaptic transmission in photoreceptors, we analyzed the spatiotemporal profile of the synaptic vesicle protein 2B (SV2B) in the control and GLE retinas at PN3, PN5, PN7, and PN10 (Figure 11). SV2B is one of three SV2 isoforms (SV2A, SV2B, and SV2C) present on presynaptic vesicles that regulates excitatory calcium-dependent vesicle release [51]. In mouse retinas, SV2B is expressed in the OPL and the IPL [52,53] and regulates rod photoreceptor neurotransmitter release [54]. In the control retinas, SV2B exhibited an age-dependent increase in extent and intensity from PN3 to PN10 (Figure 11A-D). It was detectable in the OPL at PN3 (Figure 11A). In the OPLs of GLE mice, SV2B expression was delayed and decreased such that it was barely visible at PN3 and PN5 (Figure 11M,N, respectively; Table 3). At PN7, the SV2B-IR in the GLE retinas (Figure 11O) appeared similar to that of the PN5 controls (Figure 11B). At PN10, the SV2B-IR pattern and intensity were similar in the GLE (Figure 11P) and control (Figure 11D) retinas.
Figure 11.

GLE delayed SV2B- and TRPM1-IR in the developing plexiform layers. The developing retinas from (A–L) the control and (M–X) GLE mice were double labeled with antibodies against SV2B (red: A–D and M–P) and TRPM1-L (green: E–H and Q–T), and colabeling was examined in the merged images (yellow: I–L and U–X). A–B: In the PN3 and PN5 controls, SV2B was visible in the OPL. In the PN5 retinas, punctate SV2B-IR was visible in the IPL. C: At PN7, SV2B-IR occurred more intensely in the OPL and IPL, with higher intensity in IPL-a than IPL-b. D: By PN10, SV2B-IR laminated in the OPL such that there were discrete puncta in the distal (rod spherules) and clumps in the proximal (cone pedicles) layers. The IPL laminated into IPL-a, and IPL-b separated by a layer that was not labeled. E: In the PN3 controls, TRPM1-L diffusely labeled the ISs, IPL, and GCL. F: By PN5, TRPM1-L intensely labeled the IS and weakly labeled the developing OPL. G: At PN7 TRPM1-L-IR intensified in the ISs, OPL, and IPL. Furthermore, the TRPM1-L labeled the distal somas of the BCs right below the OPL. H: This pattern of TRPM1-L-IR persisted at PN10. I–J: No colabeling was observed at PN3 and PN5. K–L: In the OPLs of the PN7 and PN10 controls, SV2B labeled just above TRPM1-L-IR with little to no overlap. No colabel was observed in the IPL. M: In the PN3 GLE retinas, SV2B-IR puncta in the developing OPL was not uniformly localized as in the age-matched controls. N–O: The OPLs of the PN5 and PN7 GLE retinas were less mature than those in the age-matched controls. Little SV2B-IR was observed in the IPL of the PN5 GLE retinas. However, by PN7, SV2B labeled the IPL; IPL-a labeled more strongly than IPL-b. P: In the PN10 GLE retinas, SV2B-IR in the OPL and IPL increased; however, IPL-a and IPL-b did not show differential labeling as in the control. Q–T: TRPM1-L-IR in the GLE retinas was similar to the controls, except at PN7, where the OPL was not labeled as brightly. U–V: SV2B and TRPM1-L did not colocalize at PN3 and PN5 in the GLE retinas as in the controls. W–X: The PN7 and PN10 GLE retinas differed from the controls; at PN7, SV2B labeled in the same plane as, rather than above, TRPM1-L. At PN10, SV2B colocalized with TRPM1-L more than in the controls. Thus, there was a two-day delay in the appearance of SV2B-IR and TRPM1 in the GLE retinas (Table 3). Scale bar = 40 μm. GLE = Gestational lead exposure; SV2B = synaptic vesicle protein 2B; TRPM1-L = transient receptor potential M1-long; IR = immunoreactivity; OPL outer plexiform layer; IPL = inner plexiform layer; PN = postnatal; IS = inner segment; GCL = ganglion cell layer.
GLE delays the expression of rod and cone BC dendrite markers in the OPL and synaptic markers in the IPL
The structural development of BC dendrites in the control and GLE retinas was observed using an antibody against the transient receptor potential melastatin 1-long isoform (TRPM1-L) protein (Figure 11). In the retina, TRPM1 is required in the light response of ON BCs; in PN14, PN21, and adult retinas, it characteristically labels ON BC dendrites, somas, and axons in the IPL and in retinal ganglion cells [54-56]. In wild-type control mice, Trpm1 gene expression is relatively low from PN2 to PN8, increases slightly at PN10 and PN14, and then markedly increases at PN21 to a steady-state level seen at PN90 [55]. In the control retinas, TRPM1-IR first appeared in the OPL at PN5 (Figure 11F) and increased its pattern and intensity of expression at PN7 (Figure 11G) and PN10 (Figure 11H). Figures 11E-H also reveals that TRPM1-IR was observed in the photoreceptor ISs, IPL, and GCL throughout early development. In the GLE retinas, TRPM1-L-IR first appeared in the OPL at PN7 (Figure 11S), although its intensity was less than that in the age-matched controls (Table 3). The pattern in PN10 GLE retinas (Figure 11T) was not markedly different than that at in the PN7 GLE retinas. These results reveal that the maturation of the ON bipolar dendrites in the GLE retinas was delayed and decreased.
To determine the structural development of BC axonal synapses, we analyzed the patterns of VGluT1 (Figure 10A-J) and SV2B (Figure 11A-D, M-P) in the IPL. In the control retinas, VGluT1 was first visible in the IPL at PN5 (Figure 10B) and exhibited an age-dependent increase in extent and intensity in IPL-a and IPL-b, consistent with the published data [7]. In the GLE retinas, the initial VGluT1 expression in the IPL was delayed until PN7 (Figure 10H; Table 3). Moreover, and in contrast to the PN10 controls (Figure 10D), VGluT1 was not expressed in the IPL-b of GLE retinas (Figure 10I), and it was lower in IPL-a. By PN60, VGluT1-IR increased in the IPL of the GLE retina (Figure 10J) relative to the controls (Figure 10E). In the controls, SV2B was first visible in the IPL at PN5 (Figure 11B) and exhibited an age-dependent increase in extent and intensity in IPL-a and IPL-b. IPL-b developed before IPL-a, consistent with the published data [53]. In the GLE retinas, the initial SV2B expression in the IPL was delayed until PN7 (Figure 11O; Table 3). At PN10, SV2B-IR was similar in the control (Figure 11D) and GLE (Figure 11P) retinas.
Discussion
The overall goal of this study was to examine the differentiation and development of rods and BCs, which increased in the adult mouse and rat GLE retinas following increased and prolonged RPC proliferation [19,21]. There were six main findings in this study. First, GLE delayed the differentiation of rod photoreceptors, as indicated by the developmental profiles of several photoreceptor-specific genes and proteins, the latter via immunocytochemistry and immunoblot studies. Second, the differentiation of rod and cone BCs was delayed, as indicated by the delayed spatiotemporal patterns of Chx10, PKCα, and SCGN protein expression. Third, GLE delayed the expression of proteins and SV2B that are associated with functional synaptic development in rod photoreceptors, such as PMCA, VGluT1. Fourth, GLE delayed the expression of several essential proteins that are associated with the dendritic and synaptic structure and function of ON and OFF BC subtypes, such as VGluT1, SV2B and TRPM1-L. Fifth, there were no treatment-related changes in the gene or protein expression of GS associated with late-born Müller glial cells. Sixth, there were no treatment-related changes in the protein expression of early-born cells in the IPL or GCL. Together, these data reveal that the development of late-born neurons, but not Müller glial cells, is delayed in GLE retinas by two to three days. We suggest that this delay in differentiation is a direct downstream consequence of an increase in and prolonged period of RPC proliferation. Moreover, the delay in the markers of functional synaptic development in rods (OPL) and BCs (OPL and IPL) reveals that the coordinated differentiation and maturation of neuronal processes are linked with cellular differentiation and are therefore similarly delayed.
These results were unexpected for three reasons. First, because we observed the increased proliferation of RPCs in GLE retinas [21], we expected to see the accelerated and/or increased, not delayed, differentiation of rods, as seen in developing retinas exposed to different mitogenic soluble factors [reviewed in 15,57] or brains exposed to drugs [58]. Second, our birth-dating experiments revealed an increased number of rods and BCs that underwent terminal mitosis during postnatal development without a phase shift in the timing [21]. Third, most delays in retinal and brain development associated with chemical or toxicant exposure (e.g., ethanol, lead, polychlorinated biphenyls, etc.) are the result of decreased neurogenesis or increased apoptosis [26,59-61]. Based on our findings, as well as on the previously published work in wild-type mice retinas described below, we conclude that RPC terminal mitosis and neuronal differentiation are separate spatiotemporal events under both normal and pathophysiological conditions. For example, in vivo and in vitro studies demonstrate that the early-born rod photoreceptors possess an intrinsically determined ability to postpone rhodopsin expression after terminal mitosis [40,62]. That is, in developing rat retinas, there are two phases of rod differentiation: 1) an early phase, where rods born before E19 exhibit a delay of 8.5–12.5 days before rhodopsin expression and 2) a late phase, where rods born after E19 exhibit a delay of only 5.5–6.5 days before rhodopsin expression [40]. Although the postnatal microenvironment cannot prematurely induce early-born rods to express rhodopsin [40,62], it is unknown whether early-born rods can further delay their expression of rhodopsin. Since the initial delay in rhodopsin expression occurred at PN2 in GLE retinas, it is likely that GLE affected the early cohort of rod photoreceptors, suggesting that the terminal commitment to rod differentiation in this early cohort of rod photoreceptors is plastic and can be delayed further.
The cellular mechanisms underlying the delay in rod and BC commitment are unknown. However, the delay in rod differentiation seen in GLE retinas is consistent with studies showing that various soluble neurotropic ligands can alter the commitment of post-mitotic late-born retinal neurons to rod differentiation. For example, in vivo and in vitro experiments show that the ciliary neurotrophic factor can delay rod differentiation and rhodopsin expression for 3–4 days [63-65]. Similarly, the Müller glial cell-derived leukemia-induced factor (LIF) can temporarily inhibit rod differentiation [63,64,66]. Ex vivo and in vitro experiments also show that mitogen retinoic acid can increase proliferation and delay rod differentiation [57,67]. Our Affymetrix data (Fox et al, unpublished) from E16.5 to PN10 control and GLE mice shows that the gene expression of the LIF receptor Lifr and its transducing subunit gp130 are transiently upregulated by 28% at E16.5 and E18.5 in the GLE retinas relative to the controls, indicating one possible mechanism of action for delay. To date, one observational report notes a delay in BC development. Wu and Chiao [68] demonstrated that dark-rearing rabbits delayed the morphological maturation of BCs by two to three days, as characterized by histology and confocal microscopy.
Although we found a decrease in rod-specific genes at PN2 in the GLE retinas, both Otx2 and Chx10 gene expression increased at this age. The Otx2 gene regulates rod photoreceptor cell fate determination, and the conditional knockout of Otx2 causes post-mitotic rod precursors to become amacrine-like cells [69,70]. The upregulation of the Otx2 gene in the retinal progenitor cell causes the cell to become a postmitotic rod or BC precursor [70]. Otx2 gene expression remains elevated in BCs but decreases in rods [70]. Therefore, the increased expression in Otx2 observed in GLE mouse retinas is likely associated with an increased number of post-mitotic rod and BC precursors at PN2.
Chx10 is also duplicitous in its expression profile. The pattern of gene expression we observed is consistent with the idea that Chx10 protein expression associates with all proliferative cells during the embryonic and early postnatal stages of retinal development and becomes specific for BCs, as their numbers increase during late postnatal ages [71,72] (Figure 5 B). Therefore, an increase in Chx10 gene expression at PN2 in GLE retinas is likely a consequence of the increase in retinal progenitor cells at this age. Interestingly, the control retinas exhibited a decreased expression in the Chx10 gene at PN2 when compared to other ages. This was not observed in GLE retinas. We postulate that this represents a general switch from a RPC-dominated retina to a microenvironment conducive for differentiation and functional development.
Finally, we showed that GLE decreased the expression of several functional proteins associated with synaptic function in both the OPL and the IPL. The photoreceptor ribbon synapse is a complex structure that is specialized for fast and sustained neurotransmitter release [73]. Critical proteins associated with proper photoreceptor synaptic function include calcium channels, ion pumps, cytoskeletal scaffolds, presynaptic endocytotic and exocytotic proteins, and postsynaptic receptors [74]. Specifically, we assessed the proteins involved in neurotransmitter uptake into VGluT1, PMCA, SV2B, and TRPM1 rather than into the scaffolding proteins structurally associated with the synaptic ribbon [75]. We showed that, although both the GLE and control animals initially expressed VGluT1 at PN3, GLE animals exhibited the decreased expression of VGluT1 in the developing OPL at PN7. Furthermore, PMCA-IR decreased in the GLE retinas at PN5. At PN3, VGluT1-IR in the OPL is indicative of cone synapses forming their first glutamatergic connections with horizontal cells, while the rod photoreceptors first form connections around PN8 in mice (Figure 2) [7]. Therefore, the pattern of the VGluT1 label in the OPL of the developing GLE retinas indicates that the cone photoreceptor terminals developed normally, whereas rod terminals were delayed in their ability to package glutamate into the vesicles. This was confirmed by the decrease in PMCA labeling observed in the OPL at PN5. PMCA localizes to the photoreceptor terminals during the first postnatal week in rats [76]. In adult mouse retinas, most PMCA is associated with rod spherules rather than cone pedicles [33,55]. Therefore, limited PMCA-IR in the OPL of PN5 GLE retinas may be a consequence of immature rod synapses. Interestingly, this delay is not seen at PN7 as with VGluT1 labeling. The fact that the delay in the PMCA label occurs two days before the major delay in the OPL VGluT1 label suggests that, in the normal maturation of the rod synapse, the mechanisms for calcium extrusion develop before those of the glutamate packaging. Therefore, the photoreceptor terminal primarily establishes a way to maintain calcium homeostasis before glutamate release is possible.
In summary, the novel GLE model enabled us to more precisely decipher and understand the coordinated and time-dependent patterns of RPC proliferation, differentiation, and maturation. Although the GLE retinas eventually caught up to the controls, the long-term retinal effects as well as the effects on central visual connectivity and processing are unknown. Whether similar changes occur in the different populations of brain cells in GLE mice is also unknown. Although the clinical and public health relevance of our findings in GLE mice has yet to be determined, neurodevelopmental delays and altered synaptic development in the brain development of children are associated with various neuropsychiatric and behavioral disorders as well as cognitive deficits [77-81].
Acknowledgments
We thank Dr. Laura J. Frishman for her support throughout this project, Dr. Weimin Xiao for his technical assistance, Dr. Roger Janz for the SV2B antibody, Dr. Anand Swaroop for help and support with the Affymetrix study, and Dr. David R. Copenhagen for his suggestions and comments on an earlier draft of this manuscript. This work was partially supported by NIH Grants RO1ES012482 to DAF, P30EY07551, T32EY07024 for SYC, an UH AGEP Grant to SYC, and an UHD ORC Grant 206400004B0303 to JEJ.
References
- 1.Young RW. Cell differentiation in the retina of the mouse. Anat Rec. 1985;212:199–205. doi: 10.1002/ar.1092120215. [DOI] [PubMed] [Google Scholar]
- 2.Rapaport DH, Wong LL, Wood ED, Yasumura D, LaVail MM. Timing and topography of cell genesis in the rat retina. J Comp Neurol. 2004;474:304–24. doi: 10.1002/cne.20134. [DOI] [PubMed] [Google Scholar]
- 3.Marquardt T, Gruss P. Generating neuronal diversity in the retina: one for nearly all. Trends Neurosci. 2002;25:32–8. doi: 10.1016/s0166-2236(00)02028-2. [DOI] [PubMed] [Google Scholar]
- 4.Turner DL, Cepko CL. A common progenitor for neurons and glia persists in rat retina late in development. Nature. 1987;328:131–6. doi: 10.1038/328131a0. [DOI] [PubMed] [Google Scholar]
- 5.Turner DL, Snyder EY, Cepko CL. Lineage-independent determination of cell type in the embryonic mouse retina. Neuron. 1990;4:833–45. doi: 10.1016/0896-6273(90)90136-4. [DOI] [PubMed] [Google Scholar]
- 6.Dyer MA, Cepko CL. Regulating proliferation during retinal development. Nat Rev Neurosci. 2007;2:333–42. doi: 10.1038/35072555. [DOI] [PubMed] [Google Scholar]
- 7.Sherry DM, Wang MM, Bates J, Frishman LJ. Expression of vesicular glutamate transporter 1 in mouse retina reveals temporal ordering in development of rod vs. cone and ON vs. OFF circuits. J Comp Neurol. 2003;465:480–98. doi: 10.1002/cne.10838. [DOI] [PubMed] [Google Scholar]
- 8.Tian N. Visual experience and maturation of retinal synaptic pathways. Vision Res. 2004;44:3307–16. doi: 10.1016/j.visres.2004.07.041. [DOI] [PubMed] [Google Scholar]
- 9.He Q, Wang P, Tian N. Light evoked synaptic activity of retinal ganglion and amacrine cells is regulated in developing mouse retina. Eur J Neurosci. 2011;33:36–48. doi: 10.1111/j.1460-9568.2010.07484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soto F, Ma X, Cecil JI, Vo BQ, Culican SM, Kerschensteiner D. Spontaneous activity promotes synapse formation in a cell-type-dependent manner in the developing retina. J Neurosci. 2012;32:5426–39. doi: 10.1523/JNEUROSCI.0194-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bilitou A, Ohnuma A. The role of cell cycle in retinal development: cyclin-dependent kinase inhibitors coordinate cell-cycle inhibition, cell fate determination and differentiation in the developing retina. Dev Dyn. 2010;239:727–36. doi: 10.1002/dvdy.22223. [DOI] [PubMed] [Google Scholar]
- 12.Bassett EA, Wallace VA. Cell fate determination in the vertebrate retina. Trends Neurosci. 2012;35:565–73. doi: 10.1016/j.tins.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Zagozewski JL. Zhang Qi, Pinto VI, Wigle JT, Eisenstat DD. The role of homeobox genes in retinal development and disease. Dev Biol. 2014;393:195–208. doi: 10.1016/j.ydbio.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 14.Brzezinski JA, Reh TA. Photoreceptor cell fate specification in vertebrates. Dev. 2015;142:3263–73. doi: 10.1242/dev.127043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levine EM, Fuhrmann S, Reh TA. Soluble factors and the development of rod photoreceptors. Cell Mol Life Sci. 2000;57:224–34. doi: 10.1007/PL00000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martins RA, Pearson RA. Control of cell proliferation by neurotransmitters in the developing vertebrate retina. Brain Res. 2008;1192:37–60. doi: 10.1016/j.brainres.2007.04.076. [DOI] [PubMed] [Google Scholar]
- 17.Lilienthal H, Kohler K, Turfeld M, Winneke G. Persistent increases in scotopic B-wave amplitudes after lead exposure in monkeys. Exp Eye Res. 1994;59:203–9. doi: 10.1006/exer.1994.1098. [DOI] [PubMed] [Google Scholar]
- 18.Rothenberg SJ, Schnaas L, Salgado-Valladares M, Casanueva E, Geller AM, Hudnell HK, Fox DA. Increased ERG a- and b-wave amplitudes in 7- to 10-year-old children resulting from prenatal lead exposure. Invest Ophthalmol Vis Sci. 2002;43:2036–44. [PubMed] [Google Scholar]
- 19.Fox DA, Kala SV, Hamilton WR, Johnson JE, O'Callaghan JP. Low-level human equivalent gestational lead exposure produces supernormal scotopic electroretinograms, increased retinal neurogenesis, and decreased retinal dopamine utilization in rats. Environ Health Perspect. 2008;116:618–25. doi: 10.1289/ehp.11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagpal AG, Brodie SE. Supranormal electroretinogram in a 10-year-old girl with lead toxicity. Doc Ophthalmol. 2009;118:163–6. doi: 10.1007/s10633-008-9144-7. [DOI] [PubMed] [Google Scholar]
- 21.Giddabasappa A, Hamilton WR, Chaney S, Xiao W, Johnson JE, Mukherjee S, Fox DA. Low-level gestational lead exposure increases retinal progenitor cell proliferation and rod photoreceptor and BC neurogenesis in mice. Environ Health Perspect. 2011;119:71–9. doi: 10.1289/ehp.1002524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fox DA, Campbell ML, Blocker YS. Functional alterations and apoptotic cell death in the retina following developmental or adult lead exposure. Neurotoxicology. 1997;18:645–64. [PubMed] [Google Scholar]
- 23.He L, Perkins GA, Poblenz AT, Harris JB, Hung M, Ellisman MH, Fox DA. Bcl-xL overexpression blocks bax-mediated mitochondrial contact site formation and apoptosis in rod photoreceptors of lead-exposed mice. Proc Natl Acad Sci USA. 2003;100:1022–7. doi: 10.1073/pnas.0333594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perkins GA, Scott R, Perez A, Ellisman MH, Johnson JE, Fox DA. Bcl-xL-mediated remodeling of rod and cone synaptic mitochondria after postnatal lead exposure: electron microscopy, tomography and oxygen consumption. Mol Vis. 2012;18:3029–48. [PMC free article] [PubMed] [Google Scholar]
- 25.Gilbert ME, Kelly ME, Samsam TE, Goodman JH. Chronic developmental lead exposure reduces neurogenesis in adult rat hippocampus but does not impair spatial learning. Toxicol Sci. 2005;86:365–74. doi: 10.1093/toxsci/kfi156. [DOI] [PubMed] [Google Scholar]
- 26.Verina T, Rohde CA, Guilarte TR. Environmental lead exposure during early life alters granule cell neurogenesis and morphology in the hippocampus of young adult rats. Neuroscience. 2007;145:1037–47. doi: 10.1016/j.neuroscience.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neal AP, Stansfield KH, Worley PF, Thompson RE, Guilarte TR. Lead exposure during synaptogenesis alters vesicular proteins and impairs vesicular release: potential role of NMDA receptor-dependent BDNF signaling. Toxicol Sci. 2010;116:249–63. doi: 10.1093/toxsci/kfq111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leasure JL, Giddabasappa A, Chaney S, Johnson JE, Jr, Pothakos K, Lau YS, Fox DA. Low-level human equivalent gestational lead exposure produces sex-specific motor and coordination abnormalities and late-onset obesity in year-old mice. Environ Health Perspect. 2008;115:355–61. doi: 10.1289/ehp.10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fox DA, Hamilton WR, Johnson JE, Xiao W, Chaney S, Mukherjee S, Miller DB, O’Callaghan JP. Gestational lead exposure selectively decreases retinal dopamine amacrine cells and dopamine content in adult mice. Toxicol Appl Pharmacol. 2011;256:258–67. doi: 10.1016/j.taap.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aleman TS, Cideciyan AV, Aguirre GK, Huang WC, Mullins CL, Roman AJ, Sumaroka A, Olivares MB, Tsai FF, Schwartz SB, Vandenberghe LH, Limberis MP, Stone EM, Bell P, Wilson JM, Jacobson SG. Human CRB1-associated retinal degeneration: comparison with the rd8 Crb1-mutant mouse model. Invest Ophthalmol Vis Sci. 2011;52:6898–910. doi: 10.1167/iovs.11-7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattapallil MJ, Wawrousek EF, Chan CC, Zhao H, Roychoudhury J, Ferguson TA, Caspi RR. The rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012;53:2921–7. doi: 10.1167/iovs.12-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson JE, Jr, Perkins GA, Giddabasappa A, Chaney S, Xiao W, White AD, Brown JM, Waggoner J, Ellisman MH, Fox DA. Spatiotemporal regulation of ATP and Ca2+ dynamics in vertebrate rod and cone ribbon synapses. Mol Vis. 2007;13:887–919. [PMC free article] [PubMed] [Google Scholar]
- 33.Graham DR, Overbeek PA, Ash JD. Leukemia inhibitory factor blocks expression of Crx and Nrl transcription factors to inhibit photoreceptor differentiation. Invest Ophthalmol Vis Sci. 2005;46:2601–10. doi: 10.1167/iovs.05-0129. [DOI] [PubMed] [Google Scholar]
- 34.Yuan JS, Wang D, Stewart CN. Statistical methods for efficiency adjusted real‐time PCR quantification. Biotechnol J. 2008;3:112–23. doi: 10.1002/biot.200700169. [DOI] [PubMed] [Google Scholar]
- 35.Palczewski K. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006;75:743–67. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hargrave PA, McDowell JH. Rhodopsin and phototransduction: a model system for G protein-linked receptors. FASEB J. 1992;6:2323–31. doi: 10.1096/fasebj.6.6.1544542. [DOI] [PubMed] [Google Scholar]
- 37.Baylor DA, Burns ME. Control of rhodopsin activity in vision. Eye (Lond) 1998;12:521–5. doi: 10.1038/eye.1998.140. [DOI] [PubMed] [Google Scholar]
- 38.Burgoyne RD, Weiss JL. The neuronal calcium sensor family of Ca2+-binding proteins. Biochem J. 2001;353:1–12. [PMC free article] [PubMed] [Google Scholar]
- 39.Treisman JE, Morabito MA. Barnstable. Opsin expression in rat retina is developmentally regulated by transcriptional activation. Mol Cell Biol. 1988;•••:1570–9. doi: 10.1128/mcb.8.4.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morrow EM, Belliveau MJ, Cepko CL. Two phases of rod photoreceptor differentiation during rat retinal development. J Neurosci. 1998;18:3738–48. doi: 10.1523/JNEUROSCI.18-10-03738.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haverkamp S, Wässle H. Immunocytochemical analysis of the mouse retina. J Comp Neurol. 2000;424:1–23. [PubMed] [Google Scholar]
- 42.Sharma RK, O'Leary TE, Fields CM, Johnson DA. Development of the outer retina in the mouse. Brain Res Dev Brain Res. 2003;145:93–105. doi: 10.1016/s0165-3806(03)00217-7. [DOI] [PubMed] [Google Scholar]
- 43.Röhlich P, van Veen T, Szél A. Two different visual pigments in one retinal cone cell. Neuron. 1994;13:1159–66. doi: 10.1016/0896-6273(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 44.Young RW. Cell differentiation in the retina of the mouse. Anat Rec. 1985;212:199–205. doi: 10.1002/ar.1092120215. [DOI] [PubMed] [Google Scholar]
- 45.Rath MF, Morin F, Shi Q, Klein DC, Møller M. Ontogenetic expression of the Otx2 and Crx homeobox genes in the retina of the rat. Exp Eye Res. 2007;85:65–73. doi: 10.1016/j.exer.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 46.Liu I, Chen J, Ploder L, Vidgen D, Kooy D, Kalnins VI, McInnes RR. Developmental expression of a novel murine homeobox gene (Chx10): evidence for roles in determination of the neuroretina and inner nuclear layer. Neuron. 1994;13:377–93. doi: 10.1016/0896-6273(94)90354-9. [DOI] [PubMed] [Google Scholar]
- 47.Burmeister M, Novak J, Liang M, Basu S, Ploder L, Hawes NL, Vidgen D, Hoover F, Goldman D, Kalnins VI, Roderick TH, Taylor BA, Hankin MH. cInnes RR. Ocular retardation mouse caused by Chx10 homeobox null allele: impaired retinal progenitor proliferation and bipolar cell differentiation. Nat Genet. 1996;12:376–84. doi: 10.1038/ng0496-376. [DOI] [PubMed] [Google Scholar]
- 48.Cepko CL, Austin CP, Yang X, Alexiades M, Ezzeddine D. Cell fate determination in the vertebrate retina. Proc Natl Acad Sci USA. 1996;93:589–95. doi: 10.1073/pnas.93.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Puthussery T, Gayet-Primo J, Taylor WR. Localization of the calcium-binding protein secretagogin in cone BCs of the mammalian retina. J Comp Neurol. 2010;518:513–25. doi: 10.1002/cne.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia M, Strehler E. Plasma membrane calcium ATPases as critical regulators o calcium homeostasis during neuronal cell function. Front Biosci. 1999;4:d869–82. doi: 10.2741/garcia. [DOI] [PubMed] [Google Scholar]
- 51.Wan Q, Zhou Z, Thakur P, Vila A, Sherry D, Janz R, Heidelberger R. SV2 acts via presynaptic calcium to regulate neurotransmitter release. Neuron. 2010;66:884–95. doi: 10.1016/j.neuron.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.von Kriegstein K, Schmitz F. The expression pattern and assembly profile of synaptic membrane proteins in ribbon synapses of the developing mouse retina. Cell Tissue Res. 2003;311:159–73. doi: 10.1007/s00441-002-0674-0. [DOI] [PubMed] [Google Scholar]
- 53.Wang MM, Janz R, Belizaire R, Frishman LJ, Sherry DM. Differential distribution and developmental expression of synaptic vesicle protein 2 isoforms in the mouse retina. J Comp Neurol. 2003;460:106–22. doi: 10.1002/cne.10636. [DOI] [PubMed] [Google Scholar]
- 54.Morgans CW, Kensel-Hammes P, Hurley JB, Burton K, Idzerda R, McKnight GS, Bejjalieh SM. Loss of the synaptic vesicle protein SV2B results in reduced neurotransmission and altered synaptic vesicle protein expression in the retina. PLoS ONE. 2009;4:e5230. doi: 10.1371/journal.pone.0005230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krizaj D, Demarco S, Johnson J, Strehler E, Copenhagen DR. Cell-specific expression of plasma membrane calcium ATPase isoforms in retinal neurons. J Comp Neurol. 2002;451:1–21. doi: 10.1002/cne.10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilliam JC, Wensel TG. TRP channel gene expression in mouse retina. Vision Res. 2011;51:2440–52. doi: 10.1016/j.visres.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Söderpalm AK, Fox DA, Karlsson JO, van Veen T. Retinoic acid produces rod photoreceptor selective apoptosis in developing mammalian retina. Invest Ophthalmol Vis Sci. 2000;41:937–47. [PubMed] [Google Scholar]
- 58.Go HS, Kim KC, Choi CS, Jeon SJ, Kwon KJ, Han SH, Lee J, Cheong JH, Ryu JH, Kim CH, Ko KH, Shin CY. Prenatal exposure to valproic acid increases the neural progenitor cell pool and induces macrocephaly in rat brain via a mechanism involving the GSK-3β/β-catenin pathway. Neuropharmacology. 2012;63:1028–41. doi: 10.1016/j.neuropharm.2012.07.028. [DOI] [PubMed] [Google Scholar]
- 59.Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108:511–33. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kashyap B, Frederickson LC, Stenkamp DL. Mechanisms for persistent microphthalmia following ethanol exposure during retinal neurogenesis in zebrafish embryos. Vis Neurosci. 2007;24:409–21. doi: 10.1017/S0952523807070423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Naveau E, Pinson A, Gérard A, Nguyen L, Charlier C, Thomé JP, Zoeller RT, Bourguignon JP, Parent AS. Alteration of rat fetal cerebral cortex development after prenatal exposure to polychlorinated biphenyls. PLoS ONE. 2014;9:e91903. doi: 10.1371/journal.pone.0091903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Watanabe T, Raff MC. Rod photoreceptor development in vitro: intrinsic properties of proliferating neuroepithelial cells change as development proceeds in the rat retina. Neuron. 1990;2:461–7. doi: 10.1016/0896-6273(90)90058-n. [DOI] [PubMed] [Google Scholar]
- 63.Kirsch M, Schulz-Key S, Wiese A, Fuhrmann S, Hofmann H. Ciliary neurotrophic factor blocks rod photoreceptor differentiation from postmitotic precursor cells in vitro. Cell Tissue Res. 1998;291:207–16. doi: 10.1007/s004410050991. [DOI] [PubMed] [Google Scholar]
- 64.Elliot J, Cayouette M, Gravel C. The CNTF/LIF signaling pathway regulates developmental programmed cell death and differentiation of rod precursor cells in mouse retina in vivo. Dev Biol. 2006;300:583–98. doi: 10.1016/j.ydbio.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 65.Ezzeddine ZD, Yang X, DeChiara T, Yancopoulos G, Cepko CL. Postmitotic cells fated to become rod photoreceptors can be respecified by CNTF treatment of the retina. Development. 1997;124:1055–67. doi: 10.1242/dev.124.5.1055. [DOI] [PubMed] [Google Scholar]
- 66.Neophytou C, Vernallis AB, Smith A, Raff MC. Müller-cell-derived leukaemia inhibitory factor arrests rod photoreceptor differentiation at a postmitotic pre-rod stage of development. Development. 1997;124:2345–54. doi: 10.1242/dev.124.12.2345. [DOI] [PubMed] [Google Scholar]
- 67.Kelley MW, Turner JK, Reh TA. Retinoic acid promotes differentiation of photoreceptors in vitro. Development. 1994;120:2091–102. doi: 10.1242/dev.120.8.2091. [DOI] [PubMed] [Google Scholar]
- 68.Wu M, Chiao C. Light deprivation delays morphological differentiation of bipolar cells in the rabbit retina. Brain Res. 2007;1170:13–9. doi: 10.1016/j.brainres.2007.06.091. [DOI] [PubMed] [Google Scholar]
- 69.Acampora D, Mazan S, Lallemand Y, Avantaggiato V, Maury M, Simeone A. Brûlet. Forebrain and midbrain regions are deleted in Otx2−/− mutants due to a defective anterior neuroectoderm specification during gastrulation. Development. 1995;121:3279–90. doi: 10.1242/dev.121.10.3279. [DOI] [PubMed] [Google Scholar]
- 70.Wang S, Sengel C, Emerson MM, Cepko CL. A gene regulatory network controls the binary fate decision of rod and bipolar cells in the vertebrate retina. Dev Cell. 2014;30:513–27. doi: 10.1016/j.devcel.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levine EM, Green ES. Cell-intrinsic regulators of proliferation in vertebrate retinal progenitors. Semin Cell Dev Biol. 2004;15:63–74. doi: 10.1016/j.semcdb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 72.Hatakeyama J, Kageyama R. Retinal cell fate determination and bHLH factors. Semin Cell Dev Biol. 2004;15:83–9. doi: 10.1016/j.semcdb.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 73.Heidelberger R, Thoreson WB, Witkovsky P. Synaptic transmission at retinal ribbon synapses. Prog Retin Eye Res. 2005;24:682–720. doi: 10.1016/j.preteyeres.2005.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morgans CW, Far OE, Berntson A, Wässle H, Taylor WR. Calcium extrusion from mammalian photoreceptor terminals. J Neurosci. 1998;18:2467–74. doi: 10.1523/JNEUROSCI.18-07-02467.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mercer AJ, Thoreson WB. The dynamic architecture of photoreceptor ribbon synapses: cytoskeletal, extracellular matrix, and intramembrane proteins. Vis Neurosci. 2011;28:453–71. doi: 10.1017/S0952523811000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rentería RC, Strehler EE, Copenhagen DR, Krizaj D. Ontogeny of plasma membrane Ca2+ ATPase isoforms in the neural retina of the postnatal rat. Vis Neurosci. 2005;22:263–74. doi: 10.1017/S0952523805223027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Majovski LV. Selected neurodevelopmental delay syndromes and their impact on adult neuromaturation and adjustment. Semin Clin Neuropsychiatry. 2000;5:171–6. doi: 10.1053/scnp.2000.6728. [DOI] [PubMed] [Google Scholar]
- 78.Waltereit R, Banaschewski T, Meyer-Lindenberg A, Poustka L. Interaction of neurodevelopmental pathways and synaptic plasticity in mental retardation, autism spectrum disorder and schizophrenia: implications for psychiatry. World J Biol Psychiatry. 2014;15:507–16. doi: 10.3109/15622975.2013.838641. [DOI] [PubMed] [Google Scholar]
- 79.Feizo MS, Magtira A, Schoenberg FP, Macgibbon K, Mullin PM. Neurodevelopmental delay in children exposed in utero to hyperemesis gravidarum. Eur J Obstet Gynecol Reprod Biol. 2015;189:79–64. doi: 10.1016/j.ejogrb.2015.03.028. [DOI] [PubMed] [Google Scholar]
- 80.Symons FJ, Tervo RC, Barney CC, Damerow J, Selim M, McAdams B, Foster S, Crabb GW, Kennedy W. Peripheral innervation in children with global developmental delay: biomarker for risk for self-injurious behavior? J Child Neurol. 2015;30:1722–7. doi: 10.1177/0883073815579704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Betts KS, Williams GM, Najman JM, Alati R. Predicting spectrums of adult mania, psychosis and depression by prospectively ascertained childhood neurodevelopment. J Psychiatr Res. 2016;72:22–9. doi: 10.1016/j.jpsychires.2015.10.013. [DOI] [PubMed] [Google Scholar]