

INTRODUCTION
Cytokines are soluble, small proteins that are produced by cells and act in a largely paracrine manner to influence the activity of other cells. Currently, the term “cytokine” describes proteins such as the tumour necrosis factor family, the interleukins, and the chemokines. Virtually every nucleated cell can produce and respond to cytokines placing these molecules at the centre of most of the body’s homeostatic mechanisms (1). Much of our knowledge of the function of cytokines has been derived from studies wherein homeostasis has been disrupted by infection and the absence of specific cytokines results in a failure to control the disease process. In this context, infection with Mycobacterium tuberculosis (Mtb) has proven to be very informative and has highlighted the role of cytokines in controlling infection without promoting uncontrolled and damaging inflammatory responses (2–4). Herein we focus on the key cytokine and chemokines that have been studied in the context of human TB using experimental medicine as well as Mtb infection of various animal models, including non-human primates, mice and rabbits. Perhaps the most important message of this chapter is that in a complex disease such as tuberculosis (TB) the role of any one cytokine cannot be designated either ‘good’ or ‘bad’ but rather that cytokines can elicit both protective and pathologic consequences depending upon context.
Why is TB such an informative probe allowing for detailed investigation of the function of cytokines and chemokines in immunity? One recent development in our understanding of TB stems from theories of co-evolution between modern humans and Mtb (5). Evolutionary patterns based on genetic analyses suggest that Mtb and humans coexisted for tens of thousands of years in Africa but that when humans left Africa and developed a more urban lifestyle TB developed into a substantial health problem (6). During co-evolution between humans and Mtb, Mtb likely evolved tools and stratagems with which to manipulate the human immune response to ensure effective transmission (7); this manipulation has been so successful that it is thought that over one third of the world’s population harbours some form of Mtb infection (8).
Two facts illustrate the focus of Mtb on manipulating the human immune response. Firstly, Mtb is the major active constituent of Complete Freund’s adjuvant, which has been used for decades to stimulate long-lived cellular immune responses in vertebrate animals. Secondly, we have exploited the strong and sensitive T cell-based inflammatory response to Mtb antigens as a skin test to indicate infection with Mtb. Thus teleologically speaking we may suggest that Mtb does not fail to induce immunity it simply manipulates it such that its need to be transmitted is met. This manipulation occurs from the start of the human Mtb interaction when immune surveillance cells of the lung recognize danger through binding of their pattern recognition receptors to exquisitely refined Mtb pathogen associated molecular molecules. It is this initial interaction that results in production of chemokines and cytokines which then recruit and activate inflammatory cells (9). Following this initial interaction, bacteria migrate to the draining lymph node where they initiate (quite effectively) antigen-specific T cells that differentiate into cytokine-producing cells capable of expressing a variety of chemokine receptors that allow them to traffic away from the lymph node and into sites of tissue inflammation (7, 9). These antigen-specific T cells must then migrate via chemokine gradients, co-locate with Mtb-infected phagocytic cells and release cytokines which activate the infected cells to kill the Mtb (7, 9). If this induction of immunity is not met by Mtb, then the host dies rapidly with no effective transmission if the bacterium to further hosts.
The need for communication between cells both for efficient migration and for specific instruction during expression of immunity is where the critical role of cytokines and chemokines in controlling TB lies. Indeed, for the majority of those infected with Mtb, the efficient expression of immunity via competent cytokine and chemokine expression results in no sign of disease other than an ability to exhibit an inflammatory response to Mtb antigen (i.e. the skin test response). However for Mtb to be efficiently transmitted, a degraded inflammatory lesion capable of delivering live bacteria to the airways must develop, and it is this evolutionary need that likely drives the development of the disease process in the lung. Mtb expresses molecules which promote inflammatory responses which then need to be regulated to avoid tissue damage. If the bacterial burden is large or if the bacteria proliferate rapidly, then the co-ordination between cells mediated by cytokines and chemokines cannot occur quickly enough and immunity cannot be expressed despite the presence of all of the required components. Understanding the functions and interactions between cytokines and chemokines is therefore critical to our attempts to limit TB. Herein we discuss the roles of specific cytokines (Table I) and chemokines (Table II) in the context of Mtb infection and how they function to stop the development of TB and also how they might contribute to the progression of disease.
Table I.
The positive and negative roles of cytokines in TB
| Cytokine | Receptor/Signal | Role in TB |
|---|---|---|
| TNFα | TNFR1, TNFR2 JNK, p38, NFκB |
Positive: Essential for survival following Mtb infection. Initiation of innate cytokine and chemokine response and phagocyte activation Negative: Mediator of tissue damage |
| IFNγ | IFNGR1, IFNGR2 JAK/STAT |
Positive: Essential for survival following Mtb infection. Coordinates and maintains mononuclear inflammation. Expressed by antigen specific T cells Negative: Potentially pathogenic |
| IFNα/IFNβ | IFNAR1, IFNAR2 JAK,TYK, ISG,ISRE |
Positive: Required for initial recruitment of phagocytes to the lung Negative: Over expression of IFNα/IFNβ results in recruitment of permissive phagocytes and regulation of T cell accumulation and function |
| IL-6 | IL-6R, gp130 JAK, STAT3, MAPK |
Positive: Potentiates early immunity – non essential unless a high dose infection. |
| IL-1α/IL-1β | IL-1R1, IL1RAcP MyD88,IRAK4,NFκB |
Positive: Essential for survival following Mtb infection. Induction of IL-17. Promotes PGE2 to limit Type I IFN |
| IL-18 | IL-18Rα, IL-18Rβ MyD88,IRAK, NFκB |
Positive: May augment IFNγ – non-essential. Regulator of neutrophil/monocyte accumulation. of neutrophil and monocyte accumulation, optimal induction of IFNγ by T-cells |
| IL-12 p40,p35 |
12Rβ1, IL-12Rβ2 JAK2, TYK2, STAT4 |
Positive: IL-12p40 and IL-12p35 essential for survival following Mtb infection. Mediate early T cell activation, polarization and survival. Negative: Over expression of IL-12p70 is toxic during Mtb infection. |
| IL-23 p40,p19 |
IL-23R, IL-12Rβ1 JAK2, TYK2, STAT3 |
Positive: Required for IL-17 and IL-22 expression during Mtb infection. Non-essential in low dose challenge required for long term control. Negative: Mediates increased pathology during chronic challenge |
| IL-27 EBI3,p28 |
IL-27Rα, gp130 JAK1/2,TYK2,STAT1/3 |
Positive: May control inflammation and reduce pathology Negative: Regulates protective immunity to Mtb infection by limiting the migration and survival of T cells at the inflamed site. |
| IL-35 p35,EBI3 |
IL-12Rβ2,gp130 STAT1/4 |
Positive? Regulate the availability of subunits of IL-12, IL-27 Negative? Potential immunoregulatory role. |
| IL-17A/F | IL-17RC, IL-17RA |
Positive: Essential for survival following infection with some strains of Mtb. Induction and maintenance of chemokine gradients for T cell migration. Negative: Drives pathology via S100A8/A9 and neutrophils |
| IL-22 | IL-22R1, IL-10R2 TYK2,JAK1,STAT3 |
Positive: Induces antimicrobial peptides and promotes epithelial repair, inhibits intracellular growth of Mtb in macrophages. |
Table II.
The positive and negative roles of chemokines in TB
| Chemokine | Receptor | Role in TB |
|---|---|---|
| CCL-3,-4,-5 | CCR1 | Positive: Upregulated during infection. Non-essential in mouse model |
| CCL-2,-7,-12 | CCR2 |
Positive: Maximizes and organizes early macrophage and T cell accumulation in the lung Negative: Mediates recruitment of permissive phagocyte accumulation into the lung |
| CCL-17,-22 | CCR4 |
Positive: Mediates optimal granuloma formation to mycobacterial antigen Negative? May limit T cell proliferation via TREG |
| CCL-3,-4,-5 | CCR5 | Positive: Regulation of pulmonary infiltrates – non-essential. May mediate early dendritic cell accumulation in the lymph node. May augment macrophage Mtb killing via CCL5? |
| CCL-20 | CCR6 |
Positive: Expression of CCR6 on T cells specific for Mtb antigens Negative? CCL-20 seen at high levels in active TB |
| CCL-19,-21, | CCR7 | Positive: Mediates efficient migration of dendritic cells and Mtb-specific T cell activation. |
| CXCL-1,-2,-3,-5,-6,-7,-8 | CXCR1 CXCR2 |
Positive: Expressed on neutrophils mediates accumulation Negative: Absence of CXCR2 or CXCL5 results in improved bacterial control and reduced neutrophil accumulation |
| CXCL-9, -10,-11 | CXCR3 | Positive: Required for optimal granuloma formation. Expressed on Mtb-specific T cells. Use of CXCL9-11 levels to indicate disease level? Required for early recruitment of T cells to lung |
| CXCL-13 | CXCR5 | Positive: Required for correct location of T cells within granulomas. Required for B cell follicle formation in Mtb infected lungs. Required for optimal protection. |
CYTOKINES
Tumour Necrosis Factor alpha (TNFα)
TNFα is a cytokine that is released following activation of the immune system. Although it is primarily produced by macrophages, TNFα can also be secreted by lymphocytes, mast cells, endothelial cells, and fibroblasts (10). Because most cells exhibit responsiveness to TNFα, it is considered a major proinflammatory mediator. It is produced as a type II transmembrane homo-trimeric protein (mTNF) which can become released into the extracellular milieu through the proteolytic action of TNFα converting enzyme (TACE) (11). Soluble TNF (sTNF) exists as a 51 kDA trimeric protein that is unstable upon reaching nanomolar concentrations (12) but which upon binding to cognate TNF receptors (TNF-R) induces activation of proinflammatory responses mediated by NFκB, JNK, and p38, as well as promotion of apoptosis (10, 13–16). There are two TNF receptors, TNF-R1 and TNF-R2. Both TNF-R1, also known as CD120a and p55/60, and TNFR-2, also known as CD120b and p75/80, can bind either the membrane or the soluble form of TNFα (17–19). An important regulator of activation is localization of receptor expression, as TNF-R1 is ubiquitously expressed, whereas TNF-R2 expression is restricted to subsets of neuronal cells, T cells, endothelial cells, microglia, oligodendrocytes, cardiac myocytes, thymocytes, and human mesenchymal stem cells (20). Signalling through TNF-R2 can only be activated through mTNF, and not sTNF. This complex interplay between positive and negative regulators of TNF activity reflects the potential disruptive power of TNFα and this regulation of immunity provides tempting targets for manipulation by Mtb.
Originally described for its ability to promote necrosis of tumours (21), TNFα has since been implicated in proliferation and differentiation of immune cells as well as multiple inflammatory processes including migration (20) apoptosis of Mtb-infected cells in vitro (22, 23). Upon initial Mtb infection, the interaction between immune surveillance cells such as phagocytes in the lung and the invading Mtb, results in the production of multiple proinflammatory cytokines, including TNFα (4) (Figure 1). Although both virulent and avirulent Mtb are able to induce comparable levels of TNFα by human alveolar macrophages, TNFα produced in response to infection with virulent Mtb strains has less bioactivity (24). This is an example of the ability of the bacterium to manipulate the host response as the decreased TNFα bioactivity has been attributed to the induction of IL-10 by the virulent strain which then results in release of soluble TNFR2 which binds the induced TNFα, thereby inhibiting its function (24). As infection progresses TNFα plays a role in co-ordinating the chemokine response within the lung and in facilitating the development of the granuloma, it is also produced by both CD4 and CD8 T cells and plays an important role in optimal macrophage activation (25).
Figure 1. The role of chemokines and cytokines in the innate response to Mtb infection.
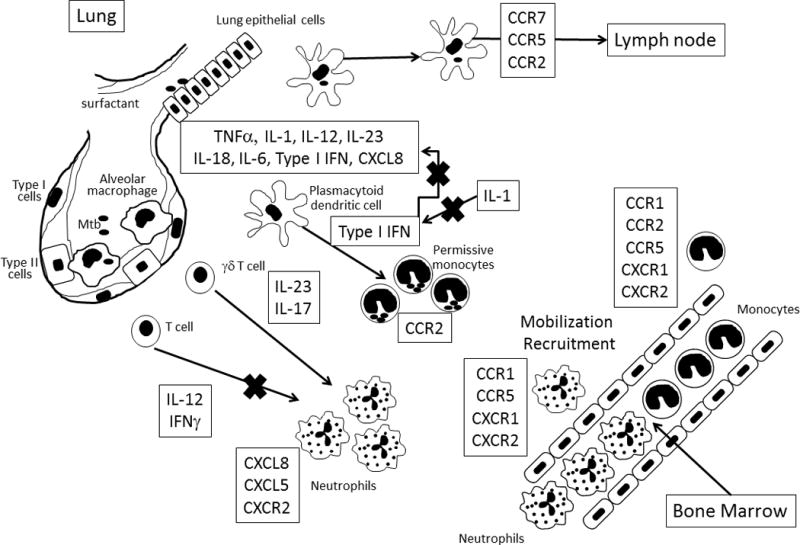
Upon early infection of the lower airways, Mtb encounters alveolar macrophages and lung epithelial cells. Alveolar macrophages are a major source of proinflammatory cytokines (TNFα), although stromal cells can produce cytokines and chemokines which will also modulate immune responses. During early infection, dendritic cell trafficking from the lungs to the lymph node via CCR7 results in primed naïve T cells and initiation of adaptive immune responses. Replicating bacteria generate a fulminant reaction which results in the mobilization and recruitment of both neutrophils and monocytes from the bone marrow via the induction of proinflammatory cytokines and chemokines. Regulation of cellular recruitment occurs via coordinated cytokine and chemokine induction. While initial recruitment of monocytes requires type I IFN, over-expression of this cytokine results in high levels of CCR2 expressing monocytes with limited ability to control bacterial growth. Type II IFN (IFNγ) regulates the recruitment of neutrophils which is promoted by IL-17. CXCL5 and CXCR2 mediate the recruitment of damaging neutrophils.
In Mtb infection models, the importance of TNFα is exemplified by mice deficient in the TNF receptor or following TNF neutralization (26). In these models, TNFα deficiency results in increased susceptibility with mice succumbing to infection within 2–3 weeks, while harbouring a high bacterial burden (26). Critically, although inflammatory cells accumulate at the site of Mtb infection in TNFα-deficient mice, they do not coalesce to form granulomas (26–29). Granulomas are considered to be a hallmark of TB and are composed of macrophages, multinucleated giants cells, CD4+ and CD8+ T cells, B cells, and neutrophils (30). In one of the earliest studies of the role of TNFα in mycobacterial disease it was shown that TNFα neutralization following BCG infection lead to the loss of granulomas (31). Neutralization of TNFα also leads to decreased expression of key chemokines such as CCL5, CXCL9 and CXCL10. CXCL9 and CXCL10 both bind to the receptor CXCR3 (32, 33), expressed on activated T cells, while CCR1 and CCR5, expressed on both innate (i.e. macrophages and neutrophils) and adaptive cells (i.e. T and B cells), bind to CCL5 (34). Thus, TNFα sits at the cross roads where innate immunity and acquired immunity as well as cytokines and chemokines interact. In the absence of TNFα, T cells expressing CXCR3 fail to encounter the ligands CXCL9 and CXCL10 required to recruit these cells into the granuloma. Thus the required communication between infected phagocytes and the instructive T cells does not occur, resulting in loss of immunity.
The importance of the granuloma in restricting the movement of Mtb to more immunoprivileged sites has long been appreciated (35). Indeed, inhibition of TNFα promotes dissemination of Mtb to sites such as the CNS (36, 37) wherein adverse events are profound and often irreversible. It is thought that Mtb migrates to the CNS secondarily from a primary site elsewhere in the host (38, 39), although how it crosses the blood-brain barrier is not clear. Although TNFα is produced in the CNS and is thought to exacerbate progression of TB related damage in the CNS in a rabbit model (40) use of neuron-specific TNFα-deficient mice, has shown that neuron-derived TNFα production is dispensable for protection against CNS-TB (41). These data support the notion that TNFα is critical for the initiation and co-ordination of cellular responses but that it has the potential to be pathogenic when expressed in the absence of immunity.
TNFα is required throughout the life of the infected host as reactivation of pulmonary TB occurs in latently-infected mice upon neutralization of TNFα (42). Upon neutralization, less defined granuloma formation is seen along with increased bacterial burden in the lung, and extra pulmonary sites such as the liver and spleen (42) (Figure 2). Enhanced histopathology is also observed in TNFα-neutralized mice supporting the importance of a regulated cellular interaction during Mtb infection. The protective role of TNFα is further highlighted in those patients with autoimmune and chronic diseases who are undergoing anti-TNFα neutralizing therapies including infliximab, adalimumab, and etanercept (43, 44). Though this therapy is successful at treating the autoimmune disease, a significant correlation between reactivation of latent TB in patients undergoing anti-TNFα therapy has been reported (45–52). Patients using infliximab and/or adalimumab have a higher incidence of TB reactivation in extra pulmonary sites compared to etanercept (50). While a mechanism for this has not been determined, mathematical modelling and bioinformatic analysis suggests that reactivation is related to drug tissue penetration, drug half-life, and relative specificity for membrane-bound TNFα or soluble TNFα (53). An intriguing mechanism for the effect of infliximab on immunity to Mtb infection has been suggested by the observation that this antibody-based drug binds to mTNF expressed on effector memory CD8 T cells thereby facilitating complement-mediated lysis and likely loss of Mtb-specific CD8 T cells (54). In a cynomolgus macaque model of TB, latently infected primates were given either soluble TNFα or adalimumab and exhibited increased reactivation and harboured higher bacterial burdens compared to their latently infected untreated counterparts (55). As would be expected due to the higher bacterial burden and the role of TNFα in limiting bacterial spread, there were more granulomas in the lungs of the treated monkey (26, 55). In the zebrafish model of M. marinum infection, TNFα is required for control of mycobacterial growth and to regulate macrophage necrosis (56).
Figure 2. The role of chemokines and cytokines in the adaptive response to Mtb infection.

Following Mtb infection of the lung, migratory cells take the bacteria to the draining lymph node likely using both cytokine (IL-12p40) and chemokine (CCR2, CCR7) pathways. Antigen is then transferred to antigen presenting cells which stimulate naïve T cells via MHC class I and class II. Antigen presenting cells make cytokines and chemokines to potentiate T cell proliferation and polarization. Activated T cells migrate from the draining lymph node through the vasculature to the inflamed site. Some T cells remain in the vasculature (CX3CR3+) while others migrate into the parenchyma (CXCR3+CCR6+). Expression of CXCR5 on antigen-specific T cells allows them to respond to IL-23- and IL-17-dependent CXCL13 and locate effectively within the granuloma, where they activate Mtb infected macrophages. T cells express a variety of cytokines in the lung including IFNγ, TNFα, IL-17, and IL-10 which have both protective and negative effects depending upon the context.
The ability to rapidly diagnose active disease from latent infection would be highly beneficial in identification and prompt treatment of TB. TNFα has recently been proposed as a biomarker to distinguish between active pulmonary disease and latently infected individuals who do not exhibit disease symptoms (57). Using polychromatic flow cytometric analysis, patients with pulmonary TB disease have a higher proportion of single positive TNFα-producing Mtb-specific CD4+ T cells compared to individuals with latent infection (57). This was further confirmed in a blinded study whereby this parameter was the sole diagnostic for pulmonary TB disease (57). TNFα lies at the crux of the TB conundrum. It is critical for control of infection with both phagocyte activating and granuloma organizing functions but too much TNFα can mediate tissue damage and promote transmission (Table I).
The Interferons
The interferon family demonstrates the potential for similar cytokines to play protective and pathological roles in TB disease. Based on receptor specificity and sequence homology, the interferons (IFNs) are classified into two types (58). IFNγ is the only type II interferon and while structurally related to the type I interferons IFNα and IFNβ, these cytokine use different receptors and have distinct chromosomal locations (58). Unlike type I IFNs that bind to a common heterodimeric receptor comprised of IFNAR1 and IFNAR2 chains, IFNγ binds to the IFNγ receptor (IFNGR) which is comprised of two ligand binding IFNGR1 chains that associate with two signal transducing IFNGR2 chains (58). In addition while IFNγ is essential for survival following Mtb infection the type I IFNs appear to be largely detrimental to the host during TB and may be co-opted by the bacterium for its own ends (Table I).
Type II Interferon (IFNγ)
IFNγ-IFNGR binding induces signalling within the cell primarily through the Janus kinase/signal transducers and activators of transcription (JAK-STAT) pathways and results in changes in both the migratory and functional capacity of multiple cell types such as macrophages, NK cells, T cells (59–61). Innate production of IFNγ by phagocytes stimulated through their pattern recognition receptors results in early pro-inflammatory responses to infection (58, 62) and unlike TNFα, which is regulated tightly by highly related molecules, production of IFNγ is regulated by cytokines such as IL-12 and IL-18, which are also secreted by immune surveillance cells upon ligation of their pattern recognition receptors (58, 63, 64).
Genetic deficiency in the IFNγ pathway in humans is associated with increased risk for mycobacterial disease and Mendelian susceptibility to mycobacterial disease (MSMD) (65, 66). Autosomal complete recessive IFNγR1-deficient patients exhibit a predisposition for mycobacterial infections manifesting early in life and with poor prognosis (67). IFNγR2 deficiency (either total protein loss or loss of function) has also been observed and results in a similar outcome to IFNγR1-deficiency (68, 69). Similarly, mice that do not express IFNγ due to targeted gene disruption are severely susceptible to both low dose aerosol (70) and intravenous infection (71, 72) and exhibit poor macrophage activation and exacerbated granulocytic inflammation (70, 71).
The classic function of IFNγ is as a phagocyte activating cytokine which instructs macrophages and other cells to change function. In particular, in the absence of IFNγ Mtb occupies an intracellular environment wherein there is little reactive radical production, the phagosome does not fuse with lysosomes and remains at neutral pH. There is also an ample supply of iron due to the location of the phagosome in the early endosomal pathway (73). While innate sources of IFNγ can activate macrophages there is very little control of Mtb growth in the absence of α/β T cells or MHC class II following aerosol infection (74) suggesting that both IFNγ and antigen-specific T cells are required for control of this infection. However, whether it is T cells producing IFNγ that is critical has not been definitively demonstrated but the strongest support of a critical need of CD4+ T cells to make IFNγ is from a transfer model in mice (75). In contrast memory T cells can mediate protection in the absence of either IFNγ or TNFα suggesting other functions need to be identified (76). Both CD4+ and CD8+ T cells produce IFNγ and accumulate with the infected lung and while absence of CD4+ T cells results in rapid susceptibility to Mtb infection, the absence of CD8+ T cells results in susceptibility later in the infection (77). The organization of the granuloma is also disrupted in the absence of CD4+ T cells with predominantly perivascular cuffing of lymphocytes observed (78), suggesting that in the absence of CD4+ T cells chemokine gradients are not established for T cell migration. It is also the case that while CD8+ T cells can make IFNγ during Mtb infection they require CD4+ T cells to do so optimally (79).
The induction of IFNγ producing T cells has been the focus of anti-TB vaccine design but has not been particularly fruitful. It is clear that humans need antigen-specific T cell responses to control TB (as those with HIV/AIDS develop TB readily) and that absence of IFNγ promotes mycobacterial disease in humans so why have we not progressed? Again we come back to the issue of the communication between the T cells and the infected phagocytes. If the T cells are unable to co-locate with the phagocytes and/or the phagocytes are unable to respond to the signals delivered by the T cells then the number of IFNγ-producing T cells circulating throughout the body is meaningless. It is therefore the case that IFNγ production by activated T cells is not a correlate of protection, rather the ability of antigen-specific T cells to penetrate and survive within the infected site may be. In this regard, recent studies demonstrate that not all cytokine producing antigen-specific T cells are able to penetrate the TB granuloma and some remain in the vasculature or cuff around the vessels and this is related to their expression of transcription factors, chemokine receptors and differentiation state (80–82); these markers should perhaps be considered as correlates of protection.
IFNγ can act on cells other than macrophages; indeed it’s most critical function in TB may not be to activate macrophages but rather to limit polymorphonuclear (PMN) inflammation (Figure 1). Most susceptible mouse strains exhibit high PMN infiltration in the lungs once infected (83–86) and inhibition of this infiltration improves survival (83). Mice which lack IFNγ exhibit high PMN infiltration as do mice lacking CD4+ T cells (70, 78). Neutrophils which lack the IFN-γR fail to undergo apoptosis and accumulate in the lungs of Mtb-infected mice and their removal improves survival without altering bacterial burden (87). Similarly chimeric mice lacking IFN-γR on their radio-resistant cells over express IL-17 and have excessive neutrophil recruitment and reduced survival (88). It is possible that the high IFNγ-producing CD4+ T cells which populate the vasculature (80, 82) are located in such a position to reduce neutrophil accumulation.
Production of IFNγ is a very useful diagnostic tool which has been developed to be more selective than the older skin test assay. In this prominent test for Mtb exposure, Mtb antigens (selected to be unique for Mtb versus other mycobacteria) are used to stimulate IFNγ release (89–91). While this test selects for those who are exposed it is not optimized to distinguish between those individuals who are infected but healthy, from those in the process of developing active disease. Recently, studies have shown that patients that have more IFNγ-producing T cells are actually more likely to progress to active disease, suggesting that this test may be optimised to identify those progressing toward disease (92).
Type I Interferon (IFN)
The Interferons (IFN) were first identified more than half a century ago for their antiviral activity (93). Type I IFNs represent the largest group, with at least thirteen gene products identified in humans and mice with IFNα and IFNβ being the best classified and the focus of this section. For clarity, IFNα and IFNβ will be collectively referred to as IFNα/β throughout this section. The innate response to pathogens occurs via Toll-like receptor (TLR) engagement resulting in a complex cytosolic cascade of signal transduction toward IFN-regulatory factor3/7 (IRF3/7)-mediated transcription of IFNα/β genes (94). Secreted IFNα/β engages IFN subunit receptors 1 and 2 (IFNAR1/2) at the cell surface, which then activate dimers of the tyrosine kinases JAK and tyrosine kinase (TYK) (94, 95). The end result is activation of IFN-stimulated gene factor (ISG) that then interacts with IFN-stimulated response elements (ISRE) at the promoters of IFNα/β regulated genes (94, 95).
The type I IFNs were not thought to play a major role in Mtb infection and indeed infection of IFNAR-deficient mice with a low dose aerosol of Mtb Erdman strain did not indicate any major impact of the loss of this receptor (96). However, use of strains with increased virulence, such as the W-Beijing strain HN878, has revealed an important strain-dependent outcome in relation to Type I IFNs. The pathogenesis of Mtb strain HN878 is associated with IFNα/β dependent reduction in the activity of the pro-inflammatory cytokines IFNγ, TNFα, IL-6 and IL-12, as well as in the anti-inflammatory IL-10 (97, 98) (Figure 1). Intranasal delivery of IFNα/β also results in increased bacterial burden and reduced survival in contrast to IFNγ treated mice (97). Further, IFNα/β signalling interferes with IFNγ-mediated killing of Mtb (99). One hypothesis regarding the role of type I IFNs during chronic Mtb infection is that the accumulation of plasmacytoid dendritic cells in the lung provides a source of excess type I IFN which then inhibits the accumulation of CD4+ and CD8+ T cells in the lung (98) (figure 1). Finally, transcriptional analysis of peripheral blood cells from those exposed to TB shows that both IFNγ and type I IFN signatures occur but that the type I IFN signature is predominantly associated with neutrophils (100).
In a mechanistic analysis of the function of IFNα/β in TB, polyinosinic-polcytidylic acid and poly-L-lysine and carboxymethylycellulose (Poly-IC) was used to induce elevated levels of IFNα/β during Mtb infection (101). This poly-IC treatment results in elevated bacterial burden and increases the recruitment of an apparently permissive CD11b+GR1int cell phenotype recruited via chemokine (C-C motif) ligand 2 (CCL2) and C-C chemokine receptor type 2 (CCR2) (101) (Figure 1). Similarly, careful analysis of the cells recruited to the lungs of mice lacking either type I or type II IFN receptors demonstrates a protective function for type I IFN signalling in that in its absence initial recruitment of target host cells for Mtb does not occur and immunity is compromised (102).
IFNα/β is another perfect example of a ‘goldilocks’ cytokine in TB (Table I). Just enough is required to initiate recruitment of phagocytes which provide activatable host cells for Mtb to invade: however, production of too much IFNα/β results in large numbers of permissive cells which cannot be effectively activated. Also too much of this cytokine can limit the activation state of the infected phagocytes and potentially limit the accumulation and function of the T cells required to regulate the mononuclear structure of the granuloma.
Interleukin-6 (IL-6)
Interleukin-6 (IL-6) is a pleiotropic cytokine produced in response to inflammatory stimuli (103) and is involved in the essential cellular processes of differentiation, proliferation, and apoptosis. Many cell types express IL-6 including those of lymphoid and non-lymphoid origin (103) and expression can be induced by other cytokines including IL-1, TNFα and IFNγ (104, 105). IL-6 signals through soluble and membrane bound IL-6R of which the glycoprotein 130 dimer (gp130) is an essential component (106). Downstream signalling is mediated by a phosphorylation cascade involving JAK, mitogen-activated protein kinase (MAPK) and STAT pathways (106, 107). The pluripotency of IL-6 warrants regulation and this is mediated by suppressor of cytokine signalling (SOCS), which inhibits STAT signalling (106, 107).
The relative importance of IL-6 during TB depends upon the route and dose of infection. As we have discussed, communication between cells is critical for successful expression of immunity and if the dose is low or bacteria are slow to grow, then the kinetics of the cellular response are not critical. However if the dose is high and systemic then the kinetics of the response becomes critical. This concept is illustrated by IL-6 as in its absence (either by antibody treatment or by gene deletion) there is increased susceptibility to intravenous challenge with a large dose of mycobacteria (108, 109). In contrast, in a low dose aerosol Mtb challenge model while modestly increased bacterial burden occurs in the lungs of IL-6 deficient mice the impact is not lethal (110). In both the low and high dose challenge models increased IL-4, as well as reduced or delayed T cell accumulation and IFNγ expression is observed suggesting that IL-6 can act to potentiate IFNγ expression at the site of infection (108, 110). It appears also that IL-6 is required for optimal induction of protective responses during vaccination as, in its absence, both BCG and a subunit vaccine are less effective (109, 111).
Interpretation of the role of IL-6 in TB is complicated by the fact that the soluble IL-6 receptor can mediate trans-signalling and is implicated in inflammatory diseases such as IBD (112). To address the role of IL-6 further, a gp130 construct capable of sequestering IL-6 in the blood (sgp130FC) was delivered to mice during Mtb infection however no impact on disease progression was seen. In contrast, when mice are made to overexpress this construct a temporary but significant increase in bacterial burden occurs during acute infection (113). This observation is consistent with an early role of IL-6 in potentiating immunity during early Mtb infection.
In vivo data support a protective role for IL-6 in the induction of early protective responses mediated through IFNγ (108, 110). Human studies also give us considerable insights into the role of IL-6 during TB. Cavitary TB is the most destructive form of caseous TB whereby necrosis liquefies cellular material and results in compromised lung function. Humans with cavitary TB express lower levels of IL-6 and the chemokine IP-10 in their bronchial alveolar lavage (BAL) fluid when compared to TB patients without cavitary disease, thereby indicating IL-6 and IP-10 as potential markers of controlled (non-cavitary) TB (114). As would be expected, elevated neutrophils were observed in BAL from cavitary TB patients while non-cavitary TB patients presented with elevated alveolar macrophages (114); interestingly there was no correlation between cytokine expression in BAL fluid and serum cytokine production (114). In contrast, an earlier study identified elevated blood plasma levels of IL-6 from TB patients with developed lung lesions (115). Based on the mechanistic data from animal studies and the human data, IL-6 appears to be associated with effective early expression of immunity in the lung via the combination of regulated mononuclear inflammation and rapid accumulation of lymphocytes. Its effects are modest but maybe critical following high dose exposure or during immunodeficiency.
IL-1 cytokines
The pro-inflammatory cytokines IL-1α, IL-1β (collectively called IL-1 here) and IL-18, are members of the IL-1 family (116). IL-1 was first identified in the 1940s as an endogenous pyrogen (117–119). IL-1 and IL-18 as well as their respective receptors (IL-1R1 and IL-18R) are widely expressed by all nucleated cells of the body including endothelial cells, monocytes, macrophages and neutrophils (116). Expression of IL-1 and IL-18 is mediated in part by the canonical pathway of inflammasome activation, which involves the sensor (e.g. Toll-like receptor, TLR), an adaptor molecule such as Myeloid differentiation primary response gene 88 (MyD88), and caspase-1 (120–122). Alternatively, IL-1β and IL-18 can also be induced by the non-canonical inflammasome pathway which is distinguished by the activation of caspase-8 and -11 on the precursor of the cytokines in the cytosol (123, 124). MyD88 is an important cytosolic mediator linking TLR signalling to the transcription of inflammasome components (122).
IL-1α is mostly associated with sterile cell injury (e.g. cigarette smoke), but is also induced during non-sterile cell injury (e.g. bacterial) where it functions locally as an alarmin (125–130). IL-1β is induced during infection and is primarily produced by monocytes, macrophages and dendritic cells (131–134). IL-1 signals through the IL-1R1 receptor present on a number of cells including endothelial cells, monocytes, macrophages and T lymphocytes (116, 128, 135, 136). IL-18 is expressed constitutively in the cytosol at low levels as a precursor, which is activated by caspase-1 activity following bacterial stimulation, stimulation by neutrophils or by IL-4 or IFNγ (137–139). IL-18 activity results from co-localization of IL-18 receptor alpha (IL-18Rα) and IL-18 receptor beta (IL-18Rβ) on host cells including monocytes and epithelial cells (140).
IL-1R/IL18R/MyD88
Signalling through MyD88 is shared between TLR, IL-1R and IL-18R (141–143). MyD88 is an essential component in innate signalling in response to TB, as MyD88 gene deficient mice exhibit profound susceptibility to Mtb infection (143). Importantly, following mycobacterial stimulation, MyD88-gene deficient macrophages and dendritic cells exhibit reduced IL-6, TNF and IL-12p40 production suggesting a critical role for MyD88 in pattern recognition responses to Mtb infection (143). Aerosol infection results in dramatically increased lung burden coinciding with increased inflammation and accumulation of neutrophils and macrophages (143). Despite the poor innate response to Mtb infection in the MyD88 gene deficient mice the accumulation of IFNγ-producing T cells was not affected. It is likely that these antigen-specific cytokine producing T cells were unable to mediate protection due to failures within the phagocytes accumulating at the site or as a result of being unable to communicate with the infected phagocytes (143). That BCG vaccination results in protection against Mtb infection in MyD88-deficient suggests that it is a failure of T cells to accumulate rapidly enough in naïve mice that contributes to their susceptibility (143).
What then is MyD88 signalling doing? Comparison of the phenotype of IL-1 deficient mice and MyD88 deficient mice is suggestive in this regard as both exhibit increased susceptibility with focal necrosis despite generation and accumulation of cytokine producing T cells (144). These observations suggest that induction of IL-1 is likely Myd88-dependent and that this pathway plays a critical role in protective immunity to TB.
IL-1
IL-1α and IL-1β are interdependent proinflammatory cytokines critical to defence against TB (145–149). Mice lacking either IL-1α or IL-1β or both are susceptible to acute and chronic infection respectively following challenge with Mtb (145–148). IL-1 α/β double deficient mice share a similar susceptibility to infection as IL-1R1KO and MyD88KO mice (143, 144, 148, 150). Deficiencies in the IL-1 pathway (IL-1 α/β or IL-1R1) have no impact on the protection against BCG delivered intravenously suggesting that virulence of the pathogen is a factor in the role of the IL-1 pathway (146). Anti-IL1α and anti-IL-1α/β antibodies delivered subcutaneously to Mtb-infected mice have also been shown to result in loss of body weight and lethality (148). Further, lung sections from anti-IL-1α-treated mice exhibit lung parenchyma consumed by cellular infiltrates (148). During sterile mediated inflammation, IL-1α appears to be involved in the expression of pro-inflammatory cytokines such as IL-6 in primary fibroblasts (151) which may be associated with mobilization of neutrophils (152). It has also recently been observed that upon activation of the inflammasome, IL-1β and IL-18 are capable of inducing expression of the neutrophil recruiting cytokine IL-17 (153–155). IL-17 responses are essential in the protection against some Mtb strains, such as HN878 and for recall responses to H37Rv (149, 156, 157). Consistent with this, IL-1R1 gene deficient mice infected with the Mtb strain HN878 produce decreased levels of IL-17 and decreased populations of IL-17-producing cells in vitro and in vivo (149).
IL-1 is produced by CD11b+Ly6G− cells following Mtb infection (145) and rescue of the lethal phenotype in IL-1α mice can be accomplished by directed viral expression of IL-1α in CD11c+ cells transplanted in IL-1α gene deficient mice (147). It would seem therefore that a primary function of the IL-1/IL-1R pathway is to mediate the recruitment and co-ordination of cellular responses by the induction of pro-inflammatory cytokines from the stroma (145, 147). One critical aspect of IL-1 function is in promotion of prostaglandin E2 (PGE2), which in turn mediates inhibition of type I IFN-induced accumulation of permissive macrophages at the site of infection (158) (Figure 1). Prostaglandins, such as PGE2 are produced by the action of cyclooxygenase (COX) enzymes on arachidonic acid and in the absence of inducible COX enzymes, mice are highly susceptible to Mtb infection and delivery of PGE2 during Mtb infection results in a partial rescue of the lethal phenotype in IL-1α/β infected mice (158). Taken together, the underlying function of IL-1 in Mtb appears to be in regulating type I IFN function and helping to maintain the balance between sufficient phagocytes to mediate control of the intracellular pathogen while inhibiting the over recruitment of permissive macrophages mediated by type I IFN (102).
IL-18
IL-18 is essential for the production of IFNγ from T cells under some conditions (159–163) and in some instances its absence can result in increased susceptibility to Mtb (150) although in other conditions, increased susceptibility to Mtb infection is not observed (162, 163). Interestingly, when susceptibility is observed the accumulation of neutrophils and inflammatory chemokines CXCL1 and CXCL2 is elevated and depletion of neutrophils and monocytes from the lung results in decreased bacterial burden (150). In Mtb-infected IL-18 deficient mice, an increased frequency of IFNγ-producing CD4+ and CD8+ T cells in the lungs is seen but total IFNγ production by these T cell populations is decreased, suggesting that IL-18 could contribute to optimal IFNγ induction during TB (150, 162, 163). It would appear therefore that IL-18 plays a role in inducing high IFNγ production in T cells but that this is not required for protection and that its more critical role (perhaps when dose or virulence of the Mtb strain is high) is that of regulator of phagocyte accumulation, possibly mirroring the role of IL-1 and MyD88.
Our working model of immunity to TB places the emphasis on rapid and correct accumulation of both phagocytes (macrophages and neutrophils) and T cells to the site of infection. This accumulation is initiated by the innate sensors within the lung and results in the induction of TNF, IFN’s and IL-1 family members. The correct ratio of these cytokines is essential for the balance of permissive and non-permissive phagocytes and for the development of a granuloma such that infected phagocytes and antigen-specific T cells can communicate effectively to stop Mtb growth. How the antigen-specific T cells develop and are regulated is covered below.
IL-12 Cytokine Family
The IL-12 family of cytokines belongs to the IL-6 super family and is the only family composed of heterodimeric cytokines (164, 165) and this unique feature bestows diverse and pleiotropic functions due to promiscuous chain pairing (166). The alpha chains of the IL-12 family (p19, p28 and p35) contain four-helix bundle structures and pair with one of two beta chains (either p40 or Epstein-Barr virus induced gene 3 (Ebi3)) (164–166). IL-12 is composed of the subunits p35/p40, IL-23 of p19/p40, IL-27 of p28/Ebi3, and IL-35 of p35/Ebi3 with expression of the distinct subunits being regulated independently (166). In addition, IL-12p40 can also be secreted both as a homodimer (IL-12p80 or IL-12p(40)2) and as a monomer (IL-12p40) (167). Both macrophages and dendritic cells are major producers of IL-12p40, IL-12, IL-23 and IL-27 (168). These cytokines are largely associated with the induction and regulation of cytokine expression within antigen-stimulated T cell populations.
IL-12
IL-12 plays an important role as a link between innate and adaptive immune responses, and is produced by and influences multiple effector cells (169, 170). Composed of IL-12p35 and IL-12/23p40, IL-12 (IL-12p70) is primarily secreted by macrophages, dendritic cells and B cells (166, 171, 172). The importance of IL-12 in TB is dramatically illustrated by several experiments of nature wherein humans with IL-12p40 deficiency display an inherent predisposition to Mtb infection (173–178). Further, patients with Mendelian susceptibility to mycobacterial disease (MSMD) harbour deficiencies in IL-12Rβ1, IFNγR1, and IL-12p40 and exhibit susceptibility to Mtb and develop BCGosis following delivery of the BCG vaccine (66, 178–181). Genetic aetiology for MSMD is associated with mutations in the autosomal genes IFNGR1, IFNGR2, STAT1, IL12B, IL12BR1, and X-linked gene IKKBG, encoding NF-κB essential modulator (NEMO) (66). All these autosomal genes are associated with IL-12/IFNγ-dependent signalling and the IFNγ-mediated activation of macrophages. Mutations in IKKBG impair CD40-dependent IL-12 production in monocytes and dendritic cells, despite normal CD40-mediated induction of costimulatory molecules on dendritic cells (182). These human data highlight the importance of this pathway to TB control.
IL-12 is expressed within the lung at the site of TB (183) and delivery of IL-12 to Mtb-infected mice decreases bacterial burden while reduction of IL-12 by antibody increases bacterial burden (184). Interestingly, delivery of IL-12 also modestly improves the outcome for mice lacking acquired cellular immunity suggesting that it can mediate immunity via direct action on innate cells (184). Mice genetically deficient for the IL-12p40 subunit are acutely susceptible to Mtb infection (185, 186) whereas those lacking IL-12p35 exhibit prolonged survival relative to the IL-12p40 deficient mice (186). This, in turn, is dependent upon the availability of the IL-23p19 subunit (187). The absence of IL-12p40 results in the substantial loss of antigen-specific IFNγ production (185, 186) (Figure 2) while the presence of IL-23p19 in the IL-12p35 deficient mice appears to promote sufficient antigen-specific IFNγ production to increase protection relative to the IL-12p40 deficient mice (187). It also appears that stable and prolonged IL-12 production is required to maintain IFNγ production and to limit bacterial growth long-term (188). This requirement for long term function may also apply in humans as absence of the IL-12R1 results in poor accumulation of IFNγ producing memory T cells (189). The innate pattern recognition receptors, TLR2 and TLR9, are necessary for optimal production of IL-12p40 in response to Mtb exposure (190) while the Mtb lipoarabinomannans have been shown to negatively regulate TLR-mediated IL-12 production by inducing an inhibitor of TLR signalling, IRAK-M (191). In contrast, mycobacterial LprA is a TLR2 agonist and promotes IL-12p40 production (192) reflecting the need for Mtb to both induce and regulate IL-12p40 for its own ends.
IL-12 signals through interactions between IL-12/23p40, and IL-12p35 with IL-12Rβ1 and IL-12Rβ2, respectively (193–195) with the IL-12p40 interacting with IL-12Rβ1 on the target cell surface thereby allowing the IL-12Rβ2 to induce JAK and STAT signalling and activate STAT4 homodimers (166). The homodimers IL-12p40, IL-12(p40)2, antagonizes IL-12-mediated immune responses though competitive binding of IL-12Rβ1 (196–198). However, in TB, it appears that IL-12(p40)2 can also function as an agonist (199), and supports dendritic cell migration to the draining lymph nodes (200, 201), to promote T cell priming and differentiation (200). Specifically, following Mtb infection, dendritic cells are thought to be the first immune cells to traffic to the draining lymph node (202) and this may occur in an IL-12p40 and IL-12Rβ1-dependent manner (200, 203). Bone marrow derived dendritic cells from mice deficient in IL-12p40 are unable to activate naïve T cells in the draining lymph node following delivery to the lung and fail to confer protective adaptive responses (200). However, treatment of the IL-12p40 deficient dendritic cells with the homodimer IL-12(p40)2, is sufficient to restore migration of the dendritic cells to the draining lymph node and for activation of naïve T cells to occur (200). Expression of IL-12Rβ1 is also required to facilitate dendritic cell migration to the draining lymph node (203) and indeed CD11c+ cells in the Mtb-infected lung express an alternative splice variant of IL-12Rβ1 that augments IL-12Rβ1-mediated effects (203). In particular, dendritic cells expressing the splice variant exhibit enhanced migration from the infected lung to the draining lymph nodes and supported activation of Mtb-specific T cells (203)(Figure 2). In an interesting example of cytokine cross talk, mice lacking the p75 receptor for TNF (TNFRp75) exhibit increased IL-12p40 and enhanced IL-12p40-mediated dendritic cell trafficking to draining lymph nodes (204). Thus, IL-12Rβ1 is important for the effector function of IL-12p40 on DCs, as well as mediating recruitment and function of CD4 T cells in response to TB.
IL-23
IL-23 utilizes the p40 beta subunit paired with the alpha chain p19 (205). Prior to the discovery of IL-23, the interpretation of data from IL-12p40 and IL-12p35 deficient models of disease had been difficult (206). Specifically, studies found that the outcome of IL-12p35 deficiency was not always the same as in IL-12p40 deficient models (207–209). The discovery of IL-23 led to the reassessment of the role of IL-12p40 (205, 206, 208, 209) and disease models previously associated with IL-12, were in fact, shown to be primarily driven by IL-23 and not IL-12 or they clarified unique disease-driving features of the two cytokines (206, 208–211). Currently, IL-23 and IL-23 pathway antagonists are in phase 2 and phase 3 clinical trials for treating patients with moderate-to-severe psoriasis (207) making determining the role of IL-23 in TB a critical undertaking. IL-23 stabilizes the induction of TH17 cell subset that produces IL-17A, IL-17F and IL-22 production and it is also required for the double expression of IL-17 and IFNγ (212). However, IL-23 alone is not sufficient to drive differentiation of TH17 cells, which require the key cytokines transforming growth factor β (TGFβ) and IL-6 (213). In addition, IL-23 can also induce IFNγ production in human T cells as well as support the proliferation of mouse memory T cells (205, 214). The interaction with its receptor, composed of IL-23R and IL-12Rβ1 subunits, activates the downstream signalling molecules, JAK and STAT, for production of its signature cytokines (166, 205, 214–216). IL-23 primarily signals through STAT3, while IL-12 can signal through STAT1, 3, 5 and 4 but preferentially signals through STAT4 (166). Upon infection with Mtb, lung DCs produce IL-23, likely mediating the induction of IL-17 production (187, 217, 218).
Although IL-23 is important for generation of Mtb-specific IL-17-producing T cells (Figure 2), mice deficient in IL-23p19 control Mtb effectively for up to 90 days whereupon bacterial growth increases relative to intact mice (187, 219). In addition, treatment of Mtb-infected mice with adenovirus-expressing IL-23 reduces Mtb burden and increases cellular responses (220). In the absence of IL-23p19 Mtb-infected mice did not develop well organized B cell follicles and this was associated with a complete absence of IL-17 and IL-22 in the lung. In addition there was very little expression of the B cell follicle associated chemokine CXCL13 resulting in increased accumulation of lymphocytes around the vessels rather than within the granulomatous regions (219)(Figure 2). Thus in support of our working model, coordinated communication between lymphocytes and infected macrophages is inefficient in the absence of specific cytokines/chemokines (in this case IL-23 dependent CXCL13) and bacterial growth occurs in the absence of this efficient communication.
IL-23 also plays a chemokine dependent role in the efficient expression of vaccine-induced mucosal immunity. This role was first highlighted in mice subcutaneously vaccinated with an adjuvant-paired I-Ab-restricted ESAT6 (1–20) peptide, which induces both IFNγ- and IL-17-producing antigen-specific CD4+ T cell responses (157). Critically, the improved kinetics of the vaccine-induced IFNγ-producing T cells is lost in the absence of IL-23 as this cytokine is required for the generation of lung resident IL-17 producing CD4+ memory T cells which generate a chemokine gradient facilitating the accelerated IFNγ response. In the absence of IL-23 vaccine-induced protection to Mtb challenge is lost (157). Co-immunization of mice with a DNA vaccine composed of Mtb antigen 85B (Ag85B) and an IL-23-expressing plasmid also confers enhanced protection through the induction of augmented T cell proliferation and IFNγ production when compared to Ag85B alone (217). Other studies also support an important role for IL-17 producing CD4+ T cell subsets in mediating mucosal vaccine-driven protection (156, 221–223). Specifically, adoptive transfer of Mtb-specific IL-17-producing T cells into unchallenged mice confers protection following exposure to Mtb (156). Further, use of adjuvants capable of driving lung-resident IL-17 producing cells are able to initiate early CXCL13 expression thereby promoting appropriate accumulation of CXCR5+ T cells within the Mtb-induced inflammatory site (221). These IL-17 producing T cells are long lived (222) and are associated with improved protection when recombinant BCG vaccines are used (223).
IL-27
IL-12 and IL-23 are pro-inflammatory cytokines with the capacity to drive cytokine production in T cells (168, 224, 225). In contrast to this clear role for IL-12 and IL-23, IL-27 is pluripotent and has a complex and sometimes apparently contradictory capacity to influence inflammation and lymphocyte function (166, 226–228) indicating a pleiotropic nature for IL-27. IL-27 is composed of the p28 alpha and Ebi3 beta chains, and signals through the IL-27Rα (WSX-1 or TCCR) and gp130 receptor subunits (166, 229, 230). IL-27 can mediate suppression of IL-17-production by T cells (231) via STAT1 signalling to promote IL-10-producing Tr1cell-like regulatory populations (232). It also promotes proliferation (229) and polarization via T-bet (233) in naïve T cells (Figure 2).
In the context of Mtb infection, IL-27R-deficient mice challenged with Mtb exhibit lower bacterial burden in the lungs and increased granuloma-localized lymphocytes (234, 235) but these mice succumb to disease earlier than control animals (235). Thus, while IL-27R activity appears to limit expression of immunity locally, it may actually protect from undue pathological damage. Due to the pleiotropic nature of IL-27 it is very difficult to dissect out its specific function in TB. The absence of the gp130 component of the IL-27R on phagocytes during Mtb infection results in loss of the increased inflammatory consequences of IL-27R deficiency but does not impact the reduced bacterial burden seen in mice lacking IL-27R on all cells suggesting that these two aspects of IL-27R deficiency are independent (236). In contrast, mice lacking IL-27R only on T cells exhibit the improved ability to control bacterial burden over the long term (82). This improvement was associated with enhanced localization and reduced differentiation (i.e. reduced T-bet expression) of IL-27R-deficient CD4+ T cells within the infected lung parenchyma. Mtb-specific CD4+ T cells lacking IL-27R are also intrinsically fitter than IL-27R-sufficient CD4+ T cells mice within the same environment (82)(Figure 2). The importance of IL-27 is further confirmed in human patients, wherein IL-27 is significantly increased in active TB patients compared to latently infected individuals (82). In our working model of TB immunity, IL-27 appears to play the role of mediator of increased inflammation within the phagocyte population while also serving to limit the efficacy of the T cell population by driving them to a state of differentiation which limits their ability to locate to, and persist within, the inflamed granuloma.
IL-35
IL-35, a dimeric protein encoded by IL-12α and IL-27β chains and has been shown to suppress CD4+ T cell responses (237). It is thought to be primarily expressed by regulatory T (TREG) cells (238) and is required for optimal function both in vivo and in vitro (239). IL-35 is important for the generation of human and mouse TREG cells, termed iTR35 cells (239), which function independently of IL-10 and transforming growth factor β (TGFβ). While a specific function for IL-35 in Mtb infection has not been directly addressed, the relative availability of IL-35 in the presence and absence of the other IL-12 family subunits makes consideration of this cytokine an important part of any interpretation of outcome in mice or humans lacking IL-12 family subunits.
IL-23 Dependent Cytokines
IL-17
The IL-17 cytokine family is composed of six members, IL-17A-F, with IL-17A and IL-17F being the most studied. Production of IL-17 is conventionally attributed T cells, however, other lymphocytes as well as innate immune cells can produce this cytokine (240). IL-17 cytokines are proinflammatory and can be protective or pathogenic depending on the nature of the challenge faced by the host (241, 242). It is at mucosal sites that IL-17 plays its most important regulatory and protective role against invading pathogens.
Following mycobacterial infection, lung-resident γδ T cells are a primary source of early IL-17 (218), and likely support early neutrophil accumulation (243) (Figure 1). Following BCG infection, IL-17 expression can be detected as early as day 1 post-infection and is dependent on IL-23 expression (243). One recently identified capacity of IL-17 is to regulate mycobacterially-induced IL-10 (244). Following vaccination with BCG, dendritic cells produce PGE2 which is required for the induction of both IL-10 and IL-23 with the IL-23 being required for IL-17 production (244). This IL-17 is then thought to down regulate IL-10 production thereby allowing increased IL-12 which subsequently promotes IFNγ production. In the absence of IL-10 the IL-23 mediated IL-17 is not required and in the absence of IL-23, BCG fails to effectively induce protective IFNγ producing T cells (244). This study was the first to show that PGE2 induction of IL-17 was sufficient to overcome the inhibitory effects of IL-10 and support the generation of antigen-specific and cytokine producing T cells during mycobacterial vaccination and challenge.
As with IL-23, low dose challenge with some strains of Mtb (i.e. H37Rv and CDC1551) in the absence of IL-17 results no obvious phenotype (149, 187, 245) until late in disease (219). In contrast, following infection with the W-Beijing strain HN878 of Mtb, IL-17R expression on radio-resistant cells (likely fibroblasts) of the lung is required to co-ordinate the rapid accumulation of cells within the lung via the induction of CXCL13 and recruitment of CXCR5+ T cells to lymphoid follicles within the tissue (149)(figure 2). Importantly, the W-Beijing HN878 Mtb strain induces high levels of IL-1β and IL-17 relative to other Mtb strains (149) and also induces excess type I IFN (97) which is capable of bringing in permissive macrophages in a CCR2 dependent manner (101)(Figure 1). HN878 induces an environment that is highly permissive for its growth and it is this environment that results in the need for the optimum expression of immunity wherein IL-17 promotes rapid accumulation and the correct localization of the T cells needed to change the permissive macrophages to ones which limit bacterial growth (149).
The role of IL-17 in initiating early co-ordination of cellular responses in naïve mice is apparent when the challenge is significant as in the case of HN878; however the concept of IL-17 as a co-ordinator of early mucosal responses in TB actually stems from vaccine work. Initial studies using a defined subunit vaccine determined that lung-resident IL-17-producing cells induced by vaccination are vital for the induction of the chemokines (CXCL9, CXCL10 and CXCL11) required for the accumulation of IFNγ-producing memory T cells (157). Further studies have shown that adoptive transfer of Mtb-specific IL-17-producing T cells into naïve mice are able to mediate protection in an aerosol challenge model thereby identifying these types of cells as valid targets for vaccine-mediated induction (156). Finally, mucosal immunization with Mtb antigens induces potent IL-17 responses which improve upon BCG vaccine-induced protection in mice (221). Interestingly, in mucosal vaccine models, IL-17 rather than IFNγ appears to most important for vaccine-induced protection against Mtb providing support for the model that it is the co-ordination of the cellular response that is the determining factor in the success of vaccination (221, 246). Critically, antigen-specific IL-17-producing memory T cells are induced by vaccination and respond up to 2 years post vaccination (222). These memory T cells appear to be metastable and become IFNγ-producers within the lung (222), probably as a result of the action of IL-23 (212). In fact, pluripotent memory T cells capable of producing not only IL-17 but also TNF and IL-2 may be the most appropriate target T cells for vaccination (247). Manipulation of BCG can also result in increased induction of IL-17-producing memory cells and this is associated with improved protection as in the case of the recombinant BCG strain rBCGΔureC:Hly (223). Thus, IL-17 drives the induction of CXCL9–11 to recruit protective antigen-specific T cells, as well CXCL-13 to localize CXCR5+ cytokine producing T cells within TB granulomas. Despite the protective outcome of IL-17 discussed above, IL-23 dependent IL-17 production is also associated with damaging neutrophil accumulation during a chronic restimulation model of TB (248). Indeed, exacerbated production of IL-17 appears to drive pathology by inducing S100A8/A9 proteins that recruit neutrophils into the lung (249). Thus, IL-17 also fits the bill as a ‘goldilocks’ cytokine in TB (Table I).
IL-22
IL-22 is primarily produced by CD4+ T cells as well as γδ T cells, natural killer (NK) cells and innate lymphoid cells following exposure to innate or infectious stimuli (250). IL-22 can have dual effects in the context of inflammation and this has been attributed to its co-expression along with IL-17 (250, 251). The major functions of IL-22 are the regeneration and survival of the intestinal, airway and external epithelium as well as stimulating the secretion of antimicrobial peptides such as lipocalin and β-defensins (245, 250, 252). In the context of Mtb, IL-22 is expressed at higher levels than IL-17 at the site of infection and within granulomas from TB patients and non-human primate models (253, 254). Furthermore, in non-human primate infected with Mtb, CD4+ T cells expressing membrane-bound IL-22 limit Mtb intracellular growth in macrophages (255). IL-22 can also inhibit intracellular growth of Mtb in human monocyte-derived macrophages by promoting phagolysosomal fusion and induction of Calgranulin A, a heterodimer of S100A8 and S100A9 proteins (256). Moreover, human NK cells cultured with Mtb-infected macrophages produce IL-22 and mediate macrophage activation (257). Finally, IL-22 increases as patients receive anti-TB treatment and this has been associated with a decrease in a regulatory B cell population (CD19+CD1d+CD5+ B cell) the in vitro depletion of which results in enhanced IL-22 production by T cells (258).
Animal studies using low dose aerosol challenge indicate that in uncomplicated infection models IL-22 producing T cells accumulate in the lung and express IFNγ (259). In the absence of this cytokine however there appears to be no significant consequences (260). However, in a BCG vaccine model, NK1.1+ cells appear to make IL-22 which contributes to protection by regulating TREG cells (261). Taken together, the current data suggest a protective role for IL-22 in TB disease progression, possibly via antimicrobial peptide production, cellular function and promotion of epithelial repair.
Regulatory Cytokines
IL-4, IL-5, IL-13
IL-4 was first described as a product of CD4+ T lymphocytes which are now known as TH2 T cells (262, 263). TH2 responses inhibit TH1 responses (264–266). IL-4, IL-5, and IL-13 are the signature cytokines associated with TH2 responses they are induced in response to helminth infections and contribute to diseases such as asthma and allergy (267–270); they mediate expulsion of multicellular parasites occupying mucosal tissues. IL-4R signaling requires hetero-dimerization of IL-4Rα (shared with IL-13) and the common gamma chain (shared with IL-2) (271). The IL-4 receptor (IL-4R) is the primary mediator action and ligation of IL-4R results in signal transduction via STAT-6 and subsequent GATA-3 transcription (272–275). Both STAT-6 and GATA-3 distinguish TH2 cells from other TH cells, including TH1 and TH17 (270). IL-4 expression is in part, regulated by IL-2 and is associated with the differentiation of TH2 cells, which then express and maintain IL-4 and IL-5 in a positive feedback loop (276, 277);. IL-4R is expressed on many cell types including lymphocytes, epithelial cells and fibroblasts (278, 279). IL-5 is primarily associated with recruitment of eosinophils (280) basophils (281) and the development of antibody producing B cells (282, 283). The IL-5 receptor comes in both low and high affinity forms whose activity is context dependent when expressed on the surface of lymphocytes, eosinophils and basophils (284).
During TB, IL-4 levels are quite variable with mRNA detectable in PBMCs (285) and IL-4-producing T cells isolatable from TB patients (286); however, PBMCs from active, Mtb-culture positive patients show decreased IL-4 expression (287–289). While a significant increase in IL-4 is observed in the plasma of TB patients compared to household contacts (290), IL-4 plasma levels are not different between HIV patients and non-HIV patients with TB (290, 291) and anti-TB treatment is associated with decreased plasma IL-4 levels (290). IL-4 mRNA has been shown to be upregulated in the necrotic areas in the lungs of HIV+ patients with pleural TB (292) and is consistent with increased CD4+ cells expressing IL-4 in TB patients exhibiting cavitary disease (293). In a non-human primate model, IL-4-expressing T cells are increased transiently at week 6 post-Mtb infection; however this population is not sustained (294). One reason for the variable association of IL-4 expression with disease profile may lie in the fact that infection with Mtb is associated with expression of the IL-4 antagonist IL-4δ2 and it may be the relative levels of IL-4 and its splice variant that define the impact of the cytokine on disease outcome (295–297)
Aerosol Mtb infection of mice deficient in IL-4, IL-4/IL-13, IL-4Rα or STAT-6 fails to result in early differences in bacterial burden (298, 299) despite increased levels of IFNγ (110); however, during chronic infection, bacterial burden increases in IL-4Rα and STAT-6 gene deficient mice (298). IL-4 can influence Mtb-induced granulomas as over-expression of this cytokine by adenovirus results in increased accumulation of monocytes and eosinophils within the granuloma (300). This demonstrates that IL-4 has the potential to deviate the Mtb-induced granuloma from its mononuclear to a more granulocytic characteristic (35, 301) but that its impact on disease is not strong.
Information regarding the role of IL-5 in TB is limited, however following intranasal infection of IL-4 or IL-5 deficient mice with BCG effective clearance is observed with no differences in bacterial burden or lung pathology among IL-4 and IL-5 deficient mice (302). One area where this cytokines may play a role however is in HIV co-infection as IL-5 is not observed in NHP monocytes infected with Mtb but during co-infection with SIV, IL-5 and IL-13 are increased (303). Non-human primate models co-infected with SIV and Mtb show disrupted CD4+ T cell levels (303) and the mechanism of loss appears to be related to monocyte-derived IL-5 that was induced following SIV infection (303).
IL-13 was originally described as a T cell-derived cytokine capable of inhibiting pro-inflammatory cytokine production (304, 305); IL-13 function has since been extended to include regulating airway restriction and anti-helminth responses (306–308). Furthermore, IL-13 is not only produced by TH2 cells, but can also be generated by invariant NK T cells (iNKT), granulocytes (e.g. basophils, eosinophils, and mast cell), murine group 2 innate lymphoid cells (ILC2s), and human ‘Chemoattractant Receptor-homologous molecule expressed on TH2 lymphocytes’ (CRTH2)-type 2 ILCs (309–313). It is structurally similar to IL-4 and signals through cell surface receptor heterodimers composed of IL-4Rα and IL-13Rα1 subunits to activate STAT6 (313). Though not much is known regarding IL-13 in Mtb infection, whole blood mRNA from latently infected children shows increased IL-13 compared to uninfected controls (314), although IL-13 levels are not different between actively and latently infected children (314). IL-13 may play a modulatory role in autophagy, which is an important homeostatic mechanism for intracellular degradation and has a protective function during mycobacterial infection (315–317). Indeed, in both murine and human macrophages, IL-13 and IL-4 are independently capable of inhibiting autophagy as well as IFNγ-induced autophagy-mediated killing of Mtb (317). Transgenic mice overexpressing IL-13 succumb to infection with Mtb sooner than control mice and have more necrotic granulomas within the lung (318) and this is associated with delayed expression of IFNγ and IL-17-producing CD4+ T cells and increased arginase production by macrophages within the necrotic granulomas (318). While this over-expression of IL-13 represents an artificial situation it highlights the potential for disruption of the T cell response to have a profound effect on TB development. Studies utilizing IL-13 gene deficient mice are necessary to truly uncover the distinct role for IL-13 in TB.
Transforming Growth Factor β (TGFβ)
TGFβ is a pleiotropic cytokine and regulates hundreds of genes (319–322) to modulate inflammation, cell proliferation and differentiation, as well as cell migration (323–325). TGFβ can be made by various cell types, including all leukocytes (e.g. lymphocytes, macrophages, monocytes, dendritic cells) (325, 326). Not surprisingly, the impact of TGFβ-on disease outcomes is dependent on cell type and stage of cellular differentiation, as well as the cytokine milieu (327).
TGFβ levels are increased in blood monocytes isolated from TB patients compared to uninfected individuals (328) and TGFβ localizes primarily to multinucleated Langhans giant cells within the granulomas of TB patients (328). TGFβ is induced in human blood monocytes by Mtb lipoarabinomannans (LAMs) (329) and human monocytes treated with TGFβ allow for increased intracellular Mtb survival, suggesting that TGFβ can play a regulatory role and potentially negative role in the context of Mtb infection (330). T cells and monocytes from TB patients co-cultured with natural inhibitors of TGFβ, such as decorin and latency associated peptide (LAP), exhibit restored T cell proliferation and monocytic control of Mtb again suggesting that TGFβ is a regulatory inhibitor of both T cell responses and anti-bacterial activity (331). TGFβ is also able to induce IL-10 and to synergize with this cytokine to suppress IFNγ production (332). The contribution of TGFβ polymorphisms to TB susceptibility is not clear (333, 334). The polymorphism +869T/C does not correlate with increased susceptibility in a Chinese population (334) whereas the same polymorphism in an Indian population reveals a significant susceptibility to Mtb in patients harboring this polymorphism. Our knowledge of the importance of context in the function of TGFβ suggests that other genetic or indeed cultural differences may mask the contribution of this polymorphism. Taken together, the data suggest that TGFβ plays an inhibitory role in host responses to Mtb infection.
IL-10
IL-10 was initially identified as a “cytokine synthesis inhibitory factor” produced by Th2 cells (335). However, IL-10 can be produced by other T cell subsets including TH1 and TH17 cells, macrophages, some dendritic cell subsets, myeloid derived suppressor cells, B cells and neutrophils (336). In addition, TREG are also a major source of IL-10 and serve to limit potentially pathogenic immune responses (336). IL-10 signals through the IL-10R which is comprised of IL-10R1 and IL-10R2 (337). IL-10R1 is induced on hematopoietic cells, while IL-10R2 is expressed constitutively on most tissues and immune cells (336). In myeloid cells, IL-10 production can occur via TLR- MyD88 dependent pathways (338), as well as TLR-independent C-type lectin receptor engagement (339).
In the context of TB, meta-analyses suggest that polymorphisms in the IL-10 gene, specifically -1082G/A polymorphisms in Europeans and -592A/C polymorphisms in Asians are significantly associated with TB risk (340). Further, antigen-specific IL-10 production is found in pulmonary TB patients (341, 342) and along with TNF-α production can be used to reliably distinguish between latent TB and pulmonary TB (342). In addition, increased accumulation of TREGS expressing IL-10 correlates with increased bacterial burden and more severe TB in an Indian population (343, 344) and a high level of IL-10 at the end of treatment in pulmonary TB patients is associated with TB recurrence (345). Finally, infection with helminths in TB patients results in decreased antigen-specific IFNγ and IL-17 responses, which is dependent on IL-10, as IL-10 blockade significantly increases frequencies of IFNγ producing cells (346, 347).
Following mycobacterial stimulation, dendritic cells and macrophages both produce IL-10 (338, 348). In macrophages, IL-10 can block phagosome maturation and macrophage activation in a STAT3-dependent manner, thus allowing a niche for Mtb to replicate and survive within the phagosome (349). In addition, IL-10 can inhibit aspects of IFNγ-mediated macrophage activation (350). In dendritic cells, mycobacterially-induced IL-10 production can inhibit antigen presentation through the down regulation of MHC Class II molecules, decreased IL-12 production and inhibition of dendritic cell trafficking to the lymph nodes for T cell priming (351, 352). In keeping with this regulatory role for IL-10, studies have shown that IL-10 gene deficient mice infected with Mtb exhibit increased TH1 and TH17 responses, and this coincides with improved Mtb control during chronic infection (336) (Figure 2). The effect is not dramatic and indeed some challenge models fail to show an impact of IL-10 gene deficiency (299, 353, 354). Interestingly, CBA mice generate significant early macrophage IL-10 production correlating with increased susceptibility to Mtb infection (355). This increased susceptibility also coincides with reduced expression of TNF-α and IFN-γ in T cells and can be reversed by blocking IL-10R signaling very early in infection (355). In CBA IL-10 gene deficient mice, Mtb infection results in development of fibrotic granulomas with similarity to lesions seen in humans (356). In vaccine models, blocking IL-10 at the time of BCG vaccination (336), or using IL-10 gene deficient mice in BCG vaccination and Mtb challenge experiments (244), demonstrates that IL-10 limits IFNγ and IL-17 responses during priming and decreases protective vaccine-induced protection against Mtb challenge. Computational modeling also highlights the pleiotropic role for IL-10 (357). Importantly, there are differences in the role of specific cytokines depending on the nature of the Mtb strains being examined. Indeed, the W-Beijing HN878 strain induces robust IL-10 production to inhibit the induction of a Th1 response (98). In the future therefore, addressing the role for IL-10 in the context of infection a variety of Mtb strains will likely provide novel insights into the function of IL-10 in TB.
THE CHEMOKINES
Limiting bacterial spread and containment of inflammation within discrete sites are hallmarks of disease control in TB and dovetails with the establishment of the TB granuloma. The TB granuloma is a multicellular immune bolus consisting of a number of cell types including macrophages, neutrophils, and lymphocytes, B cells, among others (358). Formation of the TB granuloma is governed by coordinated expression of the chemotactic cytokines referred to as chemokines. Chemokine expression establishes a chemical gradient that drives mobilization and recruitment of cells from peripheral organs to the site of infection and within the granuloma. The importance of this coordination has recently emerged to be critical in disease control with proper localization of CD4 T cells in the lung parenchyma being paramount (80–82).
Since the discovery of the first chemokine, now known as CXCL8 (i.e. IL-8), numerous chemokines have been identified, resulting in the need for a uniform nomenclature currently based on primary sequences of chemokine ligands (359–361). These chemokine ligands modulate biological processes through interactions with seven transmembrane G protein-coupled receptors (360, 362). Chemokines are divided into four families (C, CC, CXC, CX3C) based on the on the presence of cysteine(s) and the presence or absence of non-conserved amino acids between those cysteines (360, 361). The CC chemokine receptors (CCR) are involved in the recruitment of monocytes, neutrophils, lymphocytes and macrophages (34). During bacterial infection, signalling via pattern recognition receptors drive the expression of CC chemokine ligands (CCL) and the development of gradients that are responded to by specific cell surface chemokine receptors (34) with functional recruitment involving monomeric or dimeric forms the respective chemokine ligands (363). CXC chemokine ligands, denoted as CXCL, contain a non-conserved amino acid between the two cysteines, unlike CCL chemokines (360, 361). The number of chemokine ligands outnumbers the chemokine receptors suggesting redundant or highly refined roles for the receptors as in the case for CCR4 expression mediating the migration of both TH1 and TH2 responses (363). This is further exemplified by CXC receptors (CXCR) which are capable of binding multiple CXCLs to promote the migration of specific cells along a chemokine gradient (360). As development of the granuloma and communication between the cells within the granuloma are so critical for control of disease this section will explore the roles chemokines play in modulating TB disease outcome. In accordance with the most recent chemokine nomenclature, CCRs and CCLs as well as CXCRs and their ligands CXCLs, are numbered (e.g. CCR2, CCL2) in a manner that avoids the previous random naming system (364) (Table II).
CC Receptors and their Ligands
CCR1
CCR1 is expressed by T lymphocytes, neutrophils, dendritic cells, monocytes and macrophages (365–369). Under normal conditions CCR1 is constitutively expressed at low levels, but it is upregulated during stimulation of neutrophils with granulocyte-macrophage colony stimulating factor (GM-CSF), and during the differentiation of monocytes to macrophages (369, 370).
In TB patients, CD4+ T lymphocytes expressing CCR1 are elevated in the pleural fluid (371), and in the blood, increased levels of CCR1+ T lymphocytes, natural killer (NK) cells and neutrophils are seen (372). In vitro, human neutrophils express both CCR1 and produce CCL3 upon infection with Mtb (373). Infection with Mtb induces the expression of the CCR1 ligands CCL3, CCL4 and CCL5 in the lung (374, 375), however, CCR1 deficiency in mice does not impact control or disease progression following Mtb infection (35). Thus, it is likely that while CCR1 correlates with cellular activation during TB, it does not appear to play a strong role in protection.
CCR2
CCR2 has a similar distribution pattern as CCR1 on hematopoietic cells (376, 377). Expression of CCR2 on monocytes and its interaction with CCL2 and CCL7 are essential for mobilization of monocytes from the bone marrow into the circulation, and into sites of inflammation (378). Increased levels of CCL2 in the serum of pulmonary TB and TB patients with disseminated disease has been reported (379) and is associated with the damaging influx of inflammatory cells during TB pleurisy (371, 372). At a minimum, in TB patients the presence of CCR2 and its ligands are potential indicators of disease severity.
Low dose aerosol Mtb infection of mice elicits increased expression of CCL2, CCL7 and CCL12 in the lung during the acute stages of Mtb infection (380). Accordingly, mice deficient in CCR2 or CCL2 are defective in macrophage and T lymphocyte recruitment following Mtb infection (380–383) (Figure 1). Consistent with defects in recruitment of immune cells, granuloma formation in CCR2 gene deficient mice is delayed, and is associated with perivascular cuffing and loosely formed granulomas (380). Similarly, Mtb-infected CCL2 gene deficient mice also exhibit decreased granulomatous inflammation in Mtb-infected lungs (382, 383), despite these innate and adaptive immune defects, CCR2 gene deficient mice are not more susceptible to low dose Mtb infection using either the aerosol or intravenous route (380). When taken in the context of the role of the Type I IFNs in recruiting just enough (102) but not too many phagocytes (101), it may be that using a simple gene deficiency model to dissect the role of each chemokine or chemokine receptor will not be informative (Figure 1).
CCR4
CCR4 is expressed on T cell subsets including TH17, TH2, and TREG cells that mediate allergic responses and protection against extracellular pathogens (384–386). T lymphocytes expressing CCR4 are mobilized via a chemokine gradient established by dendritic cells in the context of both allergic and parasite induced inflammation (387–392). In the context of Mtb infection, TREG are increased in the peripheral blood of TB patients and inhibit production of IFNγ by Mtb-specific CD4+ T cells (393, 394) and CCR4 antagonists promote proliferation of T cells during MVA85A vaccination (395). The ligand for CCR4, CCL17 but not CCL22 has also been shown to be elevated in the serum of active TB patients (396). These data suggest that CCR4 mediated regulation of T cell responses to Mtb antigens may occur during TB and that antagonism of CCR4 maybe a target for therapeutic use or as an adjuvant during vaccination. Mechanistic analysis of CCR4 function during mycobacterially-induced granuloma responses demonstrates that despite delivery of mycobacterial and helminth antigens inducing both CCL17 and CCL22 in mouse lungs, the absence of CCR4 results in reduced granuloma formation in following mycobacterial but not helminth antigen challenge (397). Further investigation of the role of CCR4 in development of immunity and immunopathology is likely warranted.
CCR5
CCR5 is expressed on monocytes, macrophages, T lymphocytes, neutrophils and dendritic cells (34, 376, 377, 398) and responds to CCL3, CCL4 and CCL5 (374, 375). CCR5 is most notable in the study of HIV infection, where CCR5 facilitates viral entry into T lymphocytes and macrophages (398, 399). HIV infection dramatically compromises immunity to TB and this is thought to be largely as a result of compromised T cell function, however, blocking CCL5 in cultures of Mtb-infected alveolar macrophages from HIV patients results in enhanced Mtb growth (374), suggesting that CCL5 can directly promote bacterial killing by Mtb-infected macrophages. Interestingly, Analysis of single nucleotide polymorphisms in CCL5 has identified two risk haplotypes, A-C-T and G-C-C that are associated with susceptibility to TB (400). What remains unclear is whether this haplotype is associated with increased or decreased CCL5 production.
In the mouse model of Mtb infection, all three CCR5 ligands are upregulated in the lungs, with CCL5 being induced to the highest level (375, 401). Upon Mtb infection, CCL5 gene deficient mice exhibit transient early impairment in granuloma formation and delayed T cell recruitment (401) while CCR5 deficiency results in increased inflammation in the lung (375) (Figure 1). The observed delayed T cell recruitment into the lungs of CCL5 deficient mice (401) is not entirely surprising as CCR5-CCL5 are important for dendritic cell trafficking to the lymph nodes, facilitating T cell activation and accumulation during TB (375, 401). The difference between the outcome for the CCR5 and CCL5 deficient mice in terms of inflammatory outcome may reflect the action of other CCR5 ligands acting in the absence of CCL5. The increased accumulation of CD4+ and CD8+ T lymphocytes, myeloid cells, neutrophils and macrophages in the lungs of chronically infected CCR5 deficient mice suggests however that any compensating ligand is not optimal at limiting pathologic consequences (375). Whether the models are showing true redundancy in the chemokine or our failure to appreciate the subtle nature of the function of each chemokine is still an issue for debate (402).
CCR6
CCR6 has only one known ligand, CCL20 (403) and is expressed on effector and memory T cells, myeloid dendritic cells, and B cells. CCR6 is important for the recruitment of CCR6 expressing cells to mucosal surfaces and their localization at sites of inflammation in epithelial tissues (403, 404). While CCR6 plays a vital role under both homeostatic and inflammatory conditions, it has been implicated in pathologic conditions, particularly in cancer and rheumatoid arthritis models (405, 406). Although few studies have investigated the role of CCR6 in Mtb infections, emerging data supports a protective role for CCR6 (407). Specifically, memory CD4+ T cells specific for Mtb antigens co-express CXCR3 and CCR6 and these CXCR3+CCR6+ CD4+ T cells are associated with an IFNγ response (386). Following ex vivo expansion, those CD4+ T cells that produced IL-17A in response to Mtb co-expressed CXCR3 and CCR6 (408). In a detailed analysis of reactivity to mycobacterial antigens it was found that CXCR3+CCR6+ IFNγ-producing CD4+ T cells from latently Mtb-infected individuals responded to three immunodominant antigenic islands within the Mtb genome that were all associated with bacterial secretion systems (409) (Figure 2). The CCR6 ligand, CCL20 appears to at high levels in peripheral blood mononuclear cells, myeloid-derived macrophages, and bronchoalveolar lavage samples from TB patients (410, 411) and increased CCL20 mRNA is seen in murine and non-human primate lung (412, 413). It is likely therefore that CCR6 mediates the localization of memory cells to sites of Mtb-induced inflammation. Mouse studies have identified a critical role for CCR6 in the innate immune-mediated control following acute BCG infection (407). Innate cell types, such as CD1b-restricted iNKT cells, are impacted by the absence of CCR6, in that they fail to accumulate effectively in the lung and this is associated with poor bacterial control and increased susceptibility to Mtb infection (407). Collectively, the current literature suggests that there is a correlation between CCR6 expression and bacterial control, however, mechanistic studies investigating the function of CCR6 in innate immune responses as well as its role in directing memory T cell migration requires further study.
CCR7
Correct localization of dendritic cells, T and B cells, is critical for the function of secondary lymphoid tissues (414). CCR7 ligation by CCL19 and CCL21 is vital for the positioning of T cells and dendritic cells in the paracortical region of these secondary lymphoid organs (SLO) (415) (Figure 1 and 2). CCL19 and CCL21 are constitutively expressed by SLO stromal cells while CCL21, but not CCL19, is expressed on lymphatic endothelial and high endothelial venules (415–418). CCR7 has also been implicated in thymic function and development, as well as homeostatic and inflammation-induced migration of dendritic cells to draining lymph nodes via the afferent lymphatics. In Mtb infections, it is thought to be necessary for dendritic cells from the lungs to relocate in the SLOs, activate naïve T cells, and elicit recruitment of T cells into the site of infection for bacterial control. Early migration of mature dendritic cells to the mediastinal lymph node is supported by CCR7 (419) and CCR7 deficient mice exhibit impaired dendritic cell migration to this node (420)(Figure 2). Similarly, plt mutant mice, which lack expression of CCL19 and CCL21-Ser in SLO also have poor dendritic cell migration (421) and proliferation of adoptively transferred Mtb-specific CD4+ T cells is delayed in mice deficient of CCR7 (420) and plt mice (422). Interestingly, the CCR7 chemokine ligand, CCL19, is present within granulomas containing B cells aggregates that resemble lymphoid structures and in the absence of CCR7 these B cell follicles are also absent (416). While these mice display disorganized B cell aggregation, the enhanced lymphocytic infiltrations observed in the lungs of CCR7 gene deficient mice is sufficient to control Mtb to levels comparable to CCR7 sufficient mice (416). Similarly to CCR7 deficient mice, plt mutant mice fail to develop B cell follicles and have disorganized granulomas (422). However, unlike CCR7 deficient mice, plt mutant mice also have delayed accumulation of IFNγ-producing T cells and maintain higher bacterial burden (422). Thus, it seems that CCR7 plays an important role in the proper migration of dendritic cells to the draining lymph node for priming and activation of T cells as well as the migration of CD4+ T cells into lymphoid follicles following Mtb infection.
CXC Receptors and their Ligands
CXCR1 and CXCR2
CXCR1 and CXCR2 share the binding partners, CXCL6 and CXCL8, while CXCR2 can exclusively bind to CXCL1–3, CXCL5, and CXCL7 (423). All of these CXCLs contain the E-L-R motif, corresponding to a Glu-Leu-Arg tripeptide motif at the amino terminus region, adjacent to their CXC sequence. The functional role of this ELR motif is to confer angiogenic activity to the ELR-containing chemokine (424) and this motif is important for ligand-receptor binding interactions on neutrophils (425, 426). Ligand binding to CXCR1 and CXCR2 causes degranulation, intracellular calcium mobilization, and phosphorylation of MAP-kinase (427).
Although most notably expressed on neutrophils in TB patients, both CXCR1 and CXCR2 can also be expressed on NK cells, T cells, and monocytes (423, 428). Comparison of CXCR1 expression on peripheral blood from TB patients shows that individuals with pulmonary TB have increased CXCR1, whereas latent TB patients have increased CXCR2 in whole blood (429).
Moreover, increased CXCR1 correlates with impaired oxidative function in leukocytes suggesting a possible regulatory role for CXCR1 on oxidative stress (429). CXCR2 deficient mice infected intraperitoneally with M.avium have decreased neutrophil accumulation and increased bacterial burden (430). However, following pulmonary infection with M.avium, no difference in cellular infiltrate or bacterial burden is observed, which highlights the importance of route and dose on identifying the function of the highly redundant chemokines.
CXCL8 has been the most studied CXCR1/2 ligand in the context of TB and this important neutrophil chemoattractant is expressed by multiple cell types, including alveolar epithelial cells, monocytes, macrophages, and fibroblasts upon Mtb infection in vitro (431–434) (Figure 1). Following in vivo Mtb infection pulmonary granulomas containing fibroblasts are also capable of secreting CXCL8 (431) and TB sputum samples contain elevated CXCL8 levels (435). CXCL8 is present at high levels at positive tuberculin skin reaction sites and is associated with high levels of neutrophils at this site (436). Furthermore, neutrophils isolated from pulmonary TB patients have increased expression of CXCL8, which is further increased following ex vivo infection with H37Rv but not the clinical strains, S7 and S10, suggesting strain-specific regulation of CXCL8 production in neutrophils (373). Whether CXCL8 is associated with a protective or pathologic response remains controversial (437, 438). Serum CXCL8 levels from pulmonary TB patients following treatment with antibiotics decreases suggesting that CXCL8 could be a marker for treatment efficacy (439). Whether disease improvement is directly correlated to reduced CXCL8 levels or simply a reflection of reduced neutrophil accumulation is unclear (Figure 1).
CXCL5 which is also a CXCR2 ligand plays an important role in host responses against Mtb (440) as both CXCR2 and CXCL5 gene deficient mice display lower bacterial burden following Mtb infection, which correlates with reduced neutrophil accumulation to the lung (440). In this model, alveolar epithelial secretion of CXCL5, mediating the recruitment of neutrophils, is dependent on TLR2 engagement with Mtb and blocking of neutrophil recruitment improves outcome lung (440) (Figure 1). It appears that CXCR2 plays an important role in the pathological granulomatous response during TB and this could be an important target for host directed therapy.
CXCR3
CXCR3 binds to its chemokine ligand partners, CXCL9, CXCL10, and CXCL11, also known as MIG, IP-10 and I-TAC, respectively (428, 441). It is expressed primarily by activated CD4+ and CD8+ T cells, and is detected on B cells and innate lymphocytes such as NK cells and NKT cells (428). CXCR3 expression is associated with supporting localization of cells to the site of infection (442, 443). Most notably, CXCR3 expression on effector T cells plays an important role in the migration of T cells both in vitro and in vivo (33, 441, 444–446) and the transcription factor T-bet transactivates CXCR3 on T cells to promote this migration (441) (Figure 2). It is characterized as an inflammatory chemokine receptor on T cells and is significantly enhanced on T cells from inflamed tissues in human inflammatory reactions and has been implicated in various human and murine disease models, including idiopathic pulmonary fibrosis and asthma (441, 443, 447).
In response to Mtb infection, CXCR3-expressing T cells are found in the caseous, necrotic granulomas and bronchoalveolar lavage in the NHP model, as well as in the lungs of infected mice (428). The presence of these cells correlates with elevated expression of CXCL9, CXCL10, and CXCL11 localized within granulomas (428) however CXCR3 deficient mice do not exhibit differences in bacterial burden following low dose aerosol infection with Mtb (448, 449). Despite the absence of a bacterial phenotype, CXCR3 deficient mice do develop fewer granulomas of decreased size (448, 449) suggesting that CXCR3 is important for granuloma formation. The co-expression of CXCR3 with CCR6 on memory T cells in latently infected humans supports the importance of this receptor on the migration and function of human T cells during TB but the precise role is not clear (Figure 2). Human data support a protective role for CXCR3-binding chemokines as CXCL9 levels correlate with disease severity (379, 450) although the role of CXCL10 is unclear. CXCL10 has been proposed as a biomarker for improvement, as reduction of this chemokine in the serum relative to levels at recruitment is observed following TB treatment and non-responders do not show the same decrease (439, 451). Unfortunately, CXCL10 levels cannot distinguish between active and latent TB in children (452).
The mechanistic basis for CXCL10 activity in TB is not fully defined. In some cases, active TB can result in destruction of the lung parenchyma, leading to cavities. In AIDS-associated TB, increased CXCL10 is associated with non-cavitary TB whereas cavitary TB is associated with increased GM-CSF (114, 453). CXCL10 is important for modulating CXCL9 and CCL2, which promote disease, while CXCL10 is significantly increased in patients with active pulmonary tuberculosis (100) and there may be a genetic association between a single-nucleotide polymorphism within the CXCL-10 promoter (135G/A), and TB susceptibility (454).
Additional studies are needed to confirm whether CXCR3 and its ligands are protective or harmful to disease outcome. It is also unclear how CXCL9–11 are induced and whether their induction is a direct result of Mtb infection or whether upregulated expression results from expression of cytokines capable of inducing CXCL9–11 expression (455–458). Still, animal models support CXCR3 and expression of its ligands as important regulators for granuloma formation (449, 459) (Figure 2).
CXCR5
The B cell chemoattractant, CXCL13, is the only known ligand for CXCR5. It is preferentially produced in B cell follicles and through its binding with CXCR5, is important for the development of lymphoid follicles. In lymphoid organs, the main source for CXCL13 is the follicular dendritic cell (460). However, IL-17-producing T cells have also been shown to express CXCL13 in Candida albicans infection model as well as in synovial fluid from patients with rheumatoid arthritis (461). CXCL13 expression recruits CXCR5-expressing B cells and co-ordinates the correct position of these cells within SLO (462). A similar role has also been highlighted during Mtb infections. Mtb granulomas contain B cells, follicular dendritic cells and high endothelial venules and thus provide important points of entry for trafficking lymphocytes (4, 428). CXCL13 is also elevated in Mtb-infected mice and is important for the localization of CXCR5+ T cells to the lung parenchyma as well as the activation of phagocytes and control of bacterial growth (422, 459) (Figure 2). Using NHP and mouse models of Mtb, CXCR5+ T cells have been shown to traffic towards ectopic lymphoid follicles (Bronchus-Associated Lymphoid Tissues (iBALT)), adjacent to granulomas where CXCL13 is localized. In the absence of CXCR5, mice fail to develop iBALT and are more susceptible Mtb (459) with the impaired response being rescued through the adoptive transfer of CXCR5-sufficient T cells, suggesting that CXCR5 expression on T cells is important for both protection and the development of B cell follicles. Further, CXCR5 is required for the maintenance Mtb-specific CD4+ T cells during chronic infection (81). B cells from Mtb infected murine lungs are also able to migrate along a CXCL13 chemotactic gradient in vitro via their expression of CXCR5 (463). IL-23 and IL-17R dependent induction of CXCL13 within the Mtb-infected lungs appears to be important for control of this infection and CXCL-13 and CXCR5 deficient mice are the only chemokine deficient mice that show increased susceptibility upon Mtb infection, suggesting that this cytokine/chemokine axis is non-redundant in Mtb infection (Figure 2).
CONCLUSION
Chemokines and cytokines are critical for initiating and co-ordinating the organized and sequential recruitment and activation of cells into Mtb-infected lungs (Figures 1 and 2). Correct mononuclear cellular recruitment and localization are essential to ensure control of bacterial growth without the development of diffuse and damaging granulocytic inflammation. An important block to our understanding of TB pathogenesis lies in dissecting the critical aspects of the cytokine/chemokine interplay in light of the conditional role these molecules play throughout infection and disease development (Tables I and II). Much of the data highlighted in this chapter appears at first glance to be contradictory but it is the balance between the cytokines and chemokines which is critical and the ‘goldilocks’ (not too much and not too little) phenomenon is paramount in any discussion of the role of these molecules in TB. Determination of how the key chemokines/cytokines and their receptors are balanced and how the loss of that balance can promote disease is vital to understanding TB pathogenesis and to identifying novel therapies for effective eradication of this disease.
References
- 1.Dinarello CA. Historical Review of Cytokines. Eu J Immunol. 2007;37:S34–S45. doi: 10.1002/eji.200737772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cooper AM. Cell-mediated immune responses in tuberculosis. Ann Rev Immunol. 2009;27:393–422. doi: 10.1146/annurev.immunol.021908.132703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flynn JL, Chan J. Immune evasion by Mycobacterium tuberculosis: living with the enemy. Curr Opin Immunol. 2003;15:450–455. doi: 10.1016/s0952-7915(03)00075-x. [DOI] [PubMed] [Google Scholar]
- 4.Flynn JL, Chan J. Immunology of tuberculosis. Ann Rev Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 5.Brites D, Gagneux S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol Rev. 2015;264:6–24. doi: 10.1111/imr.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, Parkhill J, Malla B, Berg S, Thwaites G, Yeboah-Manu D, Bothamley G, Mei J, Wei L, Bentley S, Harris SR, Niemann S, Diel R, Aseffa A, Gao Q, Young D, Gagneux S. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45:1176–1182. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orme IM, Robinson RT, Cooper AM. The balance between protective and pathogenic immune responses in the TB-infected lung. Nat Immunol. 2015;16:57–63. doi: 10.1038/ni.3048. [DOI] [PubMed] [Google Scholar]
- 8.Dye C, Glaziou P, Floyd K, Raviglione M. Prospects for tuberculosis elimination. Annu Rev Public Health. 2013;34:271–286. doi: 10.1146/annurev-publhealth-031912-114431. [DOI] [PubMed] [Google Scholar]
- 9.Robinson RT, Orme IM, Cooper AM. The onset of adaptive immunity in the mouse model of tuberculosis and the factors that compromise its expression. Immunol Rev. 2015;264:46–59. doi: 10.1111/imr.12259. [DOI] [PubMed] [Google Scholar]
- 10.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 11.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 12.Bazan JF. Emerging families of cytokines and receptors. Curr Biol. 1993;3:603–606. doi: 10.1016/0960-9822(93)90009-d. [DOI] [PubMed] [Google Scholar]
- 13.Devin A, Lin Y, Yamaoka S, Li Z, Karin M, Liu Z. The alpha and beta subunits of IkappaB kinase (IKK) mediate TRAF2-dependent IKK recruitment to tumor necrosis factor (TNF) receptor 1 in response to TNF. Mol Cell Biol. 2001;21:3986–3994. doi: 10.1128/MCB.21.12.3986-3994.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 15.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 16.Jiang Y, Woronicz JD, Liu W, Goeddel DV. Prevention of constitutive TNF receptor 1 signaling by silencer of death domains. Science. 1999;283:543–546. doi: 10.1126/science.283.5401.543. [DOI] [PubMed] [Google Scholar]
- 17.Naismith JH, Sprang SR. Modularity in the TNF-receptor family. Trends Biochem Sci. 1998;23:74–79. doi: 10.1016/s0968-0004(97)01164-x. [DOI] [PubMed] [Google Scholar]
- 18.Banner DW, D’Arcy A, Janes W, Gentz R, Schoenfeld HJ, Broger C, Loetscher H, Lesslauer W. Crystal structure of the soluble human 55 kd TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell. 1993;73:431–445. doi: 10.1016/0092-8674(93)90132-a. [DOI] [PubMed] [Google Scholar]
- 19.Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288:2351–2354. doi: 10.1126/science.288.5475.2351. [DOI] [PubMed] [Google Scholar]
- 20.Faustman DL, Davis M. TNF Receptor 2 and Disease: Autoimmunity and Regenerative Medicine. Front Immunol. 2013;4:478. doi: 10.3389/fimmu.2013.00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Nat Acad Sci USA. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, Kornfeld H. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun. 1997;65:298–304. doi: 10.1128/iai.65.1.298-304.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000;164:2016–2020. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- 24.Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J Immunol. 1998;161:2636–2641. [PubMed] [Google Scholar]
- 25.Serbina NV, Flynn JL. Early emergence of CD8(+) T cells primed for production of type 1 cytokines in the lungs of Mycobacterium tuberculosis-infected mice. Infect Immun. 1999;67:3980–3988. doi: 10.1128/iai.67.8.3980-3988.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 27.Algood HM, Lin PL, Flynn JL. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis. 2005;41(Suppl 3):S189–193. doi: 10.1086/429994. [DOI] [PubMed] [Google Scholar]
- 28.Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol. 2002;168:4620–4627. doi: 10.4049/jimmunol.168.9.4620. [DOI] [PubMed] [Google Scholar]
- 29.Bean AG, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, Britton WJ. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J Immunol. 1999;162:3504–3511. [PubMed] [Google Scholar]
- 30.Lin PL, Plessner HL, Voitenok NN, Flynn JL. Tumor necrosis factor and tuberculosis. J Investig Dermatol Symp Proc. 2007;12:22–25. doi: 10.1038/sj.jidsymp.5650027. [DOI] [PubMed] [Google Scholar]
- 31.Kindler V, Sappino AP, Grau GE, Piguet PF, Vassalli P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell. 1989;56:731–740. doi: 10.1016/0092-8674(89)90676-4. [DOI] [PubMed] [Google Scholar]
- 32.Farber JM. Mig and IP-10: CXC chemokines that target lymphocytes. J Leukoc Biol. 1997;61:246–257. [PubMed] [Google Scholar]
- 33.Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP, Lin W, Boyd JG, Moser B, Wood DE, Sahagan BG, Neote K. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med. 1998;187:2009–2021. doi: 10.1084/jem.187.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Ann Rev Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 35.Saunders BM, Britton WJ. Life and death in the granuloma: immunopathology of tuberculosis. Immunol Cell Biol. 2007;85:103–111. doi: 10.1038/sj.icb.7100027. [DOI] [PubMed] [Google Scholar]
- 36.Lynch K, Farrell M. Cerebral tuberculoma in a patient receiving anti-TNF alpha (adalimumab) treatment. Clin Rheumatol. 2010;29:1201–1204. doi: 10.1007/s10067-010-1466-7. [DOI] [PubMed] [Google Scholar]
- 37.Seong SS, Choi CB, Woo JH, Bae KW, Joung CL, Uhm WS, Kim TH, Jun JB, Yoo DH, Lee JT, Bae SC. Incidence of tuberculosis in Korean patients with rheumatoid arthritis (RA): effects of RA itself and of tumor necrosis factor blockers. J Rheumatol. 2007;34:706–711. [PubMed] [Google Scholar]
- 38.Be NA, Kim KS, Bishai WR, Jain SK. Pathogenesis of central nervous system tuberculosis. Curr Mol Med. 2009;9:94–99. doi: 10.2174/156652409787581655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leonard JM, Des Prez RM. Tuberculous meningitis. Infect Dis Clin North Am. 1990;4:769–787. [PubMed] [Google Scholar]
- 40.Tsenova L, Bergtold A, Freedman VH, Young RA, Kaplan G. Tumor necrosis factor alpha is a determinant of pathogenesis and disease progression in mycobacterial infection in the central nervous system. Proc Natl Acad Sci U S A. 1999;96:5657–5662. doi: 10.1073/pnas.96.10.5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Francisco NM, Hsu NJ, Keeton R, Randall P, Sebesho B, Allie N, Govender D, Quesniaux V, Ryffel B, Kellaway L, Jacobs M. TNF-dependent regulation and activation of innate immune cells are essential for host protection against cerebral tuberculosis. J Neuroinflamm. 2015;12:125. doi: 10.1186/s12974-015-0345-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mohan VP, Scanga CA, Yu K, Scott HM, Tanaka KE, Tsang E, Tsai MM, Flynn JL, Chan J. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect Immun. 2001;69:1847–1855. doi: 10.1128/IAI.69.3.1847-1855.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. 2002;2:364–371. doi: 10.1038/nri802. [DOI] [PubMed] [Google Scholar]
- 44.Peyrin-Biroulet L. Anti-TNF therapy in inflammatory bowel diseases: a huge review. Minerva Gastroenterol Dietol. 2010;56:233–243. [PubMed] [Google Scholar]
- 45.Shaikha SA, Mansour K, Riad H. Reactivation of tuberculosis in three cases of psoriasis after initiation of anti-TNF therapy. Case Rep Dermatol. 2012;4:41–46. doi: 10.1159/000337145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, Braun MM. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–1104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 47.Keane J. TNF-blocking agents and tuberculosis: new drugs illuminate an old topic. Rheumatol. 2005;44:714–720. doi: 10.1093/rheumatology/keh567. [DOI] [PubMed] [Google Scholar]
- 48.Raval A, Akhavan-Toyserkani G, Brinker A, Avigan M. Brief communication: characteristics of spontaneous cases of tuberculosis associated with infliximab. Ann Intern Med. 2007;147:699–702. doi: 10.7326/0003-4819-147-10-200711200-00006. [DOI] [PubMed] [Google Scholar]
- 49.Gomez-Reino JJ, Carmona L, Valverde VR, Mola EM, Montero MD. Treatment of rheumatoid arthritis with tumor necrosis factor inhibitors may predispose to significant increase in tuberculosis risk: a multicenter active-surveillance report. Arthritis Rheum. 2003;48:2122–2127. doi: 10.1002/art.11137. [DOI] [PubMed] [Google Scholar]
- 50.Dixon WG, Watson K, Lunt M, Hyrich KL, Silman AJ, Symmons DP. Rates of serious infection, including site-specific and bacterial intracellular infection, in rheumatoid arthritis patients receiving anti-tumor necrosis factor therapy: Results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2006;54:2368–2376. doi: 10.1002/art.21978. [DOI] [PubMed] [Google Scholar]
- 51.Askling J, Fored CM, Brandt L, Baecklund E, Bertilsson L, Coster L, Geborek P, Jacobsson LT, Lindblad S, Lysholm J, Rantapaa-Dahlqvist S, Saxne T, Romanus V, Klareskog L, Feltelius N. Risk and case characteristics of tuberculosis in rheumatoid arthritis associated with tumor necrosis factor antagonists in Sweden. Arthritis Rheum. 2005;52:1986–1992. doi: 10.1002/art.21137. [DOI] [PubMed] [Google Scholar]
- 52.Tubach F, Salmon D, Ravaud P, Allanore Y, Goupille P, Breban M, Pallot-Prades B, Pouplin S, Sacchi A, Chichemanian RM, Bretagne S, Emilie D, Lemann M, Lortholary O, Mariette X. Risk of tuberculosis is higher with anti-tumor necrosis factor monoclonal antibody therapy than with soluble tumor necrosis factor receptor therapy: The three-year prospective French Research Axed on Tolerance of Biotherapies registry. Arthritis Rheum. 2009;60:1884–1894. doi: 10.1002/art.24632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fallahi-Sichani M, Flynn JL, Linderman JJ, Kirschner DE. Differential risk of tuberculosis reactivation among anti-TNF therapies is due to drug binding kinetics and permeability. J Immunol. 2012;188:3169–3178. doi: 10.4049/jimmunol.1103298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bruns H, Meinken C, Schauenberg P, Harter G, Kern P, Modlin RL, Antoni C, Stenger S. Anti-TNF immunotherapy reduces CD8+ T cell-mediated antimicrobial activity against Mycobacterium tuberculosis in humans. J Clin Invest. 2009;119:1167–1177. doi: 10.1172/JCI38482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin PL, Myers A, Smith LK, Bigbee C, Bigbee M, Fuhrman C, Grieser H, Chiosea I, Voitenek NN, Capuano SV, Klein E, Flynn JL. TNF neutralization results in disseminated disease during acute and latent M. tuberculosis infection with normal granuloma structure. Arthritis Rheum. 2010;62:340–350. doi: 10.1002/art.27271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clay H, Volkman HE, Ramakrishnan L. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity. 2008;29:283–294. doi: 10.1016/j.immuni.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harari A, Rozot V, Bellutti Enders F, Perreau M, Stalder JM, Nicod LP, Cavassini M, Calandra T, Blanchet CL, Jaton K, Faouzi M, Day CL, Hanekom WA, Bart PA, Pantaleo G. Dominant TNF-alpha+ Mycobacterium tuberculosis-specific CD4+ T cell responses discriminate between latent infection and active disease. Nat Med. 2011;17:372–376. doi: 10.1038/nm.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 59.Greenlund AC, Farrar MA, Viviano BL, Schreiber RD. Ligand-induced IFN gamma receptor tyrosine phosphorylation couples the receptor to its signal transduction system (p91) EMBO. 1994;13:1591–1600. doi: 10.1002/j.1460-2075.1994.tb06422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kovarik P, Stoiber D, Novy M, Decker T. Stat1 combines signals derived from IFN-gamma and LPS receptors during macrophage activation. EMBO. 1998;17:3660–3668. doi: 10.1093/emboj/17.13.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Frucht DM, Fukao T, Bogdan C, Schindler H, O’Shea JJ, Koyasu S. IFN-gamma production by antigen-presenting cells: mechanisms emerge. Trends Immunol. 2001;22:556–560. doi: 10.1016/s1471-4906(01)02005-1. [DOI] [PubMed] [Google Scholar]
- 62.Reed JM, Branigan PJ, Bamezai A. Interferon gamma enhances clonal expansion and survival of CD4+ T cells. J Interferon Cytokine Res. 2008;28:611–622. doi: 10.1089/jir.2007.0145. [DOI] [PubMed] [Google Scholar]
- 63.Munder M, Mallo M, Eichmann K, Modolell M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: A novel pathway of autocrine macrophage activation. J Exp Med. 1998;187:2103–2108. doi: 10.1084/jem.187.12.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Otani T, Nakamura S, Toki M, Motoda R, Kurimoto M, Orita K. Identification of IFN-gamma-producing cells in IL-12/IL-18-treated mice. Cell Immunol. 1999;198:111–119. doi: 10.1006/cimm.1999.1589. [DOI] [PubMed] [Google Scholar]
- 65.Zhang SY, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, Picard C, Abel L, Jouanguy E, Casanova JL. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev. 2008;226:29–40. doi: 10.1111/j.1600-065X.2008.00698.x. [DOI] [PubMed] [Google Scholar]
- 66.Filipe-Santos O, Bustamante J, Chapgier A, Vogt G, de Beaucoudrey L, Feinberg J, Jouanguy E, Boisson-Dupuis S, Fieschi C, Picard C, Casanova JL. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol. 2006;18:347–361. doi: 10.1016/j.smim.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 67.Sologuren I, Boisson-Dupuis S, Pestano J, Vincent QB, Fernández-Pérez L, Chapgier A, Cárdenes M, Feinberg J, García-Laorden MI, Picard C, Santiago E, Kong X, Jannière L, Colino E, Herrera-Ramos E, Francés A, Navarrete C, Blanche S, Faria E, Remiszewski P, Cordeiro A, Freeman A, Holland S, Abarca K, Valerón-Lemaur M, Gonçalo-Marques J, Silveira L, García-Castellano JM, Caminero J, Pérez-Arellano JL, Bustamante J, Abel L, Casanova J-L, Rodríguez-Gallego C. Partial recessive IFN-γR1 deficiency: genetic, immunological and clinical features of 14 patients from 11 kindreds. Hum Mol Genet. 2011;20:1509–1523. doi: 10.1093/hmg/ddr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vogt G, Chapgier A, Yang K, Chuzhanova N, Feinberg J, Fieschi C, Boisson-Dupuis S, Alcais A, Filipe-Santos O, Bustamante J, de Beaucoudrey L, Al-Mohsen I, Al-Hajjar S, Al-Ghonaium A, Adimi P, Mirsaeidi M, Khalilzadeh S, Rosenzweig S, de la Calle Martin O, Bauer TR, Puck JM, Ochs HD, Furthner D, Engelhorn C, Belohradsky B, Mansouri D, Holland SM, Schreiber RD, Abel L, Cooper DN, Soudais C, Casanova JL. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat Genet. 2005;37:692–700. doi: 10.1038/ng1581. [DOI] [PubMed] [Google Scholar]
- 69.Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest. 1998;101:2364–2369. doi: 10.1172/JCI2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 73.Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. 2001;2:569–577. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- 74.Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. The relative importance of T Cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J Exp Med. 2001;193:271–280. doi: 10.1084/jem.193.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Green AM, Difazio R, Flynn JL. IFN-gamma from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J Immunol. 2013;190:270–277. doi: 10.4049/jimmunol.1200061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gallegos AM, van Heijst JW, Samstein M, Su X, Pamer EG, Glickman MS. A gamma interferon independent mechanism of CD4 T cell mediated control of M. tuberculosis infection in vivo. PLoS Pathog. 2011;7:e1002052. doi: 10.1371/journal.ppat.1002052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Caruso AM, Serbina N, Klein E, Triebold K, Bloom BR, Flynn JL. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-gamma, yet succumb to tuberculosis. J Immunol. 1999;162:5407–5416. [PubMed] [Google Scholar]
- 78.Saunders BM, Frank AA, Orme IM, Cooper AM. CD4 is required for the development of a protective granulomatous response to pulmonary tuberculosis. Cell Immunol. 2002;216:65–72. doi: 10.1016/s0008-8749(02)00510-5. [DOI] [PubMed] [Google Scholar]
- 79.Serbina NV, Lazarevic V, Flynn JL. CD4(+) T cells are required for the development of cytotoxic CD8(+) T cells during Mycobacterium tuberculosis infection. J Immunol. 2001;167:6991–7000. doi: 10.4049/jimmunol.167.12.6991. [DOI] [PubMed] [Google Scholar]
- 80.Sakai S, Kauffman KD, Schenkel JM, McBerry CC, Mayer-Barber KD, Masopust D, Barber DL. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J Immunol. 2014;192:2965–2969. doi: 10.4049/jimmunol.1400019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moguche AO, Shafiani S, Clemons C, Larson RP, Dinh C, Higdon LE, Cambier CJ, Sissons JR, Gallegos AM, Fink PJ, Urdahl KB. ICOS and Bcl6-dependent pathways maintain a CD4 T cell population with memory-like properties during tuberculosis. J Exp Med. 2015;212:715–728. doi: 10.1084/jem.20141518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Torrado E, Fountain JJ, Liao M, Tighe M, Reiley WW, Lai RP, Meintjes G, Pearl JE, Chen X, Zak DE, Thompson EG, Aderem A, Ghilardi N, Solache A, McKinstry KK, Strutt TM, Wilkinson RJ, Swain SL, Cooper AM. Interleukin 27R regulates CD4+ T cell phenotype and impacts protective immunity during Mycobacterium tuberculosis infection. J Exp Med. 2015;212:1449–1463. doi: 10.1084/jem.20141520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Keller C, Hoffmann R, Lang R, Brandau S, Hermann C, Ehlers S. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infect Immun. 2006;74:4295–4309. doi: 10.1128/IAI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eruslanov EB, Lyadova IV, Kondratieva TK, Majorov KB, Scheglov IV, Orlova MO, Apt AS. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun. 2005;73:1744–1753. doi: 10.1128/IAI.73.3.1744-1753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Majorov KB, Eruslanov EB, Rubakova EI, Kondratieva TK, Apt AS. Analysis of cellular phenotypes that mediate genetic resistance to tuberculosis using a radiation bone marrow chimera approach. Infect Immun. 2005;73:6174–6178. doi: 10.1128/IAI.73.9.6174-6178.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mitsos LM, Cardon LR, Fortin A, Ryan L, LaCourse R, North RJ, Gros P. Genetic control of susceptibility to infection with Mycobacterium tuberculosis in mice. Genes Immun. 2000;1:467–477. doi: 10.1038/sj.gene.6363712. [DOI] [PubMed] [Google Scholar]
- 87.Nandi B, Behar SM. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208:2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Desvignes L, Ernst JD. Interferon-γ-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity. 2009;31:974–985. doi: 10.1016/j.immuni.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stefan DC, Dippenaar A, Detjen AK, Schaaf HS, Marais BJ, Kriel B, Loebenberg L, Walzl G, Hesseling AC. Interferon-gamma release assays for the detection of Mycobacterium tuberculosis infection in children with cancer. Int J Tuberc Lung Dis. 2010;14:689–694. [PubMed] [Google Scholar]
- 90.Abu-Taleb AM, El-Sokkary RH, El Tarhouny SA. Interferon-gamma release assay for detection of latent tuberculosis infection in casual and close contacts of tuberculosis cases. East Mediterr Health J. 2011;17:749–753. doi: 10.26719/2011.17.10.749. [DOI] [PubMed] [Google Scholar]
- 91.Ferrara G, Losi M, D’Amico R, Cagarelli R, Pezzi AM, Meacci M, Meccugni B, Marchetti Dori I, Rumpianesi F, Roversi P, Casali L, Fabbri LM, Richeldi L. Interferon-gamma-release assays detect recent tuberculosis re-infection in elderly contacts. Int J Immunopathol Pharmacol. 2009;22:669–677. doi: 10.1177/039463200902200312. [DOI] [PubMed] [Google Scholar]
- 92.Diel R, Loddenkemper R, Niemann S, Meywald-Walter K, Nienhaus A. Negative and positive predictive value of a whole-blood interferon-gamma release assay for developing active tuberculosis: an update. Am J Respir Crit Care Med. 2011;183:88–95. doi: 10.1164/rccm.201006-0974OC. [DOI] [PubMed] [Google Scholar]
- 93.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147:258–267. [PubMed] [Google Scholar]
- 94.McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 96.Cooper AM, Pearl JE, Brooks JV, Ehlers S, Orme IM. Expression of the nitric oxide synthase 2 gene is not essential for early control of Mycobacterium tuberculosis in the murine lung. Infect Immun. 2000;68:6879–6882. doi: 10.1128/iai.68.12.6879-6882.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, Barry CE, 3rd, Freedman VH, Kaplan G. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce TH1 type immunity and is associated with induction of IFN-alpha /beta. Proc Natl Acad Sci U S A. 2001;98:5752–5757. doi: 10.1073/pnas.091096998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ordway D, Henao-Tamayo M, Harton M, Palanisamy G, Troudt J, Shanley C, Basaraba RJ, Orme IM. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol. 2007;179:522–531. doi: 10.4049/jimmunol.179.1.522. [DOI] [PubMed] [Google Scholar]
- 99.McNab FW, Ewbank J, Howes A, Moreira-Teixeira L, Martirosyan A, Ghilardi N, Saraiva M, O’Garra A. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-gamma for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J Immunol. 2014;193:3600–3612. doi: 10.4049/jimmunol.1401088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Antonelli LR, Gigliotti Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A, Salazar AM, Feng CG, Sher A. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest. 2010;120:1674–1682. doi: 10.1172/JCI40817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Desvignes L, Wolf AJ, Ernst JD. Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J Immunol. 2012;188:6205–6215. doi: 10.4049/jimmunol.1200255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Van Snick J. Interleukin-6: an overview. Ann Rev Immunol. 1990;8:253–278. doi: 10.1146/annurev.iy.08.040190.001345. [DOI] [PubMed] [Google Scholar]
- 104.Shalaby MR, Waage A, Espevik T. Cytokine regulation of interleukin 6 production by human endothelial cells. Cell Immunol. 1989;121:372–382. doi: 10.1016/0008-8749(89)90036-1. [DOI] [PubMed] [Google Scholar]
- 105.Sanceau J, Beranger F, Gaudelet C, Wietzerbin J. IFN-gamma is an essential cosignal for triggering IFN-beta 2/BSF-2/IL-6 gene expression in human monocytic cell lines. Ann N Y Acad Sci. 1989;557:130–141. discussion 141–133. [PubMed] [Google Scholar]
- 106.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334(Pt 2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ladel CH, Blum C, Dreher A, Reifenberg K, Kopf M, Kaufmann SH. Lethal tuberculosis in interleukin-6-deficient mutant mice. Infect Immun. 1997;65:4843–4849. doi: 10.1128/iai.65.11.4843-4849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Appelberg R, Castro AG, Pedrosa J, Minoprio P. Role of interleukin-6 in the induction of protective T cells during mycobacterial infections in mice. Immunology. 1994;82:361–364. [PMC free article] [PubMed] [Google Scholar]
- 110.Saunders BM, Frank AA, Orme IM, Cooper AM. Interleukin-6 induces early gamma interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infection and Immunity. 2000;68:3322–3326. doi: 10.1128/iai.68.6.3322-3326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Leal IS, Smedegard B, Andersen P, Appelberg R. Interleukin-6 and interleukin-12 participate in induction of a type 1 protective T-cell response during vaccination with a tuberculosis subunit vaccine. Infect Immun. 1999;67:5747–5754. doi: 10.1128/iai.67.11.5747-5754.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Atreya R, Neurath MF. Involvement of IL-6 in the pathogenesis of inflammatory bowel disease and colon cancer. Clin Rev Allergy Immunol. 2005;28:187–196. doi: 10.1385/CRIAI:28:3:187. [DOI] [PubMed] [Google Scholar]
- 113.Sodenkamp J, Waetzig GH, Scheller J, Seegert D, Grotzinger J, Rose-John S, Ehlers S, Holscher C. Therapeutic targeting of interleukin-6 trans-signaling does not affect the outcome of experimental tuberculosis. Immunobiology. 2012;217:996–1004. doi: 10.1016/j.imbio.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 114.Nolan A, Condos R, Huie ML, Dawson R, Dheda K, Bateman E, Rom WN, Weiden MD. Elevated IP-10 and IL-6 from bronchoalveolar lavage cells are biomarkers of non-cavitary tuberculosis. Int J Tuberc Lung Dis. 2013;17:922–927. doi: 10.5588/ijtld.12.0610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.el-Ahmady O, Mansour M, Zoeir H, Mansour O. Elevated concentrations of interleukins and leukotriene in response to Mycobacterium tuberculosis infection. Ann Clin Biochem. 1997;34(Pt 2):160–164. doi: 10.1177/000456329703400205. [DOI] [PubMed] [Google Scholar]
- 116.Dinarello CA. Interleukin-1 and interleukin-1 antagonism. Blood. 1991;77:1627–1652. [PubMed] [Google Scholar]
- 117.Menkin V. The effect of the leukocytosis-promoting factor on the growth of cells in the bone marrow. Am J Pathol. 1943;19:1021–1029. [PMC free article] [PubMed] [Google Scholar]
- 118.Menkin V. Studies on the isolation of the factor responsible for tissue injury in inflammation. Science. 1943;97:165–167. doi: 10.1126/science.97.2511.165. [DOI] [PubMed] [Google Scholar]
- 119.Menkin V. Chemical basis of fever. Science. 1944;100:337–338. doi: 10.1126/science.100.2598.337. [DOI] [PubMed] [Google Scholar]
- 120.Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, Quadroni M, Drexler SK, Tschopp J. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 121.Sansonetti PJ, Phalipon A, Arondel J, Thirumalai K, Banerjee S, Akira S, Takeda K, Zychlinsky A. Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity. 2000;12:581–590. doi: 10.1016/s1074-7613(00)80209-5. [DOI] [PubMed] [Google Scholar]
- 122.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 124.Bossaller L, Chiang PI, Schmidt-Lauber C, Ganesan S, Kaiser WJ, Rathinam VA, Mocarski ES, Subramanian D, Green DR, Silverman N, Fitzgerald KA, Marshak-Rothstein A, Latz E. Cutting edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol. 2012;189:5508–5512. doi: 10.4049/jimmunol.1202121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 126.Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187:4835–4843. doi: 10.4049/jimmunol.1102048. [DOI] [PubMed] [Google Scholar]
- 127.Berda-Haddad Y, Robert S, Salers P, Zekraoui L, Farnarier C, Dinarello CA, Dignat-George F, Kaplanski G. Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1alpha. Proc Natl Acad Sci U S A. 2011;108:20684–20689. doi: 10.1073/pnas.1116848108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Botelho FM, Bauer CM, Finch D, Nikota JK, Zavitz CC, Kelly A, Lambert KN, Piper S, Foster ML, Goldring JJ, Wedzicha JA, Bassett J, Bramson J, Iwakura Y, Sleeman M, Kolbeck R, Coyle AJ, Humbles AA, Stampfli MR. IL-1alpha/IL-1R1 expression in chronic obstructive pulmonary disease and mechanistic relevance to smoke-induced neutrophilia in mice. PLoS One. 2011;6:e28457. doi: 10.1371/journal.pone.0028457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Freigang S, Ampenberger F, Weiss A, Kanneganti T-D, Iwakura Y, Hersberger M, Kopf M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1[alpha] and sterile vascular inflammation in atherosclerosis. Nat Immunol. 2013;14:1045–1053. doi: 10.1038/ni.2704. [DOI] [PubMed] [Google Scholar]
- 130.Barry KC, Fontana MF, Portman JL, Dugan AS, Vance RE. IL-1alpha signaling initiates the inflammatory response to virulent Legionella pneumophila in vivo. J Immunol. 2013;190:6329–6339. doi: 10.4049/jimmunol.1300100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Biondo C, Mancuso G, Midiri A, Signorino G, Domina M, Lanza Cariccio V, Mohammadi N, Venza M, Venza I, Teti G, Beninati C. The interleukin-1beta/CXCL1/2/neutrophil axis mediates host protection against group B streptococcal infection. Infect Immun. 2014;82:4508–4517. doi: 10.1128/IAI.02104-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Guo H, Gao J, Taxman DJ, Ting JP, Su L. HIV-1 infection induces interleukin-1beta production via TLR8 protein-dependent and NLRP3 inflammasome mechanisms in human monocytes. J Biol Chem. 2014;289:21716–21726. doi: 10.1074/jbc.M114.566620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rynko AE, Fryer AD, Jacoby DB. Interleukin-1beta mediates virus-induced m2 muscarinic receptor dysfunction and airway hyperreactivity. Am J Respir Cell Mol Biol. 2014;51:494–501. doi: 10.1165/rcmb.2014-0009OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shigematsu Y, Niwa T, Rehnberg E, Toyoda T, Yoshida S, Mori A, Wakabayashi M, Iwakura Y, Ichinose M, Kim YJ, Ushijima T. Interleukin-1beta induced by Helicobacter pylori infection enhances mouse gastric carcinogenesis. Cancer Lett. 2013;340:141–147. doi: 10.1016/j.canlet.2013.07.034. [DOI] [PubMed] [Google Scholar]
- 135.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Konsman JP, Vigues S, Mackerlova L, Bristow A, Blomqvist A. Rat brain vascular distribution of interleukin-1 type-1 receptor immunoreactivity: relationship to patterns of inducible cyclooxygenase expression by peripheral inflammatory stimuli. J Comp Neurol. 2004;472:113–129. doi: 10.1002/cne.20052. [DOI] [PubMed] [Google Scholar]
- 137.Marshall JD, Aste-Amezaga M, Chehimi SS, Murphy M, Olsen H, Trinchieri G. Regulation of human IL-18 mRNA expression. Clin Immunol. 1999;90:15–21. doi: 10.1006/clim.1998.4633. [DOI] [PubMed] [Google Scholar]
- 138.Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci U S A. 1999;96:2256–2261. doi: 10.1073/pnas.96.5.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sugawara S, Uehara A, Nochi T, Yamaguchi T, Ueda H, Sugiyama A, Hanzawa K, Kumagai K, Okamura H, Takada H. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J Immunol. 2001;167:6568–6575. doi: 10.4049/jimmunol.167.11.6568. [DOI] [PubMed] [Google Scholar]
- 140.Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol. 2013;4:289. doi: 10.3389/fimmu.2013.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Holscher C, Reiling N, Schaible UE, Holscher A, Bathmann C, Korbel D, Lenz I, Sonntag T, Kroger S, Akira S, Mossmann H, Kirschning CJ, Wagner H, Freudenberg M, Ehlers S. Containment of aerogenic Mycobacterium tuberculosis infection in mice does not require MyD88 adaptor function for TLR2, -4 and -9. Eur J Immunol. 2008;38:680–694. doi: 10.1002/eji.200736458. [DOI] [PubMed] [Google Scholar]
- 142.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 143.Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest. 2004;114:1790–1799. doi: 10.1172/JCI21027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, Jacobs M, Ryffel B, Quesniaux VF. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol. 2007;179:1178–1189. doi: 10.4049/jimmunol.179.2.1178. [DOI] [PubMed] [Google Scholar]
- 145.Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, Sher A. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity. 2011;35:1023–1034. doi: 10.1016/j.immuni.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bourigault ML, Segueni N, Rose S, Court N, Vacher R, Vasseur V, Erard F, Le Bert M, Garcia I, Iwakura Y, Jacobs M, Ryffel B, Quesniaux VF. Relative contribution of IL-1alpha, IL-1beta and TNF to the host response to Mycobacterium tuberculosis and attenuated M. bovis BCG. Immun Inflamm Dis. 2013;1:47–62. doi: 10.1002/iid3.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Di Paolo NC, Shafiani S, Day T, Papayannoupoulou T, Russell DW, Iwakura Y, Sherman D, Urdahl K, Shayakhmetov DM. Interdependence between Interleukin-1 and Tumor Necrosis Factor Regulates TNF-Dependent Control of Mycobacterium tuberculosis Infection. Immunity. 2015;43:1125–1136. doi: 10.1016/j.immuni.2015.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Guler R, Parihar SP, Spohn G, Johansen P, Brombacher F, Bachmann MF. Blocking IL-1alpha but not IL-1beta increases susceptibility to chronic Mycobacterium tuberculosis infection in mice. Vaccine. 2011;29:1339–1346. doi: 10.1016/j.vaccine.2010.10.045. [DOI] [PubMed] [Google Scholar]
- 149.Gopal R, Monin L, Slight S, Uche U, Blanchard E, Fallert Junecko BA, Ramos-Payan R, Stallings CL, Reinhart TA, Kolls JK, Kaushal D, Nagarajan U, Rangel-Moreno J, Khader SA. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog. 2014;10:e1004099. doi: 10.1371/journal.ppat.1004099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schneider BE, Korbel D, Hagens K, Koch M, Raupach B, Enders J, Kaufmann SH, Mittrucker HW, Schaible UE. A role for IL-18 in protective immunity against Mycobacterium tuberculosis. Eur J Immunol. 2010;40:396–405. doi: 10.1002/eji.200939583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Suwara MI, Green NJ, Borthwick LA, Mann J, Mayer-Barber KD, Barron L, Corris PA, Farrow SN, Wynn TA, Fisher AJ, Mann DA. IL-1alpha released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol. 2014;7:684–693. doi: 10.1038/mi.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fielding CA, McLoughlin RM, McLeod L, Colmont CS, Najdovska M, Grail D, Ernst M, Jones SA, Topley N, Jenkins BJ. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J Immunol. 2008;181:2189–2195. doi: 10.4049/jimmunol.181.3.2189. [DOI] [PubMed] [Google Scholar]
- 153.Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KH. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol. 2011;186:5738–5748. doi: 10.4049/jimmunol.1003597. [DOI] [PubMed] [Google Scholar]
- 154.Dunne A, Ross PJ, Pospisilova E, Masin J, Meaney A, Sutton CE, Iwakura Y, Tschopp J, Sebo P, Mills KH. Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J Immunol. 2010;185:1711–1719. doi: 10.4049/jimmunol.1000105. [DOI] [PubMed] [Google Scholar]
- 155.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Monin L, Griffiths KL, Slight S, Lin Y, Rangel-Moreno J, Khader SA. Immune requirements for protective Th17 recall responses to Mycobacterium tuberculosis challenge. Mucosal Immunol. 2015;8:1099–1109. doi: 10.1038/mi.2014.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, Locksley RM, Haynes L, Randall TD, Cooper AM. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 158.Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, Yuan X, Zhang G, Cai Y, Babu S, Catalfamo M, Salazar AM, Via LE, Barry CE, 3rd, Sher A. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014;511:99–103. doi: 10.1038/nature13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tominaga K, Yoshimoto T, Torigoe K, Kurimoto M, Matsui K, Hada T, Okamura H, Nakanishi K. IL-12 synergizes with IL-18 or IL-1beta for IFN-gamma production from human T cells. Int Immunol. 2000;12:151–160. doi: 10.1093/intimm/12.2.151. [DOI] [PubMed] [Google Scholar]
- 160.Okamura H, Kashiwamura S, Tsutsui H, Yoshimoto T, Nakanishi K. Regulation of interferon-gamma production by IL-12 and IL-18. Curr Opin Immunol. 1998;10:259–264. doi: 10.1016/s0952-7915(98)80163-5. [DOI] [PubMed] [Google Scholar]
- 161.Bohn E, Sing A, Zumbihl R, Bielfeldt C, Okamura H, Kurimoto M, Heesemann J, Autenrieth IB. IL-18 (IFN-gamma-inducing factor) regulates early cytokine production in, and promotes resolution of, bacterial infection in mice. J Immunol. 1998;160:299–307. [PubMed] [Google Scholar]
- 162.Sugawara I, Yamada H, Kaneko H, Mizuno S, Takeda K, Akira S. Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disrupted mice. Infect Immun. 1999;67:2585–2589. doi: 10.1128/iai.67.5.2585-2589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kinjo Y, Kawakami K, Uezu K, Yara S, Miyagi K, Koguchi Y, Hoshino T, Okamoto M, Kawase Y, Yokota K, Yoshino K, Takeda K, Akira S, Saito A. Contribution of IL-18 to Th1 response and host defense against infection by Mycobacterium tuberculosis: a comparative study with IL-12p40. J Immunol. 2002;169:323–329. doi: 10.4049/jimmunol.169.1.323. [DOI] [PubMed] [Google Scholar]
- 164.Jones LL, Vignali DA. Molecular interactions within the IL-6/IL-12 cytokine/receptor superfamily. Immunol Res. 2011;51:5–14. doi: 10.1007/s12026-011-8209-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Collison LW, Vignali DA. Interleukin-35: odd one out or part of the family? Immunol Rev. 2008;226:248–262. doi: 10.1111/j.1600-065X.2008.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13:722–728. doi: 10.1038/ni.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mendez-Samperio P. Role of interleukin-12 family cytokines in the cellular response to mycobacterial disease. Int J Infect Dis. 2010;14:e366–371. doi: 10.1016/j.ijid.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 168.Hunter CA. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat Rev Immunol. 2005;5:521–531. doi: 10.1038/nri1648. [DOI] [PubMed] [Google Scholar]
- 169.Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med. 1989;170:827–845. doi: 10.1084/jem.170.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gately MK, Desai BB, Wolitzky AG, Quinn PM, Dwyer CM, Podlaski FJ, Familletti PC, Sinigaglia F, Chizonnite R, Gubler U, et al. Regulation of human lymphocyte proliferation by a heterodimeric cytokine, IL-12 (cytotoxic lymphocyte maturation factor) J Immunol. 1991;147:874–882. [PubMed] [Google Scholar]
- 171.Ma X, Trinchieri G. Regulation of interleukin-12 production in antigen-presenting cells. Adv Immunol. 2001;79:55–92. doi: 10.1016/s0065-2776(01)79002-5. [DOI] [PubMed] [Google Scholar]
- 172.O’Shea JJ, Paul WE. Regulation of T(H)1 differentiation–controlling the controllers. Nat Immunol. 2002;3:506–508. doi: 10.1038/ni0602-506. [DOI] [PubMed] [Google Scholar]
- 173.Ozbek N, Fieschi C, Yilmaz BT, de Beaucoudrey L, Demirhan B, Feinberg J, Bikmaz YE, Casanova JL. Interleukin-12 receptor beta 1 chain deficiency in a child with disseminated tuberculosis. Clin Infect Dis. 2005;40:e55–58. doi: 10.1086/427879. [DOI] [PubMed] [Google Scholar]
- 174.Dorman SE, Holland SM. Interferon-gamma and interleukin-12 pathway defects and human disease. Cytokine Growth Factor Rev. 2000;11:321–333. doi: 10.1016/s1359-6101(00)00010-1. [DOI] [PubMed] [Google Scholar]
- 175.Picard C, Fieschi C, Altare F, Al-Jumaah S, Al-Hajjar S, Feinberg J, Dupuis S, Soudais C, Al-Mohsen IZ, Genin E, Lammas D, Kumararatne DS, Leclerc T, Rafii A, Frayha H, Murugasu B, Wah LB, Sinniah R, Loubser M, Okamoto E, Al-Ghonaium A, Tufenkeji H, Abel L, Casanova JL. Inherited interleukin-12 deficiency: IL12B genotype and clinical phenotype of 13 patients from six kindreds. Am J Hum Genet. 2002;70:336–348. doi: 10.1086/338625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Altare F, Ensser A, Breiman A, Reichenbach J, Baghdadi JE, Fischer A, Emile JF, Gaillard JL, Meinl E, Casanova JL. Interleukin-12 receptor beta1 deficiency in a patient with abdominal tuberculosis. J Infect Dis. 2001;184:231–236. doi: 10.1086/321999. [DOI] [PubMed] [Google Scholar]
- 177.Caragol I, Raspall M, Fieschi C, Feinberg J, Larrosa MN, Hernandez M, Figueras C, Bertran JM, Casanova JL, Espanol T. Clinical tuberculosis in 2 of 3 siblings with interleukin-12 receptor beta1 deficiency. Clin Infect Dis. 2003;37:302–306. doi: 10.1086/375587. [DOI] [PubMed] [Google Scholar]
- 178.Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Ann Rev Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- 179.Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, Mansouri N, Okada S, Bryant VL, Kong XF, Kreins A, Velez MM, Boisson B, Khalilzadeh S, Ozcelik U, Darazam IA, Schoggins JW, Rice CM, Al-Muhsen S, Behr M, Vogt G, Puel A, Bustamante J, Gros P, Huibregtse JM, Abel L, Boisson-Dupuis S, Casanova JL. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science. 2012;337:1684–1688. doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, Grant AV, Marchal CC, Hubeau M, Chapgier A, de Beaucoudrey L, Puel A, Feinberg J, Valinetz E, Janniere L, Besse C, Boland A, Brisseau JM, Blanche S, Lortholary O, Fieschi C, Emile JF, Boisson-Dupuis S, Al-Muhsen S, Woda B, Newburger PE, Condino-Neto A, Dinauer MC, Abel L, Casanova JL. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. 2011;12:213–221. doi: 10.1038/ni.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bustamante J, Picard C, Boisson-Dupuis S, Abel L, Casanova J-L. Genetic lessons learnt from X-linked Mendelian susceptibility to mycobacterial diseases. Ann N Y Acad Sci. 2011;1246:92–101. doi: 10.1111/j.1749-6632.2011.06273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Filipe-Santos O, Bustamante J, Haverkamp MH, Vinolo E, Ku CL, Puel A, Frucht DM, Christel K, von Bernuth H, Jouanguy E, Feinberg J, Durandy A, Senechal B, Chapgier A, Vogt G, de Beaucoudrey L, Fieschi C, Picard C, Garfa M, Chemli J, Bejaoui M, Tsolia MN, Kutukculer N, Plebani A, Notarangelo L, Bodemer C, Geissmann F, Israel A, Veron M, Knackstedt M, Barbouche R, Abel L, Magdorf K, Gendrel D, Agou F, Holland SM, Casanova JL. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. 2006;203:1745–1759. doi: 10.1084/jem.20060085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhang M, Gately MK, Wang E, Gong J, Wolf SF, Lu S, Modlin RL, Barnes PF. Interleukin 12 at the site of disease in tuberculosis. J Clin Invest. 1994;93:1733–1739. doi: 10.1172/JCI117157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cooper AM, Roberts AD, Rhoades ER, Callahan JE, Getzy DM, Orme IM. The role of interleukin-12 in acquired immunity to Mycobacterium tuberculosis infection. Immunology. 1995;84:423–432. [PMC free article] [PubMed] [Google Scholar]
- 185.Cooper AM, Magram J, Ferrante J, Orme IM. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with mycobacterium tuberculosis. J Exp Med. 1997;186:39–45. doi: 10.1084/jem.186.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cooper AM, Kipnis A, Turner J, Magram J, Ferrante J, Orme IM. Mice lacking bioactive IL-12 can generate protective, antigen-specific cellular responses to mycobacterial infection only if the IL-12 p40 subunit is present. J Immunol. 2002;168:1322–1327. doi: 10.4049/jimmunol.168.3.1322. [DOI] [PubMed] [Google Scholar]
- 187.Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, Ghilardi N, deSauvage F, Cooper AM. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol. 2005;175:788–795. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 188.Feng CG, Jankovic D, Kullberg M, Cheever A, Scanga CA, Hieny S, Caspar P, Yap GS, Sher A. Maintenance of pulmonary Th1 effector function in chronic tuberculosis requires persistent IL-12 production. J Immunol. 2005;174:4185–4192. doi: 10.4049/jimmunol.174.7.4185. [DOI] [PubMed] [Google Scholar]
- 189.Cleary AM, Tu W, Enright A, Giffon T, Dewaal-Malefyt R, Gutierrez K, Lewis DB. Impaired accumulation and function of memory CD4 T cells in human IL-12 receptor beta 1 deficiency. J Immunol. 2003;170:597–603. doi: 10.4049/jimmunol.170.1.597. [DOI] [PubMed] [Google Scholar]
- 190.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med. 2005;202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Pathak SK, Basu S, Bhattacharyya A, Pathak S, Kundu M, Basu J. Mycobacterium tuberculosis lipoarabinomannan-mediated IRAK-M induction negatively regulates Toll-like receptor-dependent interleukin-12 p40 production in macrophages. J Biol Chem. 2005;280:42794–42800. doi: 10.1074/jbc.M506471200. [DOI] [PubMed] [Google Scholar]
- 192.Pecora ND, Gehring AJ, Canaday DH, Boom WH, Harding CV. Mycobacterium tuberculosis LprA is a lipoprotein agonist of TLR2 that regulates innate immunity and APC function. J Immunol. 2006;177:422–429. doi: 10.4049/jimmunol.177.1.422. [DOI] [PubMed] [Google Scholar]
- 193.Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu CY, Gately MK, Gubler U. A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor subunits. Proc Natl Acad Sci U S A. 1996;93:14002–14007. doi: 10.1073/pnas.93.24.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chua AO, Chizzonite R, Desai BB, Truitt TP, Nunes P, Minetti LJ, Warrier RR, Presky DH, Levine JF, Gately MK, et al. Expression cloning of a human IL-12 receptor component. A new member of the cytokine receptor superfamily with strong homology to gp130. J Immunol. 1994;153:128–136. [PubMed] [Google Scholar]
- 195.Chua AO, Wilkinson VL, Presky DH, Gubler U. Cloning and characterization of a mouse IL-12 receptor-beta component. J Immunol. 1995;155:4286–4294. [PubMed] [Google Scholar]
- 196.Gillessen S, Carvajal D, Ling P, Podlaski FJ, Stremlo DL, Familletti PC, Gubler U, Presky DH, Stern AS, Gately MK. Mouse interleukin-12 (IL-12) p40 homodimer: a potent IL-12 antagonist. Eur J Immunol. 1995;25:200–206. doi: 10.1002/eji.1830250133. [DOI] [PubMed] [Google Scholar]
- 197.Gately MK, Carvajal DM, Connaughton SE, Gillessen S, Warrier RR, Kolinsky KD, Wilkinson VL, Dwyer CM, Higgins GF, Jr, Podlaski FJ, Faherty DA, Familletti PC, Stern AS, Presky DH. Interleukin-12 antagonist activity of mouse interleukin-12 p40 homodimer in vitro and in vivo. Ann N Y Acad Sci. 1996;795:1–12. doi: 10.1111/j.1749-6632.1996.tb52650.x. [DOI] [PubMed] [Google Scholar]
- 198.Mattner F, Fischer S, Guckes S, Jin S, Kaulen H, Schmitt E, Rude E, Germann T. The interleukin-12 subunit p40 specifically inhibits effects of the interleukin-12 heterodimer. Eur J Immunol. 1993;23:2202–2208. doi: 10.1002/eji.1830230923. [DOI] [PubMed] [Google Scholar]
- 199.Holscher C, Atkinson RA, Arendse B, Brown N, Myburgh E, Alber G, Brombacher F. A protective and agonistic function of IL-12p40 in mycobacterial infection. J Immunol. 2001;167:6957–6966. doi: 10.4049/jimmunol.167.12.6957. [DOI] [PubMed] [Google Scholar]
- 200.Khader SA, Partida-Sanchez S, Bell G, Jelley-Gibbs DM, Swain S, Pearl JE, Ghilardi N, Desauvage FJ, Lund FE, Cooper AM. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J Exp Med. 2006;203:1805–1815. doi: 10.1084/jem.20052545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Reinhardt RL, Hong S, Kang SJ, Wang ZE, Locksley RM. Visualization of IL-12/23p40 in vivo reveals immunostimulatory dendritic cell migrants that promote Th1 differentiation. J Immunol. 2006;177:1618–1627. doi: 10.4049/jimmunol.177.3.1618. [DOI] [PubMed] [Google Scholar]
- 202.Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, Ernst JD. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med. 2008;205:105–115. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Robinson RT, Khader SA, Martino CA, Fountain JJ, Teixeira-Coelho M, Pearl JE, Smiley ST, Winslow GM, Woodland DL, Walter MJ, Conejo-Garcia JR, Gubler U, Cooper AM. Mycobacterium tuberculosis infection induces il12rb1 splicing to generate a novel IL-12Rbeta1 isoform that enhances DC migration. J Exp Med. 2010;207:591–605. doi: 10.1084/jem.20091085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Keeton R, Allie N, Dambuza I, Abel B, Hsu NJ, Sebesho B, Randall P, Burger P, Fick E, Quesniaux VF, Ryffel B, Jacobs M. Soluble TNFRp75 regulates host protective immunity against Mycobacterium tuberculosis. J Clin Invest. 2014;124:1537–1551. doi: 10.1172/JCI45005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 206.Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, Robinson N, Buonocore S, Tlaskalova-Hogenova H, Cua DJ, Powrie F. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–318. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 207.Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, Cua DJ. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. 2015;21:719–729. doi: 10.1038/nm.3895. [DOI] [PubMed] [Google Scholar]
- 208.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 209.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12– and IL-23–modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 212.Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. 2011;12:255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Ann Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 214.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, To W, Wagner J, O’Farrell AM, McClanahan T, Zurawski S, Hannum C, Gorman D, Rennick DM, Kastelein RA, de Waal Malefyt R, Moore KW. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 215.Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O’Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. 2004;202:139–156. doi: 10.1111/j.0105-2896.2004.00211.x. [DOI] [PubMed] [Google Scholar]
- 216.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 217.Wozniak TM, Ryan AA, Triccas JA, Britton WJ. Plasmid interleukin-23 (IL-23), but not plasmid IL-27, enhances the protective efficacy of a DNA vaccine against Mycobacterium tuberculosis infection. Infect Immun. 2006;74:557–565. doi: 10.1128/IAI.74.1.557-565.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–4669. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- 219.Khader SA, Guglani L, Rangel-Moreno J, Gopal R, Junecko BA, Fountain JJ, Martino C, Pearl JE, Tighe M, Lin YY, Slight S, Kolls JK, Reinhart TA, Randall TD, Cooper AM. IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J Immunol. 2011;187:5402–5407. doi: 10.4049/jimmunol.1101377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Happel KI, Lockhart EA, Mason CM, Porretta E, Keoshkerian E, Odden AR, Nelson S, Ramsay AJ. Pulmonary interleukin-23 gene delivery increases local T-cell immunity and controls growth of Mycobacterium tuberculosis in the lungs. Infect Immun. 2005;73:5782–5788. doi: 10.1128/IAI.73.9.5782-5788.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gopal R, Rangel-Moreno J, Slight S, Lin Y, Nawar HF, Fallert Junecko BA, Reinhart TA, Kolls J, Randall TD, Connell TD, Khader SA. Interleukin-17-dependent CXCL13 mediates mucosal vaccine-induced immunity against tuberculosis. Mucosal Immunol. 2013;6:972–984. doi: 10.1038/mi.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lindenstrom T, Woodworth J, Dietrich J, Aagaard C, Andersen P, Agger EM. Vaccine-induced Th17 cells are maintained long-term postvaccination as a distinct and phenotypically stable memory subset. Infect Immun. 2012;80:3533–3544. doi: 10.1128/IAI.00550-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Desel C, Dorhoi A, Bandermann S, Grode L, Eisele B, Kaufmann SH. Recombinant BCG DeltaureC hly+ induces superior protection over parental BCG by stimulating a balanced combination of type 1 and type 17 cytokine responses. J Infect Dis. 2011;204:1573–1584. doi: 10.1093/infdis/jir592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Ann Rev Immunol. 2007;25:221–242. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- 225.Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev. 2004;202:96–105. doi: 10.1111/j.0105-2896.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- 226.Cox JH, Kljavin NM, Ramamoorthi N, Diehl L, Batten M, Ghilardi N. IL-27 promotes T cell-dependent colitis through multiple mechanisms. J Exp Med. 2011;208:115–123. doi: 10.1084/jem.20100410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Shimizu S, Sugiyama N, Masutani K, Sadanaga A, Miyazaki Y, Inoue Y, Akahoshi M, Katafuchi R, Hirakata H, Harada M, Hamano S, Nakashima H, Yoshida H. Membranous glomerulonephritis development with Th2-type immune deviations in MRL/lpr mice deficient for IL-27 receptor (WSX-1) J Immunol. 2005;175:7185–7192. doi: 10.4049/jimmunol.175.11.7185. [DOI] [PubMed] [Google Scholar]
- 228.Cao Y, Doodes PD, Glant TT, Finnegan A. IL-27 induces a Th1 immune response and susceptibility to experimental arthritis. J Immunol. 2008;180:922–930. doi: 10.4049/jimmunol.180.2.922. [DOI] [PubMed] [Google Scholar]
- 229.Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, Hibbert L, Churakova T, Travis M, Vaisberg E, Blumenschein WM, Mattson JD, Wagner JL, To W, Zurawski S, McClanahan TK, Gorman DM, Bazan JF, de Waal Malefyt R, Rennick D, Kastelein RA. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity. 2002;16:779–790. doi: 10.1016/s1074-7613(02)00324-2. [DOI] [PubMed] [Google Scholar]
- 230.Pflanz S, Hibbert L, Mattson J, Rosales R, Vaisberg E, Bazan JF, Phillips JH, McClanahan TK, de Waal Malefyt R, Kastelein RA. WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J Immunol. 2004;172:2225–2231. doi: 10.4049/jimmunol.172.4.2225. [DOI] [PubMed] [Google Scholar]
- 231.Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, Lee J, de Sauvage FJ, Ghilardi N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 232.Neufert C, Becker C, Wirtz S, Fantini MC, Weigmann B, Galle PR, Neurath MF. IL-27 controls the development of inducible regulatory T cells and Th17 cells via differential effects on STAT1. Eur J Immunol. 2007;37:1809–1816. doi: 10.1002/eji.200636896. [DOI] [PubMed] [Google Scholar]
- 233.Takeda A, Hamano S, Yamanaka A, Hanada T, Ishibashi T, Mak TW, Yoshimura A, Yoshida H. Cutting Edge: Role of IL-27/WSX-1 signaling for induction of T-bet through activation of STAT1 during initial Th1 commitment. The Journal of Immunology. 2003;170:4886–4890. doi: 10.4049/jimmunol.170.10.4886. [DOI] [PubMed] [Google Scholar]
- 234.Pearl JE, Khader SA, Solache A, Gilmartin L, Ghilardi N, deSauvage F, Cooper AM. IL-27 signaling compromises control of bacterial growth in mycobacteria-infected mice. J Immunol. 2004;173:7490–7496. doi: 10.4049/jimmunol.173.12.7490. [DOI] [PubMed] [Google Scholar]
- 235.Holscher C, Holscher A, Ruckerl D, Yoshimoto T, Yoshida H, Mak T, Saris C, Ehlers S. The IL-27 receptor chain WSX-1 differentially regulates antibacterial immunity and survival during experimental tuberculosis. J Immunol. 2005;174:3534–3544. doi: 10.4049/jimmunol.174.6.3534. [DOI] [PubMed] [Google Scholar]
- 236.Sodenkamp J, Behrends J, Forster I, Muller W, Ehlers S, Holscher C. gp130 on macrophages/granulocytes modulates inflammation during experimental tuberculosis. Eur J Cell Biol. 2011;90:505–514. doi: 10.1016/j.ejcb.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 237.Neurath MF. IL-12 family members in experimental colitis. Mucosal Immunol. 2008;1(Suppl 1):S28–30. doi: 10.1038/mi.2008.45. [DOI] [PubMed] [Google Scholar]
- 238.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 240.Jin W, Dong C. IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect. 2013;2:e60. doi: 10.1038/emi.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 242.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Umemura M, Yahagi A, Hamada S, Begum MD, Watanabe H, Kawakami K, Suda T, Sudo K, Nakae S, Iwakura Y, Matsuzaki G. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J Immunol. 2007;178:3786–3796. doi: 10.4049/jimmunol.178.6.3786. [DOI] [PubMed] [Google Scholar]
- 244.Gopal R, Lin Y, Obermajer N, Slight S, Nuthalapati N, Ahmed M, Kalinski P, Khader SA. IL-23-dependent IL-17 drives Th1-cell responses following Mycobacterium bovis BCG vaccination. Eur J Immunol. 2012;42:364–373. doi: 10.1002/eji.201141569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Aguilo N, Alvarez-Arguedas S, Uranga S, Marinova D, Monzon M, Badiola J, Martin C. Pulmonary but not subcutaneous delivery of BCG vaccine confers protection to tuberculosis-susceptible mice by an interleukin 17-dependent mechanism. J Infect Dis. 2015;213:831–839. doi: 10.1093/infdis/jiv503. [DOI] [PubMed] [Google Scholar]
- 247.Cruz A, Torrado E, Carmona J, Fraga AG, Costa P, Rodrigues F, Appelberg R, Correia-Neves M, Cooper AM, Saraiva M, Pedrosa J, Castro AG. BCG vaccination-induced long-lasting control of Mycobacterium tuberculosis correlates with the accumulation of a novel population of CD4(+)IL-17(+)TNF(+)IL-2(+) T cells. Vaccine. 2015;33:85–91. doi: 10.1016/j.vaccine.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 248.Cruz A, Fraga AG, Fountain JJ, Rangel-Moreno J, Torrado E, Saraiva M, Pereira DR, Randall TD, Pedrosa J, Cooper AM, Castro AG. Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J Exp Med. 2010;207:1609–1616. doi: 10.1084/jem.20100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, Fallert Junecko BA, Reinhart TA, Kolls J, Baez-Saldana R, Cruz-Lagunas A, Rodriguez-Reyna TS, Kumar NP, Tessier P, Roth J, Selman M, Becerril-Villanueva E, Baquera-Heredia J, Cumming B, Kasprowicz VO, Steyn AJ, Babu S, Kaushal D, Zuniga J, Vogl T, Rangel-Moreno J, Khader SA. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med. 2013;188:1137–1146. doi: 10.1164/rccm.201304-0803OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.McAleer JP, Kolls JK. Directing traffic: IL-17 and IL-22 coordinate pulmonary immune defense. Immunol Rev. 2014;260:129–144. doi: 10.1111/imr.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J Exp Med. 2010;207:1293–1305. doi: 10.1084/jem.20092054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Kolls JK, McCray PB, Jr, Chan YR. Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol. 2008;8:829–835. doi: 10.1038/nri2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Matthews K, Wilkinson KA, Kalsdorf B, Roberts T, Diacon A, Walzl G, Wolske J, Ntsekhe M, Syed F, Russell J, Mayosi BM, Dawson R, Dheda K, Wilkinson RJ, Hanekom WA, Scriba TJ. Predominance of interleukin-22 over interleukin-17 at the site of disease in human tuberculosis. Tuberculosis (Edinb) 2011;91:587–593. doi: 10.1016/j.tube.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Yao S, Huang D, Chen CY, Halliday L, Zeng G, Wang RC, Chen ZW. Differentiation, distribution and gammadelta T cell-driven regulation of IL-22-producing T cells in tuberculosis. PLoS Pathog. 2010;6:e1000789. doi: 10.1371/journal.ppat.1000789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Zeng G, Chen CY, Huang D, Yao S, Wang RC, Chen ZW. Membrane-bound IL-22 after de novo production in tuberculosis and anti-Mycobacterium tuberculosis effector function of IL-22+ CD4+ T cells. J Immunol. 2011;187:190–199. doi: 10.4049/jimmunol.1004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Dhiman R, Venkatasubramanian S, Paidipally P, Barnes PF, Tvinnereim A, Vankayalapati R. Interleukin 22 inhibits intracellular growth of Mycobacterium tuberculosis by enhancing calgranulin A expression. J Infect Dis. 2014;209:578–587. doi: 10.1093/infdis/jit495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Dhiman R, Indramohan M, Barnes PF, Nayak RC, Paidipally P, Rao LV, Vankayalapati R. IL-22 produced by human NK cells inhibits growth of Mycobacterium tuberculosis by enhancing phagolysosomal fusion. J Immunol. 2009;183:6639–6645. doi: 10.4049/jimmunol.0902587. [DOI] [PubMed] [Google Scholar]
- 258.Zhang M, Zeng G, Yang Q, Zhang J, Zhu X, Chen Q, Suthakaran P, Zhang Y, Deng Q, Liu H, Zhou B, Chen X. Anti-tuberculosis treatment enhances the production of IL-22 through reducing the frequencies of regulatory B cell. Tuberculosis (Edinb) 2014;94:238–244. doi: 10.1016/j.tube.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 259.Behrends J, Renauld JC, Ehlers S, Holscher C. IL-22 is mainly produced by IFNgamma-secreting cells but is dispensable for host protection against Mycobacterium tuberculosis infection. PLoS One. 2013;8:e57379. doi: 10.1371/journal.pone.0057379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Wilson MS, Feng CG, Barber DL, Yarovinsky F, Cheever AW, Sher A, Grigg M, Collins M, Fouser L, Wynn TA. Redundant and pathogenic roles for IL-22 in mycobacterial, protozoan, and helminth infections. J Immunol. 2010;184:4378–4390. doi: 10.4049/jimmunol.0903416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Dhiman R, Periasamy S, Barnes PF, Jaiswal AG, Paidipally P, Barnes AB, Tvinnereim A, Vankayalapati R. NK1.1+ cells and IL-22 regulate vaccine-induced protective immunity against challenge with Mycobacterium tuberculosis. J Immunol. 2012;189:897–905. doi: 10.4049/jimmunol.1102833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 263.Killar L, MacDonald G, West J, Woods A, Bottomly K. Cloned, Ia-restricted T cells that do not produce interleukin 4(IL 4)/B cell stimulatory factor 1(BSF-1) fail to help antigen-specific B cells. J Immunol. 1987;138:1674–1679. [PubMed] [Google Scholar]
- 264.Powrie F, Menon S, Coffman RL. Interleukin-4 and interleukin-10 synergize to inhibit cell-mediated immunity in vivo. Eur J Immunol. 1993;23:3043–3049. doi: 10.1002/eji.1830231147. [DOI] [PubMed] [Google Scholar]
- 265.Appelberg R, Orme IM, Pinto de Sousa MI, Silva MT. In vitro effects of interleukin-4 on interferon-gamma-induced macrophage activation. Immunology. 1992;76:553–559. [PMC free article] [PubMed] [Google Scholar]
- 266.Ferber IA, Lee HJ, Zonin F, Heath V, Mui A, Arai N, O’Garra A. GATA-3 significantly downregulates IFN-gamma production from developing Th1 cells in addition to inducing IL-4 and IL-5 levels. Clin Immunol. 1999;91:134–144. doi: 10.1006/clim.1999.4718. [DOI] [PubMed] [Google Scholar]
- 267.Steinke JW, Borish L. Th2 cytokines and asthma. Interleukin-4: its role in the pathogenesis of asthma, and targeting it for asthma treatment with interleukin-4 receptor antagonists. Respir Res. 2001;2:66–70. doi: 10.1186/rr40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2010;125:S73–80. doi: 10.1016/j.jaci.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.MacDonald AS, Araujo MI, Pearce EJ. Immunology of parasitic helminth infections. Infect Immun. 2002;70:427–433. doi: 10.1128/iai.70.2.427-433.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Ann Rev Immunol. 1999;17:701–738. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- 272.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 273.Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem. 1997;272:21597–21603. doi: 10.1074/jbc.272.34.21597. [DOI] [PubMed] [Google Scholar]
- 274.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 275.Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, Chu C, Quelle FW, Nosaka T, Vignali DA, Doherty PC, Grosveld G, Paul WE, Ihle JN. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630–633. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 276.Swain SL, Weinberg AD, English M, Huston G. IL-4 directs the development of Th2-like helper effectors. J Immunol. 1990;145:3796–3806. [PubMed] [Google Scholar]
- 277.Le Gros G, Ben-Sasson SZ, Seder R, Finkelman FD, Paul WE. Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells. J Exp Med. 1990;172:921–929. doi: 10.1084/jem.172.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Lowenthal JW, Castle BE, Christiansen J, Schreurs J, Rennick D, Arai N, Hoy P, Takebe Y, Howard M. Expression of high affinity receptors for murine interleukin 4 (BSF-1) on hemopoietic and nonhemopoietic cells. J Immunol. 1988;140:456–464. [PubMed] [Google Scholar]
- 279.O’Hara J, Paul WE. Receptors for B-cell stimulatory factor-1 expressed on cells of haematopoietic lineage. Nature. 1987;325:537–540. doi: 10.1038/325537a0. [DOI] [PubMed] [Google Scholar]
- 280.Coffman RL, Seymour BW, Hudak S, Jackson J, Rennick D. Antibody to interleukin-5 inhibits helminth-induced eosinophilia in mice. Science. 1989;245:308–310. doi: 10.1126/science.2787531. [DOI] [PubMed] [Google Scholar]
- 281.Phillips C, Coward WR, Pritchard DI, Hewitt CR. Basophils express a type 2 cytokine profile on exposure to proteases from helminths and house dust mites. J Leukoc Biol. 2003;73:165–171. doi: 10.1189/jlb.0702356. [DOI] [PubMed] [Google Scholar]
- 282.Hitoshi Y, Yamaguchi N, Mita S, Sonoda E, Takaki S, Tominaga A, Takatsu K. Distribution of IL-5 receptor-positive B cells. Expression of IL-5 receptor on Ly-1(CD5)+ B cells. J Immunol. 1990;144:4218–4225. [PubMed] [Google Scholar]
- 283.Rolink AG, Thalmann P, Kikuchi Y, Erdei A. Characterization of the interleukin 5-reactive splenic B cell population. Eur J Immunol. 1990;20:1949–1956. doi: 10.1002/eji.1830200912. [DOI] [PubMed] [Google Scholar]
- 284.Takatsu K. Interleukin-5 and IL-5 receptor in health and diseases. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87:463–485. doi: 10.2183/pjab.87.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Schauf V, Rom WN, Smith KA, Sampaio EP, Meyn PA, Tramontana JM, Cohn ZA, Kaplan G. Cytokine gene activation and modified responsiveness to interleukin-2 in the blood of tuberculosis patients. J Infect Dis. 1993;168:1056–1059. doi: 10.1093/infdis/168.4.1056. [DOI] [PubMed] [Google Scholar]
- 286.Surcel HM, Troye-Blomberg M, Paulie S, Andersson G, Moreno C, Pasvol G, Ivanyi J. Th1/Th2 profiles in tuberculosis, based on the proliferation and cytokine response of blood lymphocytes to mycobacterial antigens. Immunology. 1994;81:171–176. [PMC free article] [PubMed] [Google Scholar]
- 287.Zhang M, Gong J, Iyer DV, Jones BE, Modlin RL, Barnes PF. T cell cytokine responses in persons with tuberculosis and human immunodeficiency virus infection. J Clin Invest. 1994;94:2435–2442. doi: 10.1172/JCI117611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Lin Y, Zhang M, Hofman FM, Gong J, Barnes PF. Absence of a prominent Th2 cytokine response in human tuberculosis. Infect Immun. 1996;64:1351–1356. doi: 10.1128/iai.64.4.1351-1356.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Lai CK, Ho S, Chan CH, Chan J, Choy D, Leung R, Lai KN. Cytokine gene expression profile of circulating CD4+ T cells in active pulmonary tuberculosis. Chest. 1997;111:606–611. doi: 10.1378/chest.111.3.606. [DOI] [PubMed] [Google Scholar]
- 290.Mihret A, Bekele Y, Bobosha K, Kidd M, Aseffa A, Howe R, Walzl G. Plasma cytokines and chemokines differentiate between active disease and non-active tuberculosis infection. J Infect. 2013;66:357–365. doi: 10.1016/j.jinf.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 291.Mihret A, Abebe M, Bekele Y, Aseffa A, Walzl G, Howe R. Impact of HIV co-infection on plasma level of cytokines and chemokines of pulmonary tuberculosis patients. BMC Infect Dis. 2014;14:125. doi: 10.1186/1471-2334-14-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Bezuidenhout J, Roberts T, Muller L, van Helden P, Walzl G. Pleural tuberculosis in patients with early HIV infection is associated with increased TNF-alpha expression and necrosis in granulomas. PLoS One. 2009;4:e4228. doi: 10.1371/journal.pone.0004228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Mazzarella G, Bianco A, Perna F, D’Auria D, Grella E, Moscariello E, Sanduzzi A. T lymphocyte phenotypic profile in lung segments affected by cavitary and non-cavitary tuberculosis. Clin Exp Immunol. 2003;132:283–288. doi: 10.1046/j.1365-2249.2003.02121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Mattila JT, Diedrich CR, Lin PL, Phuah J, Flynn JL. Simian immunodeficiency virus-induced changes in T cell cytokine responses in cynomolgus macaques with latent Mycobacterium tuberculosis infection are associated with timing of reactivation. J Immunol. 2011;186:3527–3537. doi: 10.4049/jimmunol.1003773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Wassie L, Demissie A, Aseffa A, Abebe M, Yamuah L, Tilahun H, Petros B, Rook G, Zumla A, Andersen P, Doherty TM. Ex vivo cytokine mRNA levels correlate with changing clinical status of ethiopian TB patients and their contacts over time. PLoS One. 2008;3:e1522. doi: 10.1371/journal.pone.0001522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Fletcher HA, Owiafe P, Jeffries D, Hill P, Rook GA, Zumla A, Doherty TM, Brookes RH. Increased expression of mRNA encoding interleukin (IL)-4 and its splice variant IL-4delta2 in cells from contacts of Mycobacterium tuberculosis, in the absence of in vitro stimulation. Immunology. 2004;112:669–673. doi: 10.1111/j.1365-2567.2004.01922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Demissie A, Abebe M, Aseffa A, Rook G, Fletcher H, Zumla A, Weldingh K, Brock I, Andersen P, Doherty TM. Healthy individuals that control a latent infection with Mycobacterium tuberculosis express high levels of Th1 cytokines and the IL-4 antagonist IL-4delta2. J Immunol. 2004;172:6938–6943. doi: 10.4049/jimmunol.172.11.6938. [DOI] [PubMed] [Google Scholar]
- 298.Jung YJ, LaCourse R, Ryan L, North RJ. Evidence inconsistent with a negative influence of T helper 2 cells on protection afforded by a dominant T helper 1 response against Mycobacterium tuberculosis lung infection in mice. Infect Immun. 2002;70:6436–6443. doi: 10.1128/IAI.70.11.6436-6443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.North RJ. Mice incapable of making IL-4 or IL-10 display normal resistance to infection with Mycobacterium tuberculosis. Clin Exp Immunol. 1998;113:55–58. doi: 10.1046/j.1365-2249.1998.00636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Lukacs NW, Addison CL, Gauldie J, Graham F, Simpson K, Strieter RM, Warmington K, Chensue SW, Kunkel SL. Transgene-induced production of IL-4 alters the development and collagen expression of T helper cell 1-type pulmonary granulomas. J Immunol. 1997;158:4478–4484. [PubMed] [Google Scholar]
- 301.Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol. 2012;12:352–366. doi: 10.1038/nri3211. [DOI] [PubMed] [Google Scholar]
- 302.Erb KJ, Kirman J, Delahunt B, Chen W, Le Gros G. IL-4, IL-5 and IL-10 are not required for the control of M. bovis-BCG infection in mice. Immunol Cell Biol. 1998;76:41–46. doi: 10.1046/j.1440-1711.1998.00719.x. [DOI] [PubMed] [Google Scholar]
- 303.Diedrich CR, Mattila JT, Flynn JL. Monocyte-derived IL-5 reduces TNF production by Mycobacterium tuberculosis-specific CD4 T cells during SIV/M. tuberculosis coinfection. J Immunol. 2013;190:6320–6328. doi: 10.4049/jimmunol.1202043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Minty A, Chalon P, Derocq JM, Dumont X, Guillemot JC, Kaghad M, Labit C, Leplatois P, Liauzun P, Miloux B, et al. Interleukin-13 is a new human lymphokine regulating inflammatory and immune responses. Nature. 1993;362:248–250. doi: 10.1038/362248a0. [DOI] [PubMed] [Google Scholar]
- 305.McKenzie AN, Culpepper JA, de Waal Malefyt R, Briere F, Punnonen J, Aversa G, Sato A, Dang W, Cocks BG, Menon S, et al. Interleukin 13, a T-cell-derived cytokine that regulates human monocyte and B-cell function. Proc Natl Acad Sci U S A. 1993;90:3735–3739. doi: 10.1073/pnas.90.8.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Wynn TA. IL-13 effector functions. Ann Rev Immunol. 2003;21:425–456. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 307.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Wynn TA, Eltoum I, Oswald IP, Cheever AW, Sher A. Endogenous interleukin 12 (IL-12) regulates granuloma formation induced by eggs of Schistosoma mansoni and exogenous IL-12 both inhibits and prophylactically immunizes against egg pathology. J Exp Med. 1994;179:1551–1561. doi: 10.1084/jem.179.5.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Gessner A, Mohrs K, Mohrs M. Mast cells, basophils, and eosinophils acquire constitutive IL-4 and IL-13 transcripts during lineage differentiation that are sufficient for rapid cytokine production. J Immunol. 2005;174:1063–1072. doi: 10.4049/jimmunol.174.2.1063. [DOI] [PubMed] [Google Scholar]
- 310.Ying S, Humbert M, Barkans J, Corrigan CJ, Pfister R, Menz G, Larche M, Robinson DS, Durham SR, Kay AB. Expression of IL-4 and IL-5 mRNA and protein product by CD4+ and CD8+ T cells, eosinophils, and mast cells in bronchial biopsies obtained from atopic and nonatopic (intrinsic) asthmatics. J Immunol. 1997;158:3539–3544. [PubMed] [Google Scholar]
- 311.O’Brien TF, Bao K, Dell’Aringa M, Ang WX, Abraham S, Reinhardt RL. Cytokine expression by invariant natural killer T cells is tightly regulated throughout development and settings of type-2 inflammation. Mucosal Immunol. 2015 doi: 10.1038/mi.2015.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Bao K, Reinhardt RL. The differential expression of IL-4 and IL-13 and its impact on type-2 immunity. Cytokine. 2015;75:25–37. doi: 10.1016/j.cyto.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.McCormick SM, Heller NM. Commentary: IL-4 and IL-13 receptors and signaling. Cytokine. 2015;75:38–50. doi: 10.1016/j.cyto.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Dhanasekaran S, Jenum S, Stavrum R, Ritz C, Faurholt-Jepsen D, Kenneth J, Vaz M, Grewal HM, Doherty TM. Identification of biomarkers for Mycobacterium tuberculosis infection and disease in BCG-vaccinated young children in Southern India. Genes Immun. 2013;14:356–364. doi: 10.1038/gene.2013.26. [DOI] [PubMed] [Google Scholar]
- 315.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 316.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 317.Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, Deretic V. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity. 2007;27:505–517. doi: 10.1016/j.immuni.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 318.Heitmann L, Abad Dar M, Schreiber T, Erdmann H, Behrends J, McKenzie ANJ, Brombacher F, Ehlers S, Hölscher C. The IL-13/IL-4Rα axis is involved in tuberculosis-associated pathology. J Pathol. 2014;234:338–350. doi: 10.1002/path.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 321.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 322.Trompouki E, Bowman TV, Lawton LN, Fan ZP, Wu DC, DiBiase A, Martin CS, Cech JN, Sessa AK, Leblanc JL, Li P, Durand EM, Mosimann C, Heffner GC, Daley GQ, Paulson RF, Young RA, Zon LI. Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell. 2011;147:577–589. doi: 10.1016/j.cell.2011.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Taylor AW. Review of the activation of TGF-beta in immunity. J Leukoc Biol. 2009;85:29–33. doi: 10.1189/jlb.0708415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Roberts AB, Sporn MB. Transforming growth factor beta. Adv Cancer Res. 1988;51:107–145. [PubMed] [Google Scholar]
- 325.Massague J. The transforming growth factor-beta family. Annu Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- 326.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Ann Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 327.Sporn MB, Roberts AB. Autocrine secretion–10 years later. Ann Intern Med. 1992;117:408–414. doi: 10.7326/0003-4819-117-5-408. [DOI] [PubMed] [Google Scholar]
- 328.Toossi Z, Gogate P, Shiratsuchi H, Young T, Ellner JJ. Enhanced production of TGF-beta by blood monocytes from patients with active tuberculosis and presence of TGF-beta in tuberculous granulomatous lung lesions. J Immunol. 1995;154:465–473. [PubMed] [Google Scholar]
- 329.Dahl KE, Shiratsuchi H, Hamilton BD, Ellner JJ, Toossi Z. Selective induction of transforming growth factor beta in human monocytes by lipoarabinomannan of Mycobacterium tuberculosis. Infect Immun. 1996;64:399–405. doi: 10.1128/iai.64.2.399-405.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Hirsch CS, Yoneda T, Averill L, Ellner JJ, Toossi Z. Enhancement of intracellular growth of Mycobacterium tuberculosis in human monocytes by transforming growth factor-beta 1. J Infect Dis. 1994;170:1229–1237. doi: 10.1093/infdis/170.5.1229. [DOI] [PubMed] [Google Scholar]
- 331.Hirsch CS, Ellner JJ, Blinkhorn R, Toossi Z. In vitro restoration of T cell responses in tuberculosis and augmentation of monocyte effector function against Mycobacterium tuberculosis by natural inhibitors of transforming growth factor beta. Proc Natl Acad Sci U S A. 1997;94:3926–3931. doi: 10.1073/pnas.94.8.3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Othieno C, Hirsch CS, Hamilton BD, Wilkinson K, Ellner JJ, Toossi Z. Interaction of Mycobacterium tuberculosis-induced transforming growth factor beta1 and interleukin-10. Infect Immun. 1999;67:5730–5735. doi: 10.1128/iai.67.11.5730-5735.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Sivangala R, Ponnana M, Thada S, Joshi L, Ansari S, Hussain H, Valluri V, Gaddam S. Association of cytokine gene polymorphisms in patients with tuberculosis and their household contacts. Scand J Immunol. 2014;79:197–205. doi: 10.1111/sji.12136. [DOI] [PubMed] [Google Scholar]
- 334.Mak JC, Leung HC, Sham AS, Mok TY, Poon YN, Ling SO, Wong KC, Chan-Yeung M. Genetic polymorphisms and plasma levels of transforming growth factor-beta(1) in Chinese patients with tuberculosis in Hong Kong. Cytokine. 2007;40:177–182. doi: 10.1016/j.cyto.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 335.Vieira P, de Waal-Malefyt R, Dang MN, Johnson KE, Kastelein R, Fiorentino DF, deVries JE, Roncarolo MG, Mosmann TR, Moore KW. Isolation and expression of human cytokine synthesis inhibitory factor cDNA clones: homology to Epstein-Barr virus open reading frame BCRFI. Proc Natl Acad Sci U S A. 1991;88:1172–1176. doi: 10.1073/pnas.88.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Redford PS, Murray PJ, O’Garra A. The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol. 2011;4:261–270. doi: 10.1038/mi.2011.7. [DOI] [PubMed] [Google Scholar]
- 337.Liu Y, Wei SH, Ho AS, de Waal Malefyt R, Moore KW. Expression cloning and characterization of a human IL-10 receptor. J Immunol. 1994;152:1821–1829. [PubMed] [Google Scholar]
- 338.Jang S, Uematsu S, Akira S, Salgame P. IL-6 and IL-10 induction from dendritic cells in response to Mycobacterium tuberculosis is predominantly dependent on TLR2-mediated recognition. J Immunol. 2004;173:3392–3397. doi: 10.4049/jimmunol.173.5.3392. [DOI] [PubMed] [Google Scholar]
- 339.Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O, Schweighoffer E, Williams DL, Gordon S, Tybulewicz VL, Brown GD, Reis e Sousa C. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity. 2005;22:507–517. doi: 10.1016/j.immuni.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 340.Ke Z, Yuan L, Ma J, Zhang X, Guo Y, Xiong H. IL-10 polymorphisms and tuberculosis susceptibility: An updated meta-analysis. Yonsei Med J. 2015;56:1274–1287. doi: 10.3349/ymj.2015.56.5.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Jeong YH, Hur YG, Lee H, Kim S, Cho JE, Chang J, Shin SJ, Lee H, Kang YA, Cho SN, Ha SJ. Discrimination between active and latent tuberculosis based on ratio of antigen-specific to mitogen-induced IP-10 production. J Clin Microbiol. 2015;53:504–510. doi: 10.1128/JCM.02758-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Tebruegge M, Dutta B, Donath S, Ritz N, Forbes B, Camacho-Badilla K, Clifford V, Zufferey C, Robins-Browne R, Hanekom W, Graham SM, Connell T, Curtis N. Mycobacteria-specific cytokine responses detect tuberculosis infection and distinguish latent from active tuberculosis. Am J Respir Crit Care Med. 2015;192:485–499. doi: 10.1164/rccm.201501-0059OC. [DOI] [PubMed] [Google Scholar]
- 343.Kumar NP, Moideen K, Banurekha VV, Nair D, Sridhar R, Nutman TB, Babu S. IL-27 and TGFbeta mediated expansion of Th1 and adaptive regulatory T cells expressing IL-10 correlates with bacterial burden and disease severity in pulmonary tuberculosis. Immun Inflamm Dis. 2015;3:289–299. doi: 10.1002/iid3.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Eum SY, Jeon BY, Min JH, Kim SC, Cho S, Park SK, Cho SN. Tumor necrosis factor-alpha and interleukin-10 in whole blood is associated with disease progression in pulmonary multidrug-resistant tuberculosis patients. Respiration. 2008;76:331–337. doi: 10.1159/000113932. [DOI] [PubMed] [Google Scholar]
- 345.Lago PM, Boechat N, Migueis DP, Almeida AS, Lazzarini LC, Saldanha MM, Kritski AL, Ho JL, Lapa e Silva JR. Interleukin-10 and interferon-gamma patterns during tuberculosis treatment: possible association with recurrence. Int J Tuberc Lung Dis. 2012;16:656–659. doi: 10.5588/ijtld.11.0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.George PJ, Pavan Kumar N, Jaganathan J, Dolla C, Kumaran P, Nair D, Banurekha VV, Shen K, Nutman TB, Babu S. Modulation of pro- and anti-inflammatory cytokines in active and latent tuberculosis by coexistent Strongyloides stercoralis infection. Tuberculosis (Edinb) 2015;95:822–828. doi: 10.1016/j.tube.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.George PJ, Anuradha R, Kumar NP, Sridhar R, Banurekha VV, Nutman TB, Babu S. Helminth infections coincident with active pulmonary tuberculosis inhibit mono- and multifunctional CD4+ and CD8+ T cell responses in a process dependent on IL-10. PLoS Pathog. 2014;10:e1004375. doi: 10.1371/journal.ppat.1004375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Verreck FA, de Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, Kastelein R, Kolk A, de Waal-Malefyt R, Ottenhoff TH. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci U S A. 2004;101:4560–4565. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.O’Leary S, O’Sullivan MP, Keane J. IL-10 blocks phagosome maturation in Mycobacterium tuberculosis-infected human macrophages. Am J Respir Cell Mol Biol. 2011;45:172–180. doi: 10.1165/rcmb.2010-0319OC. [DOI] [PubMed] [Google Scholar]
- 350.Oswald IP, Wynn TA, Sher A, James SL. Interleukin 10 inhibits macrophage microbicidal activity by blocking the endogenous production of tumor necrosis factor alpha required as a costimulatory factor for interferon gamma-induced activation. Proc Natl Acad Sci U S A. 1992;89:8676–8680. doi: 10.1073/pnas.89.18.8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Ann Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 352.Richardson ET, Shukla S, Sweet DR, Wearsch PA, Tsichlis PN, Boom WH, Harding CV. Toll-like receptor 2-dependent extracellular signal-regulated kinase signaling in Mycobacterium tuberculosis-infected macrophages drives anti-inflammatory responses and inhibits Th1 polarization of responding T cells. Infect Immun. 2015;83:2242–2254. doi: 10.1128/IAI.00135-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Jung YJ, Ryan L, LaCourse R, North RJ. Increased interleukin-10 expression is not responsible for failure of T helper 1 immunity to resolve airborne Mycobacterium tuberculosis infection in mice. Immunology. 2003;109:295–299. doi: 10.1046/j.1365-2567.2003.01645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Higgins DM, Sanchez-Campillo J, Rosas-Taraco AG, Lee EJ, Orme IM, Gonzalez-Juarrero M. Lack of IL-10 alters inflammatory and immune responses during pulmonary Mycobacterium tuberculosis infection. Tuberculosis (Edinb) 2009;89:149–157. doi: 10.1016/j.tube.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 355.Beamer GL, Flaherty DK, Assogba BD, Stromberg P, Gonzalez-Juarrero M, de Waal Malefyt R, Vesosky B, Turner J. Interleukin-10 promotes Mycobacterium tuberculosis disease progression in CBA/J mice. Journal of immunology (Baltimore, Md : 1950) 2008;181:5545–5550. doi: 10.4049/jimmunol.181.8.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Cyktor JC, Carruthers B, Kominsky RA, Beamer GL, Stromberg P, Turner J. IL-10 inhibits mature fibrotic granuloma formation during Mycobacterium tuberculosis infection. J Immunol. 2013;190:2778–2790. doi: 10.4049/jimmunol.1202722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Cilfone NA, Ford CB, Marino S, Mattila JT, Gideon HP, Flynn JL, Kirschner DE, Linderman JJ. Computational modeling predicts IL-10 control of lesion sterilization by balancing early host immunity-mediated antimicrobial responses with caseation during Mycobacterium tuberculosis infection. J Immunol. 2015;194:664–677. doi: 10.4049/jimmunol.1400734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Dorhoi A, Kaufmann SH. Pathology and immune reactivity: understanding multidimensionality in pulmonary tuberculosis. Semin Immunopathol. 2015 doi: 10.1007/s00281-015-0531-3. [DOI] [PubMed] [Google Scholar]
- 359.Yoshimura T. Discovery of IL-8/CXCL8 (The Story from Frederick) Front Immunol. 2015;6:278. doi: 10.3389/fimmu.2015.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- 361.Murphy PM. International Union of Pharmacology. XXX. Update on chemokine receptor nomenclature. Pharmacol Rev. 2002;54:227–229. doi: 10.1124/pr.54.2.227. [DOI] [PubMed] [Google Scholar]
- 362.Bacon K, Baggiolini M, Broxmeyer H, Horuk R, Lindley I, Mantovani A, Maysushima K, Murphy P, Nomiyama H, Oppenheim J, Rot A, Schall T, Tsang M, Thorpe R, Van Damme J, Wadhwa M, Yoshie O, Zlotnik A, Zoon K. Chemokine/chemokine receptor nomenclature. J Interferon Cytokine Res. 2002;22:1067–1068. doi: 10.1089/107999002760624305. [DOI] [PubMed] [Google Scholar]
- 363.Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36:705–716. doi: 10.1016/j.immuni.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 365.Su SB, Mukaida N, Wang J, Nomura H, Matsushima K. Preparation of specific polyclonal antibodies to a C-C chemokine receptor, CCR1, and determination of CCR1 expression on various types of leukocytes. J Leukoc Biol. 1996;60:658–666. doi: 10.1002/jlb.60.5.658. [DOI] [PubMed] [Google Scholar]
- 366.Neote K, DiGregorio D, Mak JY, Horuk R, Schall TJ. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell. 1993;72:415–425. doi: 10.1016/0092-8674(93)90118-a. [DOI] [PubMed] [Google Scholar]
- 367.Gao JL, Kuhns DB, Tiffany HL, McDermott D, Li X, Francke U, Murphy PM. Structure and functional expression of the human macrophage inflammatory protein 1 alpha/RANTES receptor. J Exp Med. 1993;177:1421–1427. doi: 10.1084/jem.177.5.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Gao JL, Murphy PM. Cloning and differential tissue-specific expression of three mouse beta chemokine receptor-like genes, including the gene for a functional macrophage inflammatory protein-1 alpha receptor. J Biol Chem. 1995;270:17494–17501. doi: 10.1074/jbc.270.29.17494. [DOI] [PubMed] [Google Scholar]
- 369.Kaufmann A, Salentin R, Gemsa D, Sprenger H. Increase of CCR1 and CCR5 expression and enhanced functional response to MIP-1 alpha during differentiation of human monocytes to macrophages. J Leukoc Biol. 2001;69:248–252. [PubMed] [Google Scholar]
- 370.Cheng SS, Lai JJ, Lukacs NW, Kunkel SL. Granulocyte-macrophage colony stimulating factor up-regulates CCR1 in human neutrophils. J Immunol. 2001;166:1178–1184. doi: 10.4049/jimmunol.166.2.1178. [DOI] [PubMed] [Google Scholar]
- 371.Pokkali S, Das SD,RL. Expression of CXC and CC type of chemokines and its receptors in tuberculous and non-tuberculous effusions. Cytokine. 2008;41:307–314. doi: 10.1016/j.cyto.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 372.Pokkali S, Das SD. Augmented chemokine levels and chemokine receptor expression on immune cells during pulmonary tuberculosis. Hum Immunol. 2009;70:110–115. doi: 10.1016/j.humimm.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 373.Hilda JN, Narasimhan M, Das SD. Neutrophils from pulmonary tuberculosis patients show augmented levels of chemokines MIP-1alpha, IL-8 and MCP-1 which further increase upon in vitro infection with mycobacterial strains. Hum Immunol. 2014;75:914–922. doi: 10.1016/j.humimm.2014.06.020. [DOI] [PubMed] [Google Scholar]
- 374.Saukkonen JJ, Bazydlo B, Thomas M, Strieter RM, Keane J, Kornfeld H. Beta-chemokines are induced by Mycobacterium tuberculosis and inhibit its growth. Infect Immun. 2002;70:1684–1693. doi: 10.1128/IAI.70.4.1684-1693.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Algood HM, Flynn JL. CCR5-deficient mice control Mycobacterium tuberculosis infection despite increased pulmonary lymphocytic infiltration. J Immunol. 2004;173:3287–3296. doi: 10.4049/jimmunol.173.5.3287. [DOI] [PubMed] [Google Scholar]
- 376.Randolph GJ, Ochando J, Partida-Sanchez S. Migration of dendritic cell subsets and their precursors. Ann Rev Immunol. 2008;26:293–316. doi: 10.1146/annurev.immunol.26.021607.090254. [DOI] [PubMed] [Google Scholar]
- 377.Glatzel A, Wesch D, Schiemann F, Brandt E, Janssen O, Kabelitz D. Patterns of chemokine receptor expression on peripheral blood gamma delta T lymphocytes: strong expression of CCR5 is a selective feature of V delta 2/V gamma 9 gamma delta T cells. J Immunol. 2002;168:4920–4929. doi: 10.4049/jimmunol.168.10.4920. [DOI] [PubMed] [Google Scholar]
- 378.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Hasan Z, Jamil B, Khan J, Ali R, Khan MA, Nasir N, Yusuf MS, Jamil S, Irfan M, Hussain R. Relationship between circulating levels of IFN-gamma, IL-10, CXCL9 and CCL2 in pulmonary and extrapulmonary tuberculosis is dependent on disease severity. Scand J Immunol. 2009;69:259–267. doi: 10.1111/j.1365-3083.2008.02217.x. [DOI] [PubMed] [Google Scholar]
- 380.Scott HM, Flynn JL. Mycobacterium tuberculosis in chemokine receptor 2-deficient mice: influence of dose on disease progression. Infect Immun. 2002;70:5946–5954. doi: 10.1128/IAI.70.11.5946-5954.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Peters W, Scott HM, Chambers HF, Flynn JL, Charo IF, Ernst JD. Chemokine receptor 2 serves an early and essential role in resistance to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2001;98:7958–7963. doi: 10.1073/pnas.131207398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Kipnis A, Basaraba RJ, Orme IM, Cooper AM. Role of chemokine ligand 2 in the protective response to early murine pulmonary tuberculosis. Immunology. 2003;109:547–551. doi: 10.1046/j.1365-2567.2003.01680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Iellem A, Mariani M, Lang R, Recalde H, Panina-Bordignon P, Sinigaglia F, D’Ambrosio D. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2001;194:847–853. doi: 10.1084/jem.194.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 386.Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 387.Kunkel EJ, Boisvert J, Murphy K, Vierra MA, Genovese MC, Wardlaw AJ, Greenberg HB, Hodge MR, Wu L, Butcher EC, Campbell JJ. Expression of the chemokine receptors CCR4, CCR5, and CXCR3 by human tissue-infiltrating lymphocytes. Am J Pathol. 2002;160:347–355. doi: 10.1016/S0002-9440(10)64378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Hu Z, Lancaster JN, Sasiponganan C, Ehrlich LI. CCR4 promotes medullary entry and thymocyte-dendritic cell interactions required for central tolerance. J Exp Med. 2015;212:1947–1965. doi: 10.1084/jem.20150178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Cowan JE, McCarthy NI, Parnell SM, White AJ, Bacon A, Serge A, Irla M, Lane PJ, Jenkinson EJ, Jenkinson WE, Anderson G. Differential requirement for CCR4 and CCR7 during the development of innate and adaptive alphabetaT cells in the adult thymus. J Immunol. 2014;193:1204–1212. doi: 10.4049/jimmunol.1400993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Andrew DP, Ruffing N, Kim CH, Miao W, Heath H, Li Y, Murphy K, Campbell JJ, Butcher EC, Wu L. C-C chemokine receptor 4 expression defines a major subset of circulating nonintestinal memory T cells of both Th1 and Th2 potential. J Immunol. 2001;166:103–111. doi: 10.4049/jimmunol.166.1.103. [DOI] [PubMed] [Google Scholar]
- 391.Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–235. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Oliphant CJ, Barlow JL, McKenzie AN. Insights into the initiation of type 2 immune responses. Immunology. 2011;134:378–385. doi: 10.1111/j.1365-2567.2011.03499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Li L, Lao SH, Wu CY. Increased frequency of CD4(+)CD25(high) Treg cells inhibit BCG-specific induction of IFN-gamma by CD4(+) T cells from TB patients. Tuberculosis (Edinb) 2007;87:526–534. doi: 10.1016/j.tube.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 394.Roberts T, Beyers N, Aguirre A, Walzl G. Immunosuppression during active tuberculosis is characterized by decreased interferon- gamma production and CD25 expression with elevated forkhead box P3, transforming growth factor- beta, and interleukin-4 mRNA levels. J Infect Dis. 2007;195:870–878. doi: 10.1086/511277. [DOI] [PubMed] [Google Scholar]
- 395.Bayry J, Tchilian EZ, Davies MN, Forbes EK, Draper SJ, Kaveri SV, Hill AV, Kazatchkine MD, Beverley PC, Flower DR, Tough DF. In silico identified CCR4 antagonists target regulatory T cells and exert adjuvant activity in vaccination. Proc Natl Acad Sci U S A. 2008;105:10221–10226. doi: 10.1073/pnas.0803453105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Feng Y, Yin H, Mai G, Mao L, Yue J, Xiao H, Hu Z. Elevated serum levels of CCL17 correlate with increased peripheral blood platelet count in patients with active tuberculosis in China. Clin Vaccine Immunol. 2011;18:629–632. doi: 10.1128/CVI.00493-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Freeman CM, Stolberg VR, Chiu BC, Lukacs NW, Kunkel SL, Chensue SW. CCR4 participation in Th type 1 (mycobacterial) and Th type 2 (schistosomal) anamnestic pulmonary granulomatous responses. J Immunol. 2006;177:4149–4158. doi: 10.4049/jimmunol.177.6.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 399.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 400.Chu SF, Tam CM, Wong HS, Kam KM, Lau YL, Chiang AK. Association between RANTES functional polymorphisms and tuberculosis in Hong Kong Chinese. Genes Immun. 2007;8:475–479. doi: 10.1038/sj.gene.6364412. [DOI] [PubMed] [Google Scholar]
- 401.Vesosky B, Rottinghaus EK, Stromberg P, Turner J, Beamer G. CCL5 participates in early protection against Mycobacterium tuberculosis. J Leukoc Biol. 2010;87:1153–1165. doi: 10.1189/jlb.1109742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Mantovani A. The chemokine system: redundancy for robust outputs. Immunol Today. 1999;20:254–257. doi: 10.1016/s0167-5699(99)01469-3. [DOI] [PubMed] [Google Scholar]
- 403.Schutyser E, Struyf S, Van Damme J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 2003;14:409–426. doi: 10.1016/s1359-6101(03)00049-2. [DOI] [PubMed] [Google Scholar]
- 404.Ito T, Carson WFt, Cavassani KA, Connett JM, Kunkel SL. CCR6 as a mediator of immunity in the lung and gut. Exp Cell Res. 2011;317:613–619. doi: 10.1016/j.yexcr.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Nandi B, Pai C, Huang Q, Prabhala RH, Munshi NC, Gold JS. CCR6, the sole receptor for the chemokine CCL20, promotes spontaneous intestinal tumorigenesis. PLoS One. 2014;9:e97566. doi: 10.1371/journal.pone.0097566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Lee AY, Korner H. CCR6 and CCL20: emerging players in the pathogenesis of rheumatoid arthritis. 2014;92:354–358. doi: 10.1038/icb.2013.97. [DOI] [PubMed] [Google Scholar]
- 407.Stolberg VR, Chiu BC, Martin BE, Shah SA, Sandor M, Chensue SW. Cysteine-cysteinyl chemokine receptor 6 mediates invariant natural killer T cell airway recruitment and innate stage resistance during mycobacterial infection. J Innate Immun. 2011;3:99–108. doi: 10.1159/000321156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Perreau M, Rozot V, Welles HC, Belluti-Enders F, Vigano S, Maillard M, Dorta G, Mazza-Stalder J, Bart PA, Roger T, Calandra T, Nicod L, Harari A. Lack of Mycobacterium tuberculosis-specific interleukin-17A-producing CD4+ T cells in active disease. Eur J Immunol. 2013;43:939–948. doi: 10.1002/eji.201243090. [DOI] [PubMed] [Google Scholar]
- 409.Lindestam Arlehamn CS, Gerasimova A, Mele F, Henderson R, Swann J, Greenbaum JA, Kim Y, Sidney J, James EA, Taplitz R, McKinney DM, Kwok WW, Grey H, Sallusto F, Peters B, Sette A. Memory T cells in latent Mycobacterium tuberculosis infection are directed against three antigenic islands and largely contained in a CXCR3+CCR6+ Th1 subset. PLoS Pathog. 2013;9:e1003130. doi: 10.1371/journal.ppat.1003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Rivero-Lezcano OM, Gonzalez-Cortes C, Reyes-Ruvalcaba D, Diez-Tascon C. CCL20 is overexpressed in Mycobacterium tuberculosis-infected monocytes and inhibits the production of reactive oxygen species (ROS) Clin Exp Immunol. 2010;162:289–297. doi: 10.1111/j.1365-2249.2010.04168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Lee JS, Lee JY, Son JW, Oh JH, Shin DM, Yuk JM, Song CH, Paik TH, Jo EK. Expression and regulation of the CC-chemokine ligand 20 during human tuberculosis. Scand J Immunol. 2008;67:77–85. doi: 10.1111/j.1365-3083.2007.02040.x. [DOI] [PubMed] [Google Scholar]
- 412.Kang DD, Lin Y, Moreno JR, Randall TD, Khader SA. Profiling early lung immune responses in the mouse model of tuberculosis. PLoS One. 2011;6:e16161. doi: 10.1371/journal.pone.0016161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Mehra S, Pahar B, Dutta NK, Conerly CN, Philippi-Falkenstein K, Alvarez X, Kaushal D. Transcriptional reprogramming in nonhuman primate (rhesus macaque) tuberculosis granulomas. PLoS One. 2010;5:e12266. doi: 10.1371/journal.pone.0012266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol. 2008;8:362–371. doi: 10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- 415.Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, Nakano H. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Kahnert A, Hopken UE, Stein M, Bandermann S, Lipp M, Kaufmann SH. Mycobacterium tuberculosis triggers formation of lymphoid structure in murine lungs. J Infect Dis. 2007;195:46–54. doi: 10.1086/508894. [DOI] [PubMed] [Google Scholar]
- 417.Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, Williams LT. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc Natl Acad Sci U S A. 1998;95:258–263. doi: 10.1073/pnas.95.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Saeki H, Moore AM, Brown MJ, Hwang ST. Cutting edge: secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J Immunol. 1999;162:2472–2475. [PubMed] [Google Scholar]
- 419.Bhatt K, Hickman SP, Salgame P. Cutting edge: a new approach to modeling early lung immunity in murine tuberculosis. J Immunol. 2004;172:2748–2751. doi: 10.4049/jimmunol.172.5.2748. [DOI] [PubMed] [Google Scholar]
- 420.Olmos S, Stukes S, Ernst JD. Ectopic activation of Mycobacterium tuberculosis-specific CD4+ T cells in lungs of CCR7−/− mice. J Immunol. 2010;184:895–901. doi: 10.4049/jimmunol.0901230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Nakano H, Gunn MD. Gene duplications at the chemokine locus on mouse chromosome 4: multiple strain-specific haplotypes and the deletion of secondary lymphoid-organ chemokine and EBI-1 ligand chemokine genes in the plt mutation. J Immunol. 2001;166:361–369. doi: 10.4049/jimmunol.166.1.361. [DOI] [PubMed] [Google Scholar]
- 422.Khader SA, Rangel-Moreno J, Fountain JJ, Martino CA, Reiley WW, Pearl JE, Winslow GM, Woodland DL, Randall TD, Cooper AM. In a murine tuberculosis model, the absence of homeostatic chemokines delays granuloma formation and protective immunity. J Immunol. 2009;183:8004–8014. doi: 10.4049/jimmunol.0901937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Allen SJ, Crown SE, Handel TM. Chemokine: receptor structure, interactions, and antagonism. Ann Rev Immunol. 2007;25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- 424.Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A, Marriott D, et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem. 1995;270:27348–27357. doi: 10.1074/jbc.270.45.27348. [DOI] [PubMed] [Google Scholar]
- 425.Hebert CA, Vitangcol RV, Baker JB. Scanning mutagenesis of interleukin-8 identifies a cluster of residues required for receptor binding. J Biol Chem. 1991;266:18989–18994. [PubMed] [Google Scholar]
- 426.Clark-Lewis I, Dewald B, Geiser T, Moser B, Baggiolini M. Platelet factor 4 binds to interleukin 8 receptors and activates neutrophils when its N terminus is modified with Glu-Leu-Arg. Proc Natl Acad Sci U S A. 1993;90:3574–3577. doi: 10.1073/pnas.90.8.3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Jones SA, Moser B, Thelen M. A comparison of post-receptor signal transduction events in Jurkat cells transfected with either IL-8R1 or IL-8R2. Chemokine mediated activation of p42/p44 MAP-kinase (ERK-2) FEBS Lett. 1995;364:211–214. doi: 10.1016/0014-5793(95)00397-r. [DOI] [PubMed] [Google Scholar]
- 428.Slight SR, Khader SA. Chemokines shape the immune responses to tuberculosis. Cytokine Growth Factor Rev. 2013;24:105–113. doi: 10.1016/j.cytogfr.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Alaridah N, Winqvist N, Hakansson G, Tenland E, Ronnholm A, Sturegard E, Bjorkman P, Godaly G. Impaired CXCR1-dependent oxidative defence in active tuberculosis patients. Tuberculosis (Edinb) 2015;95:744–750. doi: 10.1016/j.tube.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 430.Goncalves AS, Appelberg R. The involvement of the chemokine receptor CXCR2 in neutrophil recruitment in LPS-induced inflammation and in Mycobacterium avium infection. Scand J Immunol. 2002;55:585–591. doi: 10.1046/j.1365-3083.2002.01097.x. [DOI] [PubMed] [Google Scholar]
- 431.O’Kane CM, Boyle JJ, Horncastle DE, Elkington PT, Friedland JS. Monocyte-dependent fibroblast CXCL8 secretion occurs in tuberculosis and limits survival of mycobacteria within macrophages. J Immunol. 2007;178:3767–3776. doi: 10.4049/jimmunol.178.6.3767. [DOI] [PubMed] [Google Scholar]
- 432.Friedland JS, Remick DG, Shattock R, Griffin GE. Secretion of interleukin-8 following phagocytosis of Mycobacterium tuberculosis by human monocyte cell lines. Eur J Immunol. 1992;22:1373–1378. doi: 10.1002/eji.1830220607. [DOI] [PubMed] [Google Scholar]
- 433.Zhang Y, Broser M, Cohen H, Bodkin M, Law K, Reibman J, Rom WN. Enhanced interleukin-8 release and gene expression in macrophages after exposure to Mycobacterium tuberculosis and its components. J Clin Invest. 1995;95:586–592. doi: 10.1172/JCI117702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Lin Y, Zhang M, Barnes PF. Chemokine production by a human alveolar epithelial cell line in response to Mycobacterium tuberculosis. Infect Immun. 1998;66:1121–1126. doi: 10.1128/iai.66.3.1121-1126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Kurashima K, Mukaida N, Fujimura M, Yasui M, Nakazumi Y, Matsuda T, Matsushima K. Elevated chemokine levels in bronchoalveolar lavage fluid of tuberculosis patients. Am J Respir Crit Care Med. 1997;155:1474–1477. doi: 10.1164/ajrccm.155.4.9105097. [DOI] [PubMed] [Google Scholar]
- 436.Larsen CG, Thomsen MK, Gesser B, Thomsen PD, Deleuran BW, Nowak J, Skodt V, Thomsen HK, Deleuran M, Thestrup-Pedersen K, et al. The delayed-type hypersensitivity reaction is dependent on IL-8. Inhibition of a tuberculin skin reaction by an anti-IL-8 monoclonal antibody. J Immunol. 1995;155:2151–2157. [PubMed] [Google Scholar]
- 437.Ma X, Reich RA, Wright JA, Tooker HR, Teeter LD, Musser JM, Graviss EA. Association between interleukin-8 gene alleles and human susceptibility to tuberculosis disease. J Infect Dis. 2003;188:349–355. doi: 10.1086/376559. [DOI] [PubMed] [Google Scholar]
- 438.Cooke GS, Campbell SJ, Fielding K, Sillah J, Manneh K, Sirugo G, Bennett S, McAdam KP, Lienhardt C, Hill AV. Interleukin-8 polymorphism is not associated with pulmonary tuberculosis in the gambia. J Infect Dis. 2004;189:1545–1546. doi: 10.1086/382489. [DOI] [PubMed] [Google Scholar]
- 439.Almeida Cde S, Abramo C, Alves CC, Mazzoccoli L, Ferreira AP, Teixeira HC. Anti-mycobacterial treatment reduces high plasma levels of CXC-chemokines detected in active tuberculosis by cytometric bead array. Mem Inst Oswaldo Cruz. 2009;104:1039–1041. doi: 10.1590/s0074-02762009000700018. [DOI] [PubMed] [Google Scholar]
- 440.Nouailles G, Dorhoi A, Koch M, Zerrahn J, Weiner J, 3rd, Fae KC, Arrey F, Kuhlmann S, Bandermann S, Loewe D, Mollenkopf HJ, Vogelzang A, Meyer-Schwesinger C, Mittrucker HW, McEwen G, Kaufmann SH. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J Clin Invest. 2014;124:1268–1282. doi: 10.1172/JCI72030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res. 2011;317:620–631. doi: 10.1016/j.yexcr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Thomas SY, Hou R, Boyson JE, Means TK, Hess C, Olson DP, Strominger JL, Brenner MB, Gumperz JE, Wilson SB, Luster AD. CD1d-restricted NKT cells express a chemokine receptor profile indicative of Th1-type inflammatory homing cells. J Immunol. 2003;171:2571–2580. doi: 10.4049/jimmunol.171.5.2571. [DOI] [PubMed] [Google Scholar]
- 443.Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101:746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Lu B, Humbles A, Bota D, Gerard C, Moser B, Soler D, Luster AD, Gerard NP. Structure and function of the murine chemokine receptor CXCR3. Eur J Immunol. 1999;29:3804–3812. doi: 10.1002/(SICI)1521-4141(199911)29:11<3804::AID-IMMU3804>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 445.Campanella GS, Grimm J, Manice LA, Colvin RA, Medoff BD, Wojtkiewicz GR, Weissleder R, Luster AD. Oligomerization of CXCL10 is necessary for endothelial cell presentation and in vivo activity. J Immunol. 2006;177:6991–6998. doi: 10.4049/jimmunol.177.10.6991. [DOI] [PubMed] [Google Scholar]
- 446.Loetscher M, Loetscher P, Brass N, Meese E, Moser B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol. 1998;28:3696–3705. doi: 10.1002/(SICI)1521-4141(199811)28:11<3696::AID-IMMU3696>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 447.Shields PL, Morland CM, Salmon M, Qin S, Hubscher SG, Adams DH. Chemokine and chemokine receptor interactions provide a mechanism for selective T cell recruitment to specific liver compartments within hepatitis C-infected liver. J Immunol. 1999;163:6236–6243. [PubMed] [Google Scholar]
- 448.Seiler P, Aichele P, Bandermann S, Hauser AE, Lu B, Gerard NP, Gerard C, Ehlers S, Mollenkopf HJ, Kaufmann SH. Early granuloma formation after aerosol Mycobacterium tuberculosis infection is regulated by neutrophils via CXCR3-signaling chemokines. Eur J Immunol. 2003;33:2676–2686. doi: 10.1002/eji.200323956. [DOI] [PubMed] [Google Scholar]
- 449.Chakravarty SD, Xu J, Lu B, Gerard C, Flynn J, Chan J. The chemokine receptor CXCR3 attenuates the control of chronic Mycobacterium tuberculosis infection in BALB/c mice. J Immunol. 2007;178:1723–1735. doi: 10.4049/jimmunol.178.3.1723. [DOI] [PubMed] [Google Scholar]
- 450.Hasan Z, Jamil B, Ashraf M, Islam M, Dojki M, Irfan M, Hussain R. Differential live Mycobacterium tuberculosis-, M. bovis BCG-, recombinant ESAT6-, and culture filtrate protein 10-induced immunity in tuberculosis. Clin Vaccine Immunol. 2009;16:991–998. doi: 10.1128/CVI.00091-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Azzurri A, Sow OY, Amedei A, Bah B, Diallo S, Peri G, Benagiano M, D’Elios MM, Mantovani A, Del Prete G. IFN-gamma-inducible protein 10 and pentraxin 3 plasma levels are tools for monitoring inflammation and disease activity in Mycobacterium tuberculosis infection. Microbes Infect. 2005;7:1–8. doi: 10.1016/j.micinf.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 452.Whittaker E, Gordon A, Kampmann B. Is IP-10 a better biomarker for active and latent tuberculosis in children than IFNgamma? PLoS One. 2008;3:e3901. doi: 10.1371/journal.pone.0003901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Kibiki GS, Myers LC, Kalambo CF, Hoang SB, Stoler MH, Stroup SE, Houpt ER. Bronchoalveolar neutrophils, interferon gamma-inducible protein 10 and interleukin-7 in AIDS-associated tuberculosis. Clin Exp Immunol. 2007;148:254–259. doi: 10.1111/j.1365-2249.2007.03330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Tang NL, Fan HP, Chang KC, Ching JK, Kong KP, Yew WW, Kam KM, Leung CC, Tam CM, Blackwell J, Chan CY. Genetic association between a chemokine gene CXCL-10 (IP-10, interferon gamma inducible protein 10) and susceptibility to tuberculosis. Clin Chim Acta. 2009;406:98–102. doi: 10.1016/j.cca.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Loos T, Dekeyzer L, Struyf S, Schutyser E, Gijsbers K, Gouwy M, Fraeyman A, Put W, Ronsse I, Grillet B, Opdenakker G, Van Damme J, Proost P. TLR ligands and cytokines induce CXCR3 ligands in endothelial cells: enhanced CXCL9 in autoimmune arthritis. Lab Invest. 2006;86:902–916. doi: 10.1038/labinvest.3700453. [DOI] [PubMed] [Google Scholar]
- 456.Kanda N, Shimizu T, Tada Y, Watanabe S. IL-18 enhances IFN-gamma-induced production of CXCL9, CXCL10, and CXCL11 in human keratinocytes. Eur J Immunol. 2007;37:338–350. doi: 10.1002/eji.200636420. [DOI] [PubMed] [Google Scholar]
- 457.Basset L, Chevalier S, Danger Y, Arshad MI, Piquet-Pellorce C, Gascan H, Samson M. Interleukin-27 and IFNgamma regulate the expression of CXCL9, CXCL10, and CXCL11 in hepatitis. J Mol Med (Berl) 2015;93:1355–1367. doi: 10.1007/s00109-015-1319-6. [DOI] [PubMed] [Google Scholar]
- 458.Oo YH, Banz V, Kavanagh D, Liaskou E, Withers DR, Humphreys E, Reynolds GM, Lee-Turner L, Kalia N, Hubscher SG, Klenerman P, Eksteen B, Adams DH. CXCR3-dependent recruitment and CCR6-mediated positioning of Th-17 cells in the inflamed liver. J Hepatol. 2012;57:1044–1051. doi: 10.1016/j.jhep.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, Selman M, Becerril-Villanueva E, Baquera-Heredia J, Pavon L, Kaushal D, Reinhart TA, Randall TD, Khader SA. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J Clin Invest. 2013;123:712–726. doi: 10.1172/JCI65728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Vermi W, Lonardi S, Bosisio D, Uguccioni M, Danelon G, Pileri S, Fletcher C, Sozzani S, Zorzi F, Arrigoni G, Doglioni C, Ponzoni M, Facchetti F. Identification of CXCL13 as a new marker for follicular dendritic cell sarcoma. J Pathol. 2008;216:356–364. doi: 10.1002/path.2420. [DOI] [PubMed] [Google Scholar]
- 461.Takagi R, Higashi T, Hashimoto K, Nakano K, Mizuno Y, Okazaki Y, Matsushita S. B cell chemoattractant CXCL13 is preferentially expressed by human Th17 cell clones. J Immunol. 2008;181:186–189. doi: 10.4049/jimmunol.181.1.186. [DOI] [PubMed] [Google Scholar]
- 462.Gunn MD, Ngo VN, Ansel KM, Ekland EH, Cyster JG, Williams LT. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt’s lymphoma receptor-1. Nature. 1998;391:799–803. doi: 10.1038/35876. [DOI] [PubMed] [Google Scholar]
- 463.Maglione PJ, Xu J, Chan J. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J Immunol. 2007;178:7222–7234. doi: 10.4049/jimmunol.178.11.7222. [DOI] [PubMed] [Google Scholar]