

Abstract
Objective
The gastrointestinal tract of sheep contain complex microbial communities that influence numerous aspects of the sheep’s health and development. The objective of this study was to analyze the composition and diversity of the microbiota in the gastrointestinal tract sections (rumen, reticulum, omasum, abomasum, duodenum, jejunum, ileum, cecum, colon, and rectum) of sheep.
Methods
This analysis was performed by 454 pyrosequencing using the V3-V6 region of the 16S rRNA genes. Samples were collected from five healthy, small tailed Han sheep aged 10 months, obtained at market. The bacterial composition of sheep gastrointestinal microbiota was investigated at the phylum, class, order, family, genus, and species levels.
Results
The dominant bacterial phyla in the entire gastrointestinal sections were Firmicutes, Bacteroidetes, and Proteobacteria. In the stomach, the three most dominant genera in the sheep were Prevotella, unclassified Lachnospiraceae, and Butyrivibrio. In the small intestine, the three most dominant genera in the sheep were Escherichia, unclassified Lachnospiraceae, and Ruminococcus. In the large intestine, the three most dominant genera in the sheep were Ruminococcus, unclassified Ruminococcaceae, and Prevotella. R. flavefaciens, B. fibrisolvens, and S. ruminantium were three most dominant species in the sheep gastrointestinal tract. Principal Coordinates Analysis showed that the microbial communities from each gastrointestinal section could be separated into three groups according to similarity of community composition: stomach (rumen, reticulum, omasum, and abomasum), small intestine (duodenum, jejunum, and ileum), and large intestine (cecum, colon, and rectum).
Conclusion
This is the first study to characterize the entire gastrointestinal microbiota in sheep by use of 16S rRNA gene amplicon pyrosequencing, expanding our knowledge of the gastrointestinal bacterial community of sheep.
Keywords: Pyrosequencing, Microbiota, Gastrointestinal Tract Sections, Sheep
INTRODUCTION
A huge number of various microorganisms live in the gastrointestinal tract of ruminants, about ten times their body’s own cells in number. Sheep is a ruminant animal where the gut is a fermentative chamber for a complex and dynamic microbial population. Symbiotic bacterial community is crucial for the host health in many aspects, such as in balancing the immune response, digesting the nutrients, and mediating the host physiology. In addition, symbioses between microbiota and host can facilitate the development of the latter’s gastrointestinal tract. Especially, bacteria in the ruminant gut play a major role in the biological degradation of dietary fibers. Ruminant digestion relies on the bulk of cellulose hydrolysis bacteria. However, the microbiota composition and diversity are affected by many factors, such as diet composition, host genetics, and environment, and so on. Therefore, it is now recognized that a better and sufficient understanding of the composition and diversity of the gastrointestinal microbial community is required to further enhance the growth and gut health of ruminants.
In the past, the conventional culture-based technique was used to isolate and characterize the gastrointestinal microbiota of ruminants. Indeed, even if the culture-based technique has successfully isolated key representative bacteria, it is not sufficient to characterize the entire microbial populations, because a large majority of gastrointestinal microbiota is not culturable. A recent article indicated that some unculturable microbiota were abundant in the rumen, which play an important role in the ruminal fermentation [1]. Over the last 10 years the development of high-throughput sequencing techniques has allowed for a considerable increase in knowledge of the microbial diversity of the ruminant gut. Roche 454 pyrosequencing platform provides new approaches for researchers to investigate complex microbial communities. This platform does not required a clone library, and has high throughput efficiency and sensitivity. The application of this approach enabled us to successfully analyze several samples at a time, which significantly reduced experimental cost and improved efficiency.
Although the small tail Han sheep is an important sheep breed with many favorable features, including fast growth, strong reproduction, stable genetic performance, and good adaption, data on its gut microbiota are limited. Rumen contents and feces in ruminants are often used to assess gastrointestinal microbiota [2,3], however, these sections do not represent the composition and diversity of microbiota in the entire gastrointestinal tract. Especially, the forestomach of ruminants, namely, rumen, reticulum and omasum contribute to the digestion of the cellulose substances in the diet. The small intestine is responsible for absorption of water, nutrients, and electrolytes. However, knowledge on the microbiota in the gastrointestinal tract of ruminants is limited. Recent studies explored the composition of the gastrointestinal microbiota in dairy cattle and Brazilian Nelore breed of cattle [4,5]. However, no studies to date have evaluated the entire gastrointestinal bacterial community in adult sheep using pyrosequencing, especially small tail Han sheep, which is a main sheep breed in Tianjin. The objective of this study was to characterize the gastrointestinal bacterial community of small tail Han sheep using pyrosequencing of 16S rRNA gene amplicons. Understanding the composition and structure of the microbiota in the gastrointestinal tract of sheep may be useful for developing ruminant production and management.
MATERIALS AND METHODS
Animals and sampling
The use and care of the animals used in this study were approved by the Animal Care Advisory Committee of the Tianjin Academy of Agricultural Sciences. The health of the sheep was monitored continuously before and during the experimental period. Five, healthy, small tailed Han sheep aged 10 months and weighing between 55 kg were obtained from a commercial feedlot, and fed the same feed. The diets were consisting of 57.78% green hay, 12.22% alfalfa, 16.67% corn, 8.89% soybean meal, wheat bran 3.33%, 0.11% CaHPO4, 0.89% NaCl, and 0.11% premix. One kilogram of premix contains: FeSO4·7H2O 170 g; CuSO4·5H2O 70 g; MnSO4·5H2O 290 g; ZnSO4·7H2O 240 g; CoCl2·6H2O 510 mg; KI 200 mg; NaSeO3 130 mg; vitamin A (VA) 1,620,000 IU; VD3 324,000 IU; VE 540 IU; VK3 150 mg; VB12 0.9 mg; VB5 450 mg; Calcium pantothenate 750 mg; Folic acid 15 mg. Following the standard livestock management practices, all sheep were placed in disinfected individual pens with ad libitum water and feed access. No animals showed signs of disease or ill health. Physiological and biochemical indexes of small tail Han sheep were shown in Table 1. Fresh samples (20 g) were collected from three regions of the gastrointestinal tract, namely, the stomach (rumen, reticulum, omasum, and abomasum), small intestine (duodenum, jejunum, and ileum), and large intestine (cecum, colon, and rectum). Each sample was preserved at −20°C and shipped on dry ice to the Tianjin University, Tianjin, China, for analysis of the microbiota.
Table 1.
Physiological and biochemical indexes of small tail Han sheep
| Physiological indexes | Measured values |
|---|---|
| Rectal temperature (°C) | 39.01±0.77 |
| Heart rate (times/min) | 75.20±5.38 |
| Respiration rate (times/min) | 19.07±4.92 |
| Blood physiological indexes | |
| WBC (white blood cell count) (×109/L) | 8.49±3.84 |
| RBC (red blood cell count) (×1012/L) | 11.00±0.18 |
| MCH (mean corpuscular hemoglobin) (pg) | 11.23±2.73 |
| MCV (mean corpuscular volume) (fL) | 39.30±0.31 |
| HGB (hemoglobin) (g/L) | 120.79±8.04 |
| HCT (hematocrit) (%) | 34.49±0.88 |
| PLT (platelet count) (×109/L) | 550.25±25.55 |
| Lymphocyte cell (%) | 66.53±7.16 |
| Basophil cell (%) | 0.44±0.03 |
| Acidophilic cell (%) | 3.29±1.08 |
| Monocytes (%) | 5.72±0.09 |
| Blood biochemical indexes | |
| LDH (lactate dehydrogenase) (U/L) | 433.54±45.86 |
| CA (total calcium) (mmol/L) | 2.38±0.12 |
| CHOL (total cholesterol) (mmol/L) | 3.78±0.32 |
| GLU (glucose) (mmol/L) | 2.56±0.37 |
| BUN (Blood urea nitrogen) (mmol/L) | 5.25±0.45 |
| CHE (cholinesterase) (U/L) | 640.01±2.22 |
| AKP (alkaline phosphatase) (U/L) | 325.05±69.13 |
| ALB (albumin) (g/L) | 30.18±3.00 |
| GLO (globulin) (g/L) | 23.75±3.09 |
| TP (total protein) (g/L) | 55.03±6.24 |
| GPT (glutamic-pyruvic transaminase) (U/L) | 30.15±5.06 |
| GOT (glutamic-oxalacetic transaminase) (U/L) | 81.01±15.32 |
DNA extraction
The digesta of every gastrointestinal part which contains solid and liquid was homogenized by mixing. Total DNA was extracted from the digesta samples containing solids and liquids (100 mg each) according to the instructions in E.Z.N.A stool DNA kit (Omega Bio-Tek, Doraville, CA, USA). For better analysis of gram-positive bacteria DNA, a second incubation at 95°C for 10 min was performed following the initial incubation at 70°C for 13 min in the protocol. All procedures were performed on ice. The final elution volume was 200 μL, and DNA concentration was determined by a Nano Drop spectrophotometer (Nano Drop Technologies, Wilmington, DE, USA).
PCR amplification of the V3–V6 region of bacterial 16S rRNA genes
The DNA extracted from each sample was amplified with a set of primers targeting the hypervariable V3–V6 region of the 16S rRNA gene. The forward primer was 338F: 5′-ACTCCTACGGGAGGCAGCAG-3′ and the reverse primer was 1046R: 5′-CGACAGCCATGCANCACCT-3′ [6]. The forward primer contained the sequence of the Titanium A adapter and a 9-bp barcode sequence. The reverse primer contained the sequence of the Titanium B adapter. The amplification mix contained 5 U of FastStart High Fidelity polymerase, 1×FastStart High Fidelity Reaction Buffer, 0.2 mM deoxynucleoside triphosphates, 0.4 μM of each primer, and 50 ng of template DNA in a reaction volume of 50 μL. Polymerase chain reaction (PCR) was performed on MyCyclerTM Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA) set as follows: 94°C for 4 min; 25 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 50 s; and 72°C for 10 min. The PCR amplicon products were visualized using 1% agarose gels, extracted from the gels, and then purified using a SanPrep PCR Purification Kit (Sangon Biotech, Shanghai, China) according to the manufacturer’s instructions.
454 Pyrosequencing and data analysis
The PCR products of the V3–V6 region of the 16S rRNA genes were sequenced using a Roche 454 FLX Titanium sequencer. The 16S rRNA raw sequence data were processed using the quantitative insights into microbial ecology (QIIME, University of Colorado, Boulder, CO, USA) version 1.7.0 software pipeline. The sequences were quality-filtered using default QIIME parameters, and poor-quality sequences were eliminated from the data sets (i.e. sequences where lengths were less than 200 bp or more than 1,000 bp, number of ambiguous bases exceeded a limit of six, there was a missing quality score, the mean quality score was below 25, the maximum homopolymer run exceeded a limit of six, and the number of mismatches in the primer exceeded a limit of two). Chimeras were detected and excluded using the software Black Box Chimera Check (B2C2) (Research of Testing Laboratory of the South Plains, Lubbock, TX, USA). Qualified sequences were clustered into operational taxonomic units (OTUs) with a 97% similarity threshold using the UCLUST algorithm (Tiburon, CA, USA). A representative sequence was chosen from each OTU by selecting the first sequence. The representative sequences were taxonomically classified using the ribosomal database project (RDP) classifier, and the confidence threshold was set at 0.8. The relative abundances of the phylum and class levels were plotted as bar graph. The relative abundances of the order, family and genus levels were created as heatmap. The relative abundance of the species was shown as a table. Representative sequences were aligned against the Greengenes core set using Python Nearest Alignment Space Termination (PyNAST) software (University of Colorado, Boulder, CO, USA). The minimum aligned sequence length was set at 150 bp and the minimum percent identity was set at 75%. A subsequent phylogenetic tree was built using FastTree (Lawrence Berkeley National Laboratory, Berkeley, CA, USA). To account for unequal sequencing depth across samples, subsequent analyses were performed on a randomly selected subset of 1,038 sequences per sample. For alpha diversity measurements, the estimated number of OTUs in each sample using the Chao1 index, the diversity of the populations using the Shannon index, and the amount of phylogenetic branch length observed in each sample (phylogenetic distance [PD]) were determined. For beta diversity, differences in the microbial communities between the gastrointestinal sections were investigated using the phylogeny-based unweighted UniFrac distance metric.
RESULTS
Richness and diversity analysis
Fifty samples from the 10 different gastrointestinal tract sections (rumen, reticulum, omasum, abomasum, duodenum, jejunum, ileum, cecum, colon, and rectum) of 5 sheep were used in our experiments. The pyrosequencing pipeline yielded 363,298 16S rRNA gene sequence reads from all gastrointestinal tract samples. After quality-filtering with QIIME default settings, a total of 252,030 16S rRNA sequence reads were retained from all gastrointestinal microbiota samples, including 30,350 reads from the rumen, 24,965 reads from the reticulum, 63,825 reads from the omasum, 34,825 reads from the abomasum, 11,960 reads from the duodenum, 6,035 reads from the jejunum, 31,795 reads from the ileum, 26,550 reads from the cecum, 8,840 reads from the colon, and 12,885 reads from the rectum (Table 2). The average length of the quality-checked and filtered sequences was 605 bp. The maximum length was 818 bp, and the minimum length was 200 bp.
Table 2.
Alpha diversity estimates of microbiota associated with the gastrointestinal tract sections of sheep1)
| Sample | Total reads | Total OTUs | Reads subsampled | Observed species | Chao1 | Shannon | PD |
|---|---|---|---|---|---|---|---|
| Rumen | 6,070±766 | 651±222 | 1,038 | 457.200±133.200 | 1,071.704±303.257 | 8.113±1.421 | 26.055±4.415 |
| Reticulum | 4,993±419 | 633±195 | 1,038 | 454.000±119.100 | 1,057.464±299.901 | 8.065±1.077 | 24.866±3.778 |
| Omasum | 12,765±1,222 | 789±353 | 1,038 | 572.100±203.300 | 1,819.647±401.110 | 8.525±1.609 | 32.331±4.994 |
| Abomasum | 6,965±891 | 703±347 | 1,038 | 521.500±194.600 | 1,315.026±307.458 | 8.334±1.522 | 30.340±4.636 |
| Duodenum | 2,392±432 | 362±109 | 1,038 | 326.000±79.500 | 669.954±178.903 | 7.004±0.778 | 20.650±2.330 |
| Jejunum | 1,207±367 | 322±99 | 1,038 | 316.100±77.900 | 640.548±162.316 | 6.957±0.765 | 20.924±2.421 |
| Ileum | 6,359±808 | 648±272 | 1,038 | 476.200±123.000 | 1,260.505±338.792 | 7.993±0.993 | 26.626±3.909 |
| Cecum | 5,310±776 | 644±268 | 1,038 | 447.600±108.800 | 916.780±183.394 | 8.102±1.399 | 24.127±3.111 |
| Colon | 1,768±564 | 526±190 | 1,038 | 372.000±99.100 | 721.966±137.115 | 7.585±0.820 | 20.432±2.027 |
| Rectum | 2,577±367 | 615±213 | 1,038 | 445.700±105.200 | 975.539±159.533 | 7.947±0.986 | 26.944±4.383 |
Data are expressed as average±standard deviation (n = 5 sheep). The number of observed species, Chao 1 estimator, Shannon index and phylogenetic distance (PD) index were analyzed after reads subsampled to 1,038 reads. The operational taxonomic units (OTUs) were defined with 3% dissimilarity.
To compare the bacterial species richness and diversity in the entire gastrointestinal tract sections, the bacterial richness and diversity levels were analyzed using the observed species, Chao1 estimator, Shannon index, and PD index. OTUs were grouped at the 97% similarity level. To account for unequal sequencing depth across samples, subsequent analyses were performed on a randomly selected subset of 1,038 sequences per sample. This number was chosen to avoid exclusion of samples with a lower number of sequence reads from further analysis. As shown in Table 2, the highest microbial richness of samples were found in the stomach (rumen, reticulum, omasum, and abomasum), and the average of the observed species were between 454.000 and 572.100; the average of Chao1 index varied from 1,057.464 to 1,819.647. Ileum and large intestine (cecum, colon, and rectum) samples had moderate microbial richness, and the average of the observed species were between 372.000 and 476.200; the average of Chao1 index varied from 721.966 to 1,260.505. While duodenum and jejunum samples had lowest microbial community richness (observed species 326.000±79.500 and 316.100±77.900, Chao1 index 669.954±178.903 and 640.548±162.316). Similarly, the stomach samples had the highest microbial diversity (the average of Shannon index were between 8.065 and 8.525, the average of PD index varied from 24.866 to 32.331). Ileum and large intestine samples had moderate microbial diversity, and the average of Shannon index were between 7.585 and 8.102; the average of PD index varied from 20.432 to 26.944. While duodenum and jejunum samples had lowest microbial community diversity (Shannon index 7.004±0.778 and 6.957±0.765, PD index 20.650±2.330 and 20.924±2.421).
Sheep gastrointestinal microbiota composition at the phylum level
To describe the composition of the gastrointestinal microbiota, a taxon-dependent analysis was carried out using the RDP classifier. The results, shown in Figure 1, describe the average distribution of DNA sequences into phyla (n = 5 sheep). A total of 15 bacterial phyla were identified across the entire gastrointestinal tract sections. Firmicutes and Bacteroidetes were the most abundant phyla in all gastrointestinal tract sections, and comprised 52.3%±15.7% and 32.4%±4.4% of the total sequences, followed by Proteobacteria (7.5%±2.1%). The remaining sequences were Verrucomicrobia (2.0%±0.7%), Actinobacteria (1.7%±0.5%), Tenericutes (1.0%±0.1%), and a number of low-abundance phyla. The low-abundant phyla were TM7, Spirochaetes, Synergistetes, Fibrobacteres, Cyanobacteria, Lentisphaerae, Chloroflexi, Elusimicrobia, and Sulfur River 1 (SR1). The proportion of sequences that could not be assigned to a phylum using the RDP classifier ranged from 0.3% to 1.9%. When bacterial composition was compared regionally, the bacterial composition of the each gastrointestinal tract section showed no significant differences between them. The phylum Firmicutes dominated all bacterial communities along the gastrointestinal tract except for in the omasum and abomasums, where Bacteroidetes was predominant. The phylum Bacteroidetes (7.7±1.9% and 8.9±1.3%) was less abundant in the duodenum and jejunum than other gastrointestinal tract sections. In the contrast, the phyla Proteobacteria (13.5% to 23.2%) and Actinobacteria (3.7% to 5.8%) were more abundant in the small intestine than the stomach and large intestine. Although the phyla Verrucomicrobia had the low abundance in the gastrointestinal tract sections, the Verrucomicrobia in the duodenum accounted for 13.8%±2.9% of the total sequences.
Figure 1.
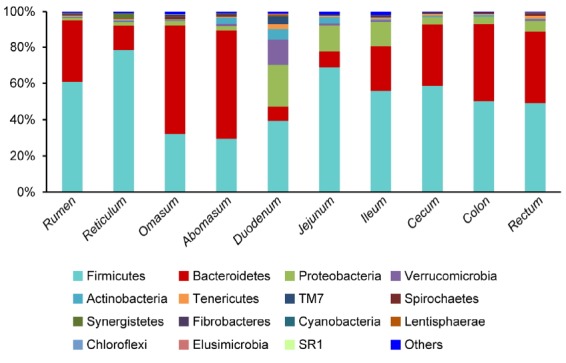
Ribosomal database project (RDP) classification of the sequence reads from different gastrointestinal tracts of sheep at the phylum level. The abscissa (X-axis) represents the different gastrointestinal tract sections, and the ordinate (Y-axis) represents the average relative abundance of the bacterial phyla (n = 5 sheep).
Sheep gastrointestinal microbiota composition at the class level
The results, shown in Figure 2, describe the average distribution of DNA sequences into classes (n = 5 sheep). A total of 25 bacterial classes were identified across the entire gastrointestinal tract sections. Clostridia and Bacteroidia were the most abundant classes in all gastrointestinal tract sections, and comprised 50.3%±13.2% and 32.3%±4.1% of the total sequences, followed by Gammaproteobacteria (6.6%±1.7%). The remaining sequences were Verruco-5 (1.9%±0.7%), Actinobacteria (1.7%±0.5%), Bacilli (1.3%±0.2%), Mollicutes (1.0%±0.1%), and a number of low-abundance classes. The results showed that several classes contributed to the differences in community composition between different gastrointestinal tract sections. In the stomach, the three most dominant classes in the sheep were Clostridia (49.2%±7.7%), Bacteroidia (41.6%±5.6%), and Gammaproteobacteria (1.7%±0.3%). It is worth mentioning that Actinobacteria (1.2%±0.4%) was also abundant in the stomach. In the small intestine, the three most dominant classes in the sheep were Clostridia (50.0%±9.9%), Gammaproteobacteria (15.8%±3.3%), and Bacteroidia (13.6%±4.1%). Classes Verruco-5 (5.2%±1.1%), Bacilli (4.0%±0.9%), and TM7-3 (1.8%±0.2%) were significantly more abundant in the small intestine than other gastrointestinal tract sections. In the large intestine, the three most dominant classes in the sheep were Clostridia (52.0%±9.7%), Bacteroidia (38.6%±6.3%), and Gammaproteobacteria (4.1%±1.6%).
Figure 2.
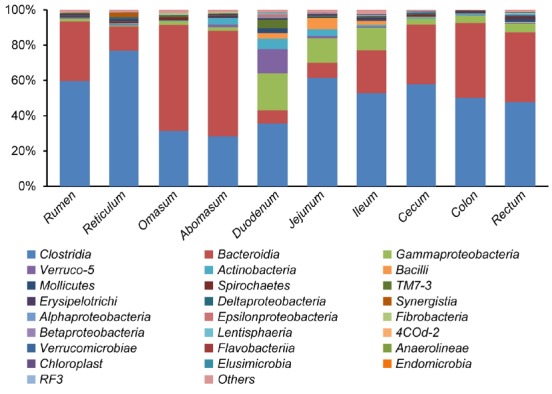
Ribosomal database project (RDP) classification of the sequence reads from different gastrointestinal tracts of sheep at the class level. The abscissa (X-axis) represents the different gastrointestinal tract sections, and the ordinate (Y-axis) represents the average relative abundance of the bacterial classes (n = 5 sheep).
Sheep gastrointestinal microbiota composition at the order level
Differences in bacterial average abundance at the order level for the sheep gastrointestinal tract sections are shown as a heatmap in Figure 3 (n = 5 sheep). Regardless of which gastrointestinal sections they occurred in, the 8 orders, including Clostridiales (24.8% to 70.2%), Bacteroidales (7.6% to 59.7%), Aeromonadales (0.2% to 3.4%), contaminated aquifer clone WCHB1-41 (0.1% to 13.8%), Coriobacteriales (0.2% to 4.6%), Bifidobacteriales (0.1% to 4.9%), RF39 (0.2% to 1.6%), and Erysipelotrichales (0.1% to 1.1%) were defined as the core orders, because they were found in all gastrointestinal tract sections of sheep. The results showed that several orders contributed to the differences in community composition between different gastrointestinal tract sections. In the stomach, the three most dominant orders in the sheep were Clostridiales (45.1%±6.6%), Bacteroidales (41.6%±5.6%), and Coriobacteriales (1.8%±0.2%). In the small intestine, the three most dominant orders in the sheep were Clostridiales (44.3%±6.9%), Bacteroidales (13.6%±4.1%), and Enterobacteriales (13.4%±3.1%). Orders contaminated aquifer clone WCHB1-41 (5.2%±1.7%), oral clone CW040 (1.8%±0.8%), and Lactobacillales (4.6%±1.8%) were significantly more abundant in the small intestine than other gastrointestinal tract sections. In the large intestine, the three most dominant orders in the sheep were Clostridiales (48.2%±6.3%), Bacteroidiales (38.6%±6.3%), and Aeromonadales (2.9%±0.4%).
Figure 3.
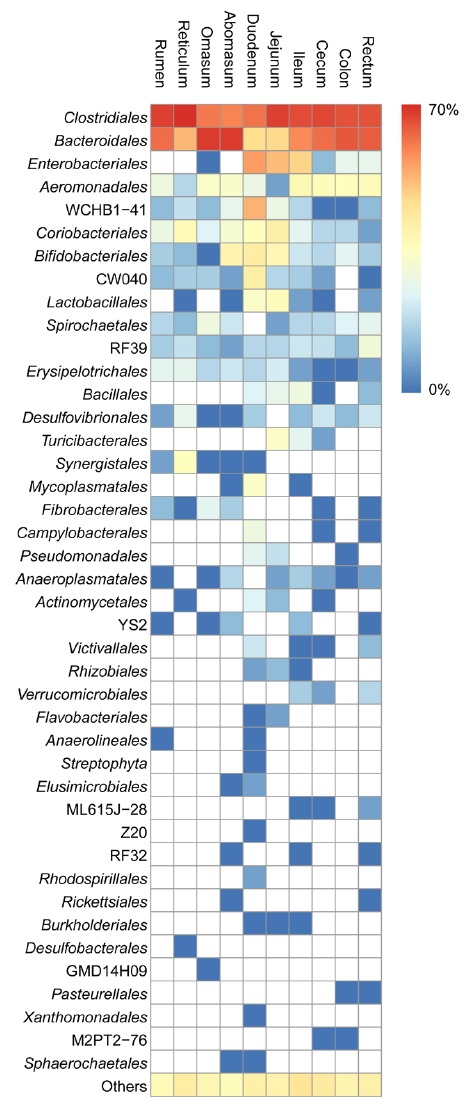
Heatmap at the order level among the sheep gastrointestinal tract sections. Heatmap columns show normalized average relative abundance (n = 5 sheep) and provide a comparison of the differences in abundance between different orders in the same section. Heatmap rows show normalized average relative abundance and provide a comparison of the differences in abundance between different sections in the same order.
Sheep gastrointestinal microbiota composition at the family level
Differences in bacterial average abundance at the family level for the sheep gastrointestinal tract sections are shown as a heatmap in Figure 4 (n = 5 sheep). Regardless of which gastrointestinal sections they occurred in, the 11 families, including Lachnospiraceae (7.1% to 37.5%), Ruminococcaceae (7.2% to 28.4%), Prevotellaceae (2.2% to 40.1%), Veillonellaceae (0.5% to 11.0%), S24-7 (a member of Bacteroidales) (0.7% to 11.0%), Paraprevotellaceae (0.3% to 6.8%), Clostridiaceae (1.4% to 8.5%), Succinivibrionaceae (0.2% to 3.4%), RFP12 (a member of WCHB1-41) (0.1% to 13.8%), Bifidobacteriaceae (0.1% to 4.9%), and Porphyromonadaceae (0.1% to 3.0%) were defined as the core families, because they were found in all gastrointestinal tract sections of sheep. The results showed that several families contributed to the differences in community composition between different gastrointestinal tract sections. In the stomach, the three most dominant families in the sheep were Prevotellaceae (26.7%±3.1%), Lachnospiraceae (22.8%±2.7%), and Ruminococcaceae (11.1%±3.3%). It is worth mentioning that Veillonellaceae (7.4%±1.6%) and S24-7 (6.5%±2.0%) were also abundant in the stomach. In the small intestine, the three most dominant families in the sheep were Lachnospiraceae (17.5%±2.9%), Enterobacteriaceae (13.4%±3.4%), and Ruminococcaceae (11.9%±2.1%). Families Peptostreptococcaeae (5.4%±0.9%), RFP12 (5.2%±0.4%), and Clostridiaceae (4.6%±1.1%) were significantly more abundant in the small intestine than other gastrointestinal tract sections. In the large intestine, the three most dominant families in the sheep were Ruminococcaceae (26.7%±3.6%), Lachnospiraceae (12.1%±2.2%), and Prevotellaceae (11.0%±2.3%). Families Bacteroidaceae (7.1%±1.5%) and Paraprevotellaceae (5.6%±0.9%) were significantly more abundant in the large intestine than other gastrointestinal tract sections.
Figure 4.
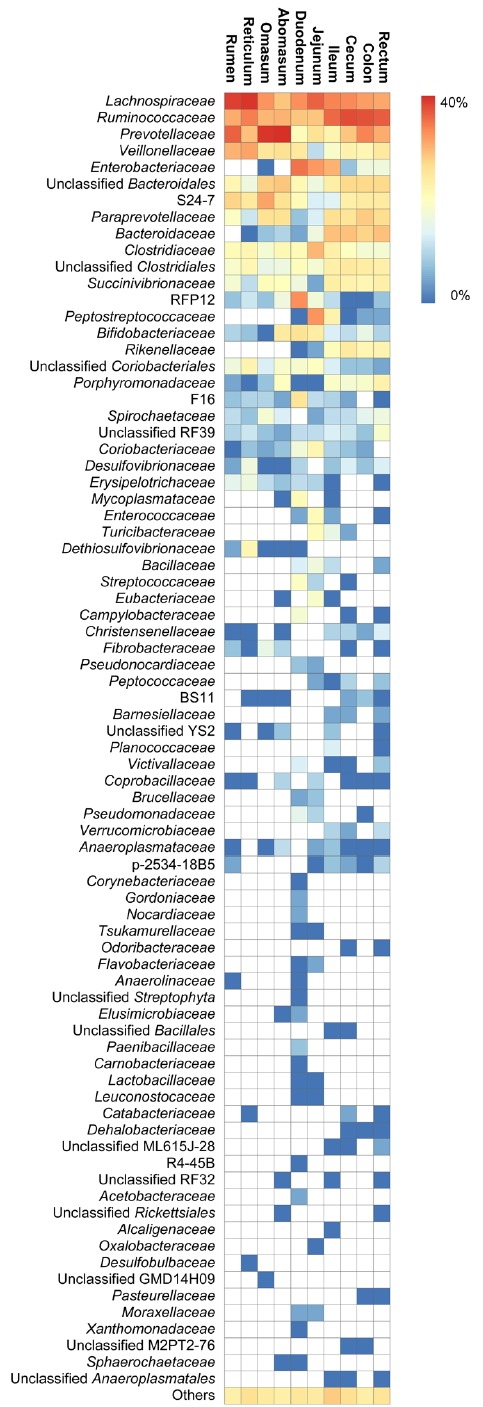
Heatmap at the family level among the sheep gastrointestinal tract sections. Heatmap columns show normalized average relative abundance (n = 5 sheep) and provide a comparison of the differences in abundance between different families in the same section. Heatmap rows show normalized average relative abundance and provide a comparison of the differences in abundance between different sections in the same family.
Sheep gastrointestinal microbiota composition at the genus level
A total of 120 genera were identified in the entire gastrointestinal tract sections of sheep (n = 5 sheep). Of these, 31 genera were found to be abundant present at ≥0.5% of gastrointestinal tract bacterial sequences (Figure 5). The 31 genera accounted for 79.0%±22.7% of the total sequences for the gastrointestinal tract sections. Regardless of which gastrointestinal sections they occurred in, the 15 genera, including Prevotella (2.2% to 40.2%), unclassified Lachnospiraceae (3.5% to 15.3%), Ruminococcus (2.0% to 14.2%), unclassified Ruminococcaceae (3.0% to 12.5%), unclassified S24-7 (0.7% to 11.0%), CF231 (a member of Paraprevotellaceae) (0.2% to 4.9%), unclassified RFP12 (0.1% to 13.8%), unclassified Clostridiaceae (0.4% to 6.1%), unclassified Bifidobacteriaceae (0.1% to 3.6%), Clostridium (0.1% to 2.3%), Oscillospira (0.1% to 3.2%), unclassified Veillonellaceae (0.1% to 4.1%), Succinivibrio (0.2% to 2.5%), Anaerovibrio (0.1% to 2.2%), and Coprococcus (0.2% to 1.9%) were defined as the core genera, because they were found in all gastrointestinal tract sections of sheep. The results showed that several genera contributed to the differences in community composition between different gastrointestinal tract sections. In the stomach, the three most dominant genera in the sheep were Prevotella (26.7%±3.1%), unclassified Lachnospiraceae (9.8%±2.0%), and Butyrivibrio (9.5%±2.1%). Genus unclassified S24-7 (6.5%±2.0%) was significantly more abundant in the stomach than other gastrointestinal tract sections. In the small intestine, the three most dominant genera in the sheep were Escherichia (12.6%±3.0%), unclassified Lachnospiraceae (10.4%±2.7%), and Ruminococcus (6.3%±1.9%). Genera unclassified Peptostreptococcaceae (5.3%±0.8%) and unclassified RFP12 (5.2%±0.4%) were significantly more abundant in the small intestine than other gastrointestinal tract sections. In the large intestine, the three most dominant genera in the sheep were Ruminococcus (13.0%±2.9%), unclassified Ruminococcaceae (12.3%±2.6%), and Prevotella (12.1%±2.2%). Genus 5–7N15 (5.6%±0.9%) was significantly more abundant in the large intestine than other gastrointestinal tract sections.
Figure 5.
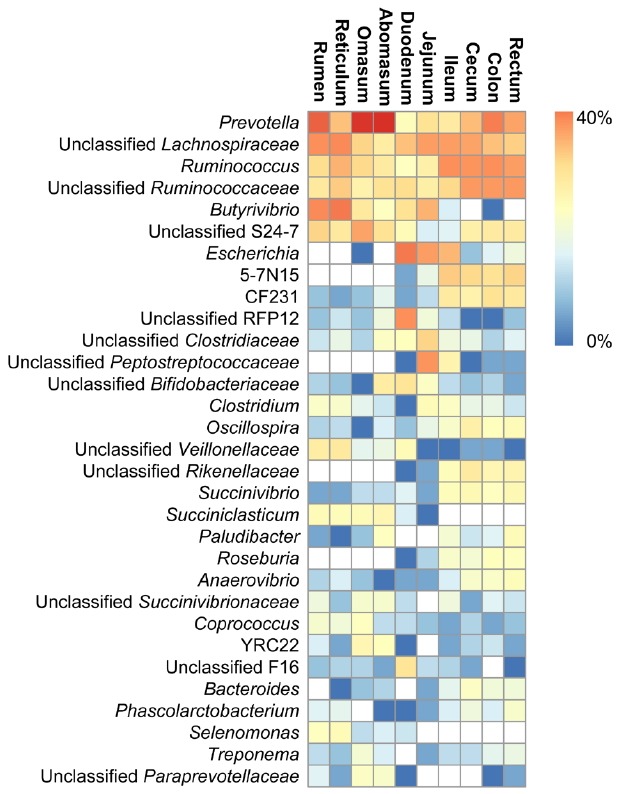
Heatmap at the genus level among the sheep gastrointestinal tract sections (more than 0.5% of the total DNA sequences). Heatmap columns show normalized average relative abundance (n = 5 sheep) and provide a comparison of the differences in abundance between different genera in the same section. Heatmap rows show normalized average relative abundance and provide a comparison of the differences in abundance between different sections in the same genus.
Sheep gastrointestinal microbiota composition at the species level
Due to the V3–V6 region of 16S rRNA gene (about 600 bp) being amplied, limited species were identified in present study. A total of 22 species are shown in Table 3, R. flavefaciens (average relative abundance was 2.5% of total sequences), B. fibrisolvens (average relative abundance was 2.3% of total sequences), and S. ruminantium (average relative abundance was 0.6% of total sequences) were three most dominant species in the sheep gastrointestinal tract. The species R. flavefaciens (2.9% to 7.9%), S. ruminantium (0.5% to 2.4%), R. bromii (0.6% to 1.6%), F. succinogenes (0.1% to 1.0%), P. ruminis (0.1% to 1.0%), P. ruminicola (0.0% to 1.0%), R. calidus (0.2% to 0.6%), Desulfovibrio D168 (0.0% to 0.7%), M. elsdenii (0.0% to 0.5%), and D. invisus (0.0% to 0.5%) were more abundant in the stomach than the other gastrointestinal sections. Meanwhile, the species B. pseudolongum (0.0% to 1.3%), C. butyricum (0.1% to 1.1%), P. copri (0.0% to 0.4%), P. fragi (0.0% to 0.4%), B. foraminis (0.0% to 0.5%), and P. stercorea (0.0% to 0.5%) were more abundant in the small intestine and large intestine than the stomach. It is worthy that P. veronii (0.5%±0.2%) has high abundance in the duodenum of sheep, and C. perfringens (0.2%±0.1% and 0.3%±0.2%) has high abundance in the jejunum and ileum of sheep.
Table 3.
RDP classification of the sequence reads from different gastrointestinal tracts of sheep at the abundant species level (%)1)
| Species | Rumen | Reticulum | Omasum | Abomasum | Duodenum | Jejunum | Ileum | Cecum | Colon | Rectum |
|---|---|---|---|---|---|---|---|---|---|---|
| Ruminococcus flavefaciens | 3.8±0.7 | 7.9±1.0 | 4.0±1.3 | 2.9±1.0 | 0.9±0.4 | 0.9±0.5 | 1.1±0.6 | 1.2±0.5 | 1.1±0.4 | 1.0±0.4 |
| Butyrivibrio fibrisolvens | 4.3±0.8 | 6.4±1.1 | 2.2±1.1 | 1.5±1.0 | 2.8±1.1 | 5.4±1.7 | 0.7±0.5 | NA | 0.1±0.1 | NA |
| Selenomonas ruminantium | 1.9±0.5 | 2.4±0.6 | 0.5±0.1 | 0.7±0.3 | 0.6±0.3 | NA | NA | NA | NA | NA |
| Ruminococcus bromii | 1.0±0.2 | 0.8±1.4 | 1.6±0.7 | 0.6±0.2 | 0.1±0.1 | 0.1±0.1 | 0.1±0.1 | 0.4±0.2 | 0.3±0.3 | 0.3±0.1 |
| Bifidobacterium pseudolongum | NA | NA | NA | 0.1±0.1 | NA | 1.3±0.6 | 0.2±0.2 | 0.3±0.1 | 0.6±0.3 | 0.3±0.2 |
| Clostridium butyricum | NA | NA | NA | 0.1±0.1 | 0.1±0.1 | 0.1±0.1 | 1.1±0.3 | 0.8±0.4 | 0.2±0.1 | 0.1±0.1 |
| Fibrobacter succinogenes | 0.3±0.1 | 0.1±0.1 | 1.0±0.6 | 0.4±0.2 | NA | NA | NA | 0.1±0.1 | NA | 0.1±0.1 |
| Pseudobutyrivibrio ruminis | 0.3±0.1 | 1.0±0.3 | 0.6±0.2 | 0.1±0.1 | NA | NA | NA | NA | NA | NA |
| Prevotella ruminicola | 0.2±0.1 | NA | 0.6±0.3 | 1.0±0.7 | NA | 0.1±0.1 | NA | NA | NA | NA |
| Prevotella copri | 0.2±0.1 | NA | 0.1±0.1 | 0.1±0.1 | 0.4±0.2 | NA | 0.4±0.2 | NA | 0.1±0.1 | 0.3±0.2 |
| Ruminococcus calidus | 0.6±0.2 | 0.3±0.2 | 0.2±0.1 | 0.2±0.1 | 0.1±0.1 | NA | NA | NA | NA | NA |
| Pseudomonas fragi | 0.1±0.1 | 0.1±0.1 | NA | 0.1±0.1 | 0.4±0.2 | 0.4±0.2 | 0.1±0.1 | 0.2±0.1 | NA | NA |
| Desulfovibrio D168 | 0.1±0.1 | 0.7±0.4 | NA | 0.1±0.1 | 0.3±0.2 | NA | NA | NA | NA | NA |
| Megasphaera elsdenii | NA | NA | 0.5±0.3 | 0.3±0.2 | NA | NA | NA | NA | NA | NA |
| Campylobacter fetus | NA | NA | NA | 0.3±0.2 | 0.3±0.1 | NA | NA | NA | 0.1±0.1 | 0.1±0.1 |
| Dialister invisus | 0.2±0.1 | 0.5±0.3 | NA | NA | NA | NA | NA | NA | NA | NA |
| Bacillus foraminis | NA | NA | NA | NA | NA | NA | 0.5±0.3 | NA | NA | 0.2±0.1 |
| Prevotella stercorea | NA | NA | NA | NA | 0.1±0.1 | NA | NA | 0.5±0.3 | NA | NA |
| Pseudomonas veronii | NA | NA | NA | 0.1±0.1 | 0.5±0.2 | NA | NA | NA | NA | NA |
| Clostridium perfringens | NA | NA | NA | NA | NA | 0.2±0.1 | 0.3±0.2 | NA | NA | NA |
| Sharpea azabuensis | 0.1±0.1 | NA | NA | 0.2±0.1 | NA | 0.2±0.1 | NA | NA | NA | NA |
| Sharpea p-3329-23G2 | 0.1±0.1 | NA | NA | 0.2±0.2 | NA | 0.2±0.2 | NA | NA | NA | NA |
Data are expressed as average±standard deviation (n = 5 sheep).
NA, not found in this gastrointestinal tract section.
Principal coordinates analysis
The diversities between samples from different gastrointestinal tract sections (i.e. beta diversity) were compared, and principal coordinates analysis (PCoA) of unweighted UniFrac distances (an evaluation of community membership that does not consider abundances) was carried out. The PCoA plot, shown in Figure 6, indicated that the microbial communities of gastrointestinal tract in sheep were separated into three groups according to similarity of community composition which include the stomach (rumen, reticulum, omasum, and abomasum), small intestine (duodenum, jejunum, and ileum), and large intestine (cecum, colon, and rectum).
Figure 6.
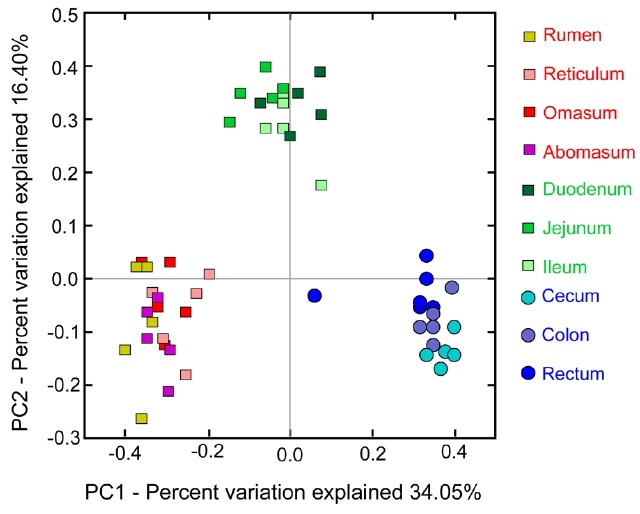
Beta diversity estimates for the gastrointestinal microbiota of sheep. Using the unweighted UniFrac distance metric to measure phylogenetic distances between samples from different gastrointestinal sections.
DISCUSSION
In our study, higher bacterial richness and diversity were observed in the stomach and large intestine than in the small intestine, which was also previously observed in the dairy cattle and the Brazilian Nelore breed of cattle [4,5]. The distal gut environment of the ruminant is more complex than the proximal gut. Most of the dietary constituents are digested in the stomach. The small intestine is much longer than the other sections, and small intestine has high concentration of bile salt and digestive enzymes so that the bacteria are difficult to grow. The partial microbial digestion that also takes place in the large intestine of ruminants could also explains the detection of high richness and diversity in the large intestine. Different oxygen tension and physiological roles in different gastrointestinal tract sections may lead to this result. The PCoA plot in the present study is in agreement with on the Nelore cattle [4] and shows that the samples from adjacent gastrointestinal tract segment (stomach, small intestine, and large intestine) harbor microbial communities more similar than from other segments.
The phylum level, the structure of bacterial community in the gastrointestinal tract was similar to that found in Chinese Mongolian sheep [7] and Holstein dairy cattle [5]. However, with regard to the abundance of these predominant phyla, there were some differences found among these studies. These differences might be due to variations in species, diets, living environment, and analysis methods. In general, the microbiota of the stomach, ileum, and large intestine exhibited greater abundances of Firmicutes and Bacteroidetes, while the duodenum and jejunum showed higher relative abundances of Firmicutes and Proteobacteria. A recent study on the dairy cattle Illumina-based method found that among the gastrointestinal tract, the high abundance of phylum Proteobacteria was found in the small intestine [5]. Firmicutes in the ruminants is known to degrade the fibre and cellulose [8]. Bacteroidetes is known to aid the digestion of complex carbohydrates, and also ferment organic matter [9]. The cause of the high abundance of Proteobacteria in the small intestine is not entirely clear, and future studies are needed to clarify this issue. To our knowledge, TM7 (0.0% to 4.6%) and SR1 (0.0% to 0.1%) phyla were found in the sheep gastrointestinal tract for the first time. TM7, which has not yet been cultivated in a laboratory, and is also reported in the feces of grass hay-fed horses [10]. SR1 has been reported in the rumen of cow using pyrosequencing method [11], however the role of this bacteria in the sheep gastrointestinal tract remains unknown.
Fifteen genera were considered as the core genera because these were existed in the all gastrointestinal tract sections. Of these 15 genera, several of unclassified genera, including S24-7, CF231, and RFP12, were detected. Therefore, this study provides detailed information regarding to both known bacteria and unclassified bacteria. A study on the rumen bacterial diversity of 80 to 110-day-old goats using 16S rRNA sequencing indicated that as the age of the goat increases, S24-7 showed an increasing trend [12]. Therefore, that the 10-months sheep in the present study had a high abundance of S24-7 in the stomach is reasonable. CF231, family Paraprevotellaceae, was the third top genus of the phylum Bacteroidetes in the rumen of steers [13]. However, in our present study, CF231 was high abundance in the ileum and large intestine, and the stomach was of low abundance. Different species could produce this discrepancy. The function of CF231 will need to be studied in the future. Family in the RFP12, order of Verrucomicrobia was the second most abundant genus among all horse feces [14]. However, in our present study, RFP12 was of high abundance in the jejunum, which was never reported before for ruminants. Therefore, the function of RFP12 will need to be characterized in the future. Of these 15 genera, a total of 11 genera, including Prevotella, unclassified Lachnospiraceae, Ruminococcus, unclassified Ruminococcaceae, CF231, unclassified RFP 12, unclassified Clostridiaceae, Clostridium, Oscillospira, unclassified Veillonellaceae, and Coprococcus, have been reported to dominate the yaks rumen using Illumina MiSeq sequencing method (Majorbio Bio-Pharm Technology Co., Ltd, Shanghai, China) [15]. Succinivibrio was previously detected in the rumen of cow using pyroseuquencing method [11]. Anaerovibrio was seen throughout the gastrointestinal tract of 3-week-old preweaned calves using pyrosequencing method [16]. Unclassified Bifidobacteriaceae was previously found in the ileum of goats using pyrosequencing method [17]. Therefore, some core genera could be shared in the most of the ruminants.
A primary finding of our study is that there are differences in microbiota composition between the segments of the gastrointestinal tract. This is in agreement with previous studies that have reported significant changes in the microbiota within the gastrointestinal tract as digesta passes from one segment to another [4]. Prevotella in the stomach or the large intestine was identified the most abundant and important genus, which was in agreement with the sequencing of rumen or cecum samples of ruminants [18,19]. Prevotella, which has a unique mucin glycoprotein degradation capability, might exploit this, resulting in the host’s increased growth and survival, and also can degrade the hemicelluloses and xylans, which promotes the digestion of the feed [20]. Butyrivibrio (average relative abundance is 9.5% of total sequences) dominated in the stomach of sheep in the present study. The abundance of Butyrivibrio was in line with the previous report on the goat rumen microbiota, using cloned 16S rRNA gene analysis, where it accounted about 10.0% [21] and dairy cattle four stomach microbiota, using Illumina MiSeq (Department of Computer Science, North Arizona University, Flagstaff, AZ, USA) platform analysis, where Butyrivibrio accounted for about 5.1% [5]. Therefore, even if the different methods and different ruminant breeds were used, Butyrivibrio dominated the stomach of ruminants. Dunne et al. demonstrated that Butyrivibrio could modulate the secretion of hemicelluloses-degrading enzymes [22], which supported the notion that this organism made an important contribution to polysaccharide degradation in the rumen. In addition, Butyrivibrio is an important butyrate producer, and promotes the stomach epithelium proliferation. Ruminococcus dominated in the small intestine and large intestine, and it account for 6.3%±1.9% and 13.0%±2.9% of total sequences, respectively. Ruminocuccus is also found in the 3-week old preweaned calves, and it dominated in the jejunum, however it is less abundant genus in the large intestine [16]. This disagreement may be due to the variance of the host age, diet and breed. Ruminococcus was found to produce carbohydrate active enzymes, and degraded the carbohydrate from the diet [23]. It is noteworthy that Escherichia (12.6%±3.0%) accounted a large amount in the small intestine, which is unexpected before. A review study [24] indicated Proteobacteria represented between 5% and 40% of the bacteria detected in the ileum of pigs, and it is well known that Escherichia was the largest genus in the Proteobacteria. Therefore, whether pigs or ruminants, Escherichia may have the same physiological roles in the small intestine. A high abundance of unclassified Lachnospiraceae was observed in the stomach and small intestine in the present study, which agrees with a review on the status of the phylogenetic diversity census of ruminal microbiomes that summarized Lachnospiraceae was one of the largest taxons [1]. A study on the ileum of goats using pyrosequencing also indicated that unclassified Lachnospiraceae dominated in the ileum [17]. Lachnospiraceae is known to be a beneficial bacterium in the human intestine, because of the role it plays in the fermentation of carbohydrates to short chain fatty acids [25]. Maybe Lachnospiraceae has the similar role in the gastrointestinal tracts of sheep. A high abundance of unclassified Ruminococcaceae (12.3%±2.6%) was observed in the large intestine from our study, which is in accordance with the report on adult goat cecal luminal content microbiota [26]. Most of the Ruminococcaceae also act as major degraders of resistant polysaccharides, such as starch and cellulose, and contributes a range of degradative enzyme systems that allow the host to break up plant cell walls [27]. In our study, the abundant genera in the large intestine does not in agree with the cattle fecal microbiota identified using pyrosequencing [28]. They suggested that Clostridium (19.7%) and Bacteroides (10.4%) predominated in the feces of cattle. However, in the present study, Clostridium and Bacteroides only accounted 0.6% and 1.3% of total sequences in the rectum, respectively. The large variation in the abundant genera could be attributed to the differences in the breed, and differences between feces and rectal samples. Therefore, the feces should not be used instead of rectal samples.
Perhaps, the most salient finding in the present study was the characterization of the microbial composition in the sheep gastrointestinal tract at the species level. As Illumina sequencing, the most popular method nowadays, would not be able to identify to the species owing to short sequencing length. However, pyrosequening has a long sequencing length, so it overcomes this problem. R. flavefaciens, R. bromii, and F. succinogenes are the most dominant cellulolytic bacteria in the rumen and feces of ruminant [1,7]. Our results demonstrated that R. flavefaciens was the most abundant species in the gastrointestinal tract. This result supports the previous finding that R. flavefaciens dominated in the rumen of goats [21]. However, the abundances of R. bromii and F. succinogenes were different in different ruminant species. The three dominant cellulolytic bacteria are consistently higher in the stomach than in the large intestine and small intestine. The stomach provides low pH and cellulosic materials, therefore is convenient for the cellulolytic bacteria growth. Another species, B. fibrisolvens, dominated in the stomach and small intestine of sheep, which is a hemicellulolytic bacterium commonly isolated from the rumen of cattle, sheep, and deer [29]. B. pseudolongum dominated in the small intestine and large intestine, which has the activity of degrading the pectin and glucose in the rabbit cecum [30]. P. ruminicola is also reported in the rumen of cattle, and it is associated with ruminal carbohydrate and protein fermentation [2].
ACKNOWLEDGMENTS
This work was funded by the Open Funding Project of the Key Laboratory of Systems Bioengineering, Ministry of Education. Thanks Docter Sheng Yang of Tianjin Agricultural University for the work of sampling.
Footnotes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
REFERENCES
- 1.Kim M, Morrison M, Yu Z. Status of the phylogenetic diversity census of ruminal microbiomes. FEMS Microbiol Ecol. 2011;76:49–63. doi: 10.1111/j.1574-6941.2010.01029.x. [DOI] [PubMed] [Google Scholar]
- 2.Dowd SE, Callaway TR, Wolcott RD, et al. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) BMC Microbiol. 2008;8:125. doi: 10.1186/1471-2180-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lettat A, Noziere P, Silberberg M, et al. Rumen microbial and fermentation characteristics are affected differently by bacterial probiotic supplementation during induced lactic and subacute acidosis in sheep. BMC Microbiol. 2012;12:142. doi: 10.1186/1471-2180-12-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Oliveira MN, Jewell KA, Freitas FS, et al. Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer. Vet Microbiol. 2013;164:307–14. doi: 10.1016/j.vetmic.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Mao S, Zhang M, Liu J, Zhu W. Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: membership and potential function. Sci Rep. 2015;5:16116. doi: 10.1038/srep16116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwambana BA, Barer MR, Bottomley C, Adegbola RA, Antonio M. Early acquisition and high nasopharyngeal co-colonisation by Streptococcus pneumoniae and three respiratory pathogens amongst Gambian new-borns and infants. BMC Infect Dis. 2011;11:175. doi: 10.1186/1471-2334-11-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeng Y, Zeng D, Zhang Y, et al. Characterization of the cellulolytic bacteria communities along the gastrointestinal tract of Chinese Mongolian sheep by using PCR-DGGE and real-time PCR analysis. World J Microbiol Biotechnol. 2015;31:1103–13. doi: 10.1007/s11274-015-1860-z. [DOI] [PubMed] [Google Scholar]
- 8.Evans NJ, Brown JM, Murray RD, et al. Characterization of novel bovine gastrointestinal tract Treponema isolates and comparison with bovine digital dermatitis treponemes. Appl Environ Microbiol. 2011;77:138–47. doi: 10.1128/AEM.00993-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spence C, Wells WG, Smith CJ. Characterization of the primary starch utilization operon in the obligate anaerobe Bacteroides fragilis: Regulation by carbon source and oxygen. J Bacteriol. 2006;188:4663–72. doi: 10.1128/JB.00125-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shepherd ML, Swecker WS, Jr, Jensen RV, Ponder MA. Characterization of the fecal bacteria communities of forage-fed horses by pyrosequencing of 16S rRNA V4 gene amplicons. FEMS Microbiol Lett. 2012;326:62–8. doi: 10.1111/j.1574-6968.2011.02434.x. [DOI] [PubMed] [Google Scholar]
- 11.Li RW, Wu S, Baldwin RL, VI, Li W, Li C. Perturbation dynamics of the rumen microbiota in response to exogenous butyrate. PLoS One. 2012;7:e29392. doi: 10.1371/journal.pone.0029392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han X, Yang Y, Yan H, et al. Rumen bacterial diversity of 80 to 110-day-old goats using 16S rRNA sequencing. PLoS One. 2015;10:e0117811. doi: 10.1371/journal.pone.0117811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao L, Meng Q, Ren L, et al. Effects of nitrate addition on rumen fermentation, bacterial biodiversity and abundance. Asian-Australas J Anim Sci. 2015;28:1433–41. doi: 10.5713/ajas.15.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steelman SM, Chowdhary BP, Dowd S, Suchodolski J, Janecka JE. Pyrosequencing of 16S rRNA genes in fecal samples reveals high diversity of hindgut microflora in horses and potential links to chronic laminitis. BMC Vet Res. 2012;8:231. doi: 10.1186/1746-6148-8-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo W, Li Y, Wang L, et al. Evaluation of composition and individual variability of rumen microbiota in yaks by 16S rRNA high-throughput sequencing technology. Anaerobe. 2015;34:74–9. doi: 10.1016/j.anaerobe.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Malmuthuge N, Griebel PJ, Guan LL. Taxonomic identification of commensal bacteria associated with the mucosa and digesta throughout the gastrointestinal tracts of preweaned calves. Appl Environ Microbiol. 2014;80:2021–28. doi: 10.1128/AEM.03864-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mao S, Huo W, Zhu W. Use of pyrosequencing to characterize the microbiota in the ileum of goats fed with increasing proportion of dietary grain. Curr Microbiol. 2013;67:341–50. doi: 10.1007/s00284-013-0371-0. [DOI] [PubMed] [Google Scholar]
- 18.Brulc JM, Antonopoulos DA, Miller ME, et al. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci USA. 2009;106:1948–53. doi: 10.1073/pnas.0806191105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pitta DW, Pinchak E, Dowd SE, et al. Rumen bacterial diversity dynamics associated with changing from bermudagrass hay to grazed winter wheat diets. Microb Ecol. 2010;59:511–22. doi: 10.1007/s00248-009-9609-6. [DOI] [PubMed] [Google Scholar]
- 20.Rho JH, Wright DP, Christie DL, et al. A novel mechanism for desulfation of mucin: identification and cloning of a mucin-desulfating glycosidase (sulfoglycosidase) from Prevotella strain RS2. J Bacteriol. 2005;187:1543–51. doi: 10.1128/JB.187.5.1543-1551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Z, Hang S, Mao S, Zhu W. Diversity of butyrivibrio group bacteria in the rumen of goats and its response to the supplementation of garlic oil. Asian-Australas J Anim Sci. 2014;27:179–86. doi: 10.5713/ajas.2013.13373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dunne JC, Li D, Kelly WJ, et al. Extracellular polysaccharide-degrading proteome of Butyrivibrio proteoclasticus. J Proteome Res. 2012;11:131–42. doi: 10.1021/pr200864j. [DOI] [PubMed] [Google Scholar]
- 23.Flint HJ, Scott KP, Duncan SH, Louis P, Forano E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes. 2012;3:289–306. doi: 10.4161/gmic.19897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isaacson R, Kim HB. The intestinal microbiome of the pig. Anim Health Res Rev. 2012;13:100–9. doi: 10.1017/S1466252312000084. [DOI] [PubMed] [Google Scholar]
- 25.Duncan SH, Louis P, Flint HJ. Cultivable bacterial diversity from the human colon. Lett Appl Microbiol. 2007;44:343–50. doi: 10.1111/j.1472-765X.2007.02129.x. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Xu T, Zhu W, Mao S. High-grain feeding alters caecal bacterial microbiota composition and fermentation and results in caecal mucosal injury in goats. Br J Nutr. 2014;112:416–27. doi: 10.1017/S0007114514000993. [DOI] [PubMed] [Google Scholar]
- 27.Collier CT, Hofacre CL, Payne AM, et al. Coccidia-induced mucogenesis promotes the onset of necrotic enteritis by supporting Clostridium perfringens growth. Vet Immunol Immunopathol. 2008;122:104–15. doi: 10.1016/j.vetimm.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 28.Callaway TR, Dowd SE, Edrington TS, et al. Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing. J Anim Sci. 2010;88:3977–83. doi: 10.2527/jas.2010-2900. [DOI] [PubMed] [Google Scholar]
- 29.Forster RJ, Gong J, Teather RM. Group-specific 16S rRNA hybridization probes for determinative and community structure studies of Butyrivibrio fibrisolvens in the rumen. Appl Environ Microbiol. 1997;63:1256–60. doi: 10.1128/aem.63.4.1256-1260.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slovakova L, Duskova D, Marounek M. Fermentation of pectin and glucose, and activity of pectin-degrading enzymes in the rabbit caecal bacterium Bifidobacterium pseudolongum. Lett Appl Microbiol. 2002;35:126–30. doi: 10.1046/j.1472-765x.2002.01159.x. [DOI] [PubMed] [Google Scholar]