

Abstract
Objective
A whole genome association study was conducted to identify single nucleotide polymorphisms (SNPs) with additive and dominant effects for growth and carcass traits in Korean native cattle, Hanwoo.
Methods
The data set comprised 61 sires and their 486 Hanwoo steers that were born between spring of 2005 and fall of 2007. The steers were genotyped with the 35,968 SNPs that were embedded in the Illumina bovine SNP 50K beadchip and six growth and carcass quality traits were measured for the steers. A series of lack-of-fit tests between the models was applied to classify gene expression pattern as additive or dominant.
Results
A total of 18 (0), 15 (3), 12 (8), 15 (18), 11 (7), and 21 (1) SNPs were detected at the 5% chromosome (genome) - wise level for weaning weight (WWT), yearling weight (YWT), carcass weight (CWT), backfat thickness (BFT), longissimus dorsi muscle area (LMA) and marbling score, respectively. Among the significant 129 SNPs, 56 SNPs had additive effects, 20 SNPs dominance effects, and 53 SNPs both additive and dominance effects, suggesting that dominance inheritance mode be considered in genetic improvement for growth and carcass quality in Hanwoo. The significant SNPs were located at 33 quantitative trait locus (QTL) regions on 18 Bos Taurus chromosomes (i.e. BTA 3, 4, 5, 6, 7, 9, 11, 12, 13, 14, 16, 17, 18, 20, 23, 26, 28, and 29) were detected. There is strong evidence that BTA14 is the key chromosome affecting CWT. Also, BTA20 is the key chromosome for almost all traits measured (WWT, YWT, LMA).
Conclusion
The application of various additive and dominance SNP models enabled better characterization of SNP inheritance mode for growth and carcass quality traits in Hanwoo, and many of the detected SNPs or QTL had dominance effects, suggesting that dominance be considered for the whole-genome SNPs data and implementation of successive molecular breeding schemes in Hanwoo.
Keywords: Hanwoo, Single Nucleotide Polymorphism, Whole Genome Association, Carcass Traits, Growth Traits
INTRODUCTION
In recent years, whole genome association study (WGAS) or genomic selection (GS) has been a main focus on livestock breeding due to the expectation of increased accuracy of selection and reduced generation interval, compared to traditional breeding approaches such as progeny testing. WGAS or GS has been enabled by high-density single nucleotide polymorphism (SNP) chips, which allows identification of causative SNPs for economic traits and improves reliability of breeding value prediction.
Single-marker association analysis with density SNP array has similar or greater power to detect quantitative trait loci (QTLs) and provides more precise QTL locations than haplotype-based or identity-by-descent based models [1]. Further, the method offers a greater flexibility of including dominance or epistatic effects in the model, enabling better characterization of the QTL in terms of inheritance mode of gene action. Indeed, some WGAS approaches have been routinely practiced in dairy and beef cattle to detect SNPs for milk production, or meat and carcass, respectively [2–4].
A Korean native cattle, Hanwoo (Bos taurus coreanae), was used as draft animals and suppliers of organic fertilizer. However, as agricultural technologies became improved and beef consumption increased, Hanwoo became more important as beef cattle. The Korean cattle had low meat production efficiency because of low milk production and slow growth rate, while having a relatively high reproductive rate and high marbling, e.g. 15% to 23% of intramuscular fat at final slaughter [5]. It has been about 30 years since Hanwoo improvement program was implemented, in order to increase meat production and quality to meet the growing demand for beef in Korea [6].
Interactions between genes at the same locus are called dominance. Dominance effect has often been ignored or treated as a nuisance parameter in genetic evaluations of livestock and quantification of variance components [7]. However, quantification of dominance effects underlying complex traits is needed due to increasing evidence of the major effects of dominant QTL on human disease and agricultural traits of economic importance [8,9]. Recently, a number of studies showing that accounting for dominance effects increased the accuracy and reduced the bias of genomically-predicted breeding values in comparison to an additive model. Su et al [10] reported that in a purebred Duroc population the dominance variance accounted for 6% of the total phenotypic variance in daily gain, emphasizing the relevance of dominance [10]. Therefore, an increasing need for WGAS methodology to routinely investigate the non-additive effects such as dominance. Herein, we performed a WGAS to identify SNPs with dominance and/or additive effects that were associated with growth and carcass traits, specifically to investigate the importance of dominance in Korean beef cattle, Hanwoo.
MATERIALS AND METHODS
Animals, phenotypes and SNPs
The Hanwoo data were collected from 486 steers that were used for progeny testing in Hanwoo Improvement Center of National Agricultural Cooperative Federation in Seosan, Korea. The steers were born between spring of 2005 and fall of 2007, sired by 61 Hanwoo bulls (paternal halfsibs). The number of steers for each sire ranged from two to 13 with the average of eight steers.
The steers were weaned at 5 or 6 months of age, and each group of 10 steers were raised in a pen. The feeding program was divided into early, middle, and late stage, each with a six months interval. In the early and middle stages, the steers were fed with concentrates with the amount of 1.8% of the body weight and ad libitum in the late stage. The concentrates were composed of 15%, 13%, and 11% of crude protein, and 71%, 72%, and 73% total digestible nutrients (TDN), in the respective feeding stages. Roughages with 4.5% crude protein and 37.5 TDN were offered ad labium with other additives such as vitamin and minerals. All steers were slaughtered at approximately 24 months of age. Traits measured were weaning weight (WWT) at approximately six months of age, 365-d yearling weight (YWT) and carcass weight (CWT) at 24 hours after slaughter. The carcasses were dissected at the last rib and the first lumber vertebra according to the Animal Product Grading System of Korea to measure backfat thickness (BFT), longissimus dorsi muscle area (LMA), and marbling score (Marb). The Marb score was numbered as 1 through 9 according to the Korean Beef Marbling Standard (1 = trace, 9 = very abundant). Details about measurement of the traits and management practices were described in Lee et al [2]. The statistics for phenotypic data used in this study are summarized in Table 1.
Table 1.
Summary statistics for 486 observations on growth and carcass traits in a Hanwoo steer population
| Trait | Average | SD | Minimum | Maximum | CV |
|---|---|---|---|---|---|
| Weaning weight (kg) | 170 | 30.7 | 84.5 | 271 | 18.0 |
| Yearling weight (kg) | 312 | 34.2 | 220 | 414 | 11.0 |
| Carcass weight (kg) | 357 | 40.0 | 158 | 481 | 11.2 |
| Backfat thickness (cm) | 1.01 | 0.41 | 0.30 | 3.50 | 40.9 |
| Longissimus dorsi muscle area (cm2) | 78.9 | 9.54 | 22 | 109 | 12.1 |
| Marbling score (1–9) | 3.38 | 1.77 | 1 | 9 | 52.5 |
SD, standard deviation; CV, coefficient of variation (%).
DNA of the sires and their steers were quantified and genotyped using the Illumina bovine SNP50K beadchip. Details on the SNP genotyping procedure were described in Lee et al [2]. Between the sires and their sons (steers), Mendelian consistency was checked, i.e. any discordance of SNP genotypes between them. If more than 1,000 SNPs were discordant, then the steer was removed. Also, the individual with more than 10% missing genotypes was discarded.
The Illumina bovine SNP50 beadchip assay contained 54,001 SNPs with an average distance interval of 51.5 kb. Criteria for SNP removal were: i) more than 5% pedigree discordant (e.g. cases where a sire was homozygous for one allele and progeny were homozygous for the other allele), ii) less than 90% call rate, iii) monomorphic SNPs or when the minor allele frequency was smaller than 0.05, iv) proportion of individuals with genotype completeness was smaller than 90%, v) markers with significant departure from Hardy-Weinberg equilibrium (p<0.00001), vi) assigned to X chromosome or not assigned to any chromosome on Bovine Genome Build 4.0.
Missing genotypes were imputed using Druet method [11]. Haplotypes were first partially reconstructed based on genotypes of relatives, i.e. halfsib steers and their sires (based on Mendelian segregation rules and linkage information) with the program, LinkPHASE (Liège, Belgium). Then, DAGPHASE (Liège, Belgium) and Beagle (Seattle, WA, USA) with scale and shift parameter set equal to 2.0 and 0.1, respectively, were iteratively used to estimate the parameters of a directed acyclic graph. The programs were run first with genotypes from animals genotyped for all markers for 10 iterations and then with genotypes from all animals for 10 more iterations. The accuracy of imputing genotypes was ranged from 0.81 to 0.96 [11].
Whole genome association analysis
A mixed-inheritance animal model was used to detect SNP with additive or dominance effects for growth and carcass quality of Hanwoo. The ‘snp_a’, ‘snp_d’, and ‘snp_ad’ options of Qxpak software (Barcelona, Spain) were applied for each SNP. For each trait, appropriate fixed factors or covariates were fitted in the models (p<0.05) using a general linear model procedure in SAS (SAS 9.1, SAS Institute Inc., Cary, NC, USA).
Firstly, the additive and dominance expression model (Add+Dom) was chosen as a base model:
Where yi is the phenotypic record of animal i, μ is the average phenotypic performance, cji is the value of the jth covariate or fixed effect for the animal i, βj is an estimate of the jth fixed effect or covariate, SNPad_i is the additive and dominance effect of SNP for animal i (e.g. individuals with marker genotypes ‘11’, ‘12’, and ‘22’ are assumed to have genetic values μkAA, μkAB, and μkBB), ui is the infinitesimal genetic effect of animal i, which is distributed as (the numerator relationship matrix A and the additive genetic variance ) and ei is a random residual for animal i, which is distributed as ( identity matrix I and residual variance ). Two fixed effects were fitted in the models: year and season of birth (5 levels) for all the traits and region where the steers were born (41 levels) for WWT, YWT, and Marb, and a covariate was fitted; weaning age for WWT, yearling age for YWT, slaughter age for CWT, BFT, and LMA, was also fitted. Pedigrees of the base population animals were traced back for two generations to form the numerator relationship matrix, and 1,033 animals were included in the pedigree analysis.
The next models are the additive (Add) and dominance (Dom) expression models, and the null model:
Where yi, μ, cji, βj, ui as described above, SNPa_i is the additive(a) effect of the SNP genotype values for animal i (e.g. individuals with marker genotypes ‘11’, ‘12’, and ‘22’ are assumed to have genetic values μkAA, 0, and μkBB), and SNPd_i is the dominance effect of the SNP for animal i (e.g. individuals with marker genotypes ‘11’, ‘12’, and ‘22’ are assumed to have genetic values 0, μkAB and 0).
A series of tests was applied to classify gene expression pattern; additive or dominant, following the decision trees (Figure 1):
Figure 1.
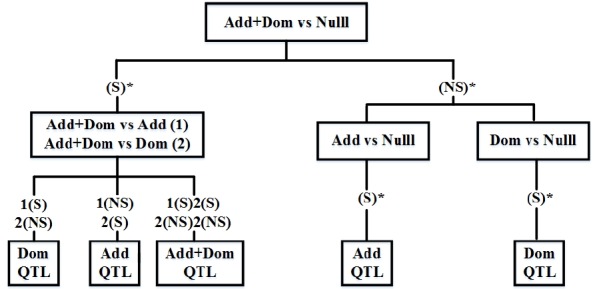
Decision trees to determine classification of QTL type using Add+Dom, Add and Dom models. S*, significant at a chromosome-wise level; NS*, non-significant at a chromosome-wise level; S, significant (p<0.05); NS, non-significant (p<0.05).
If the Add+Dom Model vs the Null model was significant at the 5% chromosome-wise (ChW) level:
If the Add+Dom model vs the Add model was significant and the Add+Dom model vs the Dom model was not significant at the 5% comparison wise level, then the SNP was defined to have dominance inheritance mode of gene action.
If the Add+Dom model vs the Add model was not significant and the Add+Dom model vs the Dom model was significant at the 5% comparison wise level, then the SNP was defined to have additive inheritance mode of gene action.
If the Add+Dom model vs the Add model and the Add+Dom model vs the Dom model were both significant or both not significant at the 5% comparison wise level, then the SNP was defined to have additive and dominance expressed.
If the Add+Dom model vs the Null model was not significant at the 5% ChW level:
If the Add model vs the Null model was significant at the 5% ChW level, then the SNP was classified as additive expressed SNP.
If the Dom model vs the Null model was significant at the 5% ChW level, then the SNP was classified as dominance expressed SNP.
All the models were fitted for each of the available SNPs. The log likelihood ratio tests (LRT) statistic was applied by comparing the log likelihoods between the full model and the reduced model:
The LRT test statistics approximately followed chi-squared distributions with degree of freedom equal to the number of extra parameters (one or two) estimated in the full model compared with the reduced model.
False discovery rate (FDR) was used to set significant thresholds to account for multiple testing, by calculating q-value based on nominal p-value from LRT test statistic for each trait using the R packages ‘qvalue’. The FDR threshold was set at 5% genome-wise (GW) or a 5% ChW level.
Marking the quantitative trait loci region
Generally, clusters of significant SNPs are expected to locate in the vicinity of a QTL, because there is linkage disequilibrium between adjacent markers that are closely located or clusters of transcription factor binding sites potentially indicating groups of co-regulated genes. In this study, the ChromoScan software (Ann Arbor, USA) was used to get statistical evidence of both a clustering of the SNP locations and a clustering of P-values from the above three mixed-inheritance animal model [12]. In the QTL region, one or more SNPs must be detected, and the variance that was explained by the QTL was estimated with the most significant SNP. The genetic variance was obtained from the estimated genotype effects from the corresponding model and the observed genotype frequencies. The percentage of the total genetic variance explained by the significant SNP was also obtained.
RESULTS AND DISCUSSION
Available single nucleotide polymorphisms
Among the 54,001 SNPs in the Illumina bovine beadchip array, 35,968 SNPs were available for GWA tests. The SNPs covered 2543.6 Mb of the bovine genome with 70.6±69.0 kb average adjacent marker interval (Table 2). The largest gap between adjacent SNPs (2,081.5 kb) was located on BTA 10. Average observed heterozygosity for SNPs was 0.37±0.12, and average minor allele frequencies (MAF) of all SNPs before (after) editing was 0.20 (0.27). The SNPs genotyped showed an almost uniform distribution across the common frequency classes.
Table 2.
SNP distributions after quality control and average distances between adjacent SNPs on Bos taurus autosomal chromosomes
| BTA | No. SNPs | Average intervals (kb) |
|---|---|---|
| 1 | 2,376 | 67.75 |
| 2 | 1,930 | 72.91 |
| 3 | 1,832 | 69.40 |
| 4 | 1,736 | 71.37 |
| 5 | 1,472 | 85.52 |
| 6 | 1,818 | 67.38 |
| 7 | 1,577 | 70.87 |
| 8 | 1,673 | 69.93 |
| 9 | 1,399 | 77.21 |
| 10 | 1,487 | 71.03 |
| 11 | 1,567 | 70.27 |
| 12 | 1,145 | 74.47 |
| 13 | 1,228 | 68.52 |
| 14 | 1,219 | 66.71 |
| 15 | 1,160 | 72.96 |
| 16 | 1,101 | 70.62 |
| 17 | 1,092 | 69.95 |
| 18 | 930 | 71.08 |
| 19 | 993 | 65.59 |
| 20 | 1,114 | 67.75 |
| 21 | 997 | 69.42 |
| 22 | 872 | 70.75 |
| 23 | 792 | 67.37 |
| 24 | 918 | 70.81 |
| 25 | 699 | 61.98 |
| 26 | 764 | 67.18 |
| 27 | 694 | 70.28 |
| 28 | 652 | 70.62 |
| 29 | 731 | 70.82 |
| Total | 35,968 |
SNP, single nucleotide polymorphism; BTA, Bos taurus autosom.
Whole genome association study
A total of 92 (37) significant SNPs were detected at 5% ChW (GW) level; 18 (0), 15 (3), 12 (8), 15 (18), 11 (7), and 21 (1) SNPs for WWT, YWT, CWT, BFT, LMA, and Marb respectively (Table 3). Among the significant SNPs, 45 (11) SNPs had additive mode of gene action, 18 (2) SNPs dominance, and 29 (24) had both additive and dominance mode of gene action. The detail description of the significant SNPs for the six growth and carcass quality traits, including their positions in the genome, additive and dominance effects, the estimated proportion of the phenotypic variance and known genes near the SNPs, are in Table 4 through 9.
Table 3.
Number of significant SNPs with additive and/or dominance effects that were detected for growth and carcass quality traits in Hanwoo1)
| Trait | Additive | Dominance | Partial | Total |
|---|---|---|---|---|
| Weaning weight (kg) | 9(0) | 2(0) | 7(0) | 18(0) |
| Yearling weight (kg) | 5(1) | 6(1) | 4(1) | 15(3) |
| Carcass weight (kg) | 6(4) | 1(0) | 5(4) | 12(8) |
| Backfat thickness (cm) | 10(6) | 1(0) | 4(12) | 15(18) |
| Longissimus dorsi muscle area (cm2) | 7(0) | 2(0) | 2(7) | 11(7) |
| Marbling score (1–9) | 8(0) | 6(1) | 7(0) | 21(1) |
| Total | 45(11) | 18(2) | 29(24) | 92(37) |
SNPs, single nucleotide polymorphism; QTL, quantitative trait loci.
The numbers were for the QTL that were detected at the 5% chromosome-wise level (genome-wise level).
Table 4.
Position, SNP alleles, estimated effects of SNPs for weaning weight (kg) that were detected at 5% chromosome-wise level
| SNP1) | Chr | Position (bp)2) | Allele3) | MAF | Estimates of SNPs effects4) | Significance (−log10P5)) | Nearest gene6) | ||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Additive | Dominance | Name | Distance (bp) | ||||||
| BTB-01747944 | 1 | 4,070,974 | T/G | G(0.19) | 12.1±2.91 | −15.2±3.33 | 4.68 | LOC785105 | 40,376 |
| BTB-02105769 | 1 | 4,149,602 | T/C | C(0.26) | 8.74±2.29 | −12.3±2.67 | 4.73 | LOC785105 | 119,004 |
| ARS-BFGL-NGS-35164 | 3 | 55,278,469 | G/A | G(0.46) | - | 8.86±2.02 | 4.78 | CDC7 | 14,260 |
| ARS-BFGL-NGS-5482 | 4 | 61,908,479 | G/A | G(0.26) | −7.65±1.74 | - | 4.79 | THAP5 | 17,976 |
| BTA-29483-no-rs | 5 | 4,963,140 | C/A | C(0.17) | −8.92±2.04 | - | 5.02 | CAPS2 | 85,827 |
| Hapmap52787-rs29024515 | 5 | 33,277,668 | T/C | C(0.21) | 11.6±2.36 | −8.63±2.9 | 4.8 | LOC505861 | within |
| BTA-73718-no-rs | 5 | 66,075,729 | T/G | G(0.07) | −12.0±3.01 | - | 4.06 | NEDD1 | 249,756 |
| ARS-BFGL-NGS-73495 | 5 | 90,096,456 | G/A | G(0.31) | −6.46±1.64 | - | 4.01 | SSPN | 53,061 |
| ARS-BFGL-NGS-34063 | 5 | 125,275,403 | G/A | G(0.35) | - | −9.06±2.03 | 5 | FAM19A5 | 324,968 |
| BTB-00317489 | 7 | 64,536,896 | G/A | G(0.27) | −8.10±1.71 | - | 5.45 | GRIA1 | 40,884 |
| ARS-BFGL-BAC-11115 | 11 | 82,938,260 | T/C | T(0.28) | −6.87±1.60 | - | 4.92 | MSGN1 | 11,224 |
| ARS-BFGL-NGS-103169 | 12 | 39,576,130 | T/C | T(0.14) | −18.6±4.29 | −21.0±4.71 | 4.05 | PCDH9 | 313,077 |
| Hapmap26937-BTA-127685 | 12 | 54,656,928 | G/A | G(0.34) | 6.49±1.56 | - | 4.64 | RBM26 | 194,009 |
| BTB-00497582 | 12 | 54,690,096 | T/G | T(0.37) | 6.31±1.51 | - | 4.65 | RBM26 | 160,841 |
| Hapmap57428-rs29016058 | 12 | 78,783,490 | T/C | C(0.11) | −21.9±4.99 | 22.7±5.44 | 3.81 | LOC787750 | 583,453 |
| ARS-BFGL-NGS-39382 | 16 | 47,648,555 | G/A | A(0.30) | 8.45±1.83 | −5.86±2.38 | 4.63 | PLCH2 | within |
| ARS-BFGL-BAC-33668 | 20 | 42,739,808 | T/G | G(0.16) | 3.58±1.56 | 6.93±2.05 | 4.4 | ADAMTS12 | within |
| Hapmap40017-BTA-65421 | 29 | 33,632,380 | T/C | T(0.05) | −14.2±3.53 | - | 4.27 | ETS1 | 86,263 |
SNP, single nucleotide polymorphism; MAF, minor allele frequency; QTL, quantitative trait loci.
SNP marker annotations and their positions were based on the bovine reference genome (btau4.0).
Nucleotides of substitution.
Estimates of additive and dominance effects with standard errors.
Negative logarithm of the comparison-wise p-value of the test-statistic against the null hypothesis of no QTL at the most likely position for the inferred QTL model.
The nearest known gene to the significant SNP.
Table 9.
Position, SNP alleles, estimated effects of SNPs for marbling score (1–9) that were detected at 5% chromosome-wise level
| SNP1) | Chr | Position (bp)2) | Allele3) | MAF | Estimates of SNPs effects4) | Significance (−log10P5)) | Nearest gene6) | ||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Additive | Dominance | Name | Distance (bp) | ||||||
| BTB-00126406 | 3 | 35,420,885 | T/C | T(0.08) | 2.04±0.41 | −1.55±0.44 | 5.78 | KCNA3 | 6,892 |
| Hapmap39048-BTA-99764 | 3 | 44,502,770 | T/G | T(0.30) | 0.53±0.12 | - | 4.81 | EDG1 | within |
| BTB-00137937 | 3 | 87,445,557 | G/A | A(0.15) | - | 0.91±0.18 | 6.32* | ROR1 | within |
| Hapmap3063-BTA-15439 | 5 | 109,235,854 | C/A | C(0.16) | - | −0.72±0.17 | 4.57 | LOC751811 | 2,138 |
| UA-IFASA-5392 | 7 | 61,523,523 | T/C | C(0.17) | −0.68±0.16 | - | 4.89 | LOC100138980 | 11,789 |
| BTB-00317097 | 7 | 65,652,720 | T/C | C(0.25) | −0.59±0.13 | - | 4.88 | LOC784642 | 39,676 |
| BFGL-NGS-114649 | 7 | 69,609,512 | T/G | T(0.37) | −0.36±0.13 | −0.32±0.16 | 3.91 | EPN4 | 409,625 |
| ARS-BFGL-NGS-63249 | 7 | 71,629,520 | T/C | C(0.47) | 0.36±0.11 | 0.49±0.15 | 4.06 | FABP6 | 6,896 |
| Hapmap40812-BTA-80049 | 7 | 80,713,755 | T/C | T(0.11) | 1.39±0.35 | −0.90±0.38 | 3.97 | ODZ2 | within |
| Hapmap25504-BTA-125586 | 10 | 53,007,997 | G/A | G(0.24) | 0.90±0.18 | −0.77±0.21 | 5.17 | GRINL1A | 113,126 |
| ARS-BFGL-NGS-73404 | 13 | 54,520,958 | T/C | C(0.10) | 0.84±0.20 | * | 4.73 | PRPF6 | within |
| BTB-00633720 | 16 | 27,646,807 | T/G | T(0.12) | 0.73±0.18 | - | 4.06 | EPB41L3 | within |
| Hapmap26379-BTA-130999 | 16 | 51,370,098 | T/C | T(0.37) | 0.50±0.12 | - | 4.71 | PDPN | 12,031 |
| Hapmap60996-rs29015760 | 17 | 25,742,715 | C/A | C(0.15) | - | 0.75±0.18 | 4.45 | LOC539187 | 72,892 |
| BTB-00689316 | 17 | 73,276,535 | T/C | C(0.43) | - | −0.62±0.15 | 4.26 | MORC2 | within |
| ARS-BFGL-NGS-107813 | 18 | 33,279,345 | C/A | A(0.32) | - | −0.64±0.15 | 4.60 | TK2 | within |
| ARS-BFGL-NGS-55014 | 18 | 39,198,464 | G/A | A(0.49) | 0.31±0.11 | −0.53±0.15 | 4.37 | HYDIN | within |
| Hapmap40994-BTA-46361 | 19 | 62,959,153 | T/C | T(0.35) | 0.50±0.12 | - | 4.18 | MAP2K6 | 78,574 |
| ARS-BFGL-NGS-36359 | 19 | 63,936,266 | T/C | C(0.13) | −0.68±0.17 | - | 4.25 | RGS9 | 38,731 |
| ARS-BFGL-NGS-16187 | 25 | 8,839,364 | G/A | A(0.45) | −0.33±0.12 | −0.59±0.15 | 4.62 | LOC538487 | 37,048 |
| BTB-111681-no-rs | 26 | 10,857,884 | G/A | G(0.13) | - | −0.75±0.19 | 4.12 | LIPK | within |
| BTB-00978135 | 28 | 12,940,047 | T/G | T(0.37) | - | −0.61±0.15 | 4.15 | BICC1 | 21,033 |
SNP, single nucleotide polymorphism; MAF, minor allele frequency; QTL, quantitative trait loci.
SNP marker annotations and their positions were based on the bovine reference genome (btau4.0).
Nucleotides of substitution.
Estimates of additive and dominance effects with standard errors.
Negative logarithm of the comparison-wise p-value of the test-statistic against the null hypothesis of no QTL at the most likely position for the inferred QTL model.
The nearest known gene to the significant SNP.
Significant at the 5% genome-wise level.
Weaning weight (WWT)
Eighteen SNPs were detected at 5% ChW level (Table 4). Of the SNPs, nine, two and seven SNPs were declared to be additive, dominant and both additive and dominant, respectively. Two, five and four SNPs were detected in BTA1, 5 and 12, respectively. Six SNPs were located within the QTL regions for WWT identified in Angus cattle (McClure et al [13]), i.e. BTB-01747944, BTB-02105769, ARS-BFGL-NGS-35164, ARS-BFGL-NGS-5482, ARS-BFGL-BAC-11115, and ARS-BFGL-NGS-39382. Among the 18 SNPs, three SNPs on BTAs 5, 16 and 20 were located within the regions of known genes, i.e. LOC505861, PLCH2, and ADAMTS12 (Table 4).
Yearling weight (YWT)
A total of 18 significant SNP were detected for YWT, of which three SNPs at the 5% GW level (Table 5). Of the SNPs, six, seven, and five were declared as additive, dominant and both additive and dominant SNP, respectively. Five SNPs were located within the QTL regions for YWT in Angus cattle [13], i.e. ARS-BFGL-NGS-105590, ARS-BFGL-NGS-17747, ARS-BFGL-NGS-38840, Hapmap50009-BTA-50200, ARS-BFGL-NGS-42226. Among the 18 significant SNPs, seven SNPs were harbored within the regions of known genes (Table 5).
Table 5.
Position, SNP alleles, estimated effects of SNPs for yearling weight (kg) that were detected at 5% chromosome-wise level
| SNP1) | Chr | Position (bp)2) | Allele3) | MAF | Estimates of SNPs effects4) | Significance (−log10P5)) | Nearest gene6) | ||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Additive | Dominance | Name | Distance (bp) | ||||||
| ARS-BFGL-NGS-98401 | 2 | 41,281,224 | G/A | G(0.39) | - | −9.54±2.47 | 4.14 | GPDM | 63,447 |
| Hapmap36520-SCAFFOLD290052_3670 | 2 | 41,325,780 | T/C | T(0.42) | - | −10.1±2.45 | 4.60 | NR4A2 | 4,484 |
| BTA-21857-no-rs | 2 | 72,889,807 | T/G | T(0.46) | - | 12.4±2.44 | 6.56* | LOC523484 | within |
| ARS-BFGL-NGS-105590 | 7 | 62,525,451 | G/A | A(0.28) | −8.19±2.00 | - | 4.66 | FAT2 | within |
| BFGL-NGS-118165 | 11 | 60,747,467 | T/C | T(0.39) | −9.56±2.06 | 6.09±2.58 | 4.90 | LRRTM4 | 489,704 |
| ARS-BFGL-BAC-11115 | 11 | 82,938,260 | T/C | C(0.28) | −8.77±1.98 | - | 5.17 | MSGN1 | 11,224 |
| ARS-BFGL-BAC-23724 | 14 | 72,289,416 | G/A | G(0.35) | −8.69±1.97 | 9.00±2.70 | 5.30* | CALB1 | 24,824 |
| ARS-BFGL-NGS-17747 | 15 | 13,517,697 | T/C | C(0.35) | 7.74±1.84 | - | 4.82 | SESN3 | 21,628 |
| ARS-BFGL-NGS-38840 | 15 | 31,874,189 | T/A | T(0.48) | 8.98±1.81 | - | 6.43* | UBASH3B | 86,347 |
| ARS-BFGL-NGS-39866 | 18 | 18,183,364 | G/A | G(0.39) | - | −11.0±2.5 | 5.29 | CYLD | within |
| Hapmap50009-BTA-50200 | 20 | 29,598,818 | G/A | G(0.09) | 35.3±9.40 | −26.3±9.77 | 4.41 | LOC100138060 | 42,816 |
| ARS-BFGL-BAC-33668 | 20 | 42,739,808 | T/G | T(0.46) | 3.26±1.92 | 9.70±2.51 | 4.49 | ADAMTS12 | within |
| ARS-BFGL-BAC-28936 | 23 | 1,664,787 | G/A | G(0.26) | - | −10.2±2.61 | 4.28 | HMGCLL1 | within |
| ARS-BFGL-BAC-30072 | 23 | 1,694,992 | T/C | T(0.33) | - | −9.68±2.53 | 4.15 | HMGCLL1 | within |
| Hapmap48951-BTA-94291 | 26 | 14,828,964 | T/C | C(0.25) | −8.86±2.21 | - | 4.28 | EXOC6 | within |
| ARS-BFGL-NGS-42226 | 26 | 31,316,453 | G/A | G(0.19) | −9.23±2.37 | - | 4.23 | DUSP5 | 18,449 |
| BTA-98192-no-rs | 27 | 32,786,807 | G/A | G(0.28) | 8.38±2.29 | −12.3±2.99 | 4.73 | LOC783570 | 184,156 |
| Hapmap36817-SCAFFOLD245829_8774 | 29 | 22,927,228 | G/A | A(0.21) | - | −11.9±2.60 | 5.59 | LOC100139055 | 185,099 |
SNP, single nucleotide polymorphism; MAF, minor allele frequency; QTL, quantitative trait loci.
SNP marker annotations and their positions were based on the bovine reference genome (btau4.0).
Nucleotides of substitution.
Estimates of additive and dominance effects with standard errors.
Negative logarithm of the comparison-wise p-value of the test-statistic against the null hypothesis of no QTL at the most likely position for the inferred QTL model.
The nearest known gene to the significant SNP.
Significant at the 5% genome-wise level.
Carcass weight (CWT)
A total of 20 significant SNPs were detected, of which eight SNPs at 5% GW level. Of the significant SNPs for CWT, ten, one and nine SNPs had additive, dominance and both additive and dominance effects, respectively (Table 6). Fourteen SNPs for CWT were located within a 17.55 Mb (between 18.65 Mb and 36.10 Mb) on BTA14. Fifteen significant SNPs were located within the chromosomal regions where QTL for CWT were reported in previous studies of beef cattle [13–17], and five SNPs for CWT were harbored within the regions of known genes (Table 6). McClure et al [13] reported a CWT QTL at 46 cM of BTA8 (95% confidence interval (CI) 41.6 to 66.0 cM) in Angus cattle, near which the SNP, BFGL-NGS-110568, was detected at 56.6 Mb in this study [13]. McClure et al [13] also found another CWT QTL at 84 cM of BTA10 (95% CI 79.0–100.0 cM), while we detected one CWT SNP, ARS-BFGL-NGS-54787, at 84.17 Mb of the same chromosome (Table 6) [13]. In the BTA14 region (18.7 to 36.1 Mb) where clusters of CWT SNP were detected, 13 SNPs that were associated with CWT were previously reported in Japanese black (Wagyu) cattle [4,15] as well as in Hanwoo [16]. Casas et al [14] reported one CWT QTL on BTA18 in a halfsib family sired by Brahman×Hereford cross, near which a GW significant SNP, ARS-BFGL-NGS-39866, was detected at 18.18 Mb (Table 6) [14].
Table 6.
Position, SNP alleles, estimated effects of SNPs for carcass weight (kg) that were detected at 5% chromosome-wise level
| SNP1) | Chr | Position (bp)2) | Allele3) | MAF | Estimates of SNPs effects4) | Significance (−log10P5)) | Nearest gene6) | ||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Additive | Dominance | Name | Distance (bp) | ||||||
| ARS-BFGL-NGS-36803 | 2 | 63,623,562 | G/A | G(0.07) | 97.8±17.6 | 101±18.0 | 5.45* | LOC100140701 | 330,224 |
| BFGL-NGS-110568 | 8 | 56,596,924 | G/A | A(0.06) | −97.8±17.6 | 100±18.0 | 5.43* | CEP78 | 14,027 |
| ARS-BFGL-NGS-59989 | 10 | 84,166,596 | T/C | C(0.46) | - | 16.1±3.17 | 4.90 | SLC8A3 | within |
| ARS-BFGL-NGS-54787 | 14 | 18,653,617 | G/A | G(0.09) | −14.3±4.14 | - | 3.32 | LOC529047 | within |
| Hapmap41993-BTA-86407 | 14 | 19,815,951 | T/C | C(0.23) | 10.3±2.70 | - | 3.76 | LOC100140381 | 40,312 |
| BTB-01143619 | 14 | 24,315,258 | T/C | C(0.06) | 22.9±5.02 | - | 5.17 | FAM110B | 79,759 |
| BTB-01143580 | 14 | 24,383,627 | G/A | A(0.06) | 27.5±5.02 | - | 7.20* | UBXN2B | 32,070 |
| Hapmap30932-BTC-011225 | 14 | 24,898,781 | T/C | T(0.06) | −24.7±5.08 | - | 5.81* | TOX | within |
| BTB-01280026 | 14 | 25,170,557 | T/C | C(0.07) | 24.0±4.51 | - | 6.75* | TOX | 96,111 |
| Hapmap27934-BTC-065223 | 14 | 25,288,714 | G/A | G(0.05) | −25.2±5.12 | - | 5.89* | LOC100140360 | 144,727 |
| UA-IFASA-9679 | 14 | 26,428,822 | G/A | G(0.49) | −9.17±2.39 | 7.08±3.20 | 4.18 | CHD7 | 101,474 |
| Hapmap49130-BTA-34437 | 14 | 28,483,105 | T/C | C(0.07) | 34.0±10.2 | −22.30±11.00 | 3.40 | YTHDF3 | 422,759 |
| BTB-00562658 | 14 | 28,536,444 | G/A | A(0.07) | 34.3±10.2 | −20.80±11.00 | 3.75 | YTHDF3 | 476,098 |
| ARS-BFGL-BAC-8496 | 14 | 30,543,605 | G/A | G(0.06) | −18.9±4.82 | - | 4.09 | RRS1 | 35,258 |
| BTB-00564066 | 14 | 30,695,187 | G/A | G(0.21) | −5.80±3.97 | 7.74±4.79 | 3.19 | MYBL1 | 24,450 |
| ARS-BFGL-NGS-6936 | 14 | 33,171,241 | G/A | A(0.05) | 18.7±5.11 | - | 3.56 | PREX2 | within |
| BTB-01640837 | 14 | 36,095,273 | T/G | G(0.43) | 8.28±2.33 | - | 3.30 | LOC509345 | 443,377 |
| ARS-BFGL-NGS-39866 | 18 | 18,183,364 | G/A | A(0.39) | −7.96±2.50 | −8.52±3.32 | 5.23* | CYLD | within |
| BTB-00752386 | 19 | 41,676,085 | T/G | T(0.13) | −26.78±6.40 | −32.3±7.19 | 4.41* | MSL1 | 7,676 |
| BTB-01392802 | 21 | 54,287,321 | G/A | A(0.11) | −67.0±13.1 | 62.9±13.3 | 4.35 | LOC787825 | 125,238 |
SNP, single nucleotide polymorphism; MAF, minor allele frequency; QTL, quantitative trait loci.
SNP marker annotations and their positions were based on the bovine reference genome (btau4.0).
Nucleotides of substitution.
Estimates of additive and dominance effects with standard errors.
Negative logarithm of the comparison-wise p-value of the test-statistic against the null hypothesis of no QTL at the most likely position for the inferred QTL model.
The nearest known gene to the significant SNP.
Significant at the 5% genome-wise level.
Backfat thickness (BFT)
Thirty three SNPs for BFT were detected at 5% ChW level, of which 18 SNPs at 5% GW level. Of the significant SNPs for BFT, 16, one and 16 SNPs had additive, dominance and both additive and dominance effects, respectively (Table 7). There were three SNP clusters for BFT, i.e. four, five and three SNPs that were located within a 16.4 Mb segment (between 21.0 Mb and 37.4 Mb) on BTA14, a 44.1 Mb segment (between 28.3 Mb and 72.4 Mb) on BTA 16, and a 10.2 Mb segment (between 26.4 and 36.6 Mb) on BTA29, respectively. Eleven SNPs were harbored within the regions of known genes (Table 7), and in the chromosomal regions where twelve SNPs that were detected in this study, BFT QTL also reported in Hanwoo, Angus or composite breeds [13,14,18,19]; Hapmap51248-BTA-51337 on BTA1; BFGL-NGS-119673, ARS-BFGL-NGS-8401, and BTB-00236217 on BTA5; BTB-00236217 and BTB-01744782 on BTA6; Hapmap34906-BES11_Contig369_1053 on BTA11; BTB-01493007 on BTA13; BTB-00566332 on BTA14;BTB-00634483 and Hapmap42533-BTA38667 on BTA16; ARS-BFGL-NGS-39535 on BTA29 (Table 7).
Table 7.
Position, SNP alleles, estimated effects of SNPs for backfat thickness (cm) that were detected at 5% chromosome-wise level
| SNP1) | Chr | Position(bp)2) | Allele3) | MAF | Estimates of SNPs effects4) | Significance (−log10P5)) | Nearest gene6) | ||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Additive | Dominance | Name | Distance (bp) | ||||||
| Hapmap24136-BTA-124014 | 1 | 55,673,837 | T/G | G(0.18) | 0.09±0.05 | 0.10±0.06 | 5.05* | CD47 | 19,151 |
| Hapmap51248-BTA-51337 | 1 | 128,511,629 | G/A | G(0.09) | −0.18±0.04 | - | 4.57 | PLSI | within |
| Hapmap27170-BTA-142780 | 5 | 6,666,948 | T/C | T(0.07) | −1.11±0.18 | −1.20±0.19 | 8.47* | ZDHHC17 | within |
| BFGL-NGS-119673 | 5 | 38,109,780 | T/C | T(0.47) | −0.10±0.03 | - | 4.29 | ANO6 | within |
| ARS-BFGL-NGS-8401 | 5 | 76,428,730 | T/G | T(0.28) | 0.13±0.03 | - | 5.00* | SYN3 | within |
| BTB-00236217 | 5 | 110,984,106 | T/C | T(0.41) | 0.11±0.03 | - | 4.13 | CD9 | 2,776 |
| BTB-01384704 | 6 | 959,692 | A/G | G(0.48) | −0.12±0.03 | - | 5.57* | LOC100139637 | 63,038 |
| BTB-01744782 | 6 | 1,452,847 | T/G | G(0.07) | 0.61±0.13 | −0.56±0.14 | 4.74* | 1-Mar | 77,282 |
| BTA-28590-no-rs | 9 | 10,295,707 | G/A | G(0.24) | −0.14±0.03 | - | 5.61* | bta-mir-30a | 406,847 |
| BTB-01374666 | 11 | 25,076,270 | T/G | G(0.32) | 0.12±0.03 | - | 4.55 | U4 | 570,076 |
| BTB-00467701 | 11 | 31,206,484 | T/C | T(0.06) | −0.21±0.05 | - | 4.14 | MSH6 | 54,643 |
| Hapmap34906-BES11_Contig369_1053 | 11 | 79,443,335 | G/A | A(0.37) | 0.11±0.03 | - | 4.26 | LOC513245 | 431,096 |
| BTB-01493007 | 13 | 52,723,213 | T/C | T(0.05) | −0.56±0.13 | −0.36±0.14 | 5.57* | PTPRA | within |
| ARS-BFGL-NGS-34390 | 14 | 20,953,071 | G/A | G(0.12) | −0.29±0.07 | −0.21±0.08 | 3.87 | ST18 | within |
| Hapmap49130-BTA-34437 | 14 | 28,483,105 | T/C | C(0.07) | 0.59±0.11 | −0.56±0.11 | 6.4* | YTHDF3 | 422,759 |
| BTB-00562658 | 14 | 28,536,444 | G/A | A(0.07) | 0.59±0.11 | −0.56±0.11 | 6.37* | YTHDF3 | 476,098 |
| BTB-00566332 | 14 | 37,352,866 | T/C | T(0.13) | - | −0.21±0.04 | 5.63 | PEX2 | 266,891 |
| ARS-BFGL-NGS-4138 | 15 | 39,970,876 | T/C | T(0.06) | −1.13±0.19 | −1.10±0.19 | 7.79* | GALNTL4 | 27,421 |
| BTB-00634483 | 16 | 28,329,540 | G/A | G(0.14) | −0.44±0.09 | −0.43±0.10 | 4.76* | SMYD3 | within |
| Hapmap42533-BTA-38667 | 16 | 31,436,094 | G/A | A(0.38) | −0.11±0.03 | - | 4.68 | CEP170 | 94,723 |
| BTB-00640968 | 16 | 40,694,131 | G/C | G(0.26) | 0.15±0.03 | - | 6.31* | CTNNBIP1 | 12,174 |
| ARS-BFGL-NGS-17466 | 16 | 53,804,655 | T/A | T(0.26) | 0.15±0.04 | 0.11±0.04 | 3.81 | MRPS14 | 10,788 |
| ARS-BFGL-NGS-85980 | 16 | 72,398,010 | T/G | G(0.08) | 0.54±0.13 | −0.57±0.14 | 3.76 | LOC100139930 | 46,502 |
| ARS-BFGL-NGS-41599 | 17 | 74,420,464 | G/A | A(0.07) | 0.49±0.11 | −0.54±0.12 | 4.82* | LOC616727 | 3,592 |
| BTA-54022-no-rs | 22 | 3,844,964 | C/A | C(0.10) | −0.41±0.09 | −0.41±0.09 | 4.81* | RBMS3 | within |
| ARS-BFGL-NGS-18665 | 22 | 7,042,486 | G/A | A(0.07) | 0.91±0.14 | −0.87±0.14 | 9.25* | DYNC1LI1 | 1,080 |
| BFGL-NGS-116395 | 23 | 11,896,040 | C/A | C(0.40) | 0.10±0.03 | - | 4.15 | MDGA1 | within |
| ARS-BFGL-BAC-30052 | 23 | 11,964,461 | G/T | T(0.45) | 0.12±0.03 | - | 5.53* | ZFAND3 | 24,327 |
| BFGL-NGS-114371 | 26 | 51,572,874 | G/A | G(0.08) | 0.19±0.05 | - | 4.2 | bta-mir-202 | 6,161 |
| ARS-BFGL-NGS-28346 | 26 | 42,712,420 | G/A | A(0.13) | 0.37±0.08 | −0.41±0.09 | 4.56* | TACC2 | within |
| ARS-BFGL-NGS-39535 | 29 | 26,364,496 | G/A | G(0.26) | 0.13±0.03 | - | 4.66 | NAV2 | 88,899 |
| BTB-01020342 | 29 | 33,014,346 | G/A | A(0.49) | −0.12±0.03 | - | 5.40* | ETS1 | 459,952 |
| ARS-BFGL-NGS-23717 | 29 | 36,574,648 | T/C | C(0.19) | 0.22±0.05 | −0.17±0.06 | 3.95 | NTM | within |
SNP, single nucleotide polymorphism; MAF, minor allele frequency; QTL, quantitative trait loci.
SNP marker annotations and their positions were based on the bovine reference genome (btau4.0).
Nucleotides of substitution.
Estimates of additive and dominance effects with standard errors.
Negative logarithm of the comparison-wise p-value of the test-statistic against the null hypothesis of no QTL at the most likely position for the inferred QTL model.
The nearest known gene to the significant SNP.
Significant at the 5% genome-wise level.
Longissimus dorsi muscle area (LMA)
A total of 18 SNPs were detected, of which seven SNPs at 5% GW level. Seven, two and nine SNPs were declared to be additive, dominance and both additive and dominance SNP, respectively (Table 8). Six LMA SNPs (BTA-99819-no-rs on BTA3; BTB-01321253 on BTA7; BFGL-NGS-110568 on BTA8; BFGL-NGS-112221 and BFGL-NGS-113227 on BTA20; ARS-BFGL-NGS-35298 on BTA24) were located within the QTL regions for LMA that were reported in a WGAS of Angus cattle [13].
Table 8.
Position, SNP alleles, estimated effects of SNPs for longissimus dorsi muscle area (cm2) that were detected at 5% chromosome-wise level
| SNP1) | Chr | Position (bp)2) | Allele3) | MAF | Estimates of SNPs effects4) | Significance (−log10P5)) | Nearest gene6) | ||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Additive | Dominance | Name | Distance (bp) | ||||||
| ARS-BFGL-NGS-96411 | 1 | 99,002,229 | G/A | A(0.08) | 15.8±3.06 | 15.9±3.20 | 5.68* | LOC100140324 | 3,400 |
| ARS-BFGL-NGS-36803 | 2 | 63,623,562 | G/A | A(0.07) | 26.4±4.29 | 27.3±4.40 | 8.13* | LOC100140701 | 330,224 |
| BTA-99819-no-rs | 3 | 84,880,188 | T/C | T(0.07) | −9.60±2.23 | 11.3±2.42 | 4.69 | PDE4B | within |
| Hapmap43716-BTA-87799 | 4 | 25,686,934 | G/A | A(0.16) | 8.32±1.78 | 9.79±1.92 | 5.54* | LOC100139029 | 144,805 |
| ARS-BFGL-NGS-11156 | 6 | 31,210,287 | T/C | C(0.06) | 15.5±3.07 | 16.8±3.22 | 5.84* | UNC5C | within |
| BTB-01321253 | 7 | 83,625,072 | G/A | G(0.17) | - | 3.88±0.90 | 4.72 | XRCC4 | 321,087 |
| BFGL-NGS-110568 | 8 | 56,596,924 | G/A | G(0.06) | −26.6±4.29 | 26.4±4.40 | 8.01* | CEP78 | 14,027 |
| ARS-BFGL-NGS-77091 | 15 | 76,834,507 | T/C | C(0.22) | - | 3.49±0.84 | 4.47 | AMBRA1 | within |
| ARS-BFGL-NGS-25982 | 20 | 4,493,745 | G/A | A(0.40) | 2.49±0.64 | - | 4.00 | NEURL1B | 9,980 |
| Hapmap53674-rs29025319 | 20 | 8,676,696 | T/C | C(0.12) | 4.69±0.96 | - | 5.84 | LOC783819 | 132,888 |
| ARS-BFGL-NGS-38482 | 20 | 37,708,167 | T/C | C(0.10) | 7.25±1.69 | 6.50±1.90 | 3.95 | RICTOR | within |
| BFGL-NGS-112221 | 20 | 75,476,187 | T/C | T(0.27) | −2.80±0.70 | - | 4.21 | ZDHHC11 | 15,990 |
| BFGL-NGS-113227 | 20 | 75,530,515 | T/C | T(0.24) | −2.82±0.72 | - | 4.02 | CEP72 | within |
| BTB-01392802 | 21 | 54,287,321 | G/A | G(0.11) | −16.5±3.21 | 14.8±3.28 | 5.95* | LOC787825 | 125,238 |
| ARS-BFGL-NGS-35298 | 24 | 13,445,705 | T/C | T(0.27) | −2.57±0.66 | - | 3.94 | LOC781724 | 3,783 |
| BTA-100770-no-rs | 24 | 40,446,177 | T/C | C(0.46) | −2.45±0.60 | - | 4.25 | EPB41L3 | within |
| ARS-BFGL-NGS-16328 | 26 | 18,231,319 | G/A | A(0.13) | −3.94±0.91 | - | 4.78 | TLL2 | within |
| ARS-BFGL-NGS-94161 | 28 | 35,084,332 | G/A | G(0.14) | −10.8±2.17 | 11.0±2.27 | 5.38* | SFTPD | 3,977 |
SNP, single nucleotide polymorphism; MAF, minor allele frequency; QTL, quantitative trait loci.
SNP marker annotations and their positions were based on the bovine reference genome (btau4.0).
Nucleotides of substitution.
Estimates of additive and dominance effects with standard errors.
Negative logarithm of the comparison-wise p-value of the test-statistic against the null hypothesis of no QTL at the most likely position for the inferred QTL model.
The nearest known gene to the significant SNP.
Significant at the 5% genome-wise level.
Marbling score (Marb)
A total of 22 SNPs were detected of which, eight, seven and seven had additive, dominance and both additive and dominance effects, respectively (Table 9). One SNP, BTB-00137937, was detected at 5% GW level. Of the significant SNPs for Marb, five SNPs were detected within a 19.2 Mb segment (between 61.5 Mb and 80.7 Mb) on BTA7. Twelve SNPs were located within the Marb QTL regions that were reported in previous studies [13,18,20]. Nine SNPs were harbored within the regions of known genes (Table 9). In particular, one SNP, Hapmap39048-BTA-99764 was located within EDG1 gene on BTA3, which was reported to be a positional function candidate gene responsible for marbling [20].
Characterization of single nucleotide polymorphism for growth and carcass quality
Many SNPs influencing growth and carcass quality in Hanwoo had dominance effects. Of the 129 significant SNPs, 45 (11) SNPs had additive expression, 18 (2) dominance expression, and 29 (24) SNPs had both additive and dominant expression at 5% ChW (GW) level, of which 27 SNPs had overdominance effects, i.e. five for WWT, three for YWT, five for CWT, five for BFT, six for LMA, three for Marb, respectively (Tables 4 to 9). Using additive plus dominance effects, rather than additive effects only, led to more significant SNPs. These results indicated that the goodness of fit was improved by including dominance. Estimates of dominance effects have not been widely used in livestock breeding because it is difficult to estimate these effects accurately based on pedigree. The development of dense SNP panels offered new opportunities for detection and use of dominance at individual loci. Boysen et al [21] reported that significant dominance effects on milk production traits in cattle were identified in a WGAS [21].
The effectiveness of selection procedures that are based on genomic information can be increased by correctly characterization of inheritance mode of desired variants [7]. Heterosis that is caused by heterozygosity in single loci is responsible for survival rate by increasing reproductive fitness [22]. Hayes and Miller (2000) showed that dominance effect was needed in mate selection to exploit previously untapped genetic variation [23], while Dekkers and Chakraborty (2004) stressed maximization of crossbred performance by incorporating information from overdominant QTL [24]. Kim et al [8] found that many QTL for growth and carcass quality had a (over) dominant mode of gene action in a cross between Angus and Brahman cattle [8]. The use of dominance effects in a scenario of genomic selection increases the accuracy of estimated breeding values and still offers the opportunity of applying mate-allocation.
These result has important implication that dominance could be routinely included the whole-genome SNPs data in Hanwoo and implementation of successive molecular breeding schemes.
Quantitative trait locus region
Thirty three QTL regions were identified on 18 BTAs for the significant SNPs for growth and carcass quality (Table 10). Of the QTL, 20, one, and 12 QTL had additive, dominance, and both additive and dominance expression modes, respectively. All the QTL explained small to moderate proportions (2.2% to 10.9%) of the phenotypic variance with an average of 4.5%±1.6%. However, the estimates may be overestimated, especially when the SNP effect was small, partly due to so called Beavis effect [25].
Table 10.
QTL regions associated with growth and carcass traits and the percentage of the total genetic variance explained
| Chr | Region type | Region start (bp)1) | Region end (bp)2) | Region size (bp) | Marker count | Region −log10P | Most significant SNP | |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| SNP | %σ2p3) | |||||||
| Weaning wight (kg) | ||||||||
| 4 | add | 61,386,842 | 61,908,479 | 521,637 | 12 | 2.98 | ARS-BFGL-NGS-5482 | 4.75 |
| 5 | add | 4,528,722 | 4,963,140 | 434,418 | 11 | 2.70 | BTA-29483-no-rs | 4.70 |
| 11 | add | 82,506,706 | 82,998,032 | 491,326 | 11 | 2.21 | ARS-BFGL-BAC-11115 | 4.07 |
| 12 | add | 54,600,867 | 54,690,096 | 89,229 | 4 | 2.17 | Hapmap26937-BTA-127685 | 4.00 |
| 20 | add+dom | 42,739,808 | 42,872,996 | 133,188 | 4 | 2.94 | ARS-BFGL-NGS-39382 | 4.33 |
| Yearling weight (kg) | ||||||||
| 11 | add+dom | 60,503,780 | 60,747,467 | 243,687 | 5 | 2.25 | BFGL-NGS-118165 | 5.67 |
| 11 | add | 82,715,271 | 82,998,032 | 282,761 | 7 | 2.07 | ARS-BFGL-BAC-11115 | 4.39 |
| 20 | add+dom | 42,739,808 | 42,819,855 | 80,047 | 3 | 2.17 | ARS-BFGL-BAC-33668 | 4.41 |
| 26 | add | 14,341,149 | 14,828,964 | 487,815 | 9 | 2.07 | Hapmap48951-BTA-94291 | 4.16 |
| Carcass weight (kg) | ||||||||
| 14 | add | 19,654,754 | 19,910,197 | 255,443 | 5 | 2.12 | Hapmap41993-BTA-86407 | 2.96 |
| 14 | add | 24,100,495 | 25,707,512 | 1,607,017 | 36 | 10.65 | BTB-01143580 | 6.25 |
| 14 | add+dom | 28,090,849 | 28,604,028 | 513,179 | 11 | 2.07 | BTB-00562658 | 3.42 |
| 14 | add | 36,023,989 | 36,095,273 | 71,284 | 3 | 2.25 | BTB-01640837 | 2.61 |
| Backfat thickness (cm) | ||||||||
| 5 | add | 37,930,354 | 38,268,770 | 338,416 | 6 | 2.09 | BFGL-NGS-119673 | 3.25 |
| 5 | add | 75,805,133 | 76,714,910 | 909,777 | 14 | 4.73 | ARS-BFGL-NGS-8401 | 4.45 |
| 6 | add+dom | 454,623 | 1,496,086 | 1,041,463 | 21 | 5 | BTB-01744782 | 5.34 |
| 9 | add | 10,080,000 | 11,411,851 | 1,331,851 | 13 | 3.31 | BTA-28590-no-rs | 4.70 |
| 14 | add+dom | 28,344,924 | 28,938,888 | 593,694 | 12 | 2.61 | Hapmap49130-BTA-34437 | 4.91 |
| 16 | add | 31,108,917 | 36,611,548 | 502,631 | 11 | 2.19 | Hapmap42533-BTA-38667 | 3.78 |
| 16 | add | 39,293,150 | 40,694,131 | 1,324,364 | 18 | 5.83 | BTB-00640968 | 5.76 |
| 16 | add+dom | 53,663,332 | 53,952,476 | 289,144 | 6 | 2.05 | ARS-BFGL-NGS-17466 | 3.49 |
| 17 | add+dom | 74,292,265 | 74,738,486 | 446,221 | 6 | 2.34 | ARS-BFGL-NGS-41599 | 3.13 |
| 23 | add | 11,627,828 | 12,207,677 | 579,849 | 9 | 2.23 | ARS-BFGL-BAC-30052 | 4.71 |
| 29 | add+dom | 35,829,053 | 36,646,213 | 817,160 | 12 | 3.38 | ARS-BFGL-NGS-23717 | 4 |
| Longissimus dorsi muscle area(cm2) | ||||||||
| 3 | add+dom | 84,750,518 | 85,176,770 | 426,252 | 11 | 2.71 | BTA-99819-no-rs | 2.97 |
| 7 | add+dom | 107,689,830 | 107,950,229 | 260,399 | 7 | 2.10 | Hapmap48901-BTA-80577 | 2.19 |
| 20 | add | 4,040,334 | 4,745,147 | 704,813 | 17 | 3.92 | ARS-BFGL-NGS-25982 | 3.65 |
| 20 | add | 8,115,195 | 8,810,833 | 695,638 | 13 | 3.12 | Hapmap53674-rs29025319 | 5.52 |
| 20 | add | 75,346,433 | 75,530,515 | 184,082 | 4 | 2.54 | BFGL-NGS-112221 | 3.82 |
| 28 | add+dom | 34,473,261 | 35,155,406 | 649,145 | 11 | 2.13 | ARS-BFGL-NGS-94161 | 10.94 |
| Marbling score (1–9) | ||||||||
| 13 | add | 54,474,756 | 54,548,692 | 73,936 | 3 | 2.24 | ARS-BFGL-NGS-73404 | 4.65 |
| 16 | add | 51,370,098 | 51,470,017 | 99,919 | 5 | 2.03 | Hapmap26379-BTA-130999 | 4.24 |
| 18 | dom | 32,990,778 | 33,279,345 | 288,567 | 9 | 2.05 | ARS-BFGL-NGS-107813 | 3.65 |
QTL, quantitative trait locus; SNP, single nucleotide polymorphism.
SNP marker annotations and their positions were based on the bovine reference genome (btau4.0).
Proportion of phenotypic variance due to the SNPs.
Some significant SNPs were located in close proximity, e.g. the two SNPs BTB-01747944 and BTB-02105769 for WWT at 4.07 Mb and 4.15 Mb on BTA1, and ARS-BFGL-BAC-28936 and ARS-BFGL-BAC-30072 for YWT at 1.66 Mb and 1.69 on BTA23, respectively (Tables 4 and 5). These SNPs might not be causal mutation, but so close to the causal SNP as to form high LD with the causal mutation for the trait, and the effect of a single QTL could be spread over multiple SNPs.
A QTL for CWT was detected at 24.1 to 25.7 Mb of BTA14 with a significant evidence (−log10P = 10.7). Also, four QTL for CWT were clustered between 19. 7 and 36.1 Mb of BTA14 (Table 10). In the region, several QTL were reported that were associated with milk production, growth and carcass traits in cattle [8,15,26]. Especially in beef cattle, a majority of the QTL were located at 18 to 36 Mb of the chromosome [27]. The QTL region corresponded to a human chromosome homo sapiens (HSA) region in which genes responsible for growth characteristics reside. Mizoshita et al [15] reported a CWT-QTL in Wagyu within a 1.1 Mb chromosomal segment flanked by DIK7012 and DIK7020 on BTA14, in which four genes, including CA8 and RAB2 resided in the corresponding HSA region or six bovine expressed sequence tag (EST) assemblies, which may be a source of additive genetic variation for carcass weight [15]. Pausch et al [3] recently reported a QTL at 24.1 to 25.4 Mb of BTA14 that was associated with polymorphisms for a polyadenylation signal of the gene encoding for ribosomal protein S20 (RPS20), explaining major genetic variation of growth-related traits in a sample of 1,800 bulls of the German Fleckvieh breed [3]. In the similar region, Karim et al [28] identified causative variations influencing bovine stature in the PLAG1-CHCHD7 inter-genic region in a Holstein-Friesian×Jersey F2 population [28]. At the same region, a QTL for carcass weight was identified in a population of 1,156 Japanese Black steers [4]. In conclusion, the evidence of CWT QTL around 25 Mb of BTA14 in this study supports the QTL or causal mutation for growth, carcass, or stature on the proximal region of BTA14 in cattle.
On BTA20, several QTL for WWT, YWT, and LMA were identified (Table 10). The QTL for WWT and YWT were located at 42.74 to 42.87 Mb, near which a gene, GHR (33.90 to 34.07 Mb) was located. Growth hormone receptor (GHR) has been proposed as a candidate gene for growth and fat metabolism [29]. Another three QTL for LMA were located at 4.04 to 4.75 Mb, 8.12 to 8.81 Mb and 75.35 to 75.53 Mb, respectively. At the 8 Mb, a QTL for LMA was reported by Garcia et al [30] in F1 crossbred steers of Bos taurus breeds (Angus, Hereford, Gelbvieh, Simmental, Charolais, Limousin, and Red Angus) comprising Cycle 7 of the Germplasm Evaluation project [30]. Also, at 75 Mb, a LMA QTL was reported in a commercial Angus population [13].
Some QTL that affected two traits were located in close distance, e.g. the QTL for WWT and YWT at 82.5 and 83.0Mb of BTA11, or the QTL for CWT and BFT at 28.1 and 28.9 Mb of BTA14 (Table 10), suggesting the nature of gene characteristics, i.e. pleiotropy or clustering of multi-genes.
IMPLICATIONS
This study described a novel approach to model additive and dominance genetic effect using information of genome-wide SNP markers. Implementation of various additive and dominance SNP models enabled characterization of inheritance mode for QTL, and we found significant evidence of additive and/or dominance QTL for growth and carcass quality in a Hanwoo steer population. The QTL regions can be fine-mapped to find causal mutations, which would provide valuable information for subsequent quantitative trait loci analyses and genomic selection schemes. Many dominant QTL influencing growth and carcass traits in Hanwoo were detected in this study. This has the important implication that dominant expression for growth and carcass composition in beef cattle is not rare and thus dominance effect needs to be considered in marker-assisted breeding schemes in Hanwoo. Validation tests for the detected SNPs (QTL) are in progress and the QTLs that are confirmed in commercial Hanwoo populations can be applied to better improve Hanwoo production.
ACKNOWLEDGMENTS
This work was supported by Program for the Outstanding Innovative Teams of Higher Learning Institutions of Shanxi (Yi Li), the National Natural Science Foundation of China No.31501002 & 31470070 (Yi Li), Shanxi Scholarship Council of China No.2013-072 (Yi Li) and the Natural Science Foundation of Shanxi No.2014011030-4 (Yi Li). Dr. Jong-Joo Kim is supported by a grant (715003-07) from the Research Center from Production Management and Technical Development for High Quality Livestock Products through Agriculture, Food and Rural Affairs Research Center Support Program (2016).
Footnotes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
REFERENCES
- 1.MacLeod IM, Hayes BJ, Savin KW, et al. Power of a genome scan to detect and locate quantitative trait loci in cattle using dense single nucleotide polymorphisms. J Anim Breed Genet. 2010;127:133–42. doi: 10.1111/j.1439-0388.2009.00831.x. [DOI] [PubMed] [Google Scholar]
- 2.Lee YM, Han CM, Li Y, et al. A Whole Genome Association Study to Detect Single Nucleotide Polymorphisms for Carcass Traits in Hanwoo Populations. Asian-Australas J Anim Sci. 2010;23:417–24. [Google Scholar]
- 3.Pausch H, Flisikowski K, Jung S, et al. Genome-wide association study identifies two major loci affecting calving ease and growth-related traits in cattle. Genetics. 2011;187:289–97. doi: 10.1534/genetics.110.124057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishimura S, Watanabe T, Mizoshita K, et al. Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet. 2012:13. doi: 10.1186/1471-2156-13-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim JB, Lee C. Historical look at the genetic improvement in Korean cattle - Review. Asian-Australas J Anim Sci. 2000;13:1467–81. [Google Scholar]
- 6.Venkata Reddy B, Sivakumar AS, Jeong DW, et al. Beef quality traits of heifer in comparison with steer, bull and cow at various feeding environments. Anim Sci J. 2015;86:1–16. doi: 10.1111/asj.12266. [DOI] [PubMed] [Google Scholar]
- 7.Rowe SJ, Pong-Wong R, Haley CS, Knott SA, De Koning DJ. Detecting dominant QTL with variance component analysis in simulated pedigrees. Genet Res. 2008;90:363–74. doi: 10.1017/S0016672308009336. [DOI] [PubMed] [Google Scholar]
- 8.Kim JJ, Farnir F, Savell J, Taylor JF. Detection of quantitative trait loci for growth and beef carcass fatness traits in a cross between Bos taurus (Angus) and Bos indicus (Brahman) cattle. J Anim Sci. 2003;81:1933–42. doi: 10.2527/2003.8181933x. [DOI] [PubMed] [Google Scholar]
- 9.Lee SH, van der Werf JHJ, Hayes BJ, Goddard ME, Visscher PM. Predicting unobserved phenotypes for complex traits from whole-genome SNP data. Plos Genet. 2008:4. doi: 10.1371/journal.pgen.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su GS, Christensen OF, Ostersen T, Henryon M, Lund MS. Estimating additive and non-additive genetic variances and predicting genetic merits using genome-wide dense single nucleotide polymorphism markers. Plos One. 2012;7:e45293. doi: 10.1371/journal.pone.0045293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Druet T, Schrooten C, de Roos APW. Imputation of genotypes from different single nucleotide polymorphism panels in dairy cattle. J Dairy Sci. 2010;93:5443–54. doi: 10.3168/jds.2010-3255. [DOI] [PubMed] [Google Scholar]
- 12.Sun YV, Jacobsen DM, Kardia SLR. ChromoScan: a scan statistic application for identifying chromosomal regions in genomic studies. Bioinformatics. 2006;22:2945–7. doi: 10.1093/bioinformatics/btl503. [DOI] [PubMed] [Google Scholar]
- 13.McClure MC, Morsci NS, Schnabel RD, et al. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim Genet. 2010;41:597–607. doi: 10.1111/j.1365-2052.2010.02063.x. [DOI] [PubMed] [Google Scholar]
- 14.Casas E, Shackelford SD, Keele JW, et al. Detection of quantitative trait loci for growth and carcass composition in cattle. J Anim Sci. 2003;81:2976–83. doi: 10.2527/2003.81122976x. [DOI] [PubMed] [Google Scholar]
- 15.Mizoshita K, Takano A, Watanabe T, Takasuga A, Sugimoto Y. Identification of a 1.1-Mb region for a carcass weight QTL onbovine Chromosome 14. Mamm Genome. 2005;16:532–7. doi: 10.1007/s00335-005-0024-0. [DOI] [PubMed] [Google Scholar]
- 16.Choi IS, Kong HS, Oh JD, et al. Analysis of microsatellite markers on bovine chromosomes 1 and 14 for potential allelic association with carcass traits in Hanwoo (Korean cattle) Asian-Australas J Anim Sci. 2006;19:927–30. [Google Scholar]
- 17.Takasuga A, Watanabe T, Mizoguchi Y, et al. Identification of bovine QTL for growth and carcass traits in Japanese Black cattle by replication and identical-by-descent mapping. Mamm Genome. 2007;18:125–36. doi: 10.1007/s00335-006-0096-5. [DOI] [PubMed] [Google Scholar]
- 18.Casas E, Keele JW, Shackelford SD, Koohmaraie M, Stone RT. Identification of quantitative trait loci for growth and carcass composition in cattle. Anim Genet. 2004;35:2–6. doi: 10.1046/j.1365-2052.2003.01067.x. [DOI] [PubMed] [Google Scholar]
- 19.Lee YM, Lee YS, Han CM, et al. Detection of quantitative trait loci for growth and carcass traits on BTA6 in a Hanwoo population. Asian-Australas J Anim Sci. 2010;23:287–91. [Google Scholar]
- 20.Sukegawa S, Miyake T, Takahagi Y, et al. Replicated association of the single nucleotide polymorphism in EDG1 with marbling in three general populations of Japanese Black beef cattle. BMC Res Notes. 2010;3:66. doi: 10.1186/1756-0500-3-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boysen TJ, Heuer C, Tetens J, Reinhardt F, Thaller G. Novel use of derived genotype probabilities to discover significant dominance effects for milk production traits in dairy cattle. Genetics. 2013;193:431–42. doi: 10.1534/genetics.112.144535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lippman ZB, Zamir D. Heterosis: revisiting the magic. Trends Genet. 2007;23:60–6. doi: 10.1016/j.tig.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 23.Hayes BJ, Miller SP. Mate selection strategies to exploit across- and within-breed dominance variation. J Anim Breed Genet-Zeitschrift Fur Tierzuchtung Und Zuchtungsbiologie. 2000;117:347–59. [Google Scholar]
- 24.Dekkers JCM, Chakraborty R. Optimizing purebred selection for crossbred performance using QTL with different degrees of dominance. Genet Sel Evol. 2004;36:297–324. doi: 10.1186/1297-9686-36-3-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beavis WD. QTL analyses: power precision and accuracy. In: Paterson AH, editor. Molecular dissection of complex traits. New York: CRC Press; 1998. pp. 145–62. [Google Scholar]
- 26.Ashwell MS, Heyen DW, Sonstegard TS, et al. Detection of quantitative trait loci affecting milk production, health, and reproductive traits in Holstein cattle. J Dairy Sci. 2004;87:468–75. doi: 10.3168/jds.S0022-0302(04)73186-0. [DOI] [PubMed] [Google Scholar]
- 27.Wibowo TA, Gaskins CT, Newberry RC, et al. Genome assembly anchored QTL map of bovine chromosome 14. Int J Biol Sci. 2008;4:406–14. doi: 10.7150/ijbs.4.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karim L, Takeda H, Lin L, et al. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat Genet. 2011;43:405–13. doi: 10.1038/ng.814. [DOI] [PubMed] [Google Scholar]
- 29.Blott S, Kim JJ, Moisio S, et al. Molecular dissection of a quantitative trait locus: a phenylalanine-to-tyrosine substitution in the transmembrane domain of the bovine growth hormone receptor is associated with a major effect on milk yield and composition. Genetics. 2003;163:253–66. doi: 10.1093/genetics/163.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia MD, Matukumalli L, Wheeler TL, et al. Markers on bovine chromosome 20 associated with carcass quality and composition traits and incidence of contracting infectious bovine keratoconjunctivitis. Anim Biotechnol. 2010;21:188–202. doi: 10.1080/10495398.2010.495012. [DOI] [PubMed] [Google Scholar]